

Abstract
α-Gal glycolipids capable of converting tumors into endogenous vaccines, have α-gal epitopes (Galα1-3Galβ1-4GlcNAc-R) and are extracted from rabbit RBC membranes. α-Gal epitopes bind anti-Gal, the most abundant natural antibody in humans constituting 1% of immunoglobulins. α-Gal glycolipids insert into tumor cell membranes, bind anti-Gal and activate complement. The complement cleavage peptides C5a and C3a recruit inflammatory cells and APC into the treated lesion. Anti-Gal further opsonizes the tumor cells and targets them for effective uptake by recruited APC, via Fcγ receptors. These APC transport internalized tumor cells to draining lymph nodes, and present immunogenic tumor antigen peptides for activation of tumor specific T cells. The present study demonstrates the ability of α-gal glycolipids treatment to prevent development of metastases at distant sites and to protect against tumor challenge in the treated mice. Adoptive transfer studies indicate that this protective immune response is mediated by CD8+ T cells, activated by tumor lesions turned vaccine. This T cell activation is potent enough to overcome the suppressive activity of Treg cells present in tumor bearing mice, however it does not elicit an autoimmune response against antigens on normal cells. Insertion of α-gal glycolipids and subsequent binding of anti-Gal are further demonstrated with human melanoma cells, suggesting that intratumoral injection of α-gal glycolipids is likely to elicit a protective immune response against micrometastases also in cancer patients.
Keywords: Tumor vaccine, α-Gal glycolipids, Anti-Gal antibody, Cancer immunotherapy, Anti-tumor immune response
Introduction
Destruction of detectable metastases by standard therapies such as resection, intratumoral injection of ethanol, radiation, or thermal ablation, provides in most cancer patients with advanced disease, only temporary relief. This is because invisible micrometastases develop within weeks or months into lethal metastases. Micrometastases destruction may be achieved by immunotherapy that stimulates the patient’s immune system to react against tumor associated antigens (TAA) on tumor cells. The high correlation between the extent of T cell infiltration within tumors and positive prognosis implies that the immune system can react against TAA in various types of tumors [6, 20, 41]. Many known common TAA are of weak immunogenicity since they are present in low amounts also on normal cells and embryonic cells [9]. An alternative group of TAA which may serve as target for immunotherapy is the wide range of TAA unique to the patient. These TAA are generated by multiple coding mutations which are the result of genomic instability in tumor cells [9, 30, 38, 39]. Some of these mutations result in changes in tumor cell proteins that provide advantageous growth, whereas other mutations are retained through tumor cell divisions with no apparent advantage to the cells [30, 39]. The mutated proteins may function as autologous TAA that can elicit a specific protective anti-tumor immune response, since they are unique to the tumor cells and absent in normal cells. Logistically, it is difficult to characterize the full range of autologous TAA in cancer patients. Therefore, tumor lesions detected visually (e.g., cutaneous melanoma), or by imaging, may be exploited in a clinical setting, as the source for autologous TAA vaccine in each patient.
Effective autologous TAA vaccines require uptake of tumor cells by antigen presenting cells (APC) such as dendritic cells (DCs) and macrophages. The APC transport TAA of internalized tumor cells to regional lymph nodes where they present processed immunogenic TAA peptides on class I and class II MHC molecules for activation of tumor specific cytotoxic and helper T cells, respectively [27, 42]. However, tumor cells in advanced stages of the disease usually evolve to evade recognition by APC [9] and thus, are “ignored” by the immune system. This is indicated by the ability of tumor cells to reside in lymph nodes without being affected by the immune system. Therefore, vaccination by autologous TAA requires effective targeting of tumor cells within lesions for uptake by APC. Clinical trials involving recruitment of APC into lesions by cytokines like GM-CSF were reported to confer only suboptimal protection [23, 29]. Since tumor cells have no markers that “label” them for uptake, recruited APC fail to identify tumor cells as cells that should be internalized and their TAA processed. Therefore, uptake of tumor cells by APC is mediated only by inefficient random endocytosis.
We have recently developed a novel immunotherapy method for converting tumors into “personalized” tumor vaccines in which autologous tumor cells are targeted for effective uptake by APC, by intratumoral injection of α-gal glycolipids [17]. These glycolipids have linear or branched carbohydrate chains with 5 to >25 units, all capped by α-gal epitopes (Galα1-3Galβ1-4GlcNAc-R). α-Gal glycolipids are extracted from rabbit red cell (RBC) membranes where they comprise the majority of glycolipids [17]. The proposed treatment exploits the natural anti-Gal antibody. Anti-Gal is the most abundant natural antibody in humans constituting 1% of immunoglobulins in healthy individuals and in cancer patients [16]. This antibody interacts specifically with α-gal epitopes [14, 18]. It should be stressed that α-gal glycolipids differ from α-galactosyl ceramide, absent in mammals, which has only one carbohydrate unit (galactose) and is presented by CD1 molecules for NKT cell activation [5].
α-Gal glycolipids dissolve in water as micelles and are injected directly into tumor lesions as a micelles suspension. The injected α-gal glycolipids bind anti-Gal and activate complement within treated lesions. The complement cleavage peptides C5a and C3a are very potent chemotactic factors that recruit inflammatory cells and APC into the lesion. Injected α-gal glycolipids further insert spontaneously into tumor cell membranes within the treated lesion because the fatty acid “tails” in the ceramide portion of these molecules are energetically much more stable when surrounded by phospholipids of the cell membrane than within micelles surrounded by water. Therefore, tumor cells in the injected lesion express α-gal epitopes on their cell membranes and bind the natural anti-Gal antibody [17]. Anti-Gal binding to the tumor cells destroys them by complement mediated cytolysis and by antibody dependent cell mediated cytolysis (ADCC). Most importantly, recruited DCs and macrophages effectively internalize tumor cells with inserted α-gal glycolipids following interaction between anti-Gal coating (opsonizing) these tumor cells and Fcγ receptors (FcγR) on APC. The Fc/FcγR interaction has been described as the most effective mechanism by which APC internalize antigens for subsequent stimulation of the immune system, since this interaction generates signals for antigen internalization as well as for maturation of DCs internalizing the antigen [3, 31, 32]. APC internalizing opsonized tumor cells, effectively transport, process and present the autologous TAA peptides for activation of TAA specific T cells in the draining lymph nodes. Therefore, we hypothesized that the natural anti-Gal antibody, present in all humans, can serve as a universal means for targeting vaccinating tumor cells to APC, provided that the tumor cells express α-gal epitopes [12, 13].
The only nonprimate mammalian experimental model in which anti-Gal mediated targeting of tumor cells to APC can be studied is the α1,3-galactosyltransferase knockout mouse (KO mouse) [36]. In contrast to other nonprimate mammals, KO mice lack α-gal epitopes (due to the disruption of the α1,3-galactosyltransferase gene) and thus, they can produce anti-Gal in titers comparable to those in humans [2, 25, 35]. In a previous study we determined the effect of α-gal glycolipids on the immune response against ovalbumin as a surrogate TAA [17]. In contrast, the present study evaluates the immune response generated against endogenous melanoma associated antigens (MAA) of melanoma B16 cells, following intratumoral injection of α-gal glycolipids. B16 mouse melanoma cells are unique among mouse tumor cells in that they do not express α-gal epitopes, a characteristic also found in human tumor cells [19]. The protective effect of α-gal glycolipids treatment is determined by analysis of tumor development in distant sites, and by identifying T cell response to distinct MAA peptides. The study further demonstrates the efficacy of the α-gal glycolipids treatment in overcoming the immunosuppressive effect of the previously reported regulatory T (Treg) cells in melanoma [37]. In addition, we demonstrate the insertion of α-gal glycolipids into human melanoma cells. This suggests that α-gal glycolipids treatment is feasible also in human melanoma.
Materials and methods
Mice
α1,3-Galactosyltransferase knockout (KO) mice on H-2bxd background [36] were bred at the UMass Medical School animal facility. The 6 week old mice received three weekly intra-peritoneal immunizations of 50 mg pig kidney membranes (PKM) homogenate for inducing anti-Gal production in titers similar to those in humans [1, 2, 17, 35]. Anti-Gal production was confirmed by ELISA with synthetic α-gal epitopes linked to bovine serum albumin (BSA) (Dextra, Reading, UK) as solid phase antigen [1, 2, 35].
Cells and materials
B16F10 mouse melanoma cells (referred to in this study as B16) were obtained from ATCC. These cells lack α-gal epitopes [17, 21, 25]. The human melanoma cell line SK-N-MC pLXSN was received from Dr. R. Welsh (UMass Medical School). DCs were produced from bone marrow cells incubated with GM-CSF and IL4, as we previously described [17]. Rabbit RBC were purchased in one liter volume of packed RBC from PelFreez (Rogers, AR). The MAA peptides TRP2180–188 and gp10025–33 and control peptide TRP2181–188 lacking two amino acids (aa) were purchased from Bio-synthesis Inc. (Lewisville, TX). The choice of immunogenic MAA peptides was based on previous studies [4].
Extraction of α-gal glycolipids from rabbit RBC
The extraction of α-gal glycolipids from rabbit RBC was performed as we previously described [17]. Briefly, packed rabbit RBC (1 l volume) were lysed by hypotonic shock in water and repeatedly washed in water for removal of hemoglobin. Extraction of RBC membranes was performed overnight in a solution of 1,000 ml chloroform and 1,700 ml methanol with constant stirring. Precipitating proteins and residual cell membranes were removed by filtration on Whatman paper, under vacuum. Distilled water (~600 ml) was gradually added to form partition of the extracting solution into a lower organic phase and an upper aqueous phase. The aqueous phase containing the α-gal glycolipids was dried in a rotary evaporator, then brought to a concentration of 10 mg/ml in water as micelles [17].
Inoculation of tumor cells and intratumoral injection of α-gal glycolipids
Mice were injected intradermally in the right abdominal flank with 106 B16 tumor cells in 0.1 ml PBS. The site of injection was shaved prior to injection in order to enable accurate measurements of tumor size. Tumors reaching the size (mean diameter) of ~5 mm (5–6 days post inoculation) were injected with 1 mg α-gal glycolipids in 0.1 ml in 2–3 sites. In some mice the tumors were injected with 0.1 ml PBS and served as control for the α-gal glycolipids treatment. Intratumoral injection was repeated once or twice in 7 day intervals. Mice with tumors reaching a size of 25 mm were euthanized. In tumor challenge studies, the mice were injected subcutaneously in the contra-lateral (left) shaven flank with 0.5 × 106 B16 cells, 1 week after the third α-gal glycolipids injection. Growth of tumors in both flanks was monitored for 30 days. In studies on protection against growth of distant metastases, the shaven left flank was injected subcutaneously with 104 cells concomitantly with the injection of 106 B16 cells in the right flank.
ELISPOT analysis of MAA specific T cells
ELISPOT studies for the identification of tumor specific T cells in KO mice bearing B16 tumors were performed 1 week after the second injection of α-gal glycolipids or of PBS, by measuring secretion of interferon-γ (IFN-γ) from the activated T cells [1, 2, 17]. ELISPOT plates (Millipore) were coated with anti-mouse IFNγ antibody. Splenocytes were incubated in these ELISPOT wells (2 × 105 cells/well) in the presence of 5 μg/ml of the following mouse immunodominant MAA peptides: tyrosinase related protein 2 (TRP2180–188-SVYDFFVWL), gp10025–33 (EGSRNQDWL) and human immunodominant peptide of gp100 (KVPRNQDWL) [4]. Truncated TRP2 peptide of seven aa lacking S and V (VYDFFWL) was used as specificity control. After overnight incubation at 37°C in 5% CO2, cells were removed by washing and anti-IFNγ-biotin (monoclonal antibody R4-6A2, Mabtech) added to each well for 2 h at room temp. The wells were then washed and Streptavidin–alkaline phosphatase added for 1 h incubation at room temperature, followed by chromogenic substrate (NBT-plus, Mabtech) for 15 min. Color reaction resulted in formation of spots that represented the location of T cells secreting IFNγ. The color reaction was stopped by addition of water. Wells were then air-dried, and spots were counted with ELISPOT Automated Reader System (performed by Zellnet, Fort Lee, NJ). Calculated frequencies were based on the average of the triplicates wells.
Identification of effector cell populations by adoptive transfer
Spleen lymphocytes were obtained from KO mice, 1 week after the second α-gal glycolipids, or after PBS injection. Naïve KO recipients were inoculated subcutaneously with 5 × 105 B16 cells and 24 h later received intravenously into the tail vein 40 × 106 lymphocytes from the tumor bearing donors. These cells were administered as total lymphocytes, lymphocytes depleted of CD8+ (from donors with α-gal glycolipids treated tumors), or lymphocytes depleted of CD4+ T cells (from donors with PBS treated tumors). Depletion of T cells subpopulations was performed by the use of magnetic microbeads coated with anti-CD8 or anti-CD4 antibodies (Miltenyi Biotech Technology), respectively. After incubation of the lymphocytes with the corresponding magnetic microbeads for 15 min at 4°C, the cell suspensions were passed through a magnetic column (Miltenyi) for the depletion of lymphocytes with attached microbeads. Lymphocytes that were not retained in the column were washed and administered intravenously as above. Tumor growth was monitored in pairs of recipient mice, one of which receiving total lymphocytes and the other receiving lymphocytes depleted of CD8+, or of CD4+ T cells from the same donor.
Flow cytometry analysis of Treg cells
CD4+ T cells were stained with the phycoerythrin (PE) labeled monoclonal anti-mouse CD4 antibodies (Pharmingen). Treg cells were identified by double staining comprised of intracellular staining for Foxp3 and cell membrane staining for CD4 [33], using a kit produced specifically for identifying these cells (Bioligand-Alexa Fluor 647 anti-mouse Foxp3).
Results
Protection against tumor challenge administered post treatment
Induction of a protective immune response following intratumoral injection of α-gal glycolipids into a tumor was determined by subsequent challenge with live tumor cells in the contra-lateral flank. KO mice were inoculated with 106 B16 in the right flank. Tumors reached the size of 5 mm ~6 days post inoculation. Such tumors received three injections of 1 mg α-gal glycolipids in 7 days intervals. One day after the third injection, the mice were inoculated in the shaven left flank with 5 × 105 B16 cells and tumor growth monitored. Ten of 15 tested mice displayed no tumor growth in the left flank (Fig. 1a). In three of the remaining five mice in which tumor developed in the left flank, growth was slowed. Mice with tumors injected with PBS could not serve as controls in this experiment since the tumors reached the protocol limiting size of 25 mm size by the time of the third intratumoral injection of PBS was administered. These mice had to be euthanized prior to tumor challenge. In all mice challenged in the left flank after two instead of three PBS injections in the “primary” lesion, tumors developed in the contra-lateral challenge site (not shown).
Fig. 1.

Induction of a protective immune response against tumor challenge by intratumoral injection of α-gal glycolipids. Primary B16 tumors received three weekly injections of 1 mg α-gal glycolipids (a), or one injection of ethanol (b). One day after last injection of α-gal glycolipids, or 3 weeks after ethanol injection, mice were challenged with 0.5 × 106 B16 cells in the contra-lateral left flank and tumor growth in the left flank monitored. Note that ten of 15 mice injected with α-gal glycolipids (a) displayed no tumor development in the left flank (one curve labeled with filled circle for all ten mice). Tumors in the remaining five mice are presented as individual curves each labeled with open circle. Mice with tumors injected with ethanol (b), all developed tumors in the left flank (n = 5)
As an alternative control to PBS treatment, KO mice received one intratumoral injection of 0.1 ml ethanol (100%) into the “primary” tumor. Ethanol injected tumors in the mice disappeared within 3 weeks post treatment, similar to ethanol induced ablation in cancer patients (not shown). At that time point, the mice were challenged in the contra-lateral left flank with 5 × 105 B16 cells. Despite the complete regression of the ethanol treated tumors, all challenging tumors in the left flank developed, reaching the size of 10–25 mm within 20 days (Fig. 1b). Thus, in contrast to the α-gal glycolipid treatment which induces a systemic protective immune response, injection of ethanol elicited no protective immune response against tumor challenge.
Protection against tumor growth at a site distant to the treated primary tumor
The ability of α-gal glycolipids to induce systemic immune protection was further studied in experiments on protection against tumor growth in a distant site. Mice were inoculated in the right flank with B16 tumor cells and at the same time in the left flank with 104 B16 cells. The treatment of the right flank tumor with the first of two α-gal glycolipids or PBS injections was initiated 5–6 days post inoculation of the tumor cells in both flanks. At that time the left flank tumors were still invisible and not palpable. These left flank tumors became visible with size of 2–7 mm, ~15 days post inoculation of control mice (Fig. 2b). In this study, the latter tumors simulate invisible micrometastases in advanced melanoma patients at the time when visible tumors are locally treated.
Fig. 2.

Prevention of distant tumor growth by intratumoral injection of α-gal glycolipids. The figure describes development of tumors in the left flank from inoculates of 104 B16 cells, administered subcutaneously at the same time as the 106 B16 cells were administered subcutaneously in the right flank. (a) Development of the distant tumor in mice receiving two α-gal glycolipids injections into the right flank “primary” tumor (n = 8). b Development of the distant tumor in mice with “primary” tumor injected twice with PBS (n = 8)
In all control mice with right flank tumor injected with PBS, the left flank tumor developed into visible tumor with measurable size (Fig. 2b). These mice had to be euthanized after 20 or 25 days because the right flank “primary” tumor reached the maximum size of 25 mm. However, in four out of eight mice with right flank tumors injected with α-gal glycolipids, no lesions developed in the left flank (Fig. 2a), indicating that this treatment prevented the growth of distant tumors into measurable lesions. One of the remaining four mice displayed only minimal tumor development in the left flank (5 mm after 30 days), whereas in the other three mice, the tumor developed, but at a growth rate slower than in the PBS treated mice. These studies strongly suggest that even with a tumor of very poor immunogenicity such as B16 melanoma, intratumoral injection of α-gal glycolipids induces a protective immune response against autologous MAA so that it destroys pre-existing micrometastases.
Analysis of T cell response to immunodominant peptides of B16 MAA by ELISPOT
The observed protection following intratumoral injections of α-gal glycolipids (Figs. 1, 2) was followed by analysis of the induction of MAA specific T cell response. This was determined by ELISPOT studies with immunodominant MAA peptides of B16 melanoma including: TRP2180–188, gp10025–33 and human gp10025–33 [4]. T cell response evaluated by IFNγ secretion in ELISPOT with TRP2180–188 (SVYDFFVWL) peptide was higher in five of the six mice with B16 tumors injected with α-gal glycolipids, than in mice with tumors injected with PBS (Fig. 3a). The mean response in the six mice treated with α-gal glycolipids was approximately sixfold higher than in the control mice treated with PBS (Fig. 3b). This effective ELISPOT response to TRP2180–188 in the former group was highly specific to the full length peptide. The truncated peptide corresponding to TRP2181–188 with only seven aa (VYDFFWL) did not stimulate lymphocytes from mice with α-gal glycolipids treated tumors (Fig. 3a, b). Stimulation by the mouse immunodominant peptide gp10025–33 (EGSRNQDWL) was also approximately sixfold higher in mice treated with α-gal glycolipids than in PBS treated mice. Human gp10025–33 (KVPRNQDWL) induced on average a twofold higher response in mice treated with α-gal glycolipids than in those treated with PBS (Fig. 3b). It should be stressed that the high standard deviation values in Fig. 3b are the result of lack of response in one of the six mice treated with α-gal glycolipids. Interestingly, this lack of response was observed in different individual mice for each of the three stimulatory peptides. Overall the ELISPOT response data support the assumption that intratumoral injection of α-gal glycolipids elicits an effective T cell immune response against various immunodominant MAA peptides of melanoma cells in the treated lesion.
Fig. 3.

ELISPOT data from mice with B16 tumors injected with PBS, or with α-gal glycolipids (GSL-glycosphingolipids), and which are stimulated in vitro with various immunodominant MAA peptides (n = 6 mice per group). a Data with spleen lymphocytes from individual mice stimulated with the following MAA peptides: TRP2180–188 (closed columns); TRP2180–188 without S180 and V187 (TRP2181–188 open columns); mouse gp10025–33 (diagonal hatched columns); human gp10025–33 (horizontal hatched columns). Data presented as number of IFNγ spots per million lymphocytes (i.e., T cells secreting IFNγ/106 cells). b Data presented as mean + standard deviation of ELISPOT response to the various MAA peptides in mice with tumors treated with PBS (open columns), or with α-gal glycolipids (closed columns)
CD8+ T cells mediate the protective anti-tumor immune response in α-gal glycolipids treated mice
Many studies have indicated that protection against tumor cells, in both mice and humans, is primarily mediated by tumor specific CD8+ T cells. In order to determine whether CD8+ T cells also mediate the systemic protection against tumor following intratumoral injection of α-gal glycolipids, we performed adoptive transfer studies. In these studies, CD8+ T cells were depleted from transferred spleen lymphocytes by the use of magnetic microbeads coated with anti-CD8 antibody. Naïve KO mice (i.e., mice lacking anti-Gal) in pairs were injected subcutaneously with 0.5 × 106 B16 cells. After 24 h, 40 × 106 total spleen lymphocytes from a treated donor mouse were injected into the tail vein of one of the two recipients in each pair. These lymphocytes were harvested 1 week after the second injection of α-gal glycolipids. The second recipient also received 40 × 106 lymphocytes from the same spleen, however, these lymphocytes were depleted of CD8+ T cells. Tumor growth was monitored and compared in each pair of recipients. Since the response pattern differed in the various mice, the tumor growth data in the individual pairs are presented in Fig. 4. Each figure is numbered according to the donor mouse.
Fig. 4.
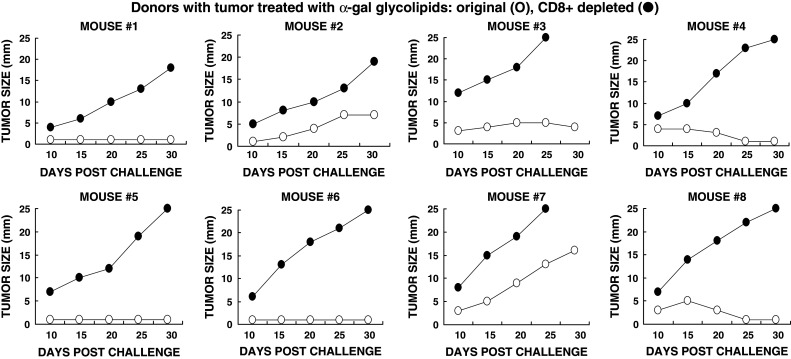
Tumor growth following adoptive transfer of lymphocytes from donor mice with tumors injected with α-gal glycolipids. Naïve recipients were challenged subcutaneously with 0.5 × 106 live B16 cells, 24 h prior to transfer of lymphocytes. Adoptive transfer of lymphocytes was performed in pairs of recipients, each receiving 40 × 106 cells. Open circle total lymphocytes, or filled circle lymphocytes depleted of CD8+ T cells. Data on tumor growth for 30 days post adoptive transfer in pairs receiving total, or CD8+ depleted lymphocytes from eight donors
In accord with our previous observations [17], adoptive transfer of total non-depleted lymphocytes from α-gal glycolipids treated donors prevented tumor growth in most naïve recipients, although the tumor cells were introduced 24 h prior to the injection of lymphocytes (Fig. 4). In four of the eight recipients no tumor growth was observed in the recipients. In three of the remaining four mice there was a minimal tumor development (<6 mm) which, either disappeared or did not continue to progress during the 30 days monitoring period. In one of the eight mice (recipient from donor mouse #7 in Fig. 4) the tumor developed, albeit slower than in control recipients of lymphocytes from mice with PBS injected tumors. Adoptive transfer of spleen lymphocytes from mice with PBS injected tumors conferred no protection against the tumor challenge in any of the naïve recipients (Fig. 5). In this group of recipients, the tumors reached a size of 15–25 mm within the 30 days period of monitoring.
Fig. 5.
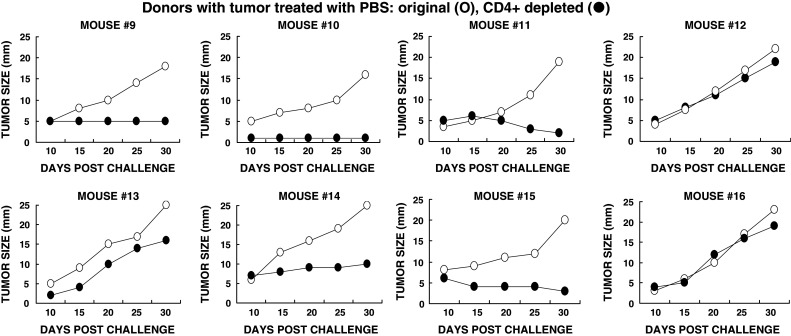
Tumor growth following adoptive transfer of lymphocytes from donor mice with tumors injected with PBS. Adoptive transfer performed as in Fig. 4. Open circle total lymphocytes; filled circle lymphocytes depleted of CD4+ T cells. Data are presented in pairs receiving total, or CD4+ depleted lymphocytes from eight donors
The protection by lymphocytes transferred from donors with α-gal glycolipids treated tumors was mediated by CD8+ T cells. This was indicated by the finding that adoptive transfer of CD8+ depleted lymphocytes from these donors, conferred no protection in the recipients. Growth of tumor lesions was observed in all eight recipients of the CD8+ depleted lymphocytes (Fig. 4). These findings imply that autologous vaccines comprised of tumors injected with α-gal glycolipids, stimulates the immune system to generate tumor specific CD8+ T cells that can detect and destroy tumor cells which express the immunizing TAA.
Involvement of Treg cells in B16 melanoma development
The observations in Fig. 4 on CD8+ T cells protecting against tumor growth in mice with α-gal glycolipids treated tumors raised the question of whether there are regulatory T (Treg) cells in tumor bearing KO mice which in the absence of this treatment may hamper the protective anti-tumor immune response. This presence of Treg cells was analyzed both by evaluating their function (Fig. 5) and by specific staining of these cells for determining their proportion among spleen lymphocytes (Fig. 6). Several studies have reported that failure of cancer immunotherapy in various types of experimental cancer in wild type (WT) mice (i.e., mice producing α-gal epitopes) is associated with the activity of Treg cells which prevent the induction of a protective anti-TAA immune response [33, 34, 37, 40].
Fig. 6.

Immunostaining of Treg cells from spleens of mice with tumors injected with PBS (a, b), or injected with α-gal glycolipids (c, d). Spleen lymphocytes were obtained 2 weeks after second injection and subjected to Foxp3 and CD4 double staining. The Treg cells are located in the upper right quadrant. Presented data are of two representatives out of five mice in each group
The presence of Treg cells in KO mice bearing B16 melanoma was first evaluated in the group of mice treated by intratumoral injections of PBS. Treg cells are characterized as CD4+, CD25+ cells that also express intracellular Foxp3 protein [33]. Thus, analysis of Treg activity was performed by adoptive transfer studies in which lymphocytes were depleted of CD4+ T cells, using magnetic microbeads coated with anti-CD4 antibodies. The total lymphocytes, or CD4+ depleted lymphocytes were infused into naive KO mice that were challenged, as in Fig. 4, with 0.5 × 106 B16 cells, 24 h prior to adoptive transfer. All recipients of total lymphocytes (i.e., not depleted of CD4+ T cells), developed tumors (Fig. 5). However, depletion of CD4+ T cells resulted in the subsequent protection against tumor growth in five out of the eight recipients of lymphocytes from donors with PBS injected tumors (Fig. 5). Since removal of Treg cells enables the subsequent protection against tumor growth, these data suggest that Treg cells suppress the development of a protective anti-melanoma immune response in KO mice that are not treated with α-gal glycolipids. The lack of protection in the remaining three recipients of CD4+ depleted lymphocytes in Fig. 5, implies that in a minority of the mice with PBS treated tumors, there is no induction of a protective immune response against the MAA on the tumor cells due to lack of sufficient activation of the immune system against these TAA. Thus, even depletion of Treg cells does not prevent tumor growth.
Immunostaining of Treg cells
The findings in Fig. 5 raised the question of whether the protective immune response in KO mice with α-gal glycolipids treated tumors is associated with a much lower number of Treg cells than in PBS treated mice. This was analyzed by measuring the proportion of Treg cells within spleens of the two groups (5 mice/group). Treg cells were identified as CD4+ T cells that were also stained for intracellular Foxp3 expression [33]. Figure 6 presents the flow cytometry data in two representative mice from each group. The proportion of Treg cells is presented in the upper right quadrant. The proportion of Treg cells in five mice with tumors injected with α-gal glycolipids (1 week post second injection) was 1.8, 2.2, 1.2, 1.4 and 2.8% (mean ± standard deviation of 2.0 ± 0.7%). The proportion of Treg cells in five mice with tumors injected with PBS was 2.6, 1.7, 3.0, 2.0 and 1.8% (mean ± standard deviation of 2.2 ± 0.6%). The average proportion of Treg cells, detected by this staining in untreated control mice that do not bear tumor, was 2.2% (not shown). Since the number of mononuclear cells was similar in all spleens (90–100 × 106 cells/spleen), these findings suggest that intratumoral injection of α-gal glycolipids does not alter the overall proportion of Treg cells in the treated mice.
Overall, the functional data in Fig. 5 indicate that KO mice bearing B16 melanoma have Treg cells which suppress the protective immune response against the tumor cells, whereas immunostaining data in Fig. 6 suggest that these Treg cells are present at similar levels in KO mice with PBS treated tumors and in mice with α-gal glycolipids treated tumors. In view of these data in Figs. 5 and 6, the data in Fig. 4 strongly suggest that the immune protection against MAA achieved by intratumoral injection of α-gal glycolipids is associated with the activation of tumor specific CD8+ T cells that overcome the immunosuppressive activity of Treg cells and prevent tumor development.
Applicability of the α-gal glycolipids treatment to human melanoma
Preliminary studies on the applicability of the α-gal glycolipids treatment to human melanoma were performed by evaluating in vitro insertion of these glycolipids into the cell membranes of human melanoma cells (the cell line SK-N-MC pLXSN). Like all other human normal and malignant cells [12, 19], these human melanoma cells lack α-gal epitope. The cells were incubated with 0.1 or 1.0 mg/ml α-gal glycolipids for 2 h at 37°C. Expression of α-gal epitopes on washed cells was evaluated by binding of mouse monoclonal anti-Gal antibody M86 [15], followed by FITC labeled anti-mouse IgM and flow cytometry analysis. Anti-Gal does not bind to the human melanoma cells since they lack α-gal epitopes, but readily binds to melanoma cells that were incubated with 0.1 or 1.0 mg/ml of α-gal glycolipids (Fig. 7a).
Fig. 7.

Insertion of α-gal glycolipids into the cell membranes of human melanoma cells (SK-N-MC pLXSN cell line). a Binding of the monoclonal anti-Gal Ab M86 to untreated melanoma cells (thin line histogram), or cells incubated for 2 h at 37°C with 0.1 or 1 mg/ml α-gal glycolipids (dashed line and thick line histograms, respectively). b Complement dependent cytolysis of the human melanoma cells with inserted α-gal glycolipids following 30 min incubation at 37°C with human serum. Open circle cells incubated with 1 mg/ml α-gal glycolipids; open square 0.1 mg/ml; open triangle no α-gal glycolipids
The human melanoma cells were further incubated for 30 min at 37°C with human serum and subsequently analyzed for cytolysis. Following the incubation with α-gal glycolipids, the melanoma cells became very sensitive to complement mediated cytolysis as a result of human anti-Gal binding (Fig. 7b). Heat inactivated serum (56°C for 30 min) induced no cytolysis due to destruction of complement (not shown). This cytolysis was proportional to the amount of inserted α-gal glycolipids, as it was higher in cells incubated with 1.0 mg/ml α-gal glycolipids than in cells incubated with 0.1 mg/ml of these glycolipids (Fig. 7b). In the absence of inserted α-gal glycolipids, no cytolysis of the melanoma cells was observed since the cells lack α-gal epitopes. These data suggest that a similar insertion of α-gal glycolipids and anti-Gal binding will occur in vivo in treated lesions of melanoma patients, ultimately resulting in in vivo conversion of the treated lesions into autologous tumor vaccines.
Discussion
The results reported here provide three novel observations on immunotherapy by intratumoral injection of α-gal glycolipids in the α1,3-galactosyltransferase knockout mice (KO mice) model: (1) The study demonstrates the effective activation of T cells specific to endogenous MAA peptides, (2) Experiments on challenge with live tumor cells prior, or post treatment, demonstrate the induction of a protective immune response that prevents tumor development in distant sites within the treated mouse, and (3) CD8+ T cells in the treated KO mice are shown to be the major effector cells which protect against tumor cells and overcome the suppressive effect of Treg cells. The study further demonstrates the insertion of α-gal glycolipids into human tumor cells and anti-Gal binding to such cells.
Immunotherapy by intratumoral injections of α-gal glycolipids includes one element that is absent in other types of immunotherapy: the active targeting of tumor cells for uptake by APC. Since tumor lesions in advance stages of the disease evolve to be “invisible” to the immune system [9], a major challenge for the in situ conversion of tumor lesions into vaccines has been the recruitment of DCs and other APC into the lesion and induction of effective uptake of the tumor cells by the recruited APC. Effective recruitment of DCs into tumors was achieved by intratumoral injection of CpG rich oligonucleotides, macrophage inflammatory protein (MIP3α) and Flt4 ligand [11], GM-CSF [8, 11, 24], or IL12 [10]. However, in tumors with weak TAA (a characteristic of most human tumors in advanced stages [9]), such recruitment of APC may not suffice [11]. This is because tumor cells have no markers identifying them as cells that should be internalized by the recruited APC. Therefore uptake by APC is insufficient since it is mediated only by random endocytosis.
Active in vitro targeting of tumor cells to APC that is mediated by Fc/FcγR interaction was demonstrated with several opsonizing monoclonal antibodies [7, 22]. By the use of ovalbumin as a surrogate TAA, we have previously shown that following intratumoral injection of α-gal glycolipids, anti-Gal in KO mice can also mediate in vivo targeting of tumor cells to APC [17]. This treatment may induce in vivo targeting of tumor cells to APC also in cancer patients. This is since all humans who are not severely immunocompromised produce large amounts of anti-Gal [12, 14, 16, 18] and since the human natural anti-Gal antibody is very effective in targeting tumor cells expressing α-gal epitopes to human DCs and macrophages [12, 28].
Intratumoral injection of α-gal glycolipids is likely to achieve multiple objectives required for conversion of human tumors into effective vaccines, including: (1) Recruitment of APC, (2) Targeting of tumor cells for effective uptake by APC via Fc/FcγR interaction, (3) Activation of APC as a result of the interaction between Fc portion of the opsonizing anti-Gal and FcγR on APC, and (4) Effective transport of TAA to the draining lymph nodes where TAA peptides are presented to cytotoxic and helper T cells, resulting in a “personalized” immune response against the multiple autologous TAA. We previously quantified in studies with viral vaccines (HIV and influenza virus), the extent of increased immunogenicity as a result of anti-Gal mediated targeting to APC by Fc/FcγR interaction. This targeting resulted in ~100-fold increase in immunogenicity of antigens within the vaccine in comparison with the same vaccines which, however, lack α-gal epitopes [1, 2]. Thus, it is probable that a similar targeting of tumor cells following intratumoral injection of α-gal glycolipids will increase immunogenicity of multiple undefined autologous TAA by many folds.
Of particular interest is the protective anti-tumor immune response demonstrated here by preventing growth of a distant tumor (Fig. 2) and of tumor cells administered post treatment (Fig. 1). In contrast to the treatment with α-gal glycolipids, tumor ablation by intratumoral injection of ethanol (a method used in the clinical setting) induced no protective immune response. This is probably because APC fail to effectively internalize and process the immunogenic MAA in the ethanol treated tumor. The observed systemic protection against distant tumor growth, following anti-Gal interaction with tumor cells expressing α-gal epitopes, is in accord with the studies demonstrating the increased numbers of MAA specific T cells in the spleen (Fig. 3). All these data strongly suggest that shortly after treatment (~1–2 weeks), tumor specific T cells are effectively activated by MAA peptides. These activated T cells can protect against autologous tumor cells by detecting and destroying distant micrometastases.
Anti-tumor protection following α-gal glycolipids treatment, is mediated primarily by CD8+ T cells as indicated by adoptive transfer studies. Depletion of CD8+ T cells from transferred lymphocytes eliminated protection against tumor development in naïve recipients (Fig. 4). Interestingly, a protective immune response was observed also with CD4+ depleted lymphocytes following adoptive transfer from the majority of mice with PBS treated tumors (Fig. 5). These findings are in accord with previous observations on the suppressive effect of Treg cells that prevent the development of a protective anti-tumor immune response [34, 37, 40]. The observations on Treg cells suggest that MAA released from the B16 lesions in control mice (i.e., mice receiving intratumoral injection of PBS) reach the lymph nodes and spleen and activate tumor specific effector T cells. However, this activation is not potent enough to overcome the suppressive activity of Treg cells. Nevertheless, the effective anti-MAA immune response elicited following intratumoral injection of α-gal glycolipids enables the immune system to overcome the suppressive effect of Treg cells to the extent that tumor cells in distant sites are destroyed and can not develop into metastases. This is further suggested by the finding that the proportion of Treg cells in spleens of α-gal glycolipids treated mice is not different from that in PBS treated mice (Fig. 6).
The present study also provides preliminary information suggesting that conversion of tumors into endogenous vaccines by intratumoral injection of α-gal glycolipids is applicable immunotherapy in cancer patients. Insertion of α-gal glycolipids and subsequent anti-Gal binding could be demonstrated with human melanoma cells (Fig. 7). The studies in mice further suggest that α-gal glycolipids have no direct toxicity. No one of the treated mice died within a period of 1 week following first, second or third injection of 1 mg injection of α-gal glycolipids. Moreover, this treatment does not seem to cause loss of immune tolerance to normal tissue antigens and does not induce autoimmunity. This is suggested from the finding that no one of the >200 mice injected with α-gal glycolipids developed vitiligo, i.e., no autoimmune response developed against normal melanocytes.
As we previously argued [17], this treatment may have even higher efficacy in humans than that observed in KO mice. This is because complement activity in humans is much higher than that in mice, thus generation of complement chemotactic factors following anti-Gal/α-gal epitope interaction is likely to be more extensive in humans [17]. In addition, tumor growth in humans is much slower than that of B16 melanoma lesions which double in size every 4–8 days. Therefore, the anti-tumor immune response elicited by intratumoral injection of α-gal glycolipids can react against micrometastases for much longer periods than in mice. Such prolonged reactivity allows for effective destruction of micrometastases before they develop into visible metastases that are resistant to immune destruction because of their size and immunosuppressive milieu [26].
Acknowledgments
This work was funded by NIH grant CA122019 (U.G.).
Abbreviations
- ADCC
Ab dependent cell mediated cytotoxicity
- α-Gal epitope
Galα1-3Galβ1-4GlcNAc-R epitope
- CDC
Complement dependent lysis
- KO
Knockout mice for the α1,3-galactosyltransferase gene
- MAA
Melanoma associated antigens
- PKM
Pig kidney membranes
- TAA
Tumor associated antigens
References
- 1.Abdel-Motal UM, Guay HM, Wigglesworth K, Welsh RM, Galili U. Increased immunogenicity of influenza virus vaccine by anti-Gal mediated targeting to antigen presenting cells. J Virol. 2007;81:9131–9141. doi: 10.1128/JVI.00647-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abdel-Motal UM, Wang S, Lu S, Wigglesworth K, Galili U. Increased immunogenicity of HIV gp120 engineered to express Galα1-3Galβ1-4GlcNac-R epitopes. J Virol. 2006;80:6943–6951. doi: 10.1128/JVI.00310-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banchereau J, Briere F, Caux C, et al. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 4.Bloom MB, Perry-Lalley D, Robbins PF, et al. Identification of tyrosinase-related protein 2 as a tumor rejection antigen for the B16 melanoma. J Exp Med. 1997;185:453–459. doi: 10.1084/jem.185.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brigl M, Brenner MB. CD1: antigen presentation and T cell function. Annu Rev Immunol. 2004;22:817–890. doi: 10.1146/annurev.immunol.22.012703.104608. [DOI] [PubMed] [Google Scholar]
- 6.Clemente CG, Mihm MC, Jr, Bufalino R, Zurrida S, Collini P, Cascinelli N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer. 1996;77:1303–1310. doi: 10.1002/(SICI)1097-0142(19960401)77:7<1303::AID-CNCR12>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 7.Dhodapkar KM, Krasovsky J, Williamson B, Dhodapkar DM. Antitumor monoclonal antibodies enhance cross-presentation of cellular antigens and the generation of myeloma-specific killer T cells by dendritic cells. J Exp Med. 2002;195:125–133. doi: 10.1084/jem.20011097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dranoff G, Jaffee E, Lazenby A, et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte–macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci USA. 1993;90:3539–3543. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunn GP, Bruce A, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 10.Egilmez NK, Jong YS, Sabel MS, Jacob JS, Mathiowitz E, Bankert RB. In situ tumor vaccination with interleukin-12-encapsulated biodegradable microspheres: induction of tumor regression and potent antitumor immunity. Cancer Res. 2000;60:3832–3837. [PubMed] [Google Scholar]
- 11.Furumoto K, Soares L, Engleman EG, Merad M. Induction of potent antitumor immunity by in situ targeting of intratumoral DCs. J Clin Invest. 2004;113:774–777. doi: 10.1172/JCI19762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galili U. Autologous tumor vaccines processed to express α-gal epitopes: a practical approach to immunotherapy in cancer. Cancer Immunol Immunother. 2004;53:935–945. doi: 10.1007/s00262-004-0524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galili U, LaTemple DC. The natural anti-Gal antibody as a universal augmenter of autologous tumor vaccine immunogenicity. Immunol Today. 1997;18:281–285. doi: 10.1016/S0167-5699(97)80024-2. [DOI] [PubMed] [Google Scholar]
- 14.Galili U, Macher BA, Buehler JS, Shohet B. Human natural anti-α-galactosyl IgG. II. The specific recognition of α(1 → 3)-linked galactose residues. J Exp Med. 1985;162:573–582. doi: 10.1084/jem.162.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galili U, LaTemple DC, Radic MZ. A sensitive assay for measuring α-gal epitope expression on cells by a monoclonal anti-Gal antibody. Transplantation. 1998;65:1129–1132. doi: 10.1097/00007890-199804270-00020. [DOI] [PubMed] [Google Scholar]
- 16.Galili U, Rachmilewitz EA, Peleg A, Flechner I. A unique natural human IgG antibody with anti-α-galactosyl specificity. J Exp Med. 1984;160:1519–1531. doi: 10.1084/jem.160.5.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galili U, Wigglesworth K, Abdel-Motal UM. Intratumoral injection of α-gal glycolipids induces xenograft-like destruction and conversion of lesions into endogenous vaccines. J Immunol. 2007;178:4676–4687. doi: 10.4049/jimmunol.178.7.4676. [DOI] [PubMed] [Google Scholar]
- 18.Galili U, Clark MR, Shohet SB, Buehler J, Macher BA. Evolutionary relationship between the anti-Gal antibody and the Galα1 → 3Gal epitope in primates. Proc Natl Acad Sci USA. 1987;84:1369–1373. doi: 10.1073/pnas.84.5.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galili U, Shohet SB, Kobrin E, Stults CLM, Macher BA. Man, apes, and Old World monkeys differ from other mammals in the expression of α-galactosyl epitopes on nucleated cells. J Biol Chem. 1988;263:17755–17762. [PubMed] [Google Scholar]
- 20.Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 21.Gorelik E, Duty L, Anaraki F, Galili U. Alterations of cell surface carbohydrates and inhibition of metastatic property of murine melanomas by α1,3-galactosyltransferase gene transfection. Cancer Res. 1995;55:4168–4173. [PubMed] [Google Scholar]
- 22.Groh V, Li YQ, Caio D, Hunder NN, et al. Efficient cross-priming of tumor antigen-specific T cells by dendritic cells sensitized with diverse anti-MICA opsonized tumor cells. Proc Natl Acad Sci USA. 2005;102:6461–6466. doi: 10.1073/pnas.0501953102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hofbauer GF, Baur T, Bonnet MC, Tartour E, Burg G, Berinstein NL, Dummer R. Clinical phase I intratumoral administration of two recombinant ALVAC canarypox viruses expressing human granulocyte–macrophage colony-stimulating factor or interleukin-2: the transgene determines the composition of the inflammatory infiltrate. Melanoma Res. 2008;18:104–111. doi: 10.1097/CMR.0b013e3282f702cf. [DOI] [PubMed] [Google Scholar]
- 24.Huang AY, Golumbek P, Ahmadzadeh M, Jaffee E, Pardoll D, Levitsky H. Role of bone marrow-derived cells in presenting MHC class I-restricted tumor antigens. Science. 1994;264:961–965. doi: 10.1126/science.7513904. [DOI] [PubMed] [Google Scholar]
- 25.LaTemple DC, Abrams JT, Zhang SU, Galili U. Increased immunogenicity of tumor vaccines complexed with anti-Gal: studies in knock out mice for α1, 3-galactosyltransferase. Cancer Res. 1999;59:3417–3423. [PubMed] [Google Scholar]
- 26.Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol. 2005;174:7516–7523. doi: 10.4049/jimmunol.174.12.7516. [DOI] [PubMed] [Google Scholar]
- 27.Maass G, Schmidt W, Berger M, et al. Priming of tumor-specific T cells in the draining lymph nodes after immunization with interleukin-2 secreting tumor cells: three consecutive stages may be required for successful tumor vaccination. Proc Natl Acad Sci USA. 1995;92:5540–5544. doi: 10.1073/pnas.92.12.5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manches O, Plumas J, Lui L, Chaperot L, Molens JP, Sotto JJ, Bensa JC, Galili U. Anti-Gal mediated targeting of human B lymphoma cells to antigen-presenting cells: a potential method for immunotherapy with autologous tumor cells. Haematologica. 2005;90:625–634. [PubMed] [Google Scholar]
- 29.Mastrangelo MJ, Maguire HC, Jr, Eisenlohr LC, et al. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther. 1999;6:409–422. doi: 10.1038/sj.cgt.7700066. [DOI] [PubMed] [Google Scholar]
- 30.Mumberg D, Wick M, Schreiber H. Unique tumor antigens redefined as mutant tumor-specific antigens. Semin Immunol. 1996;8:289–293. doi: 10.1006/smim.1996.0037. [DOI] [PubMed] [Google Scholar]
- 31.Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol. 2001;19:275–290. doi: 10.1146/annurev.immunol.19.1.275. [DOI] [PubMed] [Google Scholar]
- 32.Regnault A, Lankar D, Lacabanne V, et al. Fc-gamma receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class I-restricted antigen presentation after immune complex internalization. J Exp Med. 1999;189:371–380. doi: 10.1084/jem.189.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakaguchi S. Naturally arising Foxp3-expressing CD25+ CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–352. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- 34.Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+ CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999;163:5211–5218. [PubMed] [Google Scholar]
- 35.Tanemura M, Yin D, Chong AS, Galili U. Differential immune response to α-gal epitopes on xenografts and allografts: implications for accommodation in xenotransplantation. J Clin Invest. 2000;105:301–310. doi: 10.1172/JCI7358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thall AD, Maly P, Lowe JB. Oocyte Galα1-3Gal epitopes implicated in sperm adhesion to the zona pellucida glycoprotein ZP3 are not required for fertilization in the mouse. J Biol Chem. 1995;270:21437–21442. doi: 10.1074/jbc.270.37.21437. [DOI] [PubMed] [Google Scholar]
- 37.Turk MJ, Guevara-Patino JA, Rizzuto GA, Engelhorn ME, Sakaguchi S, Houghton AN. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J Exp Med. 2004;200:771–782. doi: 10.1084/jem.20041130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weir BA, Woo MS, Getz G, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–898. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wood LD, Parsons D, Jones WS, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 40.Yu P, Lee Y, Liu W, Krausz T, Chong A, Schreiber H, Fu YX. Intratumor depletion of CD4+ cells unmasks tumor immunogenicity leading to the rejection of late-stage tumors. J Exp Med. 2005;201:779–791. doi: 10.1084/jem.20041684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang L, Conejo-Garcia JR, Katsaros D. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–213. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 42.Zinkernagel RM, Ehl E, Aichele P, Kündig T, Hengartner H. Antigen localization regulates immune responses in a dose and time dependent fashion: a geographical view of immune reactivity. Immunol Rev. 1997;156:199–209. doi: 10.1111/j.1600-065X.1997.tb00969.x. [DOI] [PubMed] [Google Scholar]