

Abstract
We explore the diagnostic potential of imaging endogenous fluorophores using two photon microscopy and fluorescence lifetime imaging (FLIM) in human skin with two spectral detection channels. Freshly excised benign dysplastic nevi (DN) and malignant nodular Basal Cell Carcinomas (nBCCs) were excited at 760 nm. The resulting fluorescence signal was binned manually on a cell by cell basis. This improved the reliability of fitting using a double exponential decay model and allowed the fluorescence signatures from different cell populations within the tissue to be identified and studied. We also performed a direct comparison between different diagnostic groups. A statistically significant difference between the median mean fluorescence lifetime of 2.79 ns versus 2.52 ns (blue channel, 300-500 nm) and 2.08 ns versus 1.33 ns (green channel, 500-640 nm) was found between nBCCs and DN respectively, using the Mann-Whitney U test (p < 0.01). Further differences in the distribution of fluorescence lifetime parameters and inter-patient variability are also discussed.
OCIS codes: (170.1870) Dermatology; (170.6920) Time-resolved imaging; (170.6510) Spectroscopy, tissue diagnostics; (180.4315) Nonlinear microscopy; (300.6410) Spectroscopy, multiphoton; (170.3880) Medical and biological imaging
1. Introduction
Human skin contains a number of naturally occurring fluorophores that can be used as a potential source of contrast for non-invasive imaging without requiring the use of exogenous contrast agents. These fluorophores include collagen, elastin, melanin, keratin, porphyrins, NAD(P)H and flavins [1]. Fluorescence intensity can be measured using two photon excitation to provide optically sectioned images with sub-cellular resolution and allow tissue morphology to be studied and quantified in detail [1–3]. The fluorescence can also be resolved with respect to emission wavelength [4,5] and fluorescence lifetime [5–8] to provide further contrast between different tissue types. Such spectral ratiometric or lifetime measurements are insensitive to experimental measurement parameters such as laser excitation power, photon collection efficiency or detector sensitivity. This allows absolute measurements of spectroscopic parameters to be compared within and between samples.
The intracellular fluorophore NAD(P)H is integral to cellular metabolism and by measuring its fluorescence emission spectrum [9] and lifetime [10], changes in the metabolic state of tissue in vivo can be assessed. Similarly, changes in the emission spectrum of melanin [11] have been correlated with the dysplastic transformation associated with melanoma skin cancer. This ability for autofluorescence imaging to assess both the morphology and metabolic [3] state of tissue non-invasively distinguishes this technique from other non-invasive imaging modalities.
In this paper we describe the analysis of fluorescence intensity and FLIM images from freshly excised human skin samples of dysplastic nevi (DN) and nodular Basal Cell Carcinomas (nBCCs). We use a modified commercial two-photon tomograph [12] that collects the fluorescence signal into two spectrally and temporally resolved channels (‘blue channel’, 380-500 nm and ‘green channel’, 500-640 nm). For an excitation wavelength of 760 nm, we expect intracellular fluorophores in the blue channel to be dominated by fluorescence from NAD(P)H with its emission spectral peak between 450 and 470 nm [4,13] and the green channel to be dominated by melanin autofluorescence (spectral peak at 550 nm [4,14]), with potential contributions from flavins (peak at 525 nm [4,13]).
In two photon tomography of tissue the excitation beam power is restricted to prevent damage to the sample and acquisition times in vivo are minimized to avoid motion artifacts. As a consequence, the limited number of photons collected per pixel can be inadequate to reliably calculate fluorescence lifetimes per pixel using multi-exponential decay models. Although a single exponential decay model requires the fewest photons to fit reliably, it is desirable to employ more complex decay models because the fluorescence from skin originates from multiple fluorescent species, each of which are themselves likely to exhibit complex decay profiles. We address this issue by spatially binning all the photons for each cell within each image. This increases the number of photons available for fitting and allows a double exponential decay model to be fitted to the data. The work presented here demonstrates the potential to discriminate between different types of cutaneous lesions (specifically DN and nBCCs) by analyzing the tissue autofluorescence at the individual cell level with respect to both spectral and lifetime parameters. The two lesion types were selected to maximize our ability to detect differences in the spectroscopic parameters, and to determine the utility of our multi-spectral FLIM approach for future studies involving more clinically meaningful comparisons.
2. Equipment
Two photon tomographic imaging was performed using a modified DermaInspect® (JenLab GmbH, Jena, Germany), which is CE marked for clinical imaging of skin. A mode-locked 80 MHz Ti:sapphire laser (Spectra Physics®, CA, USA) equipped with a prism based chirp compensation unit (DeepSee module, Spectra Physics®) was used as the excitation laser source. The laser has a tuning range between 710 and 920 nm and the excitation power was attenuated to a maximum of 50 mW at the sample. The excitation beam passes through two galvoscanners, allowing scanning in the x-y plane, before being focused into the sample by a 40 × /1.4NA oil immersion objective. The microscope objective facilitated a working depth up to 200 µm with a lateral resolution of <1 µm and an axial resolution of <2 µm.
The emitted fluorescence passed through a 640 nm short pass dichroic filter (Chroma Technology Corp., Vermont, USA), a BG39 spectral filter (Präzisions Glas & Optik GmbH, Germany) and was spectrally separated by a long pass 500 nm dichroic beam splitter (AHF Analysentechnik AG, Germany). It was then detected by two H7722P-40 PMTs detectors (Hamamatsu Photonics K.K., Japan), each protected by another BG39 filter. The two PMTs therefore collected fluorescence in 380-500 nm (‘blue’) and 500-640 nm (‘green’) spectral channels. Fluorescence lifetimes were determined using time correlated single photon counting (TCSPC) implemented with 256 time bins using an SPC-830 module (Becker & Hickl GmbH, Germany). The gain and cooling of the PMT detectors was controlled by a DCC-100 card (Becker & Hickl GmbH).
3. Samples
The tissue was collected from patients attending the Department of Dermatology at Imperial College Healthcare NHS Trust. The study was ethically approved by the regional ethics committee (Ref 09/H0706/28) and patients gave written informed consent to participate unless they met the following exclusion criteria; age 18 years or less, in state custody, carrying a blood borne infection, having a photosensitive skin disorder, or pregnancy. Patients with clinically suspected nBCCs and DN already scheduled for excision were recruited. All clinical diagnoses were confirmed histologically following imaging.
Freshly excised lesions were rinsed with Hanks Balanced Salt Solution (HBSS) buffer without phenol red, calcium or magnesium (Gibco®, Invitrogen, CA, USA) and placed on damp gauze in an inverted glass bottomed petri dish containing a 170 µm cover slip (Matek®, MA,USA). This was attached to a metallic ring using adhesive and coupled magnetically to the DermaInspect®. A drop of water was placed between the tissue and the cover slip and a drop of Immersol 518F (Carl Zeiss Ltd, Germany) was placed between the cover slip and the objective.
4. Imaging protocol
Images were acquired as soon as possible (typically within 15 minutes) until a maximum of 2.5 hours post excision. All images were taken using an excitation wavelength of 760 nm and the excitation power was manually adjusted depending on the imaging depth. Images from several depths and often more than one field of view were taken from each lesion. Each FLIM image of 128 × 128 pixels was acquired over 25.5 s. The pixel size was between 1.38 µm and 3.28 µm, depending on the size of the scanned region. The instrument response function (IRF) was recorded each day in the blue spectral channel using the second harmonic generation (SHG) signal obtained from urea crystals (>98% Urea, Sigma-Aldrich®,USA).
5. Data analysis
5.1. Regions of interest
Regions of interest (ROIs) corresponding to each cell in every image were manually defined with the help of software written in Labview 7.1 (National Instruments®, TX, USA), see Figs. 1a and b ). The data were then batch processed to calculate the spectroscopic parameters for every ROI using an algorithm written in Matlab® (R2010b, The Mathworks Inc., USA).
Fig. 1.

a) example autofluorescence image obtained from a dysplastic nevus ex vivo. Scale bar 50 µm. b) shows the same data as a) but with the manually defined cellular regions of interest overlaid in green. c-top) shows the spatially integrated fluorescence decay profile obtained in the blue channel from a defined cellular region of interest (black dots) together with the instrument response function (blue) and double exponential fit to the data (green). c-bottom) shows the weighted residuals of the fit to the experimental data.
5.2. Fluorescence lifetime model
Photons within each ROI were first summed at each time point. The double-exponential decay model (Eq. (1) was then fitted to the fluorescence decay from each ROI (i.e. each cell) in each channel; see the example shown in Fig. 1c:
| (1) |
Hereis the total number of photons, is the pulse repetition period, (0<<1) is the fraction of photons originating from the first () decay component ([15], page 192) and = (1 −) is the proportion from the second decay component. The function is the normalized exponential decay whose integral over the measurement interval Tp equals 1 [16]. We note that the factor in Eq. (1) allows proper treatment of incomplete exponential decays [17].
Due to the Poisson nature of the photon shot noise, the fitting error function was chosen in the following form (see also [15], page 119):
| (2) |
where Nbins is the number of TCSPC time bins (Nbins = 256 for our system), is the discrete time at the center of each time bin, and Imeasured(tk) is the number of photons in the k-th time bin (ROI sum). Equation (2) is the model-weighted form of that avoids potential divisions by zero.
The predicted fluorescence decay Ipredicted(tk) in Eq. (2) can be calculated by convolving the fluorescence decay model (1) with the measured IRF ([15], page 103). In order to achieve this, we also took two further steps to ensure the comparability of the measured data to the predicted signal.
In the first step, the IRF was pre-processed by subtracting the horizontal long-time asymptote or ‘pedestal’ and normalized to have unit area. It is believed that the main contributing factors to this pedestal were a signal-independent contribution from background noise (stray light, electronic noise and cosmic rays) and a signal-dependent contribution from after-pulsing (caused by a delayed response of the PMT to a small fraction of preceding laser pulses) ([18], page 233). We compensate for these effects by calculating an offset for our decay model, which is described in more detail below.
Although the daily measurement of the IRF using SHG is only recorded in the blue channel, the IRFs of the two detection channels were sufficiently similar for us to apply the same IRF to the green channel. This was verified through separate measurements of each channel. However, it was necessary to include a temporal shift of the IRF for both channels in order to achieve a reasonable fit. This shift was determined in each channel by fitting all of the experimental data with a specific temporal shift and calculating the average across all of the ROIs. This was then repeated over a range of shift values and the shift providing the minimum (global) average was selected and applied to the IRF. The model decay Imod(tk) was then convolved with the corrected IRF ([15], page 103) and we therefore model the function to which the experimental data are fitted, Ipredicted(t), as
| (3) |
where H is the height of signal’s pedestal (defined as “per bin” value). We chose to define H as
| (4) |
where r is the after-pulsing coefficient defined as the ratio of the sum of after-pulsing counts to the total intensity of the (background-corrected) signal, pB is the power of the background noise (with dimensions counts/bin/pixel/second), texp is the exposure time used for data acquisition and F is the total number of photons in the decay. The values of pB and r used during fitting were determined experimentally in each channel from the measured IRF and corresponding dark images (acquired with the excitation power set to zero). In order to correctly include the tail of the decay from earlier excitation pulses, the IRF convolution in (3) was applied to the model signal composed of two consecutive identical model decays representing the sample’s response to all preceding laser pulses and the current pulse.
The fitting algorithm used the Matlab “lsqnonlin” function to find the four fitting parameters, where the fitting error χ2 is described by Eq. (2) and the fitting model is described by Eqs. (3) and (4). The parameters for the fluorescence intensity and lifetimes were defined for each ROI in each spectral channel. Individual decays with fewer than 900 photons per ROI per channel were excluded from the analysis. Mean fluorescence lifetimes were calculated from the fitted parameters using . The relative fluorescence signal in the blue (b) and green (g) spectral channels (RF) was calculated for each region of interest (ROI) using the total emitted fluorescence (F) in each channel, i.e.
| (5) |
We found that these parameters were useful in comparing the difference in emission spectrum between different populations of cells (Fig. 5 below).
Fig. 5.
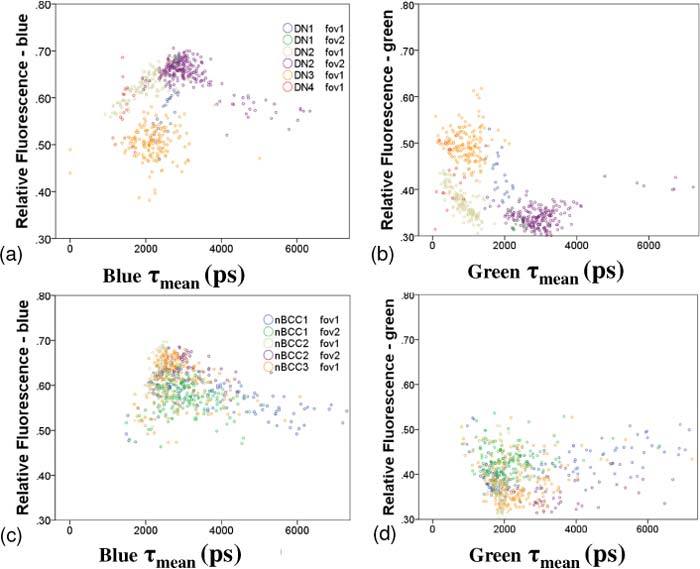
a and b) relative fluorescence emission intensity plotted against mean fluorescence lifetime in each channel respectively for the DN specimens. Different colors are used to represent each patient and each FOV. c and d) shows the same as a and b) but for the nBCC data.
5.3. Statistics
Statistical analysis of the results was performed using SPSS 18 (SPSS Inc. IL,USA).
6. Results
Excised lesions from 4 males and 3 females with a mean age of 56 (range 35 – 78 years) were included in our study. Lesions were excised from the scalp, face, chest, thigh and lower leg. From the resulting 49 FLIM images, 1180 ROIs were defined manually (614 nBCCs, 566 DN). The median (25th, 75th percentile) total photons collected per ROI were 8775 (5264, 17130) in the blue channel and 6061 (3742, 10157) in the green channel.
6.1. Fluorescence intensity imaging
Figure 2 illustrates typical image sets taken at different depths from DN and nBCCs. The images taken from the DN demonstrate normal epidermal architecture. The acellular stratum corneum, the most superficial layer of the epidermis, fluoresces brightly due to the high concentration of keratin present. Keratinocytes become clearly visible from 40 µm and cells reduce in size and increase their nuclear to cytoplasmic ratio with depth. Images taken from a depth of 60 µm show brightly fluorescent cells in the green channel, which we attribute to cells containing melanin (either melanocytes or keratinocytes) present in the basal layer of the epidermis.
Fig. 2.
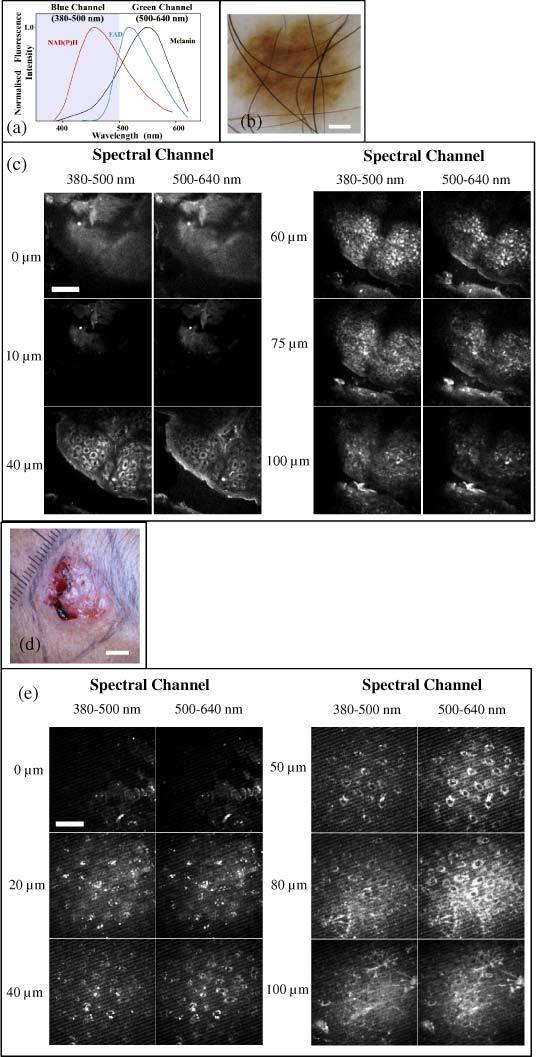
a) Schematic of the emission spectra from intracellular fluorophores. Adapted from Palero et al. [4]. b) dermatoscopic image of a dysplastic nevus (scale bar 1 mm). c) corresponding depth resolved fluorescence intensity image stacks for the two spectral channels (scale bar 50 µm). d) macroscopic image of a nodular basal cell carcinoma (scale bar 5 mm). e) corresponding depth resolved fluorescence intensity image stacks (scale bar 50 µm). We note that the feint striping artifact in the deeper images in e) is due to unavoidable stray light from the computer monitor.
Although the same cells are slightly brighter in the blue channel, the contrast is lower. This is consistent with measurements of melanin’s emission spectrum, which peaks with a broad maximum around ~550-620 nm [11,14]. Between 75 and 100 µm the reduced cellularity denotes the presence of the dermal layer in areas of the image.
The normal architecture is not present in the images taken from the ulcerated nBCC (Fig. 2 d and e). Monomorphic cells can be seen throughout all layers of the skin and there is no reduction in size and no increase in nuclear to cytoplasmic ratio with depth. Fluorescence from the collagen and elastin fibers can also be seen in the deepest image at 100 µm, with potential contributions from SHG in the blue channel
6.2. Analysis of fluorescence lifetimes
A double exponential lifetime was fitted to each ROI (cell) for each spectral channel for all images containing cells. Figure 3 shows normalized histograms for τ1 (short lifetime component), τ2 (long lifetime component) and the mean decay time τmean for each channel by diagnosis.
Fig. 3.
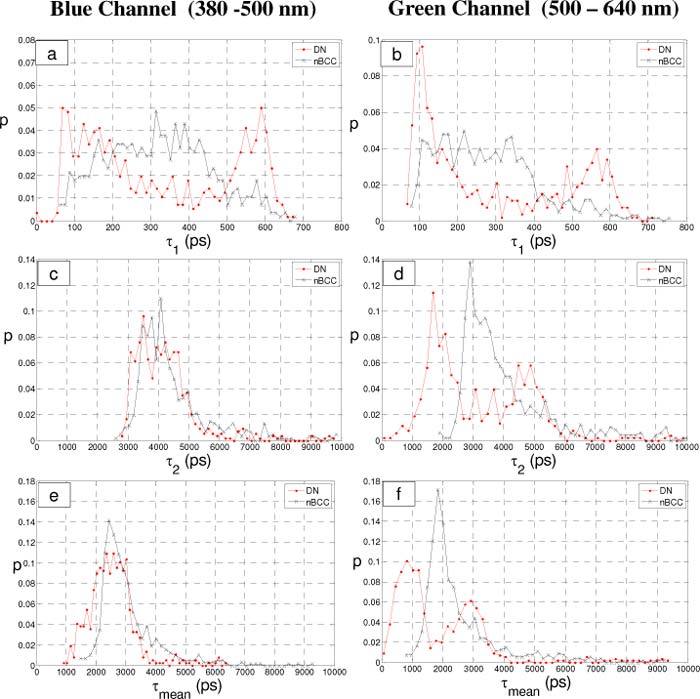
Histograms of the fluorescence decay parameters calculated on a per-cell basis for the dysplastic nevi (DN) and nodular Basal Cell Carcinoma (nBCC) specimens. a and b) histograms of τ1, c and d) histograms of τ2 and e and f) histograms of τmean. a,c,e) fluorescence lifetimes from the blue channel (left column) and b,d,f) fluorescence lifetimes from the green channel (right column).
The τ1 histograms (Figs. 3 a and b) show similar features in both channels for each diagnosis. The lifetimes from nBCC cells predominantly fall below 500 ps for both channels. The lifetimes from DN have a bimodal distribution with peaks centered around 100-150 ps and 575-600 ps. The peak with the shorter lifetime dominates in the green channel.
The histograms for the longer lifetime component, τ2, (Figs. 3 c and d) show similar distributions between the diagnostic groups in the blue channel. In the green channel however, the distribution from nBCCs cells are skewed towards longer lifetimes with a peak near 3000 ps and the DN lifetimes demonstrate a bimodal distribution with a larger peak near 1750 ps.
The histograms of mean decay time (Figs. 3 e and f) show features similar to those described above for τ2. This reflects the dominance of the longer lifetime component in the collected fluorescence (lower f1, see also Table 1 ). Since the mean decay time is not normally distributed, we have calculated the 25th, median and 75th percentile for each calculated parameter in each channel and this is shown in Table 1.
Table 1. Summary of the 25th, median and 75th percentile for all of the spectroscopic parameters for each spectral channel (blue or green) for each cell. Blue detection channel <500 nm and green detection channel >500 nm.
| Index | Channel | Diagnosis |
Percentile |
||
|---|---|---|---|---|---|
| 25 | 50 | 75 | |||
| τmean (ps) | Blue | nBCC | 2459 | 2786 | 3350 |
| DN | 2072 | 2516 | 2911 | ||
| Green | nBCC | 1783 | 2085 | 2930 | |
| DN | 810 | 1334 | 2807 | ||
| f1 | Blue | nBCC | 0.31 | 0.34 | 0.37 |
| DN | 0.37 | 0.40 | 0.44 | ||
| Green | nBCC | 0.38 | 0.42 | 0.47 | |
| DN | 0.42 | 0.46 | 0.59 | ||
| τ1 (ps) | blue | nBCC | 215 | 318 | 414 |
| DN | 147 | 279 | 534 | ||
| Green | nBCC | 173 | 263 | 354 | |
| DN | 116 | 208 | 494 | ||
| τ2 (ps) | Blue | nBCC | 3692 | 4120 | 4829 |
| DN | 3491 | 4032 | 4577 | ||
| Green | nBCC | 3044 | 3594 | 4705 | |
| DN | 1843 | 2945 | 4669 | ||
| RF | — | nBCC | 0.57 | 0.60 | 0.64 |
| DN | 0.53 | 0.62 | 0.66 | ||
The null hypotheses of “there is no difference in the median mean lifetimes between DN and nBCCs” and “there is no difference in the distribution of the mean lifetimes between DN and nBCCs” were tested for each spectral channel using a Median Test and a Mann-Whitney U test respectively. The null hypothesis was rejected in each case with p<0.01.
To explore the trend in lifetimes further, the data from the green channel (plotted in Figs. 3 b and d) can be represented as scatter plots showing τi against its fractional component fi (Figs. 4 a and b ). Figure 4a demonstrates that the shorter lifetime peak at 100-150 ps seen in Fig. 3b for DN has a large range in fractional component f1. In contrast, the longer lifetime peak between 575 and 600 ps arise from cells with similar fractional components. This same feature can also be seen comparing Fig. 3d with Fig. 4b for τ2 versus f2. The scatter plot of τ1 versus τ2 (Fig. 4c) confirms that the cells forming the short lifetime peak in the histogram of τ1 also form the short lifetime peak in the histogram of τ2. This suggests that there are two populations of cells in DN: one with consistent τ1, τ2 and f1, and the other with consistent τ1 and τ2 but with a variable f1. We tentatively attribute these two populations to melanin free keratinocytes and to melanin containing keratinocytes and melanocytes, see discussion section below. This feature was not seen for the cells from nBCCs.
Fig. 4.
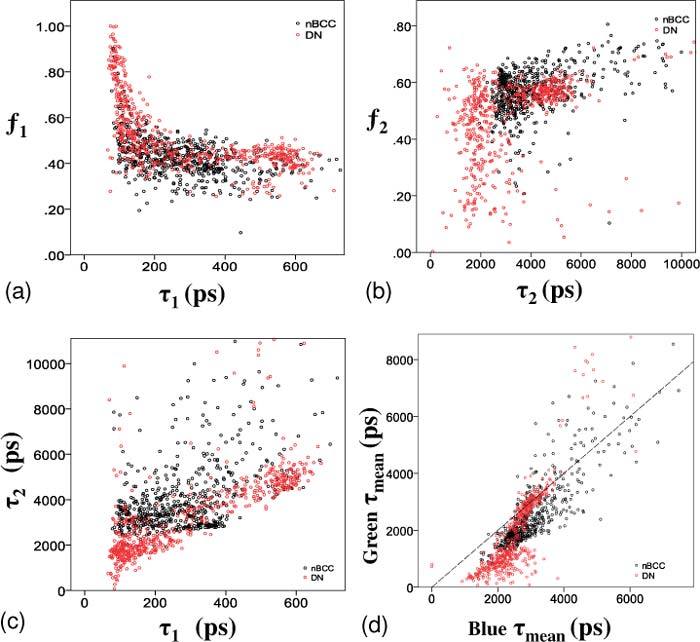
a) shows f1 correlated against τ1 and b) shows f2 correlated against τ2. c) shows τ1 correlated against τ2. a-c) show data obtained in the green channel. d) shows τmean from the blue and green spectral channels plotted against each other. This plot demonstrates that the distribution of mean lifetimes from cells for each diagnostic group are partially separated, with a tendency for shorter lifetimes in both channels for DN cells. The dashed line is x = y.
Figure 4(d) and Table 1 show that different values of τmean have been calculated for the blue and green channels. This reflects the additional spectroscopic information obtained by collecting the fluorescence signal into two channels with each channel dominated by fluorescence from different fluoropohores. The positive correlation between τmean in the two spectral channels is expected because the broad emission spectra of some tissue fluorophores emit into both detection channels.
To assess the intra- and inter-patient variability, the relative fluorescence in each channel was plotted against τmean for DN, with the color of each point representing each FOV imaged, see Figs. 5a and b. These plots show clustering of points derived from different FOV, both between different patients and between FOV within the same lesion in the same patient. The inter-FOV variability for patient DN2 is particularly evident (tan and purple points in Figs. 5a and b). This clustering was not prominent for nBCCs (see Figs. 5c and d). Our sample size was too small to assess or quantify the nature of the inter- and intra-patient variability of the fluorescence parameters.
7. Discussion
Methods of analysis of multiphoton excited TCSPC lifetime data of skin used in the literature range from using pixel sampling [5] to calculating a lifetime histogram of the entire image [7]. Such methods have potential drawbacks. Manually sampled pixels are subject to selection bias and obtaining sufficient numbers of photons in a single pixel for an accurate lifetime fit using a double exponential decay models is difficult. To address this, photon counts can be increased by binning (or smoothing) the image. However, this decreases the spatial information, and with the selected pixel potentially containing data from outside the targeted tissue component. Fitting regions larger than a single cell decreases the sensitivity for detecting intercellular changes, especially in tissue with heterogenous cell populations.
We believe that our method of binning photons into ROI corresponding to cells increases the number of photons available for accurately fitting double exponential lifetime decays whilst maintaining the ability to detect heterogenous cell populations within images. After correcting our TCSPC data for acquisition artifacts using the fitting method described in section 5, sufficient photons were available to fit double exponential lifetimes to cells decays with a mean ± s.d. reduced χ2 of 1.27 ± 0.3 in the blue channel and 1.13 ± 0.2 in the green channel.
Our results show similar morphological features to those described previously from multi-photon fluorescence intensity imaging of cutaneous malignancies [19,20]. A qualitative assessment of the morphological differences was also made between images taken from the two diagnostic groups. Although morphological differences could be seen between the two diagnostic categories (described above), a preliminary automated analysis of morphological features using the manually selected ROI boundaries was unable to highlight these differences (data not shown).
We have shown that, despite the limited sample size in this study, statistically significant differences were established between the medians and distributions of mean lifetimes between the two diagnostic groups examined. From the histograms, it can be seen that this difference is more pronounced in the green channel. Whilst the differences seen between the spectral channels provide some information as to the underlying fluorophore mix in each diagnostic category, the number of channels is insufficient to definitely attribute our findings to individual fluorophores. An exception to this is the contribution of melanin, which can be determined on the basis of its very short lifetime component [14,21] and its dominance in the green channel, see Fig. 3(b).
Recent work has shown that it is possible to distinguish malignant melanoma from benign melanocytic lesions such as naevi through changes in the fluorescence emission spectrum of melanin near to 600 nm [22,23], which is attributed to differences in the relative abundances of eumelanin and pheomelanin in these tissues. The 2-channel instrument reported here would therefore not be sensitive to these changes, as all fluorescence over 500 nm is collected into the ‘green’ channel (500-640 nm). However, future work employing an instrument with more than two spectral channels would be able to investigate this point.
The fluorescence lifetimes of cells within nBCCs overlap with the distribution observed for DN but tend to have a longer fluorescence lifetime. Since the nBCCs were not pigmented, the observed fluorescence is expected to be predominantly due to NAD(P)H and the median τ1 exponential lifetime components observed (median values: blue τ1 = 318 ps, green τ1 = 263 ps) are in reasonable agreement with measurements of NAD(P)H by other groups [10,24]. However, the measured τ2 component (median values: blue τ2 = 4120 ps, green τ2 = 3594 ps) is almost a factor of two longer than that observed in the cited previous work, which may be attributed to many differences including species, tissue type, decay fitting procedure, presence of additional fluorescent species or in vivo versus ex vivo measurement.
One of the most striking results seen is the recurrence of two distinct populations of cells seen in DN, as demonstrated by the bimodal peaks seen in many of the histograms in Fig. 3. Biologically a DN would consist of more than one cell population—keratinocytes and melanocytes. However, we believe the two populations seen in the histograms are more likely to represent melanin containing and non-melanin containing cells (mostly keratinocytes), since a melanocyte and a melanin containing keratinocyte would be very difficult to distinguish spectroscopically. Another interesting finding, shown most clearly in Fig. 4, are numerous DN cells containing a fluorophore with a similar short lifetime component but having a large spread in its fractional contribution. We speculate that this is due to the varying amounts of melanin found within the cells from DN.
We observe inter- and intra-patient variability for the fluorescence lifetime of cells as shown in Fig. 5. Part of this variability arises from the shot noise on the acquired decay data. To estimate the effect of shot noise, we simulated 300 double exponential decays using the measured IRF and the random Poisson noise generation function in Matlab (poissrnd). The parameters for the simulated decay were chosen to represent a typical skin cell and were f1 = 0.4, τ1 = 300 ps and τ2 = 3800 ps. Each of the simulated decay curves was fitted using the same code that was used to analyze the experimental data, and the resulting standard deviations were σf1 = 0.011, στ1 = 13 ps and στ2 = 123 ps. The corresponding error for the mean lifetime was 60 ps. It can be seen from Fig. 5 that the variability between cells from the same FOV are much larger than the estimated error due to shot noise alone. We attribute the variability seen in lifetimes of DN to the random sampling of typically heterogenous DN architecture by our acquisition protocol. There is a greater heterogeneity in fluorescence signature for nBCCs than for DN (Fig. 5 c and d) and this makes it harder to assess whether there is significant variation between patients or FOV.
As NADH is intimately involved in cellular metabolism, the changes in tissue oxygenation and supply of metabolites following surgical excision of skin may cause significant changes to the NAD(P)H fluorescence over the timescale of our experiments. These changes could include an increase in the NAD(P)H fluorescence intensity (measured to be up to 70% in mouse skin) and shifts in the fluorescence emission spectrum of ~5 nm [9]. Studies of the change in NAD(P)H fluorescence intensity following resection in other tissue types include breast tissue, where the fluorescence intensity decreased with time over the first 3–4 hours of tissue removal followed by stabilization of the signal [25], and colon, where the NAD(P)H signal decayed with a half-life of 118 minutes [26].
The fluorescence decay profile can also change with metabolic state and decreases in the mean fluorescence lifetime of NAD(P)H upon hypoxia of 18% have been observed in isolated hippocampal slices ex vivo [24]. In a recent study on multiphoton excited autofluorescence of skin, the same mean fluorescence lifetime (~1000 ps) was obtained from in vivo measurements as from ex vivo measurements taken on the same day but an increase in mean fluorescence lifetime from ~1000 to ~2000 ns was observed for specimens stored in culture media at room temperature over a period of three days post excision.
Therefore, care must be exercised when interpreting the results of our study. While the rate of metabolic changes in human skin will depend on a number of parameters, including e.g. temperature and sample species, it is reasonable to assume that there will still be a significant contribution from NAD(P)H in autofluorescence data acquired on the timescale of 0-2.5 hours after excision as presented here. We would expect any changes in the autofluorescence associated with the metabolic state of the tissue to be most evident in the blue spectral detection channel, which is dominated by NAD(P)H fluorescence. The green channel should be less affected as the fluorescence properties of melanin would not be expected to change following excision. Thus the NAD(P)H dominated curves in Figs. 3 (a, c and e) and the horizontal position of points in Fig. 4(d) may exhibit a broader spread than would be observed in vivo due to the range of observation time points employed in our ex vivo study. In addition, some of the inter-patient variability shown in Figs. 5(a and c) may also be due to this effect. However, we believe that the key differences observed between nBCC’s and DN remain irrespective of potential changes in NAD(P)H fluorescence with metabolic status, due to the very different spectroscopic properties of melanin and NAD(P)H.
8. Conclusions
We have presented fluorescence intensity and lifetime images obtained from two different types of skin lesion ex vivo. The data have been analyzed using an image segmentation approach of binning pixels from cellular ROI and fitting to a double exponential fluorescence decay model. Dividing the image into ROIs corresponding to individual cells is biologically logical, straightforward, relative robust and facilitates automation of the analysis thereafter. To probe the spectral-lifetime contrast between different lesion types we have used two spectral detection channels.
We have shown that, by using this methodology, changes in the fluorescence emission spectrum and fluorescence lifetime from freshly excised tissue could be observed between DN and nBCCs. Using our cellular ROI approach, we were also able to show that there are two spectroscopically different populations of cells present in DN, which we tentatively attribute to melanin and non-melanin containing cells. Within the population of melanin containing cells, there is a large variation in the relative contribution of melanin to the total fluorescence signal. Although the comparison of nBCC to DN is not clinically diagnostically useful, we believe that the data shown here demonstrates that multiphoton FLIM has significant potential to diagnose and delineate common skin tumors.
Work has already begun implementing more spectral detection channels and collecting data in vivo. We hope this will be able to quantify the effects of inter and intra-patient variability that could not be addressed by the present study and explain the longer τ2 measured for NAD(P)H compared to previous literature. As more data are collected, the next challenge is to develop the pattern recognition method(s) capable of using all three groups of cell-based quantifiers provided by FLIM (relative fluorescence intensity, lifetime and morphology), to discriminate between clinically more useful diagnostic groups and/or degrees of dysplasia.
Acknowledgments
The authors gratefully acknowledge funding from the European Commission (SKINSPECTION, FP7-HEALTH-2007-A, 201577). The assistance of the Human Biomaterials Resource Centre of Imperial College Healthcare NHS Trust in obtaining tissue for this study is gratefully acknowledged. PF acknowledges a Royal Society Wolfson Research Merit Award. The authors would like to thank Hans Georg Breunig, Marcel Höfer and Rainer Bückle of JenLab for their expert technical assistance.
References and links
- 1.König K., “Clinical multiphoton tomography,” J Biophotonics 1(1), 13–23 (2008). 10.1002/jbio.200710022 [DOI] [PubMed] [Google Scholar]
- 2.Masters B. R., So P. T., Gratton E., “Multiphoton excitation fluorescence microscopy and spectroscopy of in vivo human skin,” Biophys. J. 72(6), 2405–2412 (1997). 10.1016/S0006-3495(97)78886-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.König K., Raphael A. P., Lin L., Grice J. E., Soyer H. P., Breunig H. G., Roberts M. S., Prow T. W., “Applications of multiphoton tomographs and femtosecond laser nanoprocessing microscopes in drug delivery research,” Adv. Drug Deliv. Rev. 63(4-5), 388–404 (2011). 10.1016/j.addr.2011.03.002 [DOI] [PubMed] [Google Scholar]
- 4.Palero J. A., de Bruijn H. S., van der Ploeg van den Heuvel A., Sterenborg H. J., Gerritsen H. C., “Spectrally resolved multiphoton imaging of in vivo and excised mouse skin tissues,” Biophys. J. 93(3), 992–1007 (2007). 10.1529/biophysj.106.099457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dimitrow E., Riemann I., Ehlers A., Koehler M. J., Norgauer J., Elsner P., König K., Kaatz M., “Spectral fluorescence lifetime detection and selective melanin imaging by multiphoton laser tomography for melanoma diagnosis,” Exp. Dermatol. 18(6), 509–515 (2009). 10.1111/j.1600-0625.2008.00815.x [DOI] [PubMed] [Google Scholar]
- 6.Sanchez W. Y., Prow T. W., Sanchez W. H., Grice J. E., Roberts M. S., “Analysis of the metabolic deterioration of ex vivo skin from ischemic necrosis through the imaging of intracellular NAD(P)H by multiphoton tomography and fluorescence lifetime imaging microscopy,” J. Biomed. Opt. 15(4), 046008 (2010). 10.1117/1.3466580 [DOI] [PubMed] [Google Scholar]
- 7.Cicchi R., Sestini S., De Giorgi V., Massi D., Lotti T., Pavone F. S., “Nonlinear laser imaging of skin lesions,” J Biophotonics 1(1), 62–73 (2008). 10.1002/jbio.200710003 [DOI] [PubMed] [Google Scholar]
- 8.Benati E., Bellini V., Borsari S., Dunsby C., Ferrari C., French P., Guanti M., Guardoli D., Koenig K., Pellacani G., Ponti G., Schianchi S., Talbot C., Seidenari S., “Quantitative evaluation of healthy epidermis by means of multiphoton microscopy and fluorescence lifetime imaging microscopy,” Skin Res. Technol. 17(3), 295–303 (2011). 10.1111/j.1600-0846.2011.00496.x [DOI] [PubMed] [Google Scholar]
- 9.Palero J. A., Bader A. N., de Bruijn H. S., van der Ploeg van den Heuvel A., Sterenborg H. J. C. M., Gerritsen H. C., “In vivo monitoring of protein-bound and free NADH during ischemia by nonlinear spectral imaging microscopy,” Biomed. Opt. Express 2(5), 1030–1039 (2011). 10.1364/BOE.2.001030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Skala M. C., Riching K. M., Bird D. K., Gendron-Fitzpatrick A., Eickhoff J., Eliceiri K. W., Keely P. J., Ramanujam N., “In vivo multiphoton fluorescence lifetime imaging of protein-bound and free nicotinamide adenine dinucleotide in normal and precancerous epithelia,” J. Biomed. Opt. 12(2), 024014 (2007). 10.1117/1.2717503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leupold D., Scholz M., Stankovic G., Reda J., Buder S., Eichhorn R., Wessler G., Stücker M., Hoffmann K., Bauer J., Garbe C., “The stepwise two-photon excited melanin fluorescence is a unique diagnostic tool for the detection of malignant transformation in melanocytes,” Pigment Cell Melanoma Res 24(3), 438–445 (2011). 10.1111/j.1755-148X.2011.00853.x [DOI] [PubMed] [Google Scholar]
- 12.Koenig K., Riemann I., “High-resolution multiphoton tomography of human skin with subcellular spatial resolution and picosecond time resolution,” J. Biomed. Opt. 8(3), 432–439 (2003). 10.1117/1.1577349 [DOI] [PubMed] [Google Scholar]
- 13.Koenig K., Schneckenburger H., “Laser-induced autofluorescence for medical diagnosis,” J. Fluoresc. 4(1), 17–40 (1994). 10.1007/BF01876650 [DOI] [PubMed] [Google Scholar]
- 14.Teuchner K., Freyer W., Leupold D., Volkmer A., Birch D. J., Altmeyer P., Stücker M., Hoffmann K., “Femtosecond two-photon excited fluorescence of melanin,” Photochem. Photobiol. 70(2), 146–151 (1999). [PubMed] [Google Scholar]
- 15.J. Lakowicz, Principles of Fluorescence Spectroscopy (Kluwer Academic/Plenum, New York, 1999). [Google Scholar]
- 16.Leray A., Spriet C., Trinel D., Blossey R., Usson Y., Héliot L., “Quantitative comparison of polar approach versus fitting method in time domain FLIM image analysis,” Cytometry A 79A(2), 149–158 (2011). 10.1002/cyto.a.20996 [DOI] [PubMed] [Google Scholar]
- 17.Barber P. R., Ameer-Beg S. M., Gilbey J. D., Edens R. J., Ezike I., Vojnovic B., “Global and pixel kinetic data analysis for FRET detection by multi-photon time-domain FLIM,” Proc. SPIE 5700, 171–181 (2005). 10.1117/12.590510 [DOI] [Google Scholar]
- 18.W. Becker, Advanced Time-Correlated Single Photon Counting Techniques (Springer, Berlin, 2005). [Google Scholar]
- 19.Paoli J., Smedh M., Wennberg A. M., Ericson M. B., “Multiphoton laser scanning microscopy on non-melanoma skin cancer: morphologic features for future non-invasive diagnostics,” J. Invest. Dermatol. 128(5), 1248–1255 (2008). 10.1038/sj.jid.5701139 [DOI] [PubMed] [Google Scholar]
- 20.Dimitrow E., Ziemer M., Koehler M. J., Norgauer J., König K., Elsner P., Kaatz M., “Sensitivity and specificity of multiphoton laser tomography for in vivo and ex vivo diagnosis of malignant melanoma,” J. Invest. Dermatol. 129(7), 1752–1758 (2009). 10.1038/jid.2008.439 [DOI] [PubMed] [Google Scholar]
- 21.Ehlers A., Riemann I., Stark M., König K., “Multiphoton fluorescence lifetime imaging of human hair,” Microsc. Res. Tech. 70(2), 154–161 (2007). 10.1002/jemt.20395 [DOI] [PubMed] [Google Scholar]
- 22.Hoffmann K., Stücker M., Altmeyer P., Teuchner K., Leupold D., “Selective femtosecond pulse-excitation of melanin fluorescence in tissue,” J. Invest. Dermatol. 116(4), 629–630 (2001). 10.1046/j.1523-1747.2001.01288.x [DOI] [PubMed] [Google Scholar]
- 23.Eichhorn R., Wessler G., Scholz M., Leupold D., Stankovic G., Buder S., Stücker M., Hoffmann K., “Early diagnosis of melanotic melanoma based on laser-induced melanin fluorescence,” J. Biomed. Opt. 14(3), 034033 (2009). 10.1117/1.3155511 [DOI] [PubMed] [Google Scholar]
- 24.Vishwasrao H. D., Heikal A. A., Kasischke K. A., Webb W. W., “Conformational dependence of intracellular NADH on metabolic state revealed by associated fluorescence anisotropy,” J. Biol. Chem. 280(26), 25119–25126 (2005). 10.1074/jbc.M502475200 [DOI] [PubMed] [Google Scholar]
- 25.Gupta P. K., Majumder S. K., Uppal A., “Breast cancer diagnosis using N2 laser excited autofluorescence spectroscopy,” Lasers Surg. Med. 21(5), 417–422 (1997). 10.1002/(SICI)1096-9101(1997)21:5<417::AID-LSM2>3.0.CO;2-T [DOI] [PubMed] [Google Scholar]
- 26.Schomacker K. T., Frisoli J. K., Compton C. C., Flotte T. J., Richter J. M., Nishioka N. S., Deutsch T. F., “Ultraviolet laser-induced fluorescence of colonic tissue: basic biology and diagnostic potential,” Lasers Surg. Med. 12(1), 63–78 (1992). 10.1002/lsm.1900120111 [DOI] [PubMed] [Google Scholar]