

1. Introduction/Overview/History of PLD
Phosphatidic acid (PA) is a critical phospholipid constituent in eukaryotic cell membranes, that accounts for 1–4 % of the total lipid.1 This lipophilic glycerophospholipid has a phosphate head group, and as such serves not only a structural capacity in lipid bilayers, but also participates as an intermediate in lipid metabolism and as a signaling molecule. Because of the small head group, PA facilitates changes in lipid bilayer curvature that are important for membrane fusion events, such as vesicular trafficking and endocytosis.2 PA is also a precursor to other lipid signaling molecules including diacylglycerol (DAG) and lysophosphatidic acid (LPA). As a lipid second messenger, PA activates signaling proteins and acts as a node within the membrane to which signaling proteins translocate. Several signaling proteins, including Raf-13,4 and mTOR,5 directly bind PA to mediate translocation or activation, respectively. PA has been implicated in signaling cascades involving cell growth, proliferation, and survival. Aberrant PA signaling has been identified in multiple cancers,6 neurodegeneration,7 and platelet aggregation,8 which makes proteins that mediate cellular levels of PA attractive as potential therapeutic targets.
PA can be generated de novo9,10,11 by sequential enzyme-catalyzed acylations of glycerol-3-phosphate, or in response to cell signaling pathways (Figure 1). Every glycerophospholipid generated in eukaryotic membranes transitions through PA, a pathway characterized by Eugene Kennedy and his colleagues more than half a century ago.11,12 Signal generated PA is formed by enzymes that modify existing lipids. These enzymes include lysophosphatidic acid acyltransferase (LPAAT) which acylates LPA, DAG kinase which phosphorylates DAG at the sn-3 position, and phospholipase D (PLD) which hydrolyzes the headgroup of a phospholipid, generally phosphatidylcholine (PC), triggering the release of choline.
Figure 1.

PLD activity, an enzyme catalyzed hydrolysis of a phosphodiester bond, was first described in plants,13,14,15,16 and subsequently many enzymes from a range of viral, prokaryotic and eukaryotic organisms have been described as possessing PLD activity. To date, more than 4000 PLD enzymes have been entered in NCBI GenBank. The majority of these enzymes hydrolyze phosphodiester bonds within phospholipids such as PC (classified as EC 3.1.4.417), but there are other enzymes ascribed to having PLD activity that hydrolyze neutral lipids and even polynucleotide backbone. A large subset of enzymes with PLD activity share a conserved HxKxxxxDx6GSxN motif (HKD),18 or a variation thereof, that is responsible for catalytic activity. These enzymes are members of the PLD superfamily, and are proposed to follow a similar reaction mechanism. Non-HKD enzymes exhibiting PLD activity have divergent structures and catalytic mechanisms. These non-HKD enzymes are discussed here as a means of comparison. In this comprehensive review of the PLD superfamily, specific emphasis is given to the conventional mammalian isoforms, PLD1 and PLD2, and the tools with which these enzymes are studied. The merits of PLD as a potential therapeutic target are also reviewed, as are implications for modulation of PLD activity in cell signaling pathways, whole organisms, and aberrant or disease-related models.
2. Enzymes with Phospholipase D activity
Prior to sequencing technology or cloning of genes, enzymes were purified from the host organism and biochemically characterized. Enzymes with similar activities were described with similar nomenclature. Such is the case with PLD enzymes. Historically, many bacterial virulence factors that demonstrated the release of a choline headgroup were named PLDs for this function. Subsequent cloning and sequence analysis of these enzymes demonstrated that not all of these enzymes bear the conserved HxKxxxxD(x6GSxN) motif first described by Ponting and Kerr18 and Koonin.19 Therefore these enzymes named as PLDs are not classified as members of the PLD superfamily. At the same time, superfamily classification based on a conserved HKD motif characterized some enzymes as PLDs that were not previously considered as such solely based on biochemical analysis (ie. some endonucleases). The PLD superfamily classification based on the conserved HKD catalytic motif is useful since these enzymes are proposed to hydrolyze phosphodiester bonds via a similar reaction mechanism.
2.1. Non-HKD Enzymes
Enzymes lacking a conserved HKD motif are referred to here as non-HKD PLDs. These enzymes exhibit PLD-like activity and are no less physiologically relevant than members of the PLD superfamily. Detailed description of this class is not the focus of this review. However, brief mention of these enzymes is necessary to clarify their distinction in mechanism and enzymology from the PLD superfamily (Table 1).
Table 1.
NON-HKD PLDs
| SPECIES | ENZYME | ACTIVITY | FUNCTION | LOCALIZATION |
|---|---|---|---|---|
| Streptomyces chromofuscius | scPLD | PLD (transphosphatidylation w/M alcohol) | virulence factor | secreted into extracellular milieu |
| Corynebacterium | PLD | sphingomyelinase (releases C1P) | membrane remodeling | secreted |
| Sphingomyelinase D | LPC → LPA (in plasma) | vascular permeabilization | ||
| Arcanobacterium | PLD | sphingomyelinase (releases C1P) | bacterial adhesion | secreted |
| Sphingomyelinase D | LPC → LPA | escape from vacuole | ||
| host cell necrosis | ||||
| Loxosceles reclusa | lysoPLD | SM → C1P | hemolysis | venom |
| Sphingomyelinase D | LPC → LPA (in plasma) | platelet aggregation | ||
| inflammatory responses | ||||
| Mammalian | Autotaxin | LPC → LPA, cyclic LPA | production of lysolipids in blood | secreted into blood |
| Mammalian | cyp1A2 | monooxygenase → drug metabolism | hepatic | microsomal, membrane-bound |
| cyp2E1 | PLD (PC → PA) → unknown | microsomes/ER | ||
| Mammalian | GPI-PLD | GPI → IPG + PA, GPI-protein → protein + PA | signaling and membrane-associated protein release | secreted into serum |
| Mammalian | NAPE-PLD | NAPE → NAE + PA | endocannabinoid signaling | microsomal, membrane-associated |
2.1.1. Streptomyces chromofuscus PLD
Streptomyces chromofuscus secretes a 57 kDa phospholipase D, scPLD. This enzyme, first purified in the 1970’s20 and cloned in the early 1990’s,21 is the most well characterized non-HKD PLD.22 scPLD exhibits both phosphodiesterase as well as phosphatase activities,23 and is proposed to be secreted by the bacteria to scavenge for phosphate in the microenvironment.22 Biochemical mutagenesis analyses of scPLD demonstrate that this enzyme utilizes a metal-coordinated reaction mechanism similar to the purple-acid phosphatase family (PAP).23 A Fe3+ cation is essential for the one-step classic acid-base catalyzed reaction mechanism, whereas a Mn2+ cation is thought to be necessary for proper substrate binding.
scPLD is also able to perform transphosphatidylation, but less efficiently than HKD PLD enzymes (8–10 M primary alcohol is necessary for scPLD, compared to >95 % transphosphatidylation with 1–2 M alcohol for HKD PLD).24 scPLD also does not exhibit interfacial activation. Known as the surface dilution effect, HKD enzyme activity is affected (discussed in section 2.2.3), whereas scPLD activity is not dependent on the surface mole fraction of substrate within a lipid micelle or vesicle, hence substrate presentation does not impact scPLD activity.25 This is also referred to as the “hopping” versus “scooting” mode of activity (Figure 2). scPLD is dependent on whether the substrate is readily accessible, and therefore exhibits greater activity with monomer and mixed micelle than substrate present in a lipid vesicle.26
Figure 2.

scPLD is also the only PLD known to be activated by PA, most likely allosterically.25,27 Calcium can activate PLD by two mechanisms: calcium can directly bind the enzyme (Kd1 and Kd2), but is also able to bind to PA and make the lipid more rigid triggering product release from active site to allow new substrate to bind.25 The allosteric PA binding domain is predicted to be in the C-terminal domain, as proteolytically cleaved scPLD42/20 does not exhibit PA activation to the extent that uncleaved scPLD57 responds.28 This activation is believed to be elicited via an allosteric site secondary to the catalytic site because soluble PA can increase Vmax towards substrate present at an interface.
Despite the fact that scPLD is not a member of the PLD superfamily, many studies have used and some still use exogenous application of recombinant scPLD to rescue the deleterious effects of deletion of a HKD PLD. This is a legitimate approach as long as the results are clearly understood with regards to substrate-product relationships. Supplemental application of scPLD will hydrolyze a range of phospholipids generating PA and possibly perform phosphatase activities. Observation that scPLD rescues a phenotype following deletion of a HKD PLD enzyme suggests that PA may in fact be the functional consequence of that particular HKD PLD. However, this result or the possible lack of a “rescue” effect should not be over-interpreted. Recent studies of viral, prokaryotic, and eukaryotic PLD superfamily members demonstrate that the function of these enzymes stretches beyond generation of PA or classic catalytic product. New descriptions of protein-protein interactions and alternate catalytic products are only recently gaining an appreciation in the literature.
2.1.2. Other non-HKD PLD enzymes
2.1.2.1. Corynebacterium and Arcanobacterium PLD
Similar to Streptomyces chromofuscus, pathogenic Gram-positive Corynebacterium and Arcanobacterium secrete non-HKD enzymes, both with significant sequence identity, that are classically referred to as PLDs.29,30 These secreted enzymes exhibit divalent-cation dependent activities31 and function as virulence factors.32,33,34 Rather than hydrolyzing PC these enzymes exhibit a sphingomyelinase (SMase) activity and hydrolyze sphingomyelin (SM), present in lipid rafts in the outer leaflet of the host cell plasma membrane. However, rather than release phosphocholine to generate ceramide, the common product of mammalian SMase, these enzymes release choline to generate ceramide-1-phosphate.35,36 These enzymes are referred to as SMase D (EC 3.1.4.41) to denote the PLD-like choline headgroup release from SM. Corynebacterium and Arcanobacterium PLDs are unique unto themselves, with no conserved domains beyond a stretch with low homology to the substrate binding domain of glycero-3-phosphate dehydrogenase.29 In vivo these virulence factors function to trigger haemolysis and vascular permeabilization. This activity allows the bacteria, which infects via skin abrasions, to move into the host lymph nodes, where the infection localizes. Within lymph nodes the bacteria invade macrophages and replicate intracellularly. Generation of ceramide-1-phosphate has been proposed to remodel lipid rafts within the outer leaflet of plasma membrane, concentrating lipid raft-bound proteins and receptors, thereby enhancing protein-mediated bacterial adhesion to the macrophage plasma membrane.36,37 PLD deletion strains exhibit decreased intracellular release of bacteria from the membrane-bound vacuole.36 This suggests PLD might also trigger vacuole membrane disruption. Corynebacterium has also been shown to hydrolyze lysophosphatidylcholine (LPC) to generate LPA.38 LPA is a bioactive molecule that triggers a myriad of signaling cascades via G protein-coupled receptors (GPCR), and this activity may be important for eliciting the inflammatory response observed upon infection. This in combination with PLD-induced macrophage necrosis is likely the cause for lethal toxicity of the bacteria. This bacterial PLD protein, because it is a potent virulence factor for these two bacteria, is exploited as an effective component in vaccinations for Corynebacterium to prevent infection.39
2.1.2.2. Loxosceles PLD
The brown recluse spider, Loxosceles reclusa, and other species of the Loxosceles genus also express PLD-like SMases, called SMase Ds with significant sequence homology to the Corynebacterium and Arcanobacterium SMase D that catalyzes manganese-dependent acid-base hydrolysis of SM to ceramide-1-phosphate.40 These enzymes are the major component of spider venom, and are responsible for the dermonecrosis, haemolysis, dysregulated neutrophil activation, and other toxic physiological responses to a spider bite.41,42,43 In vitro characterization of recombinant SMase Ds demonstrates that these enzymes can be divided into two classifications based on biochemical activity.44 Class I enzymes, in addition to SMase activity, also efficiently hydrolyze lysolipids in a PLD-like fashion to generate LPA.45 As such, it has been proposed this enzyme be called a PLD, a less specific title that encompasses all activities characterized to date. LPA triggers signaling cascades in the host organism including platelet aggregation and inflammatory response.46 Lyso PLD activity is likely the cause for some of the lethal effects of Loxosceles venom. Class II enzymes exhibit SMase activity and exhibit decreased activity towards phospholipids. Subsequently, other spider and snake venoms have been characterized as having PLD or SMase D activity. The crystal structures of class I40 and class II47 SMase enzymes from Loxosceles reveal differences in the catalytic cleft that explain observed differences in substrate selectivity.
2.1.2.3. GPI-PLD
In humans, hydrolysis of the phospholipid head group to yield PA and free headgroup is generally attributed to classical HKD-containing PLD enzymes, the focus of this review. However, other human enzymes have been characterized as exhibiting PLD activities that generate bioactive signaling molecules, but are not related to the PLD superfamily. Glycosylphosphatidylinositol phospholipase D (GPI-PLD) specifically hydrolyzes the phosphodiester bond of glycosylphosphatidylinositol (GPI). This activity releases the second messengers inositolphosphoglycan (a non-N-acylated hexosamine coupled to inositol phosphate, IPG) and phosphatidic acid (EC 3.1.4.50).48 GPI-PLD is an 815 aa non-HKD enzyme that catalyzes hydrolysis at its N-terminal domain via a Zn2+ binding site coordinated by 5 conserved histidine residues.49 This enzyme shares distant homology to PI-PLC.50
GPI-PLD is the only mammalian GPI-hydrolyzing phospholipase cloned to date, and is expressed in nearly every tissue in the body, with significantly higher expression in the liver. Hepatocytes and insulin-stimulated pancreatic β-islet cells secrete GPI-PLD into serum,51 where the protein associates with HDL-like particles but remains catalytically inactive.52 Serum levels of GPI-PLD have been associated with increased TAG metabolism. Statin-induced decreases in TAG are thought to be due to a concomitant decrease in GPI-PLD serum levels,52 a beneficial off-target effect of these HMG-CoA reductase inhibitors. In vitro, catalytically active enzyme hydrolyzes GPI present in lipid rafts and caveoli. GPI often covalently anchors proteins to the plasma membrane, and GPI hydrolysis by GPI-PLD releases these proteins, often into the extracellular milieu.53 GPI that is not covalently adducted to protein can also be hydrolyzed, and yield the second messenger inositolphosphoglycan that is associated with GPI-linked signaling cascades such as insulin signaling. Although the precise in vivo function of GPI-PLD is unknown, aberrant serum levels of this enzyme have been linked with several diseases including acute hepatitis,54 nonalcoholic fatty liver,55 type1 diabetes.56,57 Decreased GPI-PLD serum levels are indirectly used as a biomarker for hepatic cell carcinoma (HCC), in that increased GPI-anchored proteins are evident.58 Exogenous supplementation of GPI-PLD increases immune clearance of the HCC cells.58 Modulation of GPI-PLD expression and activity has been suggested as a possible novel therapeutic modality for some of these diseases.52,55,58
2.1.2.4. NAPE-PLD
Another human enzyme that exhibits PLD-like phosphodiesterase activity is NAPE-PLD. This enzyme hydrolyzes N-acyl phosphatidylethanolamine species with variable N-acyl chains to generate N-acylethanolamine (NAE), an endocannabinoid, and PA. Although this enzyme is a phospholipid phosphodiesterase, it is not structurally related to scPLD or the PLD superfamily.59 Rather, NAPE-PLD is a metallo-enzyme and a member of the beta-lactamase family. This family of enzymes shares a common fold and utilizes a divalent zinc2+ active site to coordinate hydrolysis.60 NAPE-PLD is selective for NAPE substrate and does not hydrolyze PC or lyso-NAPE.60 This enzyme does not perform transphosphatidylation either, which is how it was originally identified more than 20 years ago as a unique PLD, distinct from the classic HKD PLD enzymes.59,61,62
Within the cell, NAPE-PLD is constitutively localized to intracellular membranes, mostly microsomal, and remains active. When in the NAPE headgroup N-acyl chain is arachidonate, the NAE generated is anandamide, an agonist of cannabinoid receptors CB1 and CB2, primarily expressed in central nervous system or immune and blood cells, respectively.63 Signaling through these receptors modulates cAMP levels, MAPK signaling, and ion channel activities. NAPE-PLD is expressed throughout the body, but exhibits elevated activity in the brain. Because of the critical signaling roles of NAPE-PLD hydrolytic products, and because the membrane-associated enzyme is constitutively active, this enzyme is regulated at the transcriptional level. Endocannabinoid signaling is implicated in nociception, learning and memory, and fertility. Therefore, it is not surprising that differences in NAPE-PLD expression are observed in select neuronal populations,64 or changes in uterine NAPE-PLD expression are observed in pre- and post-embryonic implantation.65,66 Some bacterial toxins, including lipopolysaccharide can also regulate enzyme expression by modulating histone acetylation which downregulates the NAPE-PLD promoter, thereby downregulating NAE production and shunting a peroxisome-proliferator activated receptor (PPAR)α-mediated host inflammatory response.67 However, endocannabinoid signaling is more complex than originally appreciated. The NAPE-PLD knockout mouse exhibits no phenotypic deficiencies.68 In these animals, levels of NAPE increased in the brain, while total NAE concomitantly decreased, as expected. Alternate NAE-generating mechanisms compensate in these KO animals because levels of polyunsaturated NAE (ie. anandamide) were not significantly altered.68 Endocannabinoid signaling regulates many in vivo functions and pharmacological manipulation of these pathways is being exploited therapeutically. Inhibitors of NAPE-PLD do not currently exist, but could be of significant therapeutic consequence in order to acutely modulate anandamide levels.69 CB1 antagonist, rimonabant, has been on the market in Europe since 2006 and is used as an anti-obesity70 and smoking cessation tool.71 Recent evidence demonstrates altered expression of cannabinoid receptors and NAPE-PLD in brain lesions of multiple sclerosis (MS) patients.72 Administration of exogenous cannabinoids have been shown to alleviate symptoms and exhibit neuroprotective effects in patients with MS.73,74
2.1.2.5. Cytochrome P450 1A2 and 2E1
Microsomal cytochrome P450 2E1 and 1A2 have been shown to have PLD-like activity and hydrolyze PC present in the ER to generate PA.75,76 Cytochrome P450 2E1 is present in the ER where it metabolizes small xenobiotics and is ethanol inducible.77 Cytochrome P450 1A2 is present predominantly in microsomes of hepatocytes where it metabolizes 15 % of all drugs, including caffeine and theophylline, and also bioactivates procarcinogens.78 This enzyme is linked to a predisposition to colon cancer. In vitro studies of hepatic microsomal and recombinant cyp 2E1 and 1A2 demonstrate these enzymes hydrolyze PC but not other lipids.75 They maintain no sequence or structural homology to the PLD superfamily and do not perform transphosphatidylation. Likewise, calcium and PI(4,5)P2, both known cofactor and activator of mammalian PLD enzymes, respectively, do not significantly modulate cytochrome P450 PLD activity. Unlike the classic monooxygenase activities of this class of enzymes, the cytochrome P450 PLD activity is not dependent on NADPH, and known P450 inhibitors, such as ketoconazole, have no effect on this activity.75,79 This led to the hypothesis that the PLD site must be separate from the monooxygenase catalytic site, and truncation mutant studies suggest the amino terminus might be critical for this unique activity.75 Later in vitro studies showed lysophosphatidylserine (LPS) is able to activate P450 PLD activity of recombinant cyp 1A2 and 2E1.79 Inclusion of LPS in the lipid vesicles results in significant conformational change in the alpha-helical content of the enzyme, as measured by circular dichroism. Overall, the PLD activity of these enzymes is quite low compared to monooxygenase activity, but PLD activity is increased >400 % in the presence of vesicles that contain a low mole fraction of LPS.79 Although the specific function of hepatic cytochrome P450 PLD activity is not clear, it is suggested that LPS acts as a molecular switch to drastically affect the activity of the enzyme. This is similar to other reported mechanisms in which local phospholipid environments modulate cytochrome P450s.76,80
2.1.2.6. Autotaxin
Lysophospholipase D activity has been described in human blood. Autotaxin (ATX or NPP2) was determined to be responsible for this lysoPLD activity and is the main source of LPA in human blood.81,82 ATX, a member of the nucleotide pyrophosphatase/phosphodiesterase family, is expressed as a preproenzyme and secreted into the extracellular milieu and serum via an N-terminal secretion signal. This enzyme does not include a conserved HKD motif and is not related to scPLD or the PLD superfamily. In vitro characterization of ATX demonstrates it has a range of activities, including phospholipase (to produce LPA and S1P),83,84,85 and nucleotide pyrophosphate hydrolysis. Lysophospholipids, including lysophosphatidylcholine (LPC), lysophosphatidylethanolamine (LPE), and LPS are high affinity substrates and predicted to be the physiologically-relevant target.86 ATX uses two Zn2+ ions in the active site for coordination and intermediate stabilization. However, unlike the other human non-HKD enzymes described above, ATX can perform both hydrolysis and transphosphatidylation.87 Depending on the divalent cation identity and salt concentration in the microenvironment, ATX will either hydrolyze LPC to form LPA, or transphosphatidylate LPC, similar to scPLD, and use the free hydroxyl group in the sn-2 position to generate cyclic LPA (cLPA).82 This difference in reactions is critical since the physiological function of LPA is distinct from cLPA. LPA is important in chemotactic cell migration and platelet aggregation, whereas cLPA inhibits cell proliferation, tumor cell invasion and metastasis. Three splice variants of ATX have been identified, ATXα, ATXβ, and ATXγ.88 ATXα and ATXβ both perform transphosphatidylation and generate cLPA. The transphosphatidylation activity of ATXγ has yet to be characterized, but is expressed in the brain where it is proposed to be responsible for the high concentrations of cLPA.88
The crystal structures of rat89 and mouse90 ATX were recently determined. Careful analysis of the structures in tandem with further biochemical characterization will be necessary to understand hydrolytic versus transphosphatidylation mechanisms and the role of divalent cations in serving as a switch between the two divergent reactions. Because of the stark contrast in signaling function of LPA versus cLPA it will be necessary to identify pharmacological agents that can be used to elicit one reaction over the other. ATX knockout mice exhibit severe phenotypic deficiencies and die around embryonic day 9.5–10.5.91,92 Much of this phenotypic response is thought to be due to the absence of ATX catalytic activity, since knock-in of a catalytic mutant elicits similar phenotypic deficiencies. However, analysis of ATX crystal structures shows two predicted LPA binding sites, and suggests that ATX may also serve as a lipid-protein carrier and deliver LPA directly to LPA receptors at the membrane via a hydrophobic tunnel.90 Recent studies also suggest that via a C-terminal MORFO (modulator of oligodendrocyte remodeling and focal adhesion organization) domain, ATX may be important for eliciting focal adhesions during oligodendrocyte maturation and myelination.93,94 Two groups have implicated ATX in regulating lymphocyte trafficking.95,96 Further structural and biochemical characterization of this enzyme is necessary, but due to its role in generating both LPA and cLPA, autotaxin appears to be a novel therapeutic target. A recent study has identified ATX as a potential therapeutic target for atherosclerosis.97
2.2 HKD Enzymes
In contrast to the various sequence, catalytic, and biochemical characteristics found in non-HKD PLDs, HKD enzymes share a conserved catalytic domain. While these enzymes do not share significant sequence or structural identity outside of this catalytic domain, conservation of this domain means these enzymes do share a similar structural core that hydrolyzes phosphodiester bonds with a similar reaction mechanism for a range of substrates. Historically there has been some dispute as to the classification of some or all of these HKD enzymes as members of the PLD superfamily. Differences in substrate (DNA backbone versus lipid) and function (endonuclease versus lipase) amongst HKD PLD enzymes have lead to discrepancies in definition of requirements for classification in the PLD superfamily. Here we propose that all phosphodiesterases with a conserved HKD or HKD-like motif are members of this diverse superfamily. Conservation of the HKD motif renders inclusion in PLD superfamily because, regardless of substrate identity, these enzymes share an SN2 ping-pong reaction mechanism that proceeds through a covalent phospho-protein intermediate in phosphodiester hydrolysis (see section 2.2.3). Members of the superfamily also perform transphosphatidylation in parallel with hydrolysis in the presence of alcohol versus water, respectively. Further subclassifications in the superfamily delineate differences in sequence, substrate and function, but superfamily classification based on the conserved HKD motif is a useful descriptor in characterizing the enzymological and mechanistic identity of an HKD enzyme. With this definition of the PLD superfamily described, this review will highlight members possessing a variety of functional and biochemical characteristics.98
2.2.1 Sequence
PLD enzymes have been identified in viruses, bacteria, plants, fungi and mammals and were classified based on biochemical activity. However, following cloning and sequencing of several PLD genes a common set of conserved motifs (I–IV) were observed.18 Conserved motifs II and IV comprise the duplicate catalytic sequence, HxKxxxxDx6G(G/S)xN (referred to here as HKD). In fact, there is significant homology between motifs I & II and III & IV. Based on this internal homology and the presence of 1-HKD motif enzymes in viruses and lower prokaryotic species, there is considerable evidence for a gene duplication event (Table 2), resulting in many PLD superfamily enzymes containing two putative HKD motifs19 (Figure 3). As discussed in section 2.2.3, the histidine residue of the HKD motif has been demonstrated to be the nucleophilic residue responsible initiating phosphodiesterase activity. Motif III is comprised of the highly conserved sequence of unknown function ‘IYIENQFF.’ In between the catalytic HKD motifs, and N-terminal to motif III, a putative polybasic phosphatidylinositol 4,5-bisphosphate PI(4,5)P2 binding domain has been described in higher eukaryotes. The C-terminus of all PLD superfamily members, despite the fact that it is not homologous, must be integral for catalysis, since activity decreases upon mutation in or truncation of this region.
Table 2.
Alignment of Catalytic motifs for PLD superfamily
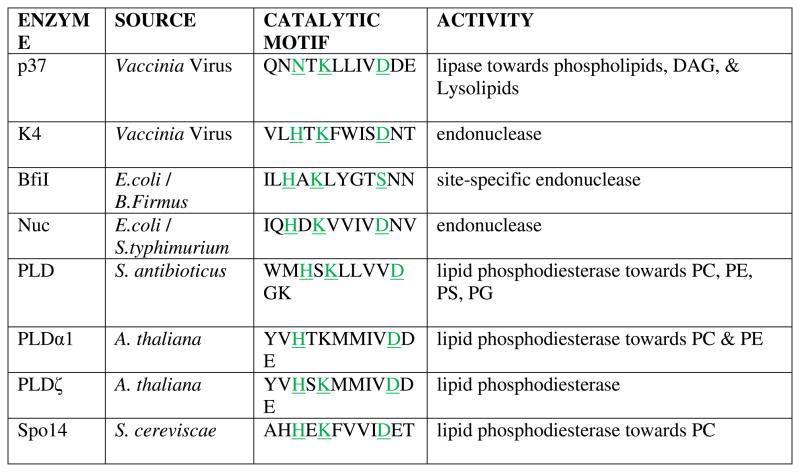
|
Figure 3.

Ponting and Kerr suggested that enzymes with these four conserved motifs were members of the PLD superfamily as described above.18 Within this superfamily, further classification was proposed based on sequence homologies. Class I comprises HKD PLDs from fungi and higher eukaryotes. Many of these enzymes have divergent N-terminal sequences that include lipid- or calcium-binding regulatory domains to allow tailored control of PLD activity in response to signaling cascades. Class II enzymes include bacterial PLDs, such as Yersinia murine toxin, YMT (see section 4.2.2) and Streptomyces sp. PMF PLD (see section 4.3) with known lipase activities. Classes III and IV include enzymes involved in lipid biosynthesis, bacterial cardiolipin synthase and phosphatidylserine synthase, respectively. The remaining classifications describe enzymes with significantly divergent functions. Class V enzymes include viral p37 and K4 (see section 3). Class VII and VIII comprise endonucleases Nuc and BfiI, respectively (see section 4.1).
2.2.2 Structure
Protein crystals of PLD superfamily members have been reported, including endonucleases and several bacterial enzymes [Nuc, BfiI, tyrosyl-DNA phosphodiesterase (tdp-1), YMT,99 cowpea,100 Streptomyces sp. PMF PLD,101 and Streptomyces antibioticus PLD, entered in PDB, (unpublished)], and tertiary crystal structures have been reported for Nuc,102 BfiI, tdp-1,103,104 Streptomyces sp. PMF PLD105 and Streptomyces antibioticus PLD. Structures for YMT and cowpea PLD were never reported. It is apparent from the available structures that a conserved fold exists for the catalytic domains of PLD superfamily members.
Nuc endonuclease from Salmonella typhimurium, a 1-HKD PLD, crystallized as a homodimer with a two-fold crystallographic axis of symmetry.102 Conserved HKD residues exist on β-strands present at the interface of the dimer and lie adjacent to one another to form the active site. Within each monomer, the β-strands connect 8 α-sheets that are sandwiched by five α-helices.
Streptomyces sp. PMF PLD was the first solved 2-HKD PLD crystal structure.105 PMF PLD consists of 35 secondary structural elements situated in repeated α-β-α-β orientation (Figure 4). In the tertiary structure, similar to the Nuc endonuclease, a common β-sandwich fold is observed, with two β-sheets connected by 8 β-strands sandwiched between 18 α-helices. This enzyme is bilobal with a pseudo two-fold axis of symmetry. Conserved HKD residues lie adjacent to one another along this axis, and at the interface exists the active site with a 30 Å aperture to allow substrate entrance. Biochemical studies with Streptomyces PLD point mutants have attributed function to specific structural elements (reviewed,106 see section 4.3 for details). Two flexible loops extend over the entrance to the active site and are thought to modulate interfacial lipid interactions and substrate specificity.107,108 The duplicate histidine and lysine residues exist on β-strands that line the active site and directly interact with substrate as it enters the active site. The aspartate residues do not directly interact with substrate, but do provide protons to the deprotonated histidine residue in the course of the reaction. The GG/GS residues line the base of the catalytic pocket and accommodate large headgroups during transphosphatidylation headgroup exchange.109
Figure 4.

In contrast to bacterial PLDs, in vitro studies of eukaryotic PLD structure and mechanism are lacking due to difficulties in expression and purification of recombinant enzyme. In the absence of a crystal structure for a higher eukaryotic PLD, much of our enzymological understanding of the PLD mechanism is based on characterization of bacterial PLDs.
2.2.3 Mechanism: hydrolysis versus transphosphatidylation
Phosphodiester hydrolysis does not commonly occur in the absence of metals.110 When it does, the mechanism must proceed through a nucleophilic attack of the substrate phosphate group, which facilitates breakage of the phosphodiester bond, and protonation via acid catalysis to enable release of the leaving group. Depending on the source of the initial nucleophile, this reaction can proceed in a single step, or in two steps, with a covalent phospho-protein intermediate. Decades of biochemical,99 structural,111 and biophysical110 research support the latter mechanism, in which a nucleophilic protein residue forms a covalent linkage to the phosphate group of the substrate (Figure 5). This covalent intermediate is subsequently destroyed via nucleophilic attack of a water molecule or alcohol, releasing the hydrolytic or transphosphatidylation product, respectively.
Figure 5.

More than four decades ago, Yang et al.112 and Stanacev and Stuhne-Sekalec et al.113 proposed that PLD catalysis proceeds through a two-step ping-pong reaction mechanism with a covalent phospho-protein intermediate. This postulation was based on analyses of cabbage PLD-induced product formation in the presence of primary alcohol. Subsequent hydrolysis and transphosphatidylation then proceed in parallel dependent on the presence water or alcohol. Early studies suggested that the sulfhydryl group of a cysteine residue may serve as the nucleophilic residue.112 This was proposed because p-chloromercuribenzoate (PCMB) treatment modified free sulfhydryl groups and disrupted catalysis, in the seven cysteine residue containing cabbage PLD enzyme.112
In the 1990’s other studies to characterize the PLD superfamily reaction mechanism attempted to identify the nucleophilic protein residue that might catalyze phosphodiesterase activity. Following Ponting & Kerr18 and Koonin’s19 observations of duplicate HxKxxxxDx6G(G/S)xN motifs in PLD superfamily members, it was suggested that the nucleophilic residue might exist in this sequence. Sung et al. proposed the conserved serine residue in the second HKD motif of yeast Spo14/PLD1 was the nucleophile.114 This conclusion was based on studies with recombinant Ser911Ala mutant. This mutation resulted in a significant drop in catalytic activity. However, subsequent studies using a 1-HKD bacterial enzyme,115 Nuc endonuclease, and a 2-HKD bacterial PLD,99 Yersinia murine toxin (YMT), demonstrated histidine residues, and not serine, are integral for catalysis. These studies used recombinant point mutants and varied pH or chemical treatments to isolate 32P-phospho-histidine intermediates. These studies proposed the reaction mechanism that is currently favored within the field, where the N-terminal histidine residue, within the HKD motif, nucleophilically attacks the phosphate group of the substrate, (step 1, Figure 5) and forms a covalent phospho-histidine intermediate. The histidine residue of the C-terminal HKD motif serves as a general acid, and donates a proton to the leaving group (step 2, Figure 5). For PLD enzymes with lipase activity, this leaving group is generally choline, and the intermediate a covalent phosphatidyl-histidine. Formation of this phospho-histidine intermediate has been proposed to be the rate limiting step, and subsequent nucleophilic attack of the hydroxyl group from either a water or a primary alcohol (steps 3 and 4, Figure 5) followed by PA or phosphatidylalcohol product release rapidly occurs in parallel.24 For most HKD enzymes, including mammalian PLDs, short chain primary alcohols are the preferred nucleophile over water (in some cases more than 1000-fold preference), allowing transphosphatidylation to occur at very low concentrations of alcohol.113 This is in contrast to the non-HKD PLD enzyme scPLD, which requires molar concentrations of alcohol to generate significant transphosphatidylation product. Some HKD enzymes, including certain bacterial, plant, and fungi PLD, are able to utilize methanol or branched alcohols in addition to other primary alcohols.24,116,117
These mechanistic conclusions were further validated when structural evidence was found to support the N-terminal histidine as the nucleophilic protein residue that forms a phospho-histidine intermediate. Histidine residues in the duplicate HKD motifs are adjacent to one another at the interface of the Salmonella typhimurium Nuc homodimer. This is also observed for the histidine residues on the duplicate HKD motifs in the crystal structure of PMF PLD. As a follow up to the first crystal structure of a 2-HKD PLD, Leiros et al. soaked PMF PLD crystals with short chain soluble PC substrate (dibutyrylphosphatidylcholine) to capture crystal structures of reaction intermediates.111 PMF PLD complexed with this substrate demonstrates that the N-terminal histidine (H170) forms a phospho-histidine intermediate (Other studies describe the C-terminal HKD histidine as the initial nucleophile and this may differ amongst PLD species118). In this structure a water molecule is positioned near the C-terminal HKD histidine (H448) and 4.02 Å from the phosphate group, an easy distance to serve as a nucleophile for completion of the hydrolytic reaction.111 Structural data lend credit to the proposed SN2 reaction mechanism, and as the catalytic cores of PLD superfamily enzymes are predicted to share a similar bilobal structure with the conserved HKD residues oriented adjacent to one another in the active site, this reaction mechanism is thought to extend to all PLD superfamily enzymes.
Finally, biophysical data also support the two-step reaction mechanism for PLD superfamily enzymes. Measurement of the changes in enthalpy and Gibbs free energy of a one-step versus a two-step mechanism demonstrates significant thermodynamic favorability for a two-step reaction proceeding through a phospho-histidine intermediate.110 In addition to the thermodynamic likelihood of the SN2 mechanism, Orth et al. used sensitive electrospray ionization mass spectrometry (ESI-MS) analysis to capture the highly unstable covalent phospho-histidine intermediate, demonstrating that it does indeed form in solution.110 Build up of covalent intermediate to levels detectable by ESI-MS was suggested to occur because the second nucleophilic reaction is the rate limiting step. This contradicts earlier studies with bacterial PLD that proposed the formation of the phospho-histidine intermediate is the rate limiting step, and hydrolysis or transphosphatidylation occur rapidly in parallel.24 Discrepancies in reaction rates require further characterization, and it is important to observe that specific activities vary depending on the biochemical reaction conditions used, including concentrations of divalent cation and substrate presentation. Such differences for in vitro activity assays are further discussed in section 2.2.5.
2.2.4 Interfacial Kinetics
Phospholipases act on substrate present in an insoluble aggregate. Many phospholipases therefore demonstrate interfacial kinetics, and do not follow classic Michaelis-Menten kinetic assumptions because the substrate is not freely diffusible in solution and is not randomly encountered dependent on soluble substrate concentration.119,120 Therefore, phospholipase activities can be described as one of two mechanisms.121 In “hopping” mode surface (Figure 2) dilution of substrate does not impact specific activity, and the interfacial component is comprised in the Km equilibrium dissociation constant. Enzymes that exhibit “hopping” mode dissociate from the interface in between hydrolytic events. In contrast, enzymes that exhibit “scooting” mode first interact with the lipid interface independent of substrate interaction, in an event described by the equilibrium dissociation constant Ks. Following interfacial binding, the enzyme laterally diffuses along the interface (in two dimensions) to encounter substrate. This is described by the equilibrium dissociation constant Km. “Scooting” enzymes exhibit processive activity, and do not dissociate from the interface between hydrolytic reactions.
The non-HKD enzyme, scPLD, does not demonstrate protein-lipid interfacial binding independent of substrate interaction.24 This enzyme functions in “hopping” mode, and directly binds substrate headgroup present at the interface.22 Following hydrolysis, scPLD falls off the substrate aggregate and the cycle recommences. scPLD activity is dependent on substrate presentation, accessibility, divalent cation concentration and cofactor binding, and positive feedback through allosteric binding of product to enhance activity24 (see section 2.1.1 for detail).
HKD enzymes often demonstrate a scooting kinetic mechanism. A lipid cofactor binds to a hydrophobic patch on the surface of the protein, at regulatory domains or within the catalytic domain, to enhance protein recruitment to the lipid interface. For many eukaryotic PLD superfamily enzymes, PI(4,5)P2 is a lipid cofactor that binds at the putative polybasic binding domain present between the catalytic HKD motifs. PI(4,5)P2 significantly enhances protein-lipid binding and decreases Ks. Once at the membrane, catalysis is controlled by multiple factors including lipid interface charge, membrane fluidity, substrate presentation or accessibility, and substrate molar fraction120,122 (or concentration of substrate present at the interfacial surface). Because of the significant impact of interfacial environment on PLD catalysis, the format of in vitro activity measurement is essential to consider (see section 2.2.5 and Figure 6).
Figure 6.

In order to study kinetic parameters for “scooting” mode enzymes, interfacial binding, Ks, must be measured separately from substrate affinity and reaction velocity. Bulk lipid binding, Ks, can be measured as described by Buser and McLaughlin.123 Following determination of Ks, Michaelis-Menten kinetic assumptions can be applied for “scooting” mode enzymes if bulk lipid concentration ⋙Ks, and interfacial binding is saturated. Molar fraction of substrate can then be varied while holding bulk lipid concentration constant by compensating for substrate molar fraction with a neutral lipid, called a neutral diluent. This format for studying kinetic parameters of an interfacial enzyme is referred to as surface dilution kinetics.122 Beyond bulk lipid composition and substrate presentation, other regulatory mechanisms control eukaryotic catalysis, including binding of calcium to the C2-domain in plant PLDs (see section 5.1), or small GTPase and PKC protein-protein interaction for mammalian PLD (see section 9.4). Elegant kinetic analyses of plant124 and mammalian PLD125 have been reported.
2.2.5 In vitro Activity Assays
Initial characterization of PLD activity monitored substrate depletion and product formation using thin layer chromatography (TLC), and co-migration of specific lipid species with purified lipid standards. Next, in vitro assays with increased precision and sensitivity have been developed that use head group release or product formation as readouts of enzyme activity. It is important to keep in mind the specific readout being measured when drawing conclusions from in vitro assays. Commercial kits are available for measuring in vitro PLD activity. However, these kits indirectly measure choline release via two subsequent enzyme-catalyzed reactions, and this method is not uniformly suitable for activity measurement. Other in vitro assays have been developed that directly measure PLD activity, and can be used directly to measure kinetic parameters.
Early studies of bacterial PLD enzymes utilized soluble small molecules with phosphodiesterase bonds to serve as substrate analogs. These small molecules have a detectable shift in light absorbance following hydrolysis, and some are capable of differentiating phosphodiester versus phosphatase activities. Soluble monomeric substrates with short acyl chains can also be used. Despite the fact that affinity for these soluble substrates is often poor, requiring higher concentrations to detect product formation, the benefit of these two options are that Michaelis-Menten kinetics can readily be performed since Ks component is omitted.
Mixed micelle and micelle assays can also be performed. Use of this format allows simple surface dilution experiments, since detergent readily compensates to adjust molar fraction of substrate (titration of increasing amounts of detergent, that will insert into mixed micelle to dilute substrate).126 In the micelle format, phospholipids and lysophospholipids are of a conical shape.127 However, many eukaryotic PLDs exhibit low activity in the absence of lipid cofactor(s) and in the presence of detergents, especially anionic detergents such as triton-x. Therefore use of pure substrate lipid micelles or mixed detergent-lipid micelles is not practical for biochemical study of eukaryotic PLD superfamily members.
Liposome assays are more complex, but closer to physiologically relevant circumstances.128,129 Higher eukaryotes demonstrate increased specific activity in the presence of lipid cofactor, PI(4,5)P2. HKD-PLD enzyme will perform processive activity if bulk lipid binding is held saturated. Separate lipid compositions can be made to vary substrate molar fraction by changing ratio of substrate to neutral diluents. Sonication is frequently used for simple liposome generation, but this makes multilamellar vesicles [(MLV), Figure 6]. These are adequate for simple measurement of activity, and comparison of different reaction conditions within an assay. However, surface concentration of substrate is not controlled for, making MLV imprecise for measurement of kinetic parameters. Extrusion is the preferred method for generating more uniform, unilamellar vesicles. The biophysical properties of the lipids in phospholipid liposomes have a significant impact on the PLD activity of scooting enzymes (Figure 6).
2.2.6 Cellular Activity Assays
It has long been appreciated that PLD enzymes perform transphosphatidylation.112,116 Stanacev and Stuhne-Sekalec demonstrated that transphosphatidylation preferentially occurs in very low concentrations of alcohol.113 This characteristic of PLD has been exploited in cellular studies of the enzyme to isolate lipid product.129 Phosphatidylalcohols (Ptd alcohol) are metabolically more stable than PA, which fluxes quickly. Historically, thin layer chromatography (TLC) has been used to visualize phosphatidylalcohols by monitoring co-migration of radioisotopically labeled lipids (on the fatty acids) with phosphatidylalcohol standards. Recently, a non-radioisotope-based cellular assay was developed.129 This assay uses ESI-MS to monitor formation of deuterated-phosphatidylbutanol following incubation of cells with low concentrations of deuterated-butanol. However, use of alcohol-treated cell preparations to identify and parse the signaling functions of PLD may have been misused (this topic is addressed extensively in sections 11 and 12). Some recent characterizations of PLD functions using RNAi and small molecule PLD inhibitors have not been able to recapitulate some of the earlier findings obtained through the use of alcohols.130,131 Small molecule inhibitors in combination with alkyne-modified lipids are powerful tools, and are being used to measure flux of specific pools of metabolic and signaling lipid.132
3. Viral PLD
Two viral proteins from the poxviridae family, p37 and K4, have been identified as PLD homologs, and are composed of four conserved domains including duplicate variants of the HKD motif.18,19 Based on sequence homology within these domains, these enzymes were originally characterized as class V members of the PLD superfamily.18 Vaccinia, arguably the most well studied member of the orthopox family which includes infamous viruses such as smallpox and cowpox, is a double-stranded DNA virus that egresses through a unique series of events.133 Unlike other DNA viruses that replicate in the nucleus and egress through budding or lysing of the host cell membrane, Vaccinia encodes for its own replication machinery. This allows the virus to replicate within cytosolic foci called “factories” prior to enveloping itself in a double membrane and being released (Figure 7).134 In the first few hours following infection, early phase genes are expressed to replicate the viral genome that is subsequently enclosed in a lipoprotein membrane, producing the intracellular mature virus (IMV). Expressed 4–6 hours post infection, late phase structural genes facilitate wrapping of the viral core in a double membrane derived from the trans-Golgi network (TGN) to produce the intracellular enveloped virus (IEV). This is trafficked to the host cell plasma membrane and released as the extracellular enveloped virus (EEV). p37 expression and lipase activity is integral to viral envelopment and egress, while recent studies have suggested a nuclease and viral DNA condensation function for the conserved but non-essential K4 protein135 (Table 3).
Figure 7.

Table 3.
BACTERIAL & VIRAL PLDs
| SPECIES | ENZYME | ACTIVITY | FUNCTION | LOCALIZATION |
|---|---|---|---|---|
| Orthopox virus | p37 | PLC, PLA, PLA2 | IMV wrapping | TGN & inner membrane wrapping of EEV |
| (Vaccinia, variola) | TAG lipase | IEV fusion & release | ||
| transphosphatidylation | ||||
| Orthopox virus | K4 | endonuclease | single strand (ss)/double strand (ds) DNA torsion release | within IMV |
| (Vaccinia, variola) | Nick-joining enzyme | |||
| Salmonella typhimurium, Escherichia coli | Nuc | nonspecific endonuclease | ssRNA | periplasm |
| breakage during DNA conjugation | ||||
| Escherichia coli | Bfil | site-specific endonuclease | Degrades dsDNA during DNA conjugation | periplasm |
| Neisseria gonorrhoeae | NgPLD | PC hydrolysis | combination of lipase & protein-protein interaction elicits bacterial invasion | host cell cytoplasm |
| transphosphatidylation | binds AKT to trigger membrane ruffling | extracellular milieu | ||
| Yersinia pestis | YMT | PLD (PC/PE lipase) | in vivo facilitates Y. pestis colonization of flea gut | bacterial cytosol |
| (formerly Pasteurella pestis) | transphosphatidylation | protects against murine plasma component | ||
| Chlamydiae | chromosomal pz PLDs | PLD | unknown | reticulate bodies |
| unknown lipase activity | lipid acquisition from LD | |||
| transphosphatidylation | ||||
| Acinetobacter baumanii | Act bau PLD | unknown | unknown function | secreted |
| enhances serum survival/host cell invasion | ||||
| Pseudomonas aeruginosa | PLDa gene | PLD (PC → PA) | increases long term infectivity/bacterial homeostasis | periplasm |
| transphosphatidylation | ||||
| Streptomyces sp | PMF PLD | PLD | unknown | periplasm |
| transphosphatidylation | secreted |
3.1 p37 lipase
F13L, the gene encoding p37 (GenBank NC_006998), encodes a 372 amino acid membrane-associated protein with duplicate variant HKD motifs (Table 2). Studies characterizing the differences in protein and phospholipid content of vaccine viral particles found a 37,000 Dalton polypeptide that comprised 5–7 % of the total EEV protein content that was absent in IMV.136 Anti-serum generated against this polypeptide was used to map the F13L gene in the Vaccinia viral genome.134 The putative initiation site of F13L is composed of a conserved ‘TAAATG’ DNA sequence, present in all except one late phase gene.137 This is consistent with evidence that p37 is expressed in the late phase to facilitate viral envelopment. Unlike the other late phase proteins, p37 does not bear an N-terminal endoplasmic reticulum (ER) localization sequence and is not glycosylated. Instead, p37 is expressed in the cytosol and post-translationally palmitoylated at two cysteine residues, positions 185 and 186, necessary for the protein to associate with the cytosolic face of the TGN and endosomal membranes.138 Disruption of ER/COPII vesicular trafficking does not perturb punctate cytosolic localization of p37.139
Viruses deficient in F13L demonstrate decreased plaque formation and are unable to generate EEV due to the inability to wrap in TGN membrane and traffic to the plasma membrane for fusion and release.140 p37 is proposed to facilitate IMV envelopment through membrane modification and protein-protein interactions with viral as well as host proteins. p37 trafficking between the TGN, plasma membrane, and subsequent recycling via the endosomal vesicles occurs via clathrin-mediated transport. p37, present on the cytosolic face of the TGN membrane, engages with viral proteins on the lipoprotein surface of the IMV to elicit double membrane wrapping (Figure 7).133,141 This results in p37 protein that is associated with the interior membrane (still in contact with the lipoprotein viral core) and the exterior cytosolic membrane of the newly-formed IEV. Cytosolic p37 is recycled via endosomal trafficking following fusion of the exterior IEV membrane with the plasma membrane upon viral egress. This mechanism requires p37 catalytic activity as well as protein-protein interactions.
Consistent with classification as a PLD, in vitro characterization of recombinantly expressed p37 demonstrates this enzyme maintains lipase activity and substrate preference for phosphatidylcholine. In vitro data demonstrate this enzyme can also hydrolyze PE, phosphatidylinositol (PI), and triacylglycerol (TAG). Unexpectedly for an enzyme classified as a PLD, p37-catalyzed hydrolysis of PC generates many products, none of which are phosphatidic acid. Instead, this enzyme exhibits PLC, PLA2, PLA1, and triacylglycerol lipase-like activity by generating diacylglycerol (DAG), LPC, and monoacylglycerol (MAG).142 Also counter to the PLD classification is the fact that recombinant p37 does not perform transphosphatidylation. Phosphatidylethanol was not detected when the enzyme was incubated with PC and ethanol. However, this in vitro data is not consistent with studies in intact cells that demonstrate PLD-like p37 lipase activity is necessary for EEV formation.143 In Vaccinia-infected HeLa cells, n-butanol, but not secondary or tertiary alcohol, inhibits viral wrapping and EEV formation, resulting in decreased plaque size. Similarly, mutation of the putative HKD variant motif (NxKxxxxD) to K314R or D319E prevents localization of p37 to the Golgi and results in decreased viral egress and plaque size.144
Discrepancies between in vitro and cellular data could be due to differences in substrate presentation or the lack of necessary conditions or activating constituents for PA generation. Alternatively, the substitution of the asparagine for the histidine residue in the putative (H)xKxxxxD motif could be responsible for the differences in lipase activity. Studies have addressed the latter by mutating the asparagine in the (H)KD motif to histidine in the p37 sequence, but this does not result in a change in the lipase activity.114,142 Likewise, mutation of the human carboxy-terminal HxKxxxxD sequence to an NxKxxxxD does not divert lipase activity, rather this simply renders the enzyme inactive. Consistent with these mutant studies and despite the fact that human PLD and p37 co-localize and facilitate Golgi vesicle formation, human PLD does not rescue viral wrapping or egress for p37-deficient virus.143 This suggests that in addition to lipase activity, p37-specific function or protein-protein interactions are required.
Cellular characterization of p37 has led to identification of two conserved protein-protein interaction domains that mediate IMV enveloping and viral egress. A tetrapeptide L domain, ‘YXXL’, is conserved among pox family p37 proteins, and is suggested to facilitate interaction with other viral proteins as well as host vesicular trafficking proteins.145 Consistent with evidence that IEV is transported via the clathrin-dependent pathway, a conserved diaromatic ‘YW’ motif was identified. In other endosomally-trafficked viruses ‘YW’ motifs facilitate protein-protein interaction with a Rab effector protein for IMV envelopment prior to trafficking.146
More notorious members of the orthopoxvirus family, including Variola virus (smallpox), remain potential threats to public health. Although eradicated for more than three decades, the possible use of smallpox virus as a biological weapon remains. There is currently no FDA-approved therapy for orthopoxvirus post exposure. Some compounds, including cidofovir, an anti-cytomegalovirus compound that inhibits DNA polymerase, have shown some efficacy in vitro but are not potent against orthopoxvirus, and have poor bioavailability with numerous adverse effects.147 Evidence that p37 function is integral to orthopox viral egress and necessary for efficient infectivity suggests this enzyme might be a good therapeutic target. Classic studies of Vaccinia and other orthopoxviruses used IMCBH (N1-Isonicotinoyl-N2-3-methyl-4-chlorobenzoylhydrazine) to block viral release and decrease plaque size.148 This compound does not disrupt IMV formation, but inhibits IMV wrapping and viral egress. IMCBH-resistant virus was identified as having a single nucleotide point mutation in F13L that conferred an amino acid change in p37 (D279T).149 This amino acid substitution is predicted to cause a conformational change in p37 that prevents IMCBH inhibition, but does not disrupt function. These studies validate p37 as a therapeutic target.
Recent small molecule screens for orthopoxvirus replication inhibitors have identified a compound that targets p37. The orally bioavailable compound, ST-246 (4-trifluoromethyl-N-(3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl)benzamide) potently inhibits replication of many orthopoxvirus species.150 The target of ST-246 has been mapped to the gene product of F13L. It is unknown whether ST-246 inhibits p37 lipase activity, but this compound does selectively disrupt p37 interaction with Rab effector proteins and thereby disrupts IMV wrapping, without perturbing p37 localization to the TGN or disrupting overall Golgi trafficking in the host cell.146 Recent studies demonstrate this compound’s utility in preventing and treating immunocompetent as well as immunodeficient host animals from orthopoxvirus, suggesting ST-246 may be an attractive prophylactic and therapeutic compound for the larger population.151 ST-246 is currently in phase II clinical trial, and the FDA has granted this compound “fast track” status for expedited review.
3.2 K4 endonuclease
The less well-characterized viral PLD family member is K4 (accession# YP_232917, gene name K4L). The Vaccinia virus K4L ORF codes for a 424 amino acid protein with two HxKxxxxD motifs, that shares 25 % sequence identity to p37.18 K4L has been described as serving a nonessential function because deletion of this gene does not affect Vaccinia virus replication or virulence.18,140 Despite initial classification in the PLD family based on sequence analysis and identification of conserved domains, the enzymatic function of this K4L gene product remained poorly understood until recently. Studies of viral extract revealed endonuclease and ligase activities that mapped to K4L.135 Subsequently, the K4L gene product was determined to be the previously-described nick-joining enzyme (NJ enzyme).152,153
The double-stranded Vaccinia virus genome is roughly 192 kilobases with continuous hairpin closures at the AT-rich termini. The covalent hairpin closures force faithful replication of the telomeres by the viral DNA polymerase to prevent base pair loss. In vitro studies of recombinantly expressed K4 protein demonstrate this enzyme can site-specifically nick single DNA strands at the apex of the terminal hairpin in an ATP and divalent cation independent manner.135 A 3′-phosphate overhang remains following endonuclease cleavage, which prevents nicked strands from serving as substrates for DNA polymerase, and precludes K4 from a role in generating initiation sites of DNA replication. Analogous to the activity of type I topoisomerases, K4 performs interstrand ligation to generate crosslinked DNA. K4 is expressed as a 50 kDa pronuclease that exhibits enhanced ligase activity upon proteolytic digestion resulting in a 44 kDa protein.152
The hydrophobic K4 protein is expressed in the late phase and localizes within the viral core. The specific role for this protein in viral replication has not been defined, due to the fact that deletion of K4L does not appear to perturb viral replication and virulence, despite the fact that endonuclease activity is lost in viral extracts of K4L deletion strains.135 This study suggests K4 protein may facilitate condensation and packaging of the supercoiled genome within the viral core, then function to alleviate torsional stress on the genome within the core during the following round of infection, gene expression, and replication. This proposed function would seem to be integral for viral replication, and therefore there may be redundancies for K4 function or host cell compensation in K4L deletion strains. The latter is more likely the case, since endonuclease activity is absent in K4L deletion viral strains, and K4L homologs are conserved in other orthopoxviruses (suggesting the function is non-redundant and somehow confers evolutionary advantage).
4. Prokaryotic PLD
Prokaryotes express PLD genes that range in function from hydrolysis of the DNA backbone, to protein-protein interactions with host signaling pathways, to the more classic lipase function. Phospholipase activity has been identified in several pathogenic bacteria. PLD enzymes are not commonly expressed among bacteria compared to other phospholipases, but bacterial PLDs have been observed in many pathogenic bacteria.154 Also, because of the ease of expressing and purifying these enzymes recombinantly, much of our structural and biochemical understanding of PLD enzymology stems from studies that utilize bacterial PLD.
4.1. Bacterial Endonucleases
4.1.1. Nuc endonuclease
Evidence that the PLD superfamily arose from a gene duplication event stems from studies of EDTA-resistant bacterial endonucleases with a single HxKxxxxD. In fact, initial characterization of the PLD superfamily was performed using Nuc, an ATP-independent, nonspecific endonuclease encoded on plasmid DNA found in Salmonella typhimurium and Escherichia coli. The crystal structure of Nuc was determined to 2.0 Å (PDB accession codes 1BYR and 1BYS, native and complexed with tungstate inhibitor, respectively), and found to contain a single HxKxxxxD motif that forms a homodimer with a crystallographic two-fold axis.102 The HKD motif within each enzyme exists on two loops held at the interface of the dimeric subunits via hydrogen bonds to form a single active site. Structural and biochemical characterization of Nuc reveals a ping-pong-like SN2 reaction mechanism that utilizes both HKD motifs within the active site. The imidazole group of one ‘HKD’ histidine residues nucleophilically attacks the phosphate atom on the substrate, breaking the phosphodiester bond within the DNA backbone and generates a covalent phospho-histidine intermediate. The histidine of the second subunit’s HKD donates a proton to the leaving group, which, in the case of an endonuclease, is the 3′ end of the DNA backbone. Hydrolysis is complete upon a water molecule nucleophilically attacking the phosphate, breaking the phospho-histidine bond, and leaving a phosphorylated 5′ terminus.115 This two-step, water-exchange reaction mechanism that proceeds through a covalent phospho-histidine intermediate is consistent with other HKD PLD enzymes, as described in section 2.2.2.
Nuc endonuclease is encoded for on the 35.4 kilobase pKM101 plasmid, a member of the broad-host range IncN plasmid classification.155 This plasmid is responsible for conjugal DNA transfer between bacterial cells via thin rigid sex pilli.156 pKM101 plasmid renders bacterial drug resistance by encoding for 15 genes that trigger spontaneous mutagenesis and error-prone DNA repair to facilitate survival.157 Nuc is expressed as a 177 amino acid (19 kDa) protein in the bacterial cytosol, but is processed to 155 amino acids (17 kDa) when the 22 amino acid signal sequence is cleaved upon secretion into the periplasmic space,158 where it is constitutively localized and never secreted into extracellular growth media. Nuc endonuclease nonspecifically hydrolyzes internal phosphodiester bonds within the backbone of single and double stranded duplex DNA and RNA (in vitro), but does not elicit exonuclease activity at terminal phosphodiester bonds. Maximal activity is observed in the presence of divalent cations, but unlike other bacterial endonucleases, Nuc remains catalytically acitve in the presence of EDTA. This unique characteristic allowed characterization of Nuc endonuclease activity in the bacterial cell background.155 Despite rigorous biochemical characterization of Nuc, its functional role remains unclear. Similar to the viral endonuclease, Nuc is nonessential for bacterial survival and does not degrade plasmid or phage DNA as it crosses the periplasmic membrane. Rather, Nuc is proposed to provide an ancillary role in DNA conjugation.
4.1.2. BfiI endonuclease
In addition to the extensive structural and biochemical characterization of Nuc, another single HKD endonuclease has been extensively studied. A homolog of E. coli helicase, BfiI, is classified as a type IIS restriction enzyme. BfiI is an EDTA-resistant endonuclease with a conserved HxKxxxxD variant (more specifically an HxKxxxxS),159,160 and based on sequence was characterized as a class VIII member of the PLD superfamily.18 However, unlike Nuc, BfiI cleaves DNA at a specific sequence. Subsequent biochemical and structural studies determined BfiI is in fact composed of two domains, an N-terminal endonuclease domain that consists of a single HKD variant, and a C-terminal DNA binding domain that specifically binds the sequence 5′-ACTGGG-3′.159 The N-terminal endonuclease forms a homodimer, similar to Nuc with 1.4 Å rms deviation from the Nuc tertiary structure; while the DNA-binding domains remain at opposite ends of the dimer and do not interact.161 Hence, BfiI is able to bind two double-stranded DNA sequences simultaneously, and the endonuclease activity, via the SN2 reaction mechanism of the conserved HKD homodimer, hydrolyzes the phosphodiester bond in a single DNA backbone 4 and 5 nucleotides downstream of the binding sequence, top then bottom strand, respectively.159 Ultimately, four phosphodiester bonds are hydrolyzed in the single active site, resulting in two double stranded DNA breaks.
BfiI catalytic activity is significantly enhanced upon occupancy of both DNA-binding domains. In the absence of bound DNA, the negatively-charged linker region between the DNA-binding domain and the N-terminal HKD domain binds in the catalytic pocket of the HKD homodimer to sterically hinder endonuclease activity. However, upon occupancy of the C-terminal DNA binding domain, the linker region takes on an extended conformation and allows DNA to access the catalytic pocket for endonuclease activity.161 Also, studies using recombinant truncation mutants of BfiI demonstrate that the N-terminal nuclease nonspecifically cleaves phosphodiester bonds in the DNA backbone in the absence of the DNA-binding domain. Unlike other restriction enzymes that dimerize upon interaction with specific substrate, BfiI is constitutively dimerized via the N-terminal HKD motif. Based on these studies, it is proposed that BfiI is the product of a gene fusion event where the gene for a nonspecific, HKD-containing endonuclease, similar to Salmonella typhimurium Nuc endonuclease, fused with a gene encoding a DNA-binding domain.161 Similar to other restriction enzymes, BfiI is thought to serve a protective function by site-specifically degrading foreign DNA that enters the bacterial cell.
4.2. Bacterial PLD as virulence factors
Phospholipases are common toxins and virulence factors for pathogenic bacteria. These enzymes facilitate bacterial infection and replication through several functions, including penetration of basal cell membranes (i.e. mucus layer or blood vessel wall), triggering engulfment of the bacterium by the host cell, or cytolysis to release intracellular bacteria from host cells such as macrophages. Phospholipase C and Phospholipase A are the most common class of bacterial phospholipases that serve as virulence factors. These enzymes are capable of destabilizing or destroying host cell membranes directly, through lipid hydrolysis or indirectly, through upregulation of host cell signaling pathways via lipid product formation.162,163,164,165 Although less common, some bacterial PLDs have also been identified as virulence factors. The localization and functions of these enzymes in eliciting virulence are divergent, and the unifying theme amongst these enzymes is the conserved HKD motif responsible for catalytic activity.
Bacterial PLDs that function as virulence factors are generally expressed by Gram-negative pathogenic bacteria that are obligately intracellular, and require plant or mammalian host cell invasion in order to replicate. These enzymes are often secreted by the bacteria into the extracellular milieu or directly injected into the host cell cytosol via one of several known secretion mechanisms. Several of these PLD genes have been proposed to be acquired by lateral gene transfer from other bacteria or host cells.166 Acquisition of these bacterial PLDs can enable immune evasion, expand potential host colonization, and can provide pathogenic advantage.
4.2.1. Neisseria gonorrhoeae PLD
Neisseria gonorrhoeae is an exclusively human bacterial pathogen that associates with, and invades cervical and urethral epithelial cells. Intracellularly this pathogen is able to replicate as well as evade the host immune response. Recently, a 2-HKD PLD was identified as a virulence factor for gonococcal Neisseria gonorrhoeae infection of human cervical epithelium.167 This enzyme was first identified as a 55 kDa protein in the growth media of N. gonorrhoeae-infected primary human cervical epithelial cells. Proteomic analysis identified this protein, NgPLD (GenBank accession number AY307929) as sharing significant homology to a hypothetical PLD enzyme in the Neisseria meningitidis genome. NgPLD sequence bears a predicted N-terminal signal sequence that is likely processed following secretion into the extracellular milieu, and two HxKxxxxD motifs. Growth medium containing secreted enzyme demonstrates classic PLD catalysis in a choline release assay, compared to PLD-null growth medium which yields no activity.
Primary cell-based studies of NgPLD demonstrate that this enzyme is specifically secreted upon infection of primary human cervical and urethral epithelial cells. NgPLD is necessary for efficient bacterial association and invasion of human cervical epithelial cells. This association and invasion is largely the result of complement-receptor type 3 (CR3)-mediated membrane ruffling and endocytosis.168 The I-domain within the host CR3 receptor binds gonorrheal porin and pilus structures to elicit macropinocytosis of the gonococcal bacteria.169,170 Host cell surface recruitment of CR3 and membrane ruffling is dependent on NgPLD activity.167 NgPLD is proposed to elicit receptor translocation and membrane ruffling through several mechanisms. NgPLD product formation is necessary for gonococcal association and invasion. NgPLD expression and activity, dispersed between cytosolic and membrane fractions of the host cell, increase upon prolonged gonococcal infection, whereas neither expression nor activity of human PLD isoforms is modulated upon infection. Also, NgPLD appears to be capable of transphosphatidylation since primary alcohol (but not secondary or tertiary) can block bacterial association and invasion in the same way knocking out the NgPLD gene does. This suggests NgPLD product formation may drive CR3 translocation, membrane curvature alteration, or cytoskeletal rearrangement to trigger membrane ruffling and engulfment of the bacteria. However, NgPLD product alone is not likely enough to render successful bacterial invasion, since addition of Streptomyces sp. PLD did not rescue the decreased invasion of NgPLD knockout strains. This suggests NgPLD may itself be capable of modulating host cell signaling pathways through protein-protein mechanisms not conserved amongst other bacterial PLD.
This is indeed the case, as more recent studies have demonstrated the ability of NgPLD to bind human Akt and compete for PI(3,4,5)P3 binding at the Akt PH domain.171 Based on data that neomycin, a small molecule that partitions into PI(4,5)P2-containing membranes and blocks phospho-headgroup access, disrupts Akt activity and bacterial association and invasion, the NgPLD-human Akt signaling pathway is suggested to be PI3K independent. This interaction is proposed to facilitate Akt translocation to PI(4,5)P2-enriched membrane ruffles where Akt is phosphorylated and activated by PDK1. This subsequently leaves activated Akt in close proximity to interact with CR3 and polymerized actin. This is intriguing evidence that bacterial PLDs function as virulence factors in both the capacity to hydrolyze and generate specific lipid products, but also can exploit host cell signaling pathways through protein-protein interactions with critical signaling proteins. NgPLD is proposed to be the phospholipase virulence factor that was originally characterized as a PC-dependent PLC,172 since a putative PLC sequence has not been identified in pathogenic Neisseria genomes.167
4.2.2. Yersinia murine toxin
Yersinia pestis, the causative agent of bubonic plague, is an enteric gram-negative bacterium that is genomically very closely related to Yersinia pseudotuberculosis. There are very few genomic differences between these two Yersinia species, but the few differences that do exist allow Y. pestis extensive adaptability to infect and thrive in the gut of an arthropod (flea) as well as infect higher eukaryotes including mice and humans.173 The Yersinia murine toxin (Ymt) is one such advantageous gene encoded for by Y. pestis. Ymt is a 61 kDa protein that is present within the bacterial cytosol, rather than secreted into the extracellular milieu. Sequence analysis reveals Ymt is a class II member of the PLD superfamily and consists of two HxKxxxxD motifs.18 In vitro characterization of this enzyme demonstrates it is capable of hydrolyzing PC, PE, and other phospholipids, and like other members of the PLD family is able to perform transphosphatidylation with primary alcohols. Biochemical studies of Ymt were among the first to demonstrate that the reaction mechanism proceeds through a phospho-histidine covalent intermediate.99
The specific function of this protein was originally unclear, but it was designated a toxin because it was found to be lethal in mice and rats.174 However, studies with recombinantly expressed Ymt demonstrate that this protein alone is not lethal, rather this protein likely acts in synergy with other Y. pestis proteins to elicit vascular collapse and subsequent cardiac failure.175,176 Despite rigorous in vitro biochemical characterization of the catalytic activity of this enzyme, the native substrate is unknown. Whole animal studies in mice and rats demonstrate that this gene product is not necessary for Yersinia virulence, rather it plays a role in transmission. Yersinia pestis colonizes the midgut of the flea, ultimately blocking the ability of the flea to feed. Continued attempts of the flea to feed dislodges bacteria from the flea gut and delivers Yersinia to the blood stream of the flea bite on the host.177 Ymt-deletion strains are unable to grow and survive in flea midgut,173,177 decreasing the ability to transmit bacterial infection to the host. Yersinia with the Ymt gene deleted, several hours after infection lose the outer membrane and form spheroblasts prior to being eliminated from the flea gut. Ymt is protective against a mouse blood plasma or digested mouse blood plasma component that the bacterium comes into contact with in the flea gut during feeding.177 Ymt, predicted to have been acquired through horizontal gene transfer, is evolutionarily advantageous to the pathogenic bacteria because it broadens the range of hosts in which Yersinia pestis is able to colonize.178
4.2.3. Other less well characterized bacterial PLDs
With a greater number of bacterial genomes being sequenced and an increased understanding of the protein sequences and motifs that confer phospholipase activity, other bacterial PLDs continue to be identified and biochemically characterized. Some of these enzymes function as virulence factors, while others appear to be integral to bacterial homeostasis and replication.
4.2.3.1 Chlamydia PLD
Chlamydia is an obligate intracellular Gram-negative bacterium that is a significant health risk in the greater population, whose genome encodes for one or more putative PLD. This intracellular bacterium goes through a biphasic life cycle (reviewed).179 Briefly, host cell-associated bacteria, called elementary bodies (EB), are endocytosed. Within this membrane-bound inclusion the EB is transformed to the reticulate body (RB) and divides via binary fission. Following replication, the RB transitions back to EB, which are released into the extracellular milieu by exocytosis or cell lysis. The genomes of all Chlamydia species encode for at least one PLD enzyme.180,181,182 Conserved PLD genes exist within a chromosomal genome that does not vary significantly between Chlamydia species (chromosomal PLD). PLD genes have also been identified in variable stretches of the genome called plasticity zones (PZ), these enzymes are referred to as pzPLD. Genes encoded in the PZ are strain-specific, and many are thought to function as virulence factors and be protective for host immune responses or engender an environment-specific advantage to the bacterium. Limited characterization has been done for PLD proteins in the Chlamydia trachomatis species. This strain expresses several putative ‘HKD’ PLD proteins- 2 chromosomal PLDs (CT284 and CT084) and five pzPLDs (CT154, CT155, CT156- truncated prior to conserved HKD, CT157, and CT158).183 Unlike the other PLDs, which are expressed in later phases of replication, chromosomal CT284 and PZ CT156 are expressed early in infection, suggesting a unique function. Studies in HeLa cells demonstrate that the enzymatic activities of the pzPLDs are necessary for inclusion formation, as n-butanol only decreases the infectious yield of Chlamydia strains that encode for pzPLD.183 Although the precise in vivo function and substrate specificity of Chlamydia PLD is unknown, it is predicted that these enzymes facilitate intrainclusion acquisition and lipolysis of lipid droplets, which are a major source of phospholipid content necessary for bacterial replication.184 Other studies have proposed pzPLDs function as nucleases, similar to Nuc endonuclease from Salmonella typhimurium.183 In the absence of complete biochemical characterization, it is unclear whether chromosomal PLDs or pzPLDs function as classic virulence factors.
4.2.3.2 Acinetobacter baumanii PLD
PLD genes have recently been identified in pathogenic Acinetobacter baumanii. This Gram-negative bacterium is a frequent cause of nosocomial infections, particularly in trauma patients, and antibiotic resistant strains have emerged with high mortality rates. A recent study characterized the genomic distinctions between Acinetobacter strains that exhibit differences in serum resistance.185 Two genes were identified that encode for putative PLD enzymes. The first, a two-HKD PLD at locus position A1S_2989, was identified and ultimately determined to encode for a 543 amino acid protein with a predicted type III secretion signal (despite the fact that this secretion mechanism has not been described in Acinetobacter). In vitro experiments demonstrate this protein facilitates serum survival and bacterial engulfment by cultured host epithelial cells. Deletion strains at locus A1S_2989 were unable to effectively invade. However, in vivo studies in a mouse model demonstrate no difference in infectivity or colonization. A second PLD was also identified at locus A1S_2891.185 Further characterization is necessary to determine whether this enzyme can compensate for deletion at locus A1S_2989 in vivo. Confirmation of PLD enzymatic activity in a well-validated assay has not as yet been reported for these enzymes.
4.2.3.3 Pseudomonas PLD
Bacterial PLD enzymes more commonly share greater homology with plant PLDs, however, some bacterial PLDs appear to be a more recent acquisition and demonstrate greater homology to fungi and mammalian PLD. Pseudomonas aureginosa is an opportunistic Gram-negative bacterium. Roughly 30 % of the Pseudomonas strains identified in nature express a plda gene that encodes for an 116 kDa 2-HKD PLD.186 Unlike two Pseudomonas PLC enzymes, this enzyme is localized to the periplasm and does not possess a type II secretion signal. However, the possibility of type III secretion directly into host cell cytosol or in vivo secretion into extracellular milieu has not been rigorously determined. In vitro characterization of recombinant Pseudomonas PLD demonstrates it is capable of PC hydrolysis to generate PA and performs transphosphatidylation in the presence of primary alcohols. In vitro Pseudomonas PLD activity is significantly enhanced by mM concentrations of calcium, but does not require PI(4,5)P2 for activity. It is unclear as to whether this enzyme functions as a virulence factor or plays a role in bacterial homeostasis, as PC has recently been identified in the inner and outer membrane leaflet of Pseudomonas aureginosa bacteria.186 Infectivity is not diminished in plda deletion strains, however, long-term survival is decreased. Finally, it is proposed that this gene was acquired through horizontal gene transfer from a higher organism, potentially of eukaryotic origin. NCBI database BLASTN sequence analysis of plda reveals greatest sequence homology to mouse PLD2.
4.3 Streptomyces PLD
Gram-positive Streptomyces encompass the largest genus within the Actinomycetes class of bacteria that includes Corynebacterium and Mycobacterium. Streptomyces bacteria flourish in soil and secrete secondary metabolites and enzymes, including phospholipases, able to scavenge the environment for nutrients. Streptomyces are rarely pathogenic to humans.187 In fact, many Streptomyces species are of immense commercial and industrial value for several reasons. More than two thirds of all clinically relevant natural antibiotics are derived from these bacteria, including vancomycin, chloramphenicol, and rapamycin.187,188 Also, enzymes secreted by Streptomyces species are used as biocatalysts in industrial manufacturing of foods, cosmetics, and pharmaceuticals.106,189
Enzymes belonging to the PLD superfamily have been isolated in secretions from Streptomyces species including S. antibioticus, S. cinnanoneus, S. halstedii, and S. septatus. These enzymes share significant sequence homology (>70 %) and are some of the most rigorously biochemically and structurally characterized members of the PLD superfamily.106 In contrast to scPLD from Streptomyces chromofuscus, these Streptomyces enzymes maintain the conserved domains I–IV and are class II members of the PLD superfamily, similar to YMT, as characterized by Ponting and Kerr.18 These enzymes are robustly expressed and secreted into the extracellular milieu, but their exact function is unknown.
Robust expression and secretion of Streptomyces PLD, coupled with the observation that many of these enzymes display the highest transphosphatidylation activity of any bacterial PLD make these enzymes useful tools for industrial production of natural and synthetic phospholipids.189 These enzymes exhibit broad substrate specificity that is exploited to facilitate headgroup exchange with natural and unnatural nucleophiles. In fact, use of these enzymes in industry has spurred rigorous enzymological characterization in order to engineer Streptomyces PLD with enhanced activities or altered substrate specificities for tailored use.190,191
The crystal structures of Streptomyces sp. PMF PLD105,111 and Streptomyces antibioticus (deposited in PDB, unpublished) have been determined without substrate (PDB code: 1F0I, 1V0S, 2ZE4), in complex with short acyl chain substrate (1V0W, 2ZE9) or complexed with phosphate analog, tungstate (1V0R). These structures are reviewed in detail in section 2.2.2. These structures, in addition to biochemical data, further validate the proposed two-step SN2 reaction mechanism. The Streptomyces PLD structures share a common fold to that of the Nuc endonuclease homodimer102 and the endonuclease domain of BfiI.161 The bilobal structure has an apparent crystallographic two-fold axis of symmetry that cuts through the cone-shaped active site at the interface of the two lobes. The conserved HKD motifs exist on loops that lie adjacent to one another within the active site pocket. Crystal structures with complexed substrate or phosphate analogs demonstrate there is significant hydrophobic bonding between residues of the active site holding the substrate in place. The reaction mechanism proceeds via a covalent intermediate that is formed following N-terminal histidine, of the HKD motif, nucleophilic attack on the phosphate of the substrate headgroup. Biochemical analysis suggests formation of this covalent intermediate is the rate limiting step of catalysis, and that subsequent nucleophilic attack of the lone pair of electrons on the oxygen from either the water or primary alcohol molecule, for hydrolysis or transphosphatidylation reaction, respectively, can proceed in parallel with similar rates.24 Crystal structures of PMF PLD suggest that Streptomyces PLD can also perform a second round of hydrolysis of PA, to release DAG and a covalently-bound phosphate to active site, referred to as the dead end reaction,106,111 although this reaction appears to be much slower (product observed for crystals soaked with substrate for a week reproduced this finding of DAG formation using an in vitro biochemical assay, Selvy and Brown, unpublished observation).
Because these enzymes are stably secreted into the extracellular growth medium, rigorous in vitro biochemical characterization of Streptomyces PLD has been possible. These enzymes possess a signal sequence that facilitates secretion from the bacterial cytosol into the non-reducing environment of the periplasmic domain. Some Streptomyces PLD have been reported to possess a critical disulfide bond that is thought to form in conjunction with proper folding only in the non-reducing environment of the periplasm.192 Historically, much of the biochemical and structural studies have used secreted enzyme purified from the growth media of native Streptomyces cultures. Efforts to recombinantly express these enzymes in Gram-negative E. coli has proven difficult, and required use of secretion signal sequences193 (to elicit periplasmic localization and secretion), or vectors with thioredoxin tags194 (to enhance cytosolic disulfide bond formation).
Following structural characterization, biochemical studies of Streptomyces PLD homologs with significant sequence identity have subsequently been performed to further probe the function of different components of the Strepotmyces PLD structure. S. septatus TH-2PLD has the highest specific activity and transphosphaditylation rates of any bacterial PLD identified to date,195 while pldp exhibits quite low activity. These differences in PLD activity between these two enzymes exist, despite the fact that these enzymes share significant sequence identity. This suggests that critical differences in a small number of residues elicit major differences in PLD enzymatic activity. Uesugi et al. used these two PLD genes to generate a series of chimeric constructs.196 Using a random repeat-length independent and broad spectrum, RIBS, in vivo DNA shuffle technique chimeric mutants were generated composed of stretches of TH-2PLD and pldp.196,197 Biochemical characterization of these constructs identified residues that were critical in modulating substrate specificity, interfacial activity, transphosphatidylation, and thermostability. The tertiary locations of these residues were then mapped in the Streptomyces PLD structure (models of TH-2PLD based on the PMF PLD structure) to further clarify their mechanistic function.
The Streptomyces PLD structures show two flexible loops that gate the 30 Å wide entrance to the active site cleft. The N-terminal loop is located between beta-strand 7 and alpha-helix 7. The C-terminal loop is between beta-strand 13 and beta-strand 14. Chimeric analysis identified two residues in the N-terminal loop, (Gly188 and Asp191 for TH-2PLD) that dictate interfacial activity and sensitivity to substrate presentation.196 Streptomyces PLD prefer substrate presented as monomer or mixed micelles, and demonstrate lower activity towards phospholipid vesicles.24 Computer modeled docking of phospholipids into the Streptomyces PLD structure suggests these residues in the N-terminal loop might serve as a second phospholipid binding site for PA, PE, or PS.198 The C-terminal loop, specifically residues Ala426 and Lys438 of TH-2PLD, are involved in enhancing the specific hydrolase and transphosphatidylation activity, regardless of substrate presentation. These residues also participate in phospholipid head group specificity, and enhance thermostability of the enzyme.108 Uesugi et al. used surface plasmon resonance and inactive mutants to measure substrate binding affinities.107 The specificity for zwitterionic phospholipids over anionic phospholipids was narrowed down to the same residues, Ala426 and Lys438, in the C-terminal loop that are proposed to act as a gate at the entrance to the active site cleft.107 Substrate specificity can be altered by point mutation of residues in this loop. Masayama et al. have exploited this characteristic by mutating residues in the C-terminal loop to facilitate production of phosphatidylinositols via head group exchange, an activity that is not observed with the wildtype enzyme.190
Other studies have characterized the function of the conserved GG and GS residues that lie downstream of the HKD motifs, N-terminal and C-terminal motifs, respectively, in most PLD superfamily enzymes. Ogino et al. showed that the GG/GS residues, specifically the serine residue, downstream of the putative HKD motifs are critical for dictating the transphosphatidylation activity of the enzyme.109 These residues line the base of the active site and are proposed to control active site conformation and stability, and subsequently modulate substrate specificity and ability to transphosphatidylate. Deletion of the serine residue decreases overall activity by a third compared to wildtype enzyme.109
5. Plant PLD
Plant PLDs make up the largest family of HKD enzymes, with more than 80 genes identified and several dozen cloned. These enzymes are more complex than bacterial PLD, because they encode regulatory domains that facilitate differential activities under various signaling environments (reviewed199,200,201,202). Plant PLD enzymes contribute to the rich history of the PLD superfamily, in that the first description of a PLD enzyme was made from carrot.13 The PLD hydrolytic and transphosphatidylation activities were originally described in plants, in 194714 and 1967,112,116 respectively. Also, the first PLD enzyme was cloned from the castor bean in 1994.203 Cloning of the castor bean PLD by the Xuemin Wang lab subsequently facilitated identification and cloning of fungi117,204 and animal205 homologs. The Arabidopsis thaliana genome has been sequenced, making identification of PLD superfamily members and genetic manipulation of this model organism feasible. The bulk of the plant PLD literature focuses on Arabidopsis, therefore this model organism will be the focus of this section with a few noteworthy exceptions from other organisms (Table 4).
Table 4.
Plant PLD Enzymes
| ENZYME | REGULTORY DOMAIN | CATALYTIC REQUIREMENTS | SUBSTRATE | SIGNALING |
|---|---|---|---|---|
| PLDα | C2-domain | mM Ca2+ | PC>PE | hormone/stress response, senescence, nutrient sensing |
| PLDβ | C2-domain | μM Ca2+, PI(4,5)P2 | PC=PE=PS=NAPE | actin polymerization |
| PLDγ | C2-domain | μM Ca2+, PI(4,5)P2 | PE=NAPE>PC | hormone/stress response (?) |
| PLDδ | C2-domain | μM Ca2+, oleate, PI(4,5)P2 | PE>PC | cell viability, ROS response, binds microtubules |
| PLDε | C2-domain | μM Ca2+, oleate, PI(4,5)P2 | PE>PC | root growth, elongation |
| PLDζ | PX-PH | PI(4,5)P2 | PC | root growth, elongation |
PA makes up less than 1 % total lipid in plants, but is an important second messenger.201,206 Several pathways have been characterized that generate PA, but in plants the two main signaling mechanisms for generating PA involve PLC-DAGK tandem activity, or PLD activity. Lipidomic analyses have been performed and characterized the major PA species in Arabidopsis as having long polyunsaturated fatty acids [34:2(16:0–18:2); 34:3(16:0–18:3); 36:4(18:2–18:2); 36:5(18:2–18:3); 36:6(18:3–18:3)].206 Different PA species change in response to different stimuli and environmental conditions. Drought and soil salinity are common environmental stresses, and are a major focus of plant research because these conditions affect crop production worldwide.206 Plant PLD enzymes have variant regulatory mechanisms to respond to extracellular stimuli such as these, and mediate intracellular responses via PA production and protein-protein interactions.
5.1 Classes of plant PLD enzymes
Plant PLD enzymes consist of two-conserved HxKxxxxD motifs separated by roughly 320 aa, which include the conserved region III (IYIENQFF). The function of this region is unknown, but is present in every PLD superfamily member with true phospholipase activity. Most plant PLD region III sequences encode ‘IYIENQYF’, while two enzymes more closely related to the mammalian PLDs encode for ‘IYIENQFF’.207 Plant PLD enzymes can be divided into two subdomains, C2-PLDs and PXPH-PLDs, based on the presence of amino-terminal regulatory domains upstream of the catalytic domain.202,208 C2-PLDs have an N-terminal C2 calcium binding domain that is distinct to plant PLD enzymes.206 This domain is not found in other higher order PLDs. PXPH-PLDs are more closely related to mammalian PLDs, and have amino-terminal phox homology and pleckstrin homology domains important for specific lipid interactions.206,207 At least 12 Arabidopsis genes have been identified, of which ten are classified as C2-PLD genes and two are classified as PXPH PLD genes.209 Within these classes specific isoforms have been identified that exhibit differential genetic architecture, sequence identity, catalytic activities, and regulatory requirements.199,207
In contrast to the multiple crystal structures available for bacterial enzymes, a crystal structure for the more complex plant PLD does not exist, despite reported crystallization of cowpea PLD over a decade ago.100 Therefore, the current model of proposed tertiary structure of the catalytic domain and reaction mechanism are based on the structure and characterization of the bacterial PLDs (discussed in section 2.2). The limited structural analysis of plant PLD that does exist has used non-crystallographic analytical tools. One such study used mass spectrometry analysis to characterize the sulfhydryl groups on cabbage PLD.210 Increasing numbers of plant PLDs of both C2 and PXPH subfamilies have been cloned and recombinantly expressed in bacteria,124,207,211,212 which has lead to a greater understanding of the individual biochemical characteristics of different plant PLD isoforms.
5.1.1 C2-PLD
In the mid to late 1990’s following cloning of the castor bean PLD,203 a surge of plant PLD enzymes were identified, sequenced, and characterized by genetic and biochemical approaches.202 Comparisons within this growing pool of plant PLDs led to observations of clusters of similar enzymes based on genetic architectures, sequences, and biochemical characteristics. Members of the C2-PLD subdomain were subsequently categorized as PLDα, PLDβ, PLDγ, PLDδ, PLDε. It is important to note that as sequence and biochemical characterization improved, some initial cluster designations have changed (PLDα4 is no longer included as a PLDα isoform, and PLDδ1 was reclassified PLDβ2).207
Regardless of cluster classification, all members of the C2-PLD subfamily encompass a conserved 130 aa C2 domain at the amino terminus that is important in calcium sensing and phospholipid binding.200,202 More than 4000 consensus sequences have been reported for the C2 domain and are commonly present in proteins involved in lipid metabolism, signal transduction, and membrane trafficking.213 The crystal structure for several C2 domains has been determined and a common antiparallel 8-β strand sandwich fold is conserved.214,215,216,217 Two or three calcium ions are known to bind at 4–5 acidic residues in the loops between the beta strands.213 The β-strand sandwich fold is predicted to be conserved in plant PLD, but structural characterization of this domain from several C2-PLD isoforms demonstrates that a significant conformation change occurs upon calcium ion binding, which is not observed in C2 domains from other proteins or species.218 This suggests plant C2 domains may be a variant of those previously characterized.
In addition to the divergent protein conformations upon calcium binding, some plant PLD isoforms have substitutions in the C2 domain acidic residues.218 This results in isoform-selective differences in calcium binding affinities and catalytic responses. C2 domains also bind lipids dependent on calcium concentration, therefore cytoplasmic calcium levels are thought to modulate C2-domain conformation and lipid binding affinity.212 C2-domains also demonstrate lipid binding specificity. Arabidopsis C2 domains bind PI(4,5)P2 and PC in a calcium-dependent manner.218,219 C2 truncation mutants bind lipid vesicles but with lower affinity and these PLD enzymes display decreased activity.220 Many C2 domains elicit constitutive binding to the lipid membrane (ie. no stimulus-induced translocation) therefore these enzymes are proposed to function in the scooting mode with processive catalytic activity. All C2-PLD enzymes characterized to date require some level of calcium for catalysis and can perform transphosphatidylation.220 This plant PLD subfamily is responsible for the majority of the PA produced in response to environmental stress signaling.
5.1.1.1 PLDα
PLDα is the first classification within the C2-PLD subfamily, and these enzymes are the most predominant PLD in plants.206 PLDα from Arabidopsis exhibits similar genetic architecture to PLD1 from castor bean and rice (ie. 4 exons and 3 introns), which suggests this class of PLD enzyme stems from a common ancestor. Three PLDα genes have been identified, PLDα1, PLDα2, and PLDα3,209 which exist on separate chromosomes (α1 chromosome III, α2 chromosome I, α3 chromosome V).207 The original PLD enzyme cloned from castor bean was classified as a PLDα enzyme. Subsequently, the Arabidopsis PLDα was cloned by BLAST database search using castor bean cDNA sequence, which gave an incomplete cDNA.221 Therefore, later attempts to clone the Arabidopsis PLDα required nested primers and PCR be used. The Arabidopsis PLDα enzyme is 809 aa, with a 20 aa leader sequence of unknown function that is removed during maturation.202 The conserved C2-domain lies 30 aa into the sequence; however, in a conserved region of this domain two of the four acidic residues necessary for calcium binding are substituted for neutral or positively charged residues.218 This may be the cause of the observed shift in calcium binding affinity and the requisite millimolar (mM) concentrations of calcium necessary for catalytic activity under some in vitro conditions.218
PLDα hydrolyzes PC and PE, and is unable to hydrolyze PI, PS, NAPE, or cardiolipin.202,211 In vitro characterization of this activity was originally performed at physiological pH with vesicles composed of a single phospholipid species. Under these conditions, mM calcium concentrations (20–100 mM) were required for catalytic activity.16 Structural and biochemical characterization of the PLDα C2 domain using circular dichroism and isothermal titration calorimetry demonstrates a significant shift in protein conformation upon calcium binding. Between one and three calcium ions bind to the PLDα C2 domain in the 470–590 μM calcium range.218
The cellular significance of such high concentrations of calcium actually regulating PLDα was suspect. PLDα activity comprises the majority of PLD activity in the plant. Subsequent in vitro studies demonstrate lower micromolar (μM) concentrations of calcium are conducive to activity in the presence of an acidic pH and when substrate is presented in a vesicle with mixed phospholipid composition.212,222 Calcium is proposed to bind at the acidic residues within the C2 domain and trigger a conformational change that exposes hydrophobic residues able to bind neutral lipids such as PC. In vitro characterization of PLDα demonstrates protein-lipid binding to PC-only vesicles occurs in a calcium concentration-dependent manner.218
In addition to calcium, PI(4,5)P2 has also been shown to regulate PLDα activity in vitro. Conserved polybasic PI(4,5)P2 binding motifs flank the 2nd HKD, but in PLDα enzymes four of the five critical basic residues are substituted with acidic or neutral residues.202,218 Therefore, PI(4,5)P2 likely binds PLDα at the C2 domain only. Competition binding studies demonstrate PI(4,5)P2 binding at the C2 domain is displaced with increasing calcium concentrations. This is proposed to be the reason that the PLDα binding affinity for PI(4,5)P2 is significantly decreased from that of other C2-PLDs with intact polybasic regions, and the likely reason that PLDα catalytic activity is not highly responsive to PI(4,5)P2 stimulation under optimal calcium concentrations (20–100 mM).202
The fact that any in vitro stimulation of PLDα catalytic activity is observed in response to PI or PI(4,5)P2 is likely due to an indirect effect, such as a change in interfacial lipid environment. Supporting this indirect activation theory, is the fact that in the presence of suboptimal calcium concentrations, PI(4,5)P2 appears to stimulate PLDα activity.212 This was demonstrated by Qin et al., who showed that PI(4,5)P2 activation of PLDα is insensitive to neomycin treatment, which partitions into PI(4,5)P2 headgroups and prevents lipid species-specific interaction; whereas PLDβ and PLDγ activity was significantly and dose dependently inhibited by neomycin.212 In a similar fashion, PLDα enzymes are stimulated by detergents, such as sodium dodecyl sulfate (SDS). This likely occurs by changing access to substrate at the interface of the mixed micelle. LysoPE and N-acyl ethanolamine have been shown to inhibit PLDα activity.202,223 Under signaling conditions, it has also been noted that PLDα is inhibited by Gα in absence of GTP.199,224
Specific PLDα isoforms contribute to the bulk of the signaling PA response in plants, and as such have been implicated in a broad range of signaling pathways.199,202 PLDα1, the major plant PLD, is involved in the hyperosmotic stress response via a bifurcating pathway (discussed in 5.3.2). PLDα1 has also been implicated in membrane proliferation necessary for rapid growth and wound repair.225 PLDα2 is the constitutive form present in all tissues,226 while PLDα3 is more highly regulated and involved in senescence and hormone/stress stimulation.227,228
5.1.1.2 PLDβ & PLDγ
PLDβ and PLDγ classifications of the C2-PLD subfamily share 40 % sequence identity and 60 % similarity to PLDα.212 These enzymes share common catalytic and regulatory requirements, but are distinct genes with 65 % sequence identity. PLDβ and PLDγ encode 2-HKD enzymes with an N-terminal C2 domain, and putative polybasic PI(4,5)P2 binding motifs flanking the 2nd HKD (RxxxxKxRR or inverse).
PLDβ was more difficult to clone than PLDα. Nested primers, PCR, and screening of cDNA library were used to identify the full sequence, while 5′-RACE was used to determine the N-terminal start site.221 PLDβ is968 aa (109 kDa) and exhibits more sequence homology to yeast and human PLDs than PLDα. Two isoforms have been identified in Arabidopsis, PLDβ1 (present on chromosome II) and PLDβ2 (present on chromosome IV), which share 89 % sequence similarity. Of this class PLDβ1, formerly known as PLDβ, is the best characterized. PLDβ enzymes hydrolyze PC, PE, PS, and NAPE without preference, but do not hydrolyze PI, PIP2, or cardiolipin.211
PLDγ classified enzymes, of which there are three isoforms with 95 % sequence similarity, PLDγ1, PLDγ2, and PLDγ3, are all encoded for in tandem on chromosome IV.202 These enzymes contain a putative myristoylation site at the amino terminus (MGxxxS) 30 aa upstream of the first C2-domain β-loop.212 The specific function of this predicted lipid modification has yet to be determined, but may serve to facilitate protein-membrane interaction as is observed for other myristoylated proteins. PLDγ preferentially hydrolyzes PE and NAPE over PC, but cannot hydrolyze PI, PI(4,5)P2, or cardiolipin.211
PLDβ and PLDγ catalysis requires μM calcium concentrations for optimal activity (less than 100 μM), whereas high mM calcium concentrations inhibit enzyme activity.202 Three calcium ions bind with different affinities (Kd1 = 0.8 μM, Kd2 = Kd3 = 24 μM) at the conserved acidic residues within the calcium binding loops of the C2 domain.218 The ability of calcium to both activate and inhibit at different concentrations is likely due to disruption of requisite PI(4,5)P2. Similar to PLDα, PI(4,5)P2 binds the C2 domain under low calcium concentrations. PI(4,5)P2 binding at the C2 domain is displaced with increasing calcium concentrations, where maximal calcium binding is observed near 100 μM for PLDβ and PLDγ versus >1 mM for PLDα.218 However, in contrast to PLDα, increased calcium concentrations concomitantly trigger PI(4,5)P2 binding at putative polybasic binding motifs that flank the 2nd HKD.
This binding is requisite for PLDβ and PLDγ activity. At these polybasic motifs, PI(4,5)P2 directly and dose-dependently enhances lipid binding and catalysis whereas for PLDα, PI(4,5)P2 indirectly stimulates activity by modifying fluidity and substrate access at the lipid interface.212,218 Synergistic activation of PLDβ and PLDγ by PI(4,5)P2 and calcium impacts Ks and allows the enzyme to processively scoot along the lipid interface without dissociation (see section 2.2.3 for details on interfacial kinetics and Figure 2).219 Mass spectrometry proteomic analysis was used to demonstrate that reversible phosphorylation occurs within the first putative polybasic PI(4,5)P2 binding motif, and decreases PI(4,5)P2 binding, thereby decreasing PLDγ catalytic activity.229 These phosphorylated serine and threonine residues are conserved amongst other plant PLDs, and it has been suggested that post-translational modification may be yet another mechanism for regulating PLD activity, in addition to divalent cation concentration and polyphosphoinositide binding.
In vitro biochemical characterization of these enzymes has demonstrated that PE must also be present in the vesicle for catalysis to occur (optimal ratio of 3:1, PE:PC).202,211 Analogous to PI(4,5)P2 effects on PLDα, the apparent PE-induced activation is likely due to local interfacial lipid changes rather than direct interaction with the enzyme.230 Some studies have shown PA and cardiolipin can partially substitute for the PE effect.202
PLDβ and PLDγ have been shown to bind 14-3-3 proteins involved in cell signaling and metabolism.231 Due to their ability to hydrolyze NAPE and release endocannabinoids, PLDβ and PLDγ have been implicated in defense signaling pathways, such as pathogenic infection. PLDβ1 binds actin to modulate actin polymerization. Monomeric G-actin inhibits PLDβ activity while polymerized F-actin promotes PLD activity. PLD-produced PA binds the actin heterodimeric capping protein and inhibits its interaction with actin,232 essentially, promoting actin polymerization.233 However, this actin binding motif in PLDβ1 is conserved in other plant PLD enzymes, so it may be a broader plant PLD trait not limited to this enzyme.
5.1.1.3 PLDδ
C2-PLD subfamily members classified as PLDδ enzymes show divergent catalytic regulation to other plant PLDs.124,234,235 In addition to calcium and PI(4,5)P2, these enzymes are activated by unsaturated fatty acids, specifically oleic acid [(18:1) less so by linoleic acid (18:2), not at all by saturated fatty acids].234 Mutagenesis studies have identified distinct oleate and PI(4,5)P2 binding sites on PLDδ. PLDδ retains a putative polybasic PI(4,5)P2 binding motif also present in PLDβ and PLDγ catalytic domains, but not observed in PLDα.234 Mutation of the residues within this polybasic motif decrease PI(4,5)P2 stimulation by 80 %. The oleate binding site was identified when PLDδ sequence was compared to human PLD2, which is also oleate activated. The oleate binding site is located 30 aa after 1st HKD. When this region is mutated a 70 % decrease in oleate stimulation was observed.
Similar to PLDβ and PLDγ, PLDδ activity requires μM calcium concentrations, and is activated by PI(4,5)P2.234 However, PI(4,5)P2 interaction is not integral to catalysis, and PI(4,5)P2 does not affect Ks. Rather, PI(4,5)P2 lowers Km and increases the affinity of the active site for PC binding and hydrolysis.124 At a distinct location, PLDδ binds oleate. Again, this specific interaction is not requisite for activity, and oleate can be replaced with detergent triton-x 100 for in vitro study.124 Similar to the function of PE for PLDβ and PLDγ, oleic acid is proposed to modulate substrate exposure and enhance presentation to enzyme. As such, PLDδ mutated at the oleate binding motif still retains catalytic activity, albeit at a decreased level.234
PLDδ hydrolyzes PE and PC. In vitro study demonstrates the bulk lipid binding affinity (1/Ks) for PE- or PC- micelles is similar, but Km for PE is lower than PC, suggesting PE binds with higher affinity to the active site, and therefore PE is the preferred substrate.124 To date, one PLDδ isoform has been identified on chromosome IV, which results in two splice variants: PLDδa and PLDδb. These enzymes are tightly associated with the microsomal membranes, and detergent is required to solubilize and isolate these enzymes. PLDδ also localizes with tubulin and cortical microtubules and is predicted to function as a signaling communication bridge between the plasma membrane and microtubule infrastructure.236
In cells, PLDδ expression is regulated by signaling pathways that are triggered in response to cell stress, including soil salinity and drought. PLDδ activity is stimulated by H2O2.237 The subsequent PA response protects cells from reactive oxygen species (ROS), and promotes freeze tolerance in plants.
5.1.1.4 PLDε
The most recently identified and least well characterized C2-PLD is PLDε.238 The catalytic requirements for this enzyme are highly promiscuous, and phylogenetic analysis demonstrates that this enzyme is the most closely related C2-PLD subfamily member to the plant PXPH-PLD, PLDζ. PLDε retains the C2-domain but none of the conserved acidic calcium binding residues. Regardless, PLDε is active under a broad range of catalytic conditions, including in the presence of mM calcium concentrations, similar to PLDα, as well as μM calcium concentrations, in the presence of PI(4,5)P2 and/or oleate, similar to PLDβ and PLDγ or PLDδ, respectively.238 In vitro PLDε exhibits catalytic activity towards PC-, PE-, PG-, or PS- only vesicles, with preference towards PC>PE>PG or PS.
In cells, PLDε localizes to microsomal membranes, and is not observed in the cytosol.238 Low basal expression of PLDε exists in every tissue, except pollen where expression exceeds PLDα1. However, expression is upregulated in response to cell stress including nitrogen deprivation.206 PLDε subsequently elicits root elongation, primary and lateral root growth. Root growth functions to increase water and nutrient uptake, and enhanced nitrogen sensing. PLDε knockouts yield smaller plants than wildtype, while overexpression mutants yield larger plants.238 Treatment with primary alcohol also yields smaller plants, and suggests PLDε-generated PA and not the enzyme itself supports plant growth.
5.1.2 PXPH-PLD- PLDζ
In contrast to C2-PLD enzymes, two plant PLDs have been identified that encode for phox homology (PX) and pleckstrin homology (PH) lipid binding domains at the amino-terminus, PLDζ1 and PLDζ2.207,239 These genes are both located on chromosome III. PLDζ1 and PLDζ2 do not require calcium for catalysis, rather these enzymes selectively cleave PC in a PI(4,5)P2 dependent manner.207 As such, these plant PLDs are more closely related to the mammalian PLD enzymes PLD1 and PLD2. In mammalian PLDs, the PX domain has been shown to bind PI(3,4,5)P3, and anionic lipids, while the PH domain binds PI and PIPn species. PLDζ1 and PLDζ2 also retain four of the five basic residues in the conserved PI(4,5)P2 binding motifs that flank the 2nd HKD.207 These polybasic motifs may serve to regulate PLDζ catalysis in response to PI(4,5)P2, similar to mammalian PLDs.
Cellular characterization of this subfamily of plant PLD enzymes remains sparse, but some recent studies have shown PLDζ enzymes are involved in environmental stress responses. PLDζ2 is transcriptionally regulated in response to phosphate starvation and auxin levels.239,240 Exogenous auxin supplementation can stimulate PLDζ2 transcription. Plant PXPH PLDs have also been shown to mediate vesicular trafficking, phosphate recycling and root gravitropism.241
5.2 Signaling
Plant PLD enzymes are structurally more diverse and complex than bacterial homologs. As in other higher eukaryotes, PA is largely involved in stress-mediated signaling pathways in plants.201 As such, plant PLD enzymes have evolved diverse regulatory mechanisms to respond to specific extracellular stimuli. As detailed in section 5.1, plant PLD enzymes can be regulated at the level of transcription or translation, via post-translational modification (lipidation or phosphorylation), or via cytosolic and membrane cofactors and conditions (calcium, PI(4,5)P2, substrate presentation/membrane fluidity, and pH). PA signaling can also be regulated and attenuated post production by phosphorylation to generate DAG pyrophosphate (DGPP).242 It is currently unknown whether DGPP is itself also a signaling molecule.
Despite the historical precedent in plant studies of PLD, development of pharmacological tools to modulate the activities of these enzymes has lagged behind that of other eukaryotes. To this day, the use of knockout models and primary alcohols remain the only known tools with which plant PLD can be studied.243 Using a primary alcohol, product formation can be diverted to the transphosphatidylation product phosphatidylalcohol (Figure 5). However, as detailed in section 11, alcohols are imprecise tools because of their lack of specificity and potency. While only primary alcohols are able to serve as nucleophiles in the PLD reaction mechanism, both primary and secondary alcohols activate plant PLD activity.244 Off target activation of heterotrimeric G proteins also occurs in response to alcohols, making it difficult to delineate the specific role of PLD in receptor-mediated stress induced signaling pathways. A few plant stress response cascades are briefly described here to demonstrate a few of the numerous roles in which plant PLD enzymes have been implicated.
5.2.1 Growth and biomass accumulation
Plant PLD-generated PA has been implicated in regulation of plant development and growth through multiple signaling pathways including the target of rapamycin (TOR), a homolog of the mammalian target of rapamycin (mTOR).245,246 Plant TOR, a large protein belonging to the PI3K-related kinases, is a protein kinase involved in seed germination, embryonic development, meristem driven cell growth, and hyperosmotic stress response.247 This large protein regulates protein expression and cell growth in response to nutrients, mitogens and growth factors by forming protein complexes and activating other proteins via phosphorylation. In mammals, insulin and insulin growth factor signaling activate mTOR, which phosphorylates S6K to trigger phosphorylation of S6 ribosomal protein (S6rp) on the 40S ribosomal subunit (see section 9.7 for further details on mTOR signaling). TOR kinase activity is inhibited by rapamycin, an antiproliferative agent secreted by Streptomyces hygroscopius. Rapamycin, binds FKBP12 (peptidyl-prolyl isomerase 12 kDa FK506 binding protein) in the requisite first step of inhibitory complex formation, followed by binding at the FRB (FKBP12-rapamycin binding) domain on TOR. NMR studies have shown that PA competitively blocks formation of the rapamycin-FKBP12 inhibitory complex by directly binding mTOR at Arg2109 in a hydrophobic pocket at the FRB domain.248
TOR signaling in plants is poorly characterized, but several studies have shown different species of plants are have differential sensitivity to rapamycin. Initial TOR studies performed in Arabidopsis thaliana (AtTOR) demonstrate this organism is resistant to rapamycin.246,249 cDNA sequence analysis shows this rapamycin insensitivity is due to substitution of conserved residues in the FKBP12-rapamcyin drug binding site, while the FRB domain is similar between AtTOR and mTOR. This suggests that the rapamycin/PA binding at AtTOR might be conserved. In fact, subsequent studies have shown that transgenic expression of Sacchormyces cerevisceae FKBP12 rescues sensitivity to rapamycin.250 However, equivalents of insulin signaling pathways have not been identified in Arabidopsis, making it difficult to determine whether PA might serve a similar regulatory role in AtTOR signaling to that of mTOR.
More recent studies demonstrate that other plant species, including maize251 (Zea mays) and algae249 (Chlamydomonas reinhardtii) are in fact responsive to rapamycin. Maize S6K activity and S6rp phosphorylation increased during seed germination in response to insulin, and this activity decreased upon rapamycin treatment. Consistent with this observation and proposed activation of Zea mays TOR (ZmTOR) in response to insulin stimulation, exogenous addition of PA also increased ZmTOR activity.251 These studies suggest that PA and similar TOR signaling pathways extensively characterized in mammalian systems may be conserved in some plant species.
The major source of the PA in plants is predicted to be derived from PLDα3 or PLDε. PLDα3 is known to promote root growth, as knocking out PLDα3 decreases rootgrowth, and PLDα3 overexpression increases root growth in response to dehydration.227 Likewise, PLDε promotes lateral root growth in response to drought, high salt or nitrogen deprivation.238
Plant PLD activity has also been implicated in ethylene signaling response. Ethylene is a plant stress hormone that activates PLD and subsequently increases transcription and translation.252 PLD-produced PA binds to CTR1, a plant homolog to mammalian raf kinase, and a negative regulator of ethylene response.253 This binding triggers CTR1 translocation away from ETR1 (ethylene receptor) allowing other ETR1 downstream signaling to occur.
5.2.2 Hyperosmotic stress/Abscisic acid stimululation
Terrestrial plants perform gas exchange through stomata, openings in leaves bordered by guard cells. Water loss occurs through transpiration of open stomata, therefore intricate signaling pathways have evolved to sense water levels and regulate stomatal closure. Under low water concentrations, cells release abscisic acid (ABA), a phytohormone, to promote stomatal closure.206,254 ABA triggers translocation of cytosolic PLDα1 to microsomal membranes. PLDα1 KO plants exhibit a higher rate of transpirational water loss,206 while overexpression of PLDα1 increases sensitivity to ABA, resulting in stomata closure and decreased water loss. Exogenous PA supplement promotes stomatal closure.206 PLDα1 involvement in ABA response199 was first observed in castor bean where PLDα1 gene expression increased with ABA stimulation. PLDα1 activity increases ROS formation and NADPH oxidase activity to facilitate stomatal closure.255 PLDα1 is constitutively expressed in most plant tissues, while PLDδ1 expression is only induced in response to high salt and dehydration. PLDδ1 makes PA in response to H2O2 (ROS) activation. Double α1/δ1 knockout plants are less tolerant to high salt.206,225 In addition to ROS generation, PLDα1-generated PA binds ABI1 (PP2C) to trigger stomatal closure.255
GDP-bound Gα blocks ABA stimulation. In the absence of GTP, Gα binds PLDα and inhibits phospholipase activity.256 Therefore ABA-mediated response occurs via Gα-PLDα signaling. Overall PLD activity increases in response to hyperosmotic stress.
5.2.3 Defense and wound healing
Plants upregulate the expression of a host of genes in response to pathogenesis or wounding via immune signaling cascades. PA has been shown to serve dual functions in these signaling pathways as both an integral lipid second messenger and precursor to a lipidic hormone (reviewed199,200,201). PA produced immediately upon wounding has been shown to be produced by PLDα, the major plant PLD, as it translocates from the cytosol to the plasma membrane upon increased calcium concentration and wound response signaling.257 Downstream of this activation expression of lipoxygenase 2 (LOX2), an enzyme involved in wound response, is induced.258 A second delayed PA response is observed due to upregulated transcription of PLDβ and PLDγ1 and PLDγ2. Increased PA has been shown to indirectly activate a wound-induced MAPK.259
PA changes the lipid interface microenvironment and has been suggested to activate PLA2 hydrolysis of phospholipids at the sn-2 position, releasing fatty acids.260 PA is itself a substrate for PLA2. PLA2-catalyzed hydrolysis of PA releases linolenic acid (18:3), a precursor for LOX2-catalyzed production of jasmonic acid. Jasmonic acid is a 12-carbon prohormone that is activated upon conjugation to an amino acid (ie. L-isoleucine).261 JA-Ile triggers transcription factor activation and increased expression of genes involved in growth and plant repair as well as defense.
Jasmonic acid analogs are secreted as virulence factors during pathogenic infection to manipulate plant signaling pathways or to downregulate and attenuate immune responses.262,263 Jasmonic acid and Jasmonate analogs are of interest commercially. Efforts to characterize the signaling pathways that generate these secondary metabolites are underway in attempts to exploit and/or engineer pathways to produce industrial quantities for use in pharmaceuticals, cosmetics, and perfumes.
6. Fungal PLD
Fungal PLD, identified in yeast and slime mold, regulate critical developmental functions. Similar to plants, PLD activity was first described in yeast using biochemical methods. Nearly four decades ago glucose-stimulated PLD activity was measured for a species of budding yeast, Saccharomyces cerevisiae, grown in low (1 %) glucose content.264,265 These growth conditions induce glucose repression that triggers low oxygen uptake. Yeast harvested from these growth conditions demonstrated 14C-lecithin hydrolysis and PA production in mitochondrial fractions.265 This activity was increased in response to glucose repression during aerobic growth and decreased oxygen uptake. The increased activity was determined to be due to induction of an unknown cytosolic enzyme rather than a protein of mitochondrial origin since cyclohexamide blockage of cytosolic protein synthesis perturbed the PLD activity, and chloramphenicol inhibition of mitochondrial protein synthesis did not.265 This observation was largely ignored until a series of parallel studies decades later identified specific PLD enzymes in different yeast species. Spo14, a PLD superfamily member also known as PLD1, was identified in the budding yeast Saccharomyces cerevisiae.117,204,266,267,268 Other groups have identified similar Spo14-like enzymes in pathogenic budding yeast,269 Candida albicans,270 and in fission yeast, Schizosaccharomyces pombe.266 In addition a biochemically distinct enzyme, PLD2,269 has been described in Saccharomyces cerevisiae. Subsequent studies have demonstrated that the PLD activity initially observed in budding yeast (in the 1970’s) is distinct from the PLD superfamily, and this activity has been attributed to PLD2.271,272 Yeast PLD1 enzymes, including Spo14, share sequence and biochemical similarities to plant and other eukaryotic PLDs. These enzymes have been shown to function in yeast sporulation,204,267 vesicular trafficking,273 mating,274 and virulence for the pathogenic species.275,276
6.1 Budding Yeast Spo14/PLD1
Spo14 was originally identified during phenotypic studies of fission and budding yeast deficient in meiosis and sporulation.266,267 The most extensive follow up studies of this gene and gene product have been performed using the budding yeast Saccharomyces cerevisiae. S. cerevisae have distinct regulatory pathways for mitosis separate from those observed for meiosis I and meiosis II during sporulation. Early studies observed sporulation defects in mutagenized yeast as a means of identifying genes that might be involved in meiotic signaling pathways.267 In the first meiotic step, parental cells replicate genomic DNA and homologous chromosomes perform recombination as they align near the spindle pole bodies (SPB) in preparation for meiosis I. During meiosis I, similar chromosomes move to opposite poles of the nucleus and two diploid daughter nuclei are generated by separation of the chromosomes with the SPB. Reversal of meiosis is possible through meiosis I. In fact, cells with fully formed SPB are able to instead perform mitosis in response to changes in extracellular conditions and remain diploid. However, upon entry into meiosis II, the cell is committed to meiosis and unable to reverse to mitosis despite changes in extracellular growth conditions. During meiosis II, sister chromatids move to opposite poles of the nucleus to generate four haploid nuclei. These haploid nuclei are packaged into spores with prespore membrane (PSM), double layer membrane generated de novo, within the mother cell. This packaging is akin to acrosomes generated during spermatogenesis.271
Spo14 was identified as a gene involved in S. cerevisiae sporulation by Honigberg et al.267 In this study, mutagenized S. cerevisiae were subjected to various growth conditions, including changes in temperature, in order to observe phenotypic sporulation deficiencies. Cells with disrupted Spo14 genes showed 1.5-fold less yeast transition through meiosis I, and 10-fold fewer cells complete meiosis II.204,267 The cells that did complete meiosis I and meiosis II had degraded nuclei and were not viable. It was also observed that cells with disrupted Spo14 did not commit to meiosis at meiosis II, a phenomenon in wildtype yeast referred to as “commitment to meiosis.277 Rather, cells in later stages of sporulation with irregular nuclear composition were observed to reverse and mitotically divide.277
In parallel, Ella et al. subjected S. cerevisiae to different growth medium and measured changes in PLD activity.117 This group demonstrated that PLD activity is induced under nitrogen deprivation when yeast are grown in a medium containing a non-fermentable carbon source, ie. acetate.117 Supplemental application of glucose to these growth conditions decreased PLD activity. Sporulation, more specifically meiosis I, is triggered under nutrient deprivation conditions but cells can be reversed and induced to mitotically divide if nutrients are supplemented prior to transition into meiosis II.267,277 These studies suggest PLD activity is increased during sporulation, and the activity measured by this group is the same as that characterized by Rose et al.204 Spo14, called PLD1 by this group268 and others,278 is the enzyme responsible for the observed sporulation-induced activity. Spo14 is capable of PC hydrolysis and can perform transphosphatidylation with primary alcohols.117 These activities suggested that this newly-identified enzyme was indeed a PLD similar to PLDs identified in plants.
6.1.1 Sequence, catalysis, and regulation
Earlier cloning of castor bean PLD sequence facilitated cloning of Spo14,267 also known as PLD1,268,278 which later lead to cloning of the human PLD homolog.205 Genomic sequencing of Saccharomyces cerevisiae identified Spo14 on chromosome XI. Spo14 is predicted to be the only HKD PLD in this organism, and is a member of the PLD superfamily. The gene for PEL1/PGS1, a phosphatidylglycerol phosphate synthase, is the only other gene encoding for an HKD enzyme in S. cerevisiae.271
Spo14 protein sequence is 1683 amino acids, with a molecular weight of 195.2 kDa. A stretch of 440 amino acids in the middle of the sequence are 21 % identical to castor bean PLD, demonstrating conservation of the catalytic domain observed for members of the PLD superfamily.204 Separate groups cloned this enzyme, naming it either Spo14,204 based on function in the initial sporulation defects study, or PLD1268,278 to delineate this activity from an apparently separate PLD activity described in the 1970’s. Spo14 sequence analysis shows this enzyme retains two conserved HKD catalytic motifs, present in the majority of eukaryotic PLD superfamily members. A putative polybasic PI(4,5)P2 binding domain, found in other PLD superfamily members and originally described in Spo14, exists between these HKD motifs.279
Unique to yeast PLD, the amino-terminus contains a regulatory LOCO/phos domain encompassing residues 1–313.280 This region is hyperphosphorylated at serine and threonine residues upon meiotic initiation.280 Hyperphosphorylation shifts the molecular weight of Spo14 from 195 kDa to roughly 220 kDa. Hyperphosphorylation is a necessary regulatory mechanism for Spo14 function in meiosis, but not for other cellular functions of Spo14 or in vitro catalytic activity (detailed in 6.1.2). Downstream of the LOCO/phos domain, the amino terminus also possesses PX and PH domains. The PH domain binds PI(4,5)P2 to facilitate basal protein-membrane localization as well as protein translocation within the cell.281,282,283 As such, amino-terminal LOCO/phos and lipid binding domains are not integral to in vitro catalytic activity.
In vitro biochemical characterization of Spo14 has been performed using recombinant protein heterologously expressed in either insect204 or bacterial268 systems. Similar to other eukaryotic PLD enzymes, PI(4,5)P2 binding at the putative polybasic motif, but not the PH domain, is requisite for catalytic activity.279 Similar to some eukaryotic PLD enzymes, oleate (5mM) was shown to stimulate activity seven-fold.269 However, Spo14 is unique from plant or mammalian PLD in that it is insensitive to calcium, and inhibited by magnesium.
Spo14 catalytic activity is substrate-specific to PC, and little to no PI or PE is hydrolyzed.117 Spo14 can catalyze transphosphatidylation reactions with a broader range of alcohols than other eukaryotic PLDs. Although preference is given for primary alcohols, such as n-butanol, branched-chain alcohols, such as 3-methyl-1-butanol can also be used as nucleophilic substrates.117 Spo14 appears to be less effective at transphosphatidylation than mammalian homologs. This is postulated to be due to Spo14 potentially hydrolyzing phosphatidylalcohols271 shortly after production, but this remains to be demonstrated. Also, in vitro catalytic activity is stimulated in the presence of alcohol.117
In vitro, Spo14 catalytic activity is regulated by access to lipid cofactor PI(4,5)P2 and substrate, PC. In contrast to other eukaryotic PLD enzymes, Spo14 activity is not modulated by small GTPases, such as ADP-ribosylation factor (Arf).284 In contrast to in vitro regulation, cellular regulation of Spo14 is more complex and is dependent on the specific functional pathway, such as sporulation or mating (see section 6.1.2). Cellular Spo14 is not directly regulated by Arf, but Arf GTP/GDP cycling via Arf GAP, Gcs1, does modulate Spo14 activity during sporulation.285 Arf cycling is also is critical for sporulation.284 In general, cellular Spo14 is transcriptionally and translationally regulated in most functional pathways in which it has been implicated. Induction of Spo14 RNA and protein is observed, 7-fold and 3-fold, respectively, in late meiosis. Post-translational modification such as phosphorylation has been shown to regulate Spo14 localization. Finally, access to the lipid cofactor PI(4,5)P2 regulates both localization via the PH domain as well as activity via polybasic binding domain. Vegetative cells demonstrate PLD activity in both soluble and particulate fractions, likely localized to intracellular endosomal membranes, while Spo14 translocation to specific membranes, such as the PSM, has been demonstrated for specific functional responses.269
In the 1970s, Dharmalingam et al.264 and Grossman et al.265 described glucose-stimulated PLD activity in S. cerevisiae. More recent characterization of yeast PLD activities suggests this observed PLD activity is due to a separate class of enzyme, likely that of PLD2.271,272,286,287 PLD2 was described, but not cloned, as a calcium-dependent enzyme and does not require PI(4,5)P2 for activity. This activity was observed in Spo14 deletion mutants in the absence of EGTA or EDTA. This enzyme does not perform transphosphatidylation and preferentially hydrolyzes PE and PS rather than PC. This demonstrates PLD2 activity is distinct from that of Spo14/PLD1. The fact that Spo14 is the only HKD PLD present in the S. cerevisiae genome, and that PLD2 does not perform transphosphatidylation suggests this enzyme is likely a PLD-like enzyme distinct from the PLD superfamily with a unique reaction mechanism.
6.1.2 Function
Spo14/PLD1 deletion mutants do not demonstrate any phenotypic disruption in vegetative growth. Similar to the exocytic and vesicular function of HKD PLD enzymes in other higher eukaryotes, Spo14 appears to be integral for specific functional processes involving membrane formation, fusion, and secretion. In response to nitrogen deprivation and non-fermentable carbon sources, Spo14 responds by translocating in preparation for sporulation.281 Spo14 activity is integral for rescuing vesicular trafficking in a mechanism that responds to loss of PI-transfer protein Sec14. 282,288 Finally, Spo14/PLD1 has recently been shown to participate in mating and pheromone signaling pathways,274 and is a virulence factor integral for pathogenic yeast Candida albicans.275,276
6.1.2.1 Meiosis and prospore membrane formation
Nitrogen deprivation and exposure to non-fermentable carbon sources trigger sporulation in budding yeast of opposite mating type (a/α).289 Sporulation generates haploid daughter nuclei via meiosis I and meiosis II (see details in section 6.1). Following meiosis II, haploid nuclei are wrapped in newly synthesized membrane, the PSM, to generate spores.278 PSM formation and nuclei wrapping occurs near the spindle pole body (SPB, akin to the mammalian centrosome), and is dependent on Spo14. In fact, the Spo14 gene was originally identified in a screen for sporulation mutants.267 A strain of S. cerevisiae harboring a Spo14 point mutant, Spo14-3, resulted in early protein truncation at residue 313. Yeast homozygous for Spo14-3 resulted in loss of PLD catalytic activity and lack of sporulation, while yeast heterozygous for Spo14-3/Spo14 retained PLD activity and did not demonstrate any disruption in sporulation.204 These findings strongly suggest that Spo14 mutations are recessive, and that a single copy of a functional Spo14 gene is all that is necessary for its function in sporulation.
Spo14 is regulated via multiple mechanisms during meiosis and sporulation including increased protein expression, translocation, and post-translational modification. Spo14 activity is crucial during late meiosis. Spo14 RNA and protein induction is maximal at 12–20 hours post meiotic initiation, and Spo14 deletion strains demonstrate a ten-fold decrease in cells completing meiosis II compared to 1.5-fold drop in meiosis 1 completion. However, the initial increase in PLD activity during meiosis is due to translocation of cytosolic Spo14 to PSM near SPB, rather that protein induction. Translocation to PSM is likely due to PH-domain interaction with locally enriched pools of PI(4,5)P2, as PH-null Spo14 mutants fail to localize to PSM.283 The importance of translocation of basal PLD versus induction of Spo14 was shown using cyclohexamide, an agent that prevents new protein synthesis. Upon cyclohexamide treatment, changes in PLD activity and sporulation were not observed, demonstrating basal PLD translocation is sufficient for PSM formation. Later studies showed that Spo14 translocates and is subsequently hyperphosphorylated in response to growth conditions (ie. nitrogen deprivation and non-fermentable carbon source) rather than responding to induction of sporulation pathways.280 This was demonstrated using non-sporulating/non-mating yeast (a/a or α/α mating types), where Spo14 translocation and phosphorylation occur in response to growth conditions and independently of meiosis.280
PSM is synthesized de novo separate from the mother cell plasma membrane. Formation of these membranes is initiated and subsequently elongated at SPB by separate Spo14-dependent processes. PSM is not synthesized in the absence of functional Spo14. Lack of successful PSM formation and sporulation is a similar phenotype to that of yeast deficient for Spo20, a sporulation-specific SNAP-25 SNARE homolog.279 Spo20 is a downstream target of Spo14, and binds PA at a N-terminal amphiphathic helix.273,290 In Spo14 deletion mutants, Spo20 does not localize to PSM to facilitate prospore membrane fusion. However, generation of a chimeric Spo20 that is constitutively membrane-anchored, essentially bypassing the need for PSM-PA production, does not rescue vesicle fusion or PSM formation. From these studies it was concluded that Spo14, in addition to PA, participates in membrane trafficking and vesicle fusion.
While PA mediates protein localization and lowers the activation energy necessary for membrane fusion, Spo14 interacts with proteins implicated in meiosis. Immunoprecipitation and mass spectrometry proteomic analysis reportedly identified Mum2, a meiotic regulator of DNA replication machinery,291 as a Spo14 interacting protein.292 Another study found Sma1, a meiotic protein essential for sporulation, interacts with Spo14.293,294 Sma1 facilitates Spo14 localization,294 and this protein complex functions in PSM enlargement.293 Sma1-deficient yeast initiate PSM formation, but fail to enlarge PSM or form haploid daughter cells. Finally the Arf GTPase activating protein, Gsc1, regulates Spo14 activity during sporulation.285 Unlike PLD enzymes in higher eukaryotes, Spo14 is not activated by small GTPases including Arf. However, in cells, Arf cycling via Gsc1 is necessary for Spo14 activity during sporulation.285
PA identified as a transcription factor that regulates genes involved in phospholipid metabolism.295 In addition to PA generation and newly characterized protein interactions, Spo14 may also generate other lipid products. It is currently unclear whether Spo14 can utilize DAG as a nucleophile to generate bisphosphatidic acid (or subsequently semilysobisphosphatidic acid) via transphosphatidylation.294 Bisphosphatidic acid and semi-lysobisphosphatidic acid are proposed to induce membrane curvature and are components of the Golgi membrane that may be utilized during PSM formation and vesicular trafficking.
6.1.2.2 Sec14 bypass and vesicular transport
Spo14 is a nonessential gene in S. cerevisiae vegetative growth. However, as discussed in section 6.1.2.1 certain cellular functions require intact Spo14 catalytic activity. In 1998, Xie et al. described the requirement of Spo14 activity for the Sec14 bypass mechanism.282 Sec14p is the main PI-transfer protein in yeast, and is essential for in vitro transfer of monomeric PI or PC between lipid membranes296 (fungal/plant PITP and metazoan PTIP share no sequence identity, but are functionally indistinguishable).297 Through regulation of PI and PC content, Sec14p indirectly maintains the DAG content in the Golgi and thereby regulates trans-Golgi vesicle secretion pathways.298,299 Mutations in one of seven genes involved in PC synthesis and metabolism allows the cell to bypass the need for Sec14p, in a mechanism called Sec14 bypass.300 Under these circumstances, Spo14 activity is necessary but not sufficient for Sec14 bypass pathway.282 In SEC14 deficient mutants the bypass mechanism requires Spo14 activity to rescue TGN secretion. Spo14 activity maintains the PC to PI ratio in the Golgi in the absence of functional Sec14p by hydrolyzing PC to generate PA which is later hydrolyzed to DAG.
In contrast to the regulation of Spo14 during meiosis, phosphorylation at the LOCO/phos domain is not necessary for participation in the Sec14 bypass mechanism. Also, Arf GAP, Gcs1p, is regulated by DAG, and therefore is downstream of PLD in this pathway, rather than upstream of Spo14 as in sporulation. During Sec14 bypass, Spo14 activity is instead regulated by access to PI(4,5)P2 lipid cofactor. At the Golgi, production of PI(4,5)P2 is dependent on PI4-kinase, Stt4, and PI4P5-kinase, MSS4. Further studies have identified other non-stereotypical PI transfer proteins (PITPs) that also regulate Spo14 activity both during vegetative and Sec14 bypass conditions. For example, Sfh2p, a PITP that binds PI but not PC, colocalizes to endosomal membranes and is required for vegetative Spo14 activity in addition to Sec14 bypass catalysis.301
6.1.2.3 Mating
PLD activity has been implicated in signaling pathways involved in S. cerevisiae mating, including pheromone response274 and mating projection formation.302 Haploid spores generate small soluble peptidic pheromones that bind G-protein coupled receptors, Ste2 and Ste3, in the outer membrane of neighboring spores.303 Pheromones trigger MAPK and MAPK-independent pathways via these receptors to elicit changes in protein expression and cell polarization. Cdc42, a downstream target of active receptor, activates both MAPK-dependent and independent pathways. In combination with the dissociated βγ G-protein subunits Cdc42 binds Ste20p, a PAK (p21cdc42-activated) kinase, to activate Ste11p in the MAPK pathway and ultimately trigger changes in transcription. In a parallel but separate pathway, Cdc42 activation of Ste20p triggers formation of a protein complex called the polarisome that facilitates polarized cell growth (PCG) and actin cytoskeletal rearrangement. PCG occurs prior to mating projection (schmoo) formation. Unlike the transcriptional response, PCG is dependent on Spo14 catalysis. In mammalian cells Cdc42 is a well-characterized activator of PLD activity.304 Likewise, yeast Cdc42 activates Spo14 to generate PA that facilitates Ste20p kinase activation. Spo14 deletion mutants slow PCG and results in distorted cell and mating projection morphology.274
6.1.2.4 Pathogenic yeast virulence
The opportunistic human pathogen, Candida albicans, is troublesome in immune-compromised individuals and often results in lethal septic shock. C. albicans is a dimorphic fungus that transitions through several morphogenic states in response to extracellular stimuli.305 Dimorphic fungi reproduce in either a yeast or a hyphal form. The yeast form divides via daughter cell budding from a mother cell, whereas pathogenic hyphal cells elongate and divide by a septum that pinches off the daughter cell. PLD catalysis has been implicated in regulating C. albicans morphogenic transition from the yeast to hyphal form.275,306 Similar to Spo14, caPLD1 activity is increased in response to non-fermentable carbon source.306 While vegetative yeast growth is unchanged for caPLD1 deletion mutants, C. albicans deficient for this PLD activity (due to either mutant caPLD1 or the presence of alcohol results in transphosphatidylation product rather than hydrolytic product) do not form hyphae and are not capable of tissue invasion and epithelial cell penetration during host organism colonization.276,306 As such, caPLD1 has been referred to as a virulence factor.276 Kanoh et al. cloned caPLD1 from this pathogenic fungus, and demonstrate this enzyme shares significant sequence identity and biochemical properties to that of Spo14.270
6.2 Fission Yeast: PLD1
Similar to Spo14/PLD1 in budding yeast, a 157 kDa 2-HKD PLD enzyme has also been identified in fission yeast, Schizosaccharomyces pombe.269 This enzyme, called PLD1, bears significant sequence homology to Spo14, but with unique biochemical characteristics that have been linked to amino acid differences between the two enzymes. S. pombe PLD1 is oleate-stimulated (maximal activity at 5 mM), and PI(4,5)P2-independent. Harkins et al. postulate the absence of requisite PI(4,5)P2 activation may be due to proline to glycine point mutation in the putative polybasic PI(4,5)P2 binding domain.269 The proline is highly conserved in this polybasic region in other PI(4,5)P2-dependent PLD enzymes, including Spo14. Similar to requisite PLD activity during budding yeast sporulaiton, S. pombe PLD1 catalysis is integral for fission yeast sporulation and is involved in mating. Also similar to Spo14, this enzyme is capable of performing transphosphatidylation with branched chain alcohols as well as primary.269 However, calcium insensitive S. pombe PLD1 is differentially regulated and cannot compensate for nor rescue PLD-dependent functions in Spo14-deficient S. cerevisiae. This is likely due to differences in PI(4,5)P2 binding requirements.269
6.3 Dictyostelium PLD
Another type of fungi that is extensively studied is the unique slime mold Dictyostelium discoidium. This model organism possesses PLD activity, and similar to budding yeast Spo14/PLD1, this activity has proven integral for critical developmental processes. This slime mold is found in soil of Eastern North America and Eastern China, and is studied as a model organism because it exhibits several distinct life cycles dependent on environmental growth conditions.307 Also, Dictyostelium bears many similar signaling pathways and mechanisms to eukaryotes in which PLD participates.
Slime mold grows in monolayer or suspension cultures and feeds on bacteria. In the presence of ample nutrients, Dictyostelium exists as a haploid unicellular form that mitotically replicates. In response to low nutrients or high density, unicellular cells replicate in one of two cycles: sexual or assexual. These replication cycles are the reason that Dictyostelium are so intensely studied. Dictyostelium are referred to as social amoeba that exhibit social cooperation or altruism by sacrificing some individual cells for the benefit of the species.307
Sexual replication occurs upon contact with a haploid cell of opposite mating type, and the cells and nuclei fuse to form a diploid zygote.308 The zygote secretes cAMP and other chemoattractant molecules to coercively draw other cells near, whereupon the zygote cannibalizes them to harvest nutrients and form the cellulose-bound macrocyst structure.307 The macrocyst replicates via meiosis and then germinates. Signaling pathways and mechanisms in the sexual reproduction pathways have not been characterized, but this has been because the emphasis has been on the asexual cycle.
In the absence of fusion with opposite mating type cells, Dictyostelium respond to low nutrient and high cell density by secreting chemoattractant molecules. This facilitates quorum sensing and triggers cell signaling responses in neighboring cells. Unicellular forms constitutively express a glycoprotein, conditioned medium factor (CMF), which is only secreted in response to low nutrient starvation conditions. In response to quorum sensing molecule CMF, heterotrimeric G-protein signaling pathways ensue. Under low nutrient and high density conditions Dictyostelium secrete waves of cAMP.308 The waves of cAMP bind cyclin AMP-receptors (cAR), GPCRs at the surface of neighboring cells. Binding elicits signaling pathways that trigger cell migration and aggregation towards one another. A mound of cells forms, and continued waves of cAMP and bioactive molecule secretion, such as differentiation inducing factor (DIF-1), generate a molecular gradient of small molecules. This gradient elicits polarization of the cooperative cells into anterior and posterior regions, and DIF-1 induces nonuniform cell differentiation into one of two types of pre-cells that ultimately generate either a stalk or spore-harboring fruiting body. The polarized cell aggregate, called a slug, is able to migrate greater distances than unicellular forms and is protective from predatory consumption (e.g., C. elegans). Once a new location is selected, the pre-stalk and pre-spore forms further differentiate into mature stalk and spore, destined for cell death, or dispersal and germination, respectively.
PLD activity has been described in Dictyostelium for the different growth stages of the three distinct reproductive cycles. Three PLD transcripts were identified, plda, pldb, and pldc.309,310 The plda is constitutively expressed at unaltered levels in vegetative and reproductive cell types, whereas pldb mRNA message and protein levels fluctuate with changes in growth or reproductive cycle. As such, pldb is the most extensively studied isoform, and has been shown to participate in quorum sensing and facilitate polarized cell migration. This 867 aa enzyme is 32 % similar and 21 % identical to human PLD1, with conserved PH and CRI–IV domains, and loop and tail regions. The pldb is PI(4,5)P2-dependent and performs transphosphatidylation with primary alcohols. However, in contrast to human PLD, Dictyostelium pldb preferentially hydrolyzes ether-containing PE species.311
The pldb negatively regulates quorum sensing in two ways. First, PLD-generated PA counteracts cell responses to CMF by modulating heterotrimeric G-protein signaling responses and RGS (regulatory of G-protein signaling) regulation.309,312 Also, pldb-generated PA is suggested to facilitate cAR receptor internalization and recycling.309,310,312 Unicellular Dictyostelium treated with primary alcohol, or pldb deletion mutants aggregate at lower densities independently of CMF. This is likely due to enhanced cAR levels at the plasma membrane (lack of receptor internalization or recycling) and increased G-protein signaling in the absence of RGS modulation. The pldb overexpression mutants increase the density and CMF signaling threshold necessary to trigger unicellular aggregation.309
The pldb is also necessary for actin localization and actin-based motility in two ways. Pldb localizes to the leading pseudopodia extensions of the slug, and PLD-generated PA levels are highest at the leading edge, with a decreasing gradient towards the posterior.313 PA facilitates membrane curvature necessary for pseudopodia formation, but PA also activates PI4P5K, which generates PI(4,5)P2 in a positive feedback loop on pldb.311 In addition to activating PLD, PI(4,5)P2 localizes actin nucleating factors (Arp2/3 complex) to the leading edge of pseudopodia for F-actin polymerization.311 As a result of primary alcohol treatment, actin assembles in the nucleus and results in aberrant morphologies. In light of the importance of PA in specific signaling and structural capacities, pldb activity is integral to asexual reproduction in Dictyostelium. Further study will determine the role of PLD in vegetative or sexual reproduction cycles.
7. C. elegans PLD
A single PLD isoform has been cloned from the nematode Caenorhabditis elegans (C. elegans). This large 1427 aa enzyme maintains the conserved domains I–IV, similar to other PLD superfamily members, but the HKD motifs are separated by an exceptionally long loop region (300 aa). This PLD enzyme is ubiquitously expressed in pharyngeal muscles and neurons.314 However a functional role for this enzyme has not been demonstrated. C. elegans PLD is not necessary for development or sensory neuron signaling, and PLD knockout animals do not demonstrate a visible phenotype.315,316 Recent studies suggest this enzyme may participate in dynamic changes in phagosome structure.317 The function of this enzyme is the subject of ongoing investigation in whole animal studies.
8. Drosophila PLD
Similar to C. elegans, the fruit fly Drosophila melanogaster expresses a single dPLD protein (see a recent thorough review310) that maintains domain architecture similar to mammalian PLD enzymes: 2-HKD catalytic domains separated by an extended loop region (170aa), PX and PH domains, and requisite PI(4,5)P2 lipid cofactor for lipid hydrolysis. dPLD activity was initially described in larvae grown on ethanol-containing food. GTPγS and calcium stimulated phosphatidylethanol formation was attributed to the transphosphatidylation activity of a drosophila PLD, which was later cloned from the complete Drosophila cDNA library. In zygotes, PLD transcript is maternally derived, and native dPLD expression is subsequently observed throughout embryonic development and in adult tissues.318 In addition to the classic role of PLD in Drosophila Golgi secretory vesicle trafficking,318 dPLD has been implicated in rhabdomeres formation, which is important for phototransduction.319,320 In Drosophila, photoreceptors present at the apex (ie. the rhabodomere region) of polarized cells transduce sensory signals to the intracellular domain. dPLD deficient animals demonstrate aberrant rhabdomere formation,319 and decreased signal transduction rendering the animal less sensitive to light stimulus.320
9. Zebrafish PLD
Seminal work in characterizing the function of PLD in the context of a whole vertebrate was recently performed using zebrafish, Danio rerio. In 2003, Ghosh et al. partially cloned a PLD enzyme (aa 380–916) from zebrafish embryos and determined it was expressed during gastrulation.321 Zeng et al. followed up this study with cloning the complete zPld1 sequence.322 This 1042 aa enzyme contains the two HKD motifs present in most eukaryotic PLDs, and is 64–68 % and 50 % homologous to mammalian PLD1 and PLD2, respectively. zPld1 regulation is also similar to mammalian PLD, with conserved PKC (1–314 aa) and Rho (859–1010 aa) binding domains. In vitro characterization shows this enzyme is activated by Arf1 and PKCα. A second zebrafish PLD isoform, 927 aa zPld2, has been partially cloned.
Zebrafish are uniquely suited to whole organism studies of PLD activity and function because, as Zeng et al. demonstrated, PLD activity can be stimulated and measured with whole organism treatment of phorbol ester (PMA) and deuterated n-butanol. zPld1 activity was monitored using MS by monitoring deuterated phosphatidylbutanol formation (cell based MS assay described in section 2.2.6, and detailed129). Similar to other zebrafish phospholipases, in whole animal studies, zPld11 was determined to be involved in vascular development. This was determined using two parallel methods: (1) zPld11 was either knocked down using targeted morpholinos to disrupt zPld11 translation and mRNA splicing, or (2) zebrafish were treated with n-butanol to divert zPld11 activity to transphosphatidylation. Unlike the development of other systems including motor neuron organization, there was a severe deficiency in intersegmental blood vessel formation. More recent studies have observed zPld11 mediates Golgi secretory vesicle formation.323 Aberrant zPld11 activity due to unregulated Arf-stimulation results in decreased lipid absorption in the intestine. Utility of zebrafish in measuring PLD activity and monitoring substrate and product localization in a whole vertebrate animal will facilitate determination of the function of PLD with respect to the whole organism.
10. Mammalian PLD
While PLD was first identified in plants in 1947,14 PLD activity was not described in mammalian tissues until 1973 by Kanfer and colleagues.324 Subsequently, multiple mammalian PLD enzymes and isoforms have been cloned, rigorous biochemical characterization performed, and extensive cell signaling studies undertaken. From this, mammalian PLD enzymes have been implicated in critical cell signaling pathways involved in development and cell death. These pathways modulate cell growth, proliferation, survival, and migration. As such, aberrant PLD activity has been detected in disease states, including cancer, inflammation, pathogenic infection, and neurodegeneration.
10.1 Isoforms
Cloning of plant and yeast PLD enzymes facilitated cloning of a full length PLD enzyme from HeLa cell cDNA205 and rat325 PLD1. Shortly thereafter, a second mammalian PLD enzyme, PLD2, was cloned.326,327,328 These two isoforms share 50 % sequence homology, mostly at the catalytic domain that includes two conserved HxKxxxxDxxxxxxG(G/S)xN catalytic motifs separated by variable length of sequence predicted to form a thermolabile loop. N-terminal to a conserved polybasic PI(4,5)P2 binding domain,279 the loop region differs for these two PLD isoforms. PLD1 harbors an extended thermolabile loop prone to proteolytic cleavage.329 The length of this loop region is variable dependent on the splice variant330 (PLD1a = 116 aa versus PLD1b = 78 aa), while PLD2 does not possess a significant loop region (4 aa predicted loop). Shortened splice variants of both PLD1 and PLD2 have been identified that compose catalytically inactive enzyme.328 Expression of these inactive enzymes is observed in different tissues, including the brain, but their function is unknown (see section 10.3.2.4).
At the amino-terminus, PLD1 and PLD2 share similar regulatory domains to PLDζ and Spo14, including PX and PH lipid binding domains. The PX domain binds polyphosphoinositides with high specificity, and anionic lipids with lower specificity331 (see section 10.3.3 for details), but this domain has also been implicated in protein interactions with regulatory proteins, including Dynamin and Grb2 (see section 10.3.4 for details). Tyrosine residues in the PLD2 PX domain can be phosphorylated. The PH domain binds anionic phospholipids with low specificity. This domain is palmitoylated at two conserved cysteine residues that facilitate protein localization and do not impact catalytic activity (Figure 8).
Figure 8.

Despite similarities between the regulatory domain architecture of the classic PLD isoforms, the majority of the sequence divergence between these two mammalian PLD isoforms exists at the amino-terminus. Deletion of the PX domain enhances PLD1 activity. Truncation of the PLD1 PX domain and a portion of the PH domain further increases activity. However, conserved residues in a predicted α-helix at the C-terminal end of the PH domain are necessary for catalysis in the liposome activity assay332 (unpublished data, Henage, Selvy and Brown). Cell-based studies demonstrate that N-terminally truncated PLD1 enzymes maintain high activity levels upon cellular stimulation (see section 10.3.4 for details). This suggests, similar to the extended loop region of PLD1, the amino terminus of PLD1 is autoinhibitory, whereas deletion of the amino-terminus of PLD2 decreases activity and suggests PLD2 amino-terminus might facilitate increased basal activity.
PLD1 and PLD2 share homologous C-terminal sequences. The specific identity of the residues in this sequence must be maintained for mammalian PLD activity. Non-conserved point mutation or deletion impairs catalytic activity.333 The C-terminal residues are suggested to interact with the catalytic core.333 Studies by Steed et al. support this with identification of naturally occurring PLD2 splice variants with truncated C-termini that result in significantly decreased activity.328
The bulk of mammalian PLD activity is attributed to these classical PLD isoforms. These two isoforms, and subsequent splice variants, hydrolyze phospholipids to generate phosphatidic acid, and readily perform transphosphatidylation in the presence of low concentrations of alcohol to perform headgroup exchange and phosphatidylalcohol formation. Both isoforms are capable of hydrolyzing PC, PE, PS, LPC, and LPS, but are not capable of hydrolyzing PI, PG or cardiolipin. Although PA is the major hydrolytic product, hydrolysis of a lyso-lipid generates LPA. Recently, mammalian PLD was proposed to generate cLPA from lyso-lipids.334 cLPA could be formed similar to the transphosphatidylation of LPC observed with autotaxin, where the internal sn-2 hydroxyl group serves as the secondary nucleophile to cyclize the product (see section 2.1.2.6).
In addition to PLD1 and PLD2, two mammalian enzymes have been identified with significant sequence homology to viral and prokaryotic PLD. PLD3, also called Hu-K4, bears significant sequence homology to viral PLD enzymes K4 (48 %) and p37 (25–30 %)335 (see section 3 for details on viral PLDs). This enzyme has two HxKxxxxD/E motifs (in one motif the aspartate is mutated to glutamate) and was recently shown to harbor a predicted N-terminal type II transmembrane domain.336 This facilitates protein insertion into the ER, with 38 aa exposed to the cytosol, and the large C-terminus, including the HKD motifs and multiple glycosylation motifs, exposed to the ER lumen. Catalytic activity has not been detected for this PLD isoform, but it has been postulated that this enzyme might hydrolyze lipids at the lumenal phase of the ER, or may not bear lipase activity, similar to the endonuclease activity of viral K4.336 The murine homolog of this enzyme, Sam9, is expressed in the forebrain during late neural development.337 Catalytic activity has yet to be defined for this enzyme as well.
A single-HKD enzyme with homology to Nuc endonuclease, called mitoPLD, was described.338 This enzyme bears an N-terminal mitochondrial localization sequence (MLS) in place of PX or PH lipid binding domains. However, this localization sequence is not processed, and instead may facilitate insertion or anchoring into the outer mitochondrial membrane. This enzyme is predicted to homodimerize, similar to Nuc. This is not the first description of PLD activity localized at the mitochondria,339 but previous reports suggested mitochondrial PLD hydrolyzed PE to generate PA. Instead, mitoPLD hydrolyzes cardiolipin, an abundant mitochondrial lipid, to generate PA. This product facilitates mitochondrial fusion events, since overexpression of mitoPLD results in formation of a single large perinuclear mitochondrion, whereas expression of a catalytically inactive mutant resulted in fragmented mitochondria.340
10.2 Tissue expression and subcellular localization
The classic PLD isoforms, PLD1 and PLD2 are expressed in nearly all mammalian tissues. Due to the lack of clean, specific antibodies northern blot analysis has routinely been used to characterize PLD expression patterns. PLD1 and PLD2 are both robustly expressed in heart, brain, and spleen. PLD1 exhibits low expression in peripheral blood leukocytes and PLD2 is poorly expressed in liver and skeletal muscle.
While classic PLD isoforms, PLD1 and PLD2, catalyze the same reaction, and utilize similar substrates to generate PA or transphosphatidylation species, these enzymes are differentially localized within the cell. There has been some discrepancies in reports of subcellular localization of each PLD isoform, but this could be due to differences in the cellular systems, growth conditions, or the methods of detection (ie. subcellular fractionation or immunofluroescence of native versus tagged proteins; note that tags can impact localization).
10.2.1 PLD1 subcellular localization
It is generally accepted that PLD1 is localized to perinuclear membranes, including early endosomes, and Golgi under basal conditions,326,341 with no reported difference in localization for splice variants PLD1a and PLD1b.341
Different regions of the protein contribute to basal subcellular localization. Truncation and point mutations have been used to identify the contribution of these different regions. Sugars et al. determined PLD1 basal localization is dependent on the PH domain, specifically an acidic region (residues 252 and 253) thought to be important for IP3 binding, and conserved tryptophan residues in a predicted α-helix that is critical for catalytic activity342 (see section 10.4 for details). PLD1 is palmitoylated on two cysteine residues in the PH domain (see section 10.3.3 for details), and this lipid modification supports basal protein localization at intracellular membranes.332 Point mutation of these cysteine residues impairs palmitoylation and results in aberrant protein localization. In the presence of serum protein basally localized to plasma membrane,342 whereas in the absence of serum these palmitoylation mutants are dispersed in the cytosol and translocation is triggered to the plasma membrane only upon serum stimulation.343 Hughes and Parker suggested the C-terminal residues of PLD1 might also be necessary for endosomal localization.341 This region of the enzyme is certainly necessary for catalytic activity, and native splice variants of PLD1a and PLD1b that lack these C-terminal residues do not basally localize to endosomes. However, it has been suggested that the C-terminus is integral for catalysis because it supports the structure of the active site.333 Therefore enzymes lacking this region may not in fact be folded properly, and this could result in aberrant localization rather than the C-terminus itself directly participating in protein localization. Catalytic activity is not requisite for protein localization. Catalytically inactive point mutants (PLD1b K466E and K860E) localize to perinuclear endosomes similar to wildtype enzyme.341 It should be noted that individual domains of PLD1 expressed in isolation do not localize similar to the full enzyme.342,343,344 This suggests that multiple components and regions of the enzyme participate in basal localization.
Upon cell stimulation, PLD1 translocates to the plasma membrane or late endosomes. However, the type of stimulation results in differences in translocation, for example serum stimulation in Cos7 cells results in translocation to late endosomes and plasma membrane, whereas PMA stimulation triggers translocation to the plasma membrane343 (unpublished data Selvy and Brown). PLD1 translocation to the plasma membrane in response to cell stimulation is thought to be due to PI(4,5)P2 binding at the polybasic binding region between the HKD motifs.343 Point mutations in this polybasic region, including mutation of highly conserved arginine residues 691 and 695, impair PLD1 translocation to the plasma membrane upon stimulation. These data are supported by evidence that production of PI(4,5)P2 positively increases PLD activity. Finally, N-terminal PX and PH domains facilitate recycling to specific intracellular membranes.343
Nuclear PLD activity that responds to GTPγS via Rho GTPase, but not Arf activation, has been described.345 A recent report suggests this activity is due to nuclear import of PLD1 via direct protein interaction with importin-β.346 A highly conserved putative nuclear localization sequence (NLS) was identified between residues 553 and 564 for PLDb (KxRKxxKxxxxK). Importin-β binds the NLS and facilitates active transport into the nucleus. The NLS sequence exists in the loop region between the catalytic HKD motifs, and is not present in PLD2. Mutation of any or all of the conserved residues in this NLS sequence impairs nuclear localization. Similar to the plasma membrane, PC is the major phospholipid present in nuclear membrane. Nuclear PLD activity generates PA that is rapidly metabolized to DAG. Jang et al. report this PLD activity stimulates nuclear PKCα and ERK phosphorylation and activation.346 Catalytically-inactive PLD1 point mutants and PLD-selective small molecule inhibitors (section 11) disrupt nuclear PKCα and ERK activation, supporting the lipase-dependent activation mechanism. Immunofluoresence microscopy and subcellular fractionation analysis have also identified a nuclear PLD2 population, however a putative NLS has not been identified in PLD2 sequence,346 and further study is necessary to determine the mechanism for PLD2 nuclear import. The intriguing report of a PLD1 nuclear import mechanism begs further investigation to determine the potential signaling pathways modulated by nuclear PA production.
10.2.2 PLD2 subcellular localization
In contrast to the intricate regulation of PLD1 via protein translocation, PLD2 is generally observed to be constitutively localized to the plasma membrane under basal conditions and translocates to recycled vesicles with agonist-stimulated and desensitized receptors.326 PLD2 also binds to and localizes with β-actin,347 and in response to EGF-stimulation localizes at membrane ruffles.348 Instead of translocation upon cell stimulation, PLD2 activity and protein interactions are modulated via phosphorylation at multiple residues (see section 10.3.2.2 for details).
PLD activity has also been described for crude preparations of mitochondria. Biochemically, this PLD activity differs from that attributed to MitoPLD. In these mitochondrial fractions, calcium-stimulation of an unknown enzyme hydrolyzes PE to generate PA. This enzyme may not be a member of the PLD superfamily since it is unable to perform transphosphatidylation.349
In some studies, PLD1 and PLD2 were observed to colocalize at perinuclear and plasma membrane under basal conditions. The finding that PLD isoforms may form intracellular complexes might explain why introducing catalytic point mutants results in dominant negative effects and reduces basal PLD activity.114,350
10.3 Regulation
PA is a critical lipid second messenger for a range of signaling cascades, but makes up 1-4 % total lipid in the cell.351 PLD contributes to signaling pools of PA, and therefore this enzyme is under tight regulation by elaborate mechanisms including cofactor availability, signal induced subcellular translocation, post-translational modifications, and protein-protein interactions.
10.3.1 Divalent cations
Similar to other PLD superfamily members, mammalian PLD catalysis responds to divalent cation concentrations. However, in contrast to other superfamily members, including many plant enzymes, mammalian PLD catalysis is largely unresponsive to calcium concentration in vitro.330 In vivo, however, PLD activity is mediated by cellular calcium fluctuations, which suggests calcium facilitates protein-activator activation, such as PKC, and indirectly modulates PLD activity. In contrast, optimal catalysis levels require the presence of magnesium. In vitro PLD activity responds to changes in magnesium concentration, with half maximal Arf-activated PLD activity at 100 μM magnesium. This concentration of magnesium may facilitate catalysis directly, because this divalent cation does not impact in vitro protein-lipid binding (unpublished data Selvy and Brown).
10.3.2 Post-translational modification
Shortly after cloning of the first mammalian PLD enzymes, reports emerged that these enzymes were post-translationally modified in response to specific signaling pathways. Further characterization highlights lipid modification, phosphorylation, ubiquitination, and proteolytic mechanisms of PLD regulation.
10.3.2.1 Lipid modification
PLD1332 and PLD2352 are post-translationally palmitoylated at two cysteine residues in the PH domain. In vitro, this modification does not significantly impact catalytic activity, suggesting palmitoylation serves to regulate protein localization.332,342 In the cell, however, this lipid modification facilitates protein sorting into specific intracellular and plasma membrane domains including lipid rafts (as mentioned in section 10.2). In PLD1, Cys240 and Cys241 are palmitoylated, with Cys241 the dominant modification site. As determined by modeling the PLD1 PH domain onto the crystal structure of PLCδ PH domain, these residues exist in a region predicted to be an extended loop of the PH domain.332,342 Lipid modification requires expression of full length, catalytically-competent PLD1. Expression of the PH domain in isolation, or of severely truncated constructs of the enzyme do not result in modification.342 ΔPX PLD1 construct, lacking first 210 amino acids, is the shortest truncation that can be expressed that yields similar localization and catalytic activity to wildtype PLD1, and this truncation construct is lipid modified.
10.3.2.2 Phosphorylation
Mammalian PLD isoforms PLD1 and PLD2 are phosphorylated in response to signal transduction as a regulatory mechanism. PLD was originally determined to be phosphorylated when it was immunoprecipitated with polyclonal phospho-tyrosine antibodies.353 Since this initial discovery, rigorous biochemical and molecular biology techniques have been employed to determine specific residues that are modified and the resulting impact on PLD activity and signal transduction.
PLD1 regulatory mechanisms reported to date largely center on protein translocation, while multiple PLD2 phosphorylation sites have been described. Therefore, few reports of PLD1 phosphorylation exist. Early studies used sequence analysis to identify two putative tyrosine phosphorylation sites [RK](x)2/3[DE](x)2/3Y in PLD1 (aa 288–295 and aa 807–815). Evidence of phosphorylation at these residues does not exist. PLD1 phosphorylation occurs in response to H2O2 stimulation, and increased phosphorylation has been shown to correlate to increased lipase activity.354 c-Src has also been reported to phosphorylate PLD1, but this does not modulate lipase activity, rather modulates c-Src activity for downstream protein substrate.355 PKC isoforms are also known to modulate PLD1 activity.356 Despite the evidence that PKC activation of PLD1 is phosphorylation-independent, three residues are phosphorylated by PKC (Ser2, Thr147, Ser 561).357 In vitro catalytic analysis demonstrates that PKC phosphorylation of PLD1 likely serves as an inhibitory mechanism.358 Maximal PKC-stimulated PLD activity is observed roughly one minute following PLD-PKC mixing. The timecourse of this activation suggests protein-protein interactions induce PLD activation (further discussed in section 10.3.4.2). Maximal PLD1 phosphorylation at threonine 147, however, occurs nearly 60 minutes after PLD-PKC mixing.358 Maximal PLD1 localization to the membrane also occurs at 60 minutes.
Multiple PLD2 residues are reportedly capable of being phosphorylated by numerous kinases. Gomez-Cambronero and colleagues have characterized tyrosine residues in the PLD2 PX domain that mediate lipase activity and binding with SH2 domains. Tyr169 is highly conserved in all eukaryotic PLD and is proposed to be important for high PLD2 basal activity.359 Tyr179 is present only in mammalian PLD and has been proposed to negatively regulate Ras signaling359 (Ras/MAPK signaling is increased nearly two-fold with Y179F mutation). Phosphorylation at these residues recruits the SH2 domain of Grb2, which binds the Ras GEF, Sos, via its SH3 domain, to activate MAPK pathway.359 The kinase responsible for phosphorylation of these residues has not been identified. However, kinases responsible for phosphorylation at other PLD2 residues have been identified. Tyr175 exists in a consensus Akt phosphorylation site, and was identified using a polyclonal antibody for tyrosine phosphorylation at these consensus sequences.360 Phosphorylation at Tyr175 reportedly increases DNA synthesis via MEK activation.
Recently, a better understanding of the regulation of PLD2 activity via phosphorylation was reported.361 Cycling of phosphorylation and dephosphorylation of PLD2 results in differences in lipase activity and downstream signaling consequences. PLD2 binding Grb2 via phosphorylated tyrosine residues in the PX domain results in increased lipase activity (see section 10.3.4.3 for details), while dephosphorylation of these residues by tyrosine phosphatase, CD45, increases cell proliferation.361 Further studies have used MS-based proteomic analysis to identify other modified residues.362 Epidermal growth factor receptor (EGFR) negatively regulates PLD2 lipase activity via phosphorylation at Tyr296. In contrast, JAK3 increases PLD2 lipase activity via Tyr415 phosphorylation. Finally, Src, also shown to modify PLD1, phosphorylates Tyr511 on PLD2. The latter modification does not directly modulate lipase activity, instead likely impacts protein interaction with Src and facilitates downstream events, similar to Src interaction with PLD1. Multiple phosphorylation modifications can be integrated to finely tune the activity level of PLD2 dependent on signaling requirements.
10.3.2.3 Ubiquitination
A recent report demonstrated a previously uncharacterized post-translational modification of PLD1, but not PLD2, important for modulating both protein localization and curbing lipase activity.363 PLD1 is multi-monoubiquitinated at the PH domain in a catalytic and palmitoylation-dependent manner. Catalytically-inactive point mutants are not ubiquitinated, and treatment with PLD-selective pharmacological inhibitors (see section 11) but not primary alcohol, disrupts PLD1 ubiquitination. Also, disruption of PLD1 palmitoylation impairs ubiquitination. Taken together, this suggests that properly localized and catalytically-competent PLD1 allows ubiquitination, and this modification is not a substrate-product feedback mechanism. The precise E3 ubiquitin ligase responsible for this modification is unknown, but following ubiquitination PLD1 is shuttled to the proteasome for degradation rather than the lysosome. Also, this modification translocates protein from endosomal membranes to an enlarged vesicle structure present in all cells transfected with stably ubiquitinated PLD. These stably-ubiquitinated constructs are not processed by de-ubiquitinating enzymes. As this modification results in changes in PLD1 localization and marks PLD1 for proteosomal degradation, ubiquitination of PLD1 is likely an important regulatory mechanism to change or curb lipase activity.363
10.3.2.4 Proteolysis
Classic mammalian PLD isoforms PLD1 and PLD2 have been implicated in pro- and anti-apoptotic signaling mechanisms, and were recently reported to be substrates for proteolytic caspase cleavage. Caspase cleavage of the PLD isoforms appears to divergently regulate these enzymes during apoptotic signaling. In vitro364 and in vivo329,365 studies demonstrate PLD1 is cleaved in multiple locations by activated caspase 3, 7, and 8, while PLD2 is cleaved at several sites by caspase 3, and 8. During apoptosis initiation, caspase 8 cleaves pro-caspase 3 to generate active caspase 3. Caspase 3 cleaves amyloid β4a precursor protein, making this enzyme the dominant caspase in neuronal cell death mechanisms in Alzheimer’s disease. Caspase-3 cleavage of PLD2 occurs at two or three sites near the N-terminus (aa13–28, a region N-terminal to the PX domain) and does not result in significant changes to molecular weight, catalytic activity, localization, or apoptotic signaling.329,364 PLD2 renders an anti-apoptotic response, likely via induction of anti-apoptotic protein expression (Bcl-2 and Bcl-XI) and down-regulation of pro-apoptotic proteins (Egr-1 and PTEN). Inhibition or RNAi knockdown of PLD2 increases apoptotic signaling.
In contrast, caspase proteolysis appears to be a significant regulatory mechanism for PLD1. In vitro, PLD1 is cleaved by caspase 3 in three positions (Asp41, Asp545, Asp581).364 In vivo, position 545 is the dominant cleavage site.329 This residue lies in the PLD1 loop region that separates the two catalytic HKD motifs. Cleavage at this position produces a 56 kDa C-terminal fragment (CF-PLD1) which localizes to the nucleus via an exposed nuclear localization sequence (see section 10.2.1 for details), and a 60 kDa N-terminal fragment (NF-PLD1) that remains in the cytosol.329 Full length PLD1 activity is protective against apoptosis by suppressing p53 signaling. NF-PLD1 acts as a dominant negative for full length PLD1 (via hydrophobic interactions), inhibiting PLD1 activity, and resulting in de-repression of p53.365 Therefore, caspase cleavage of PLD1 decreases in vivo activity, and induces p53-dependent apoptotic signaling. Steed et al. identified a PLD1 splice variant, PLD1c, that expresses a PLD1 enzyme with an early stop codon at residue 513.328 This protein is expressed in human brain, and may function in a pro-apoptotic mechanism, similar to NF-PLD1. Further study of this truncated splice-variant and NF-PLD1 induced signaling is necessary. Jang et al. demonstrated PLD1 proteolytic processing is pathologically relevant.329 Analysis of post-mortem brain tissue from Alzheimers patients demonstrated increased active caspase 3 and evidence for caspase-proteolyzed PLD1 fragments, compared to age-matched brain tissue.
10.3.3 Lipid cofactors
PLD localization and subsequent post-translational modification have a significant impact on lipase activity. In cells, lipid cofactors are thought to mediate subcellular localization through directly interacting with lipid binding domains of the enzyme (see section 10.2.2), as deletion or mutation of these domains changes subcellular localization. In some cases the mutant constructs change the ability of the enzyme to interact with membranes basally or change translocation of the enzyme to membranes upon cell stimulation. Recruitment of PLD upon PIP2, or PIP3 production allows upstream lipid kinases or phosphatases to mediate PLD lipase activity. It has been observed that when PLD fails to localize properly or be recruited to the proper membrane substrate upon stimulation total lipase activity is impaired.
In vitro, phospholipids directly and indirectly modulate lipase activity. Many of the observed in vitro effects of specific lipid species must be rigorously confirmed, because, as discussed in section 2.2.4, the properties of the lipid substrate presentation can modulate PLD activity in ways that may or may not be physiologically relevant. For example, inclusion of high concentrations of negatively charged phospholipid may impair the ability of the enzyme to interact with the lipid interface or with substrate head group. Also, lipase activity on lysolipid substrates is significantly enhanced when presented in a lyso-lipid micelle when compared to more complex presentations, such as lyso-lipids in a diacyl phospholipid liposome (unpublished observations Spencer, Scott and Brown). As discussed in section 2.2.4, this is likely due to headgroup access, rather than a direct allosteric modulatory affect on the enzyme.
The presence of some lipid species can directly affect protein-interface binding (Ks) by directly binding the enzyme. Separate from the active site, three other allosteric lipid binding sites have been described for PLD1 and PLD2, including the PX, PH, and polybasic PI(4,5)P2 lipid binding motif. The PX domain binds polyphosphoinositides [PI(3,4,5)P3≫PI(3)P>PI(5)P>other PIs] with high specificity at the putative primary binding pocket composed of conserved lysine and arginine residues.331 At a secondary site, likely in the form of an exposed protein surface rather than a binding pocket including a conserved arginine (present in PLD1 and not PLD2), anionic lipids including PA and PS also bind. However, in comparison to the other two lipid binding domains, the PLD PX domain binds lipids with poor affinity. This suggests the PX domain likely acts as a tertiary regulatory domain, to fine tune protein-lipid interactions initiated by another lipid binding site.
The PH lipid binding domain binds PI(3,4)P2 and PI(4,5)P2 with specificity over other phosphoinositides.283,366 However, as discussed in section 10.2.2, this domain is lipid modified, and many of the observed effects of deletion of this domain may be due to the absence of this palmitoylation. In vitro, the entire PH domain is not requisite for PLD activity, although deletion of a conserved alpha-helix at the C-terminus of the PH domain does impair lipase activity.
Finally, the polybasic PI(4,5)P2 binding motif binds PI(4,5)P2 with high specificity and affinity.279 Lipid binding at this motif facilitates interfacial lipid interaction and enhances catalytic activity. Human PLD1 bulk lipid binding constant (Ks = 10 μM) for PE:PC:PI(4,5)P2 lipid vesicles (87:8:5 mol %) is more than 7-fold higher than bulk lipid binding constant for PE:PC vesicles (unpublished data Selvy and Brown). Optimal PI(4,5)P2 mol %, 5–8 %, in a phospholipid vesicle enhances stimulation by regulatory proteins including Arf GTPase125 (see section 10.3.4.1). Some reports of in vitro lipase activity can be measured for full length PLD in the absence of PI(4,5)P2 with the addition of molar concentrations of ammonium sulfate (optimal activity at 1–1.6 M).367,368
Other reports of modulatory phospholipids are scattered in early PLD literature, but have not been followed up on. An intriguing observation by Nakayama et al. suggested that PE, including dioleoyl and plasmalogen-rich species but not dipalmitoyl-containing species, enhances PC hydrolytic activity of PLD isolated from bovine kidney.367 Another report suggested that that PI, LPI, and LPS, but not PS, negatively impact PLD activity.369 It is unknown whether these effects are direct or indirect and whether they were specific to the in vitro assay format.
10.3.4 Regulatory proteins
With increased ease of recombinant PLD expression (see section 10.4) and measurement of in vitro PLD activity (see section 2.2.5), a growing number of proteins have been reported to modulate PLD activity. Some of these proteins have been shown to directly modulate mammalian PLD activity through a protein-protein interaction; those are described here, whereas others may indirectly regulate PLD and participate in PLD signaling pathways, these proteins are detailed in section 10.5 and Table 5.
Table 5.
Mammalian PLD regulatory proteins
| CLASS | ACTIVATOR | PLD ISOFORM | CONSEQUENCE |
|---|---|---|---|
| small GTPase | Arf | PLD1, PLD2 | activate (kcat) |
| RhoA | PLD1 | activate (Km) | |
| Rac1 | PLD1 | activate (Km) | |
| Rac2 | PLD2 | activate | |
| Cdc42 | PLD1 | activate (Km) | |
| Kinase | PKC | PLD1 (PLD2) | activate (kcat & Km) |
| Src | PLD2 | phosphorylate | |
| Other | Gβγ | inhibit | |
| Grb2 | PLD2 | activate | |
| F-actin | activate | ||
| G-actin | inhibit | ||
| Amphyphysin II | inhibit | ||
| AP3/AP180 | inhibit |
10.3.4.1 Small GTPases
Small GTPases were the first proteins demonstrated to directly modulate PLD activity through allosterically binding PLD. These enzymes are conformationally-activated upon binding GTP in place of constitutively-bound GDP, sometimes with the aid of guanine exchange factors (GEF) proteins, in response to signal transduction (see section 10.5 for example of functional consequences of small GTPase activation). GTPase activating proteins (GAPs) functionally inactivate the GTPases through facilitating intrinsic GTP hydrolysis. Subfamily members of the Ras GTPase superfamily, including Arf128,370 and Rho family of GTPases, 356,371,372 stimulate PLD activity in vitro.
Arf GTPases, including Arf1 and Arf6, stimulate PLD activity.128 These were the first proteins demonstrated to activate mammalian PLD in an in vitro reconstitution system. Early in vitro characterization of PLD1 and PLD2 suggested that PLD1 alone was stimulated by Arf.326 Subsequent studies have shown that PLD2, while not activated to the same extent, can be stimulated 2-fold over the already high basal activity with GTPγS-activated Arf.327,373 Henage et al. demonstrated that Arf1 increases total maximal activity (kcat) in a concentration dependent manner. At 150 nM Arf1, PLD1 activity increased 4 to 6-fold over basal levels.125 Arf stimulation is strongly dependent on the PI(4,5)P2 mol %. This has lead some to speculate that Arf may indirectly activate PLD by rearranging the phospholipid head groups at the interface in a PI(4,5)P2 dependent fashion.374 This may be true, but we have recently demonstrated that Arf activates PLD in the absence of PI(4,5)P2375,376 (unpublished data Selvy and Brown), suggesting possibly a second mechanism of activation for Arf. Intriguingly, synergistic stimulation of PLD1 activity is observed when Arf is combined with PKCα or Rho family GTPases.125 This demonstrates that Arf acts in concert with other modulatory enzymes to titrate the PLD response, and this finding could be of immense consequence in vivo. Some groups have attempted to identify the precise PLD binding site for Arf,377 but to this date the site has not been unambiguously determined. In vitro, Arf activates N-terminally truncated PLD1125 and PLD2,373 therefore the site likely exists somewhere in the catalytic domain. Arf is myristoylated at its amino-terminus. Arf-activation of PLD does not require this lipid modification, but stimulation is enhanced with N-myristolated Arf. In fact, the N-terminus Arf is the specific region implicated in PLD interactions.378
The Rho family of GTPases, including RhoA, Cdc42, Rac1 and Rac2 directly activate mammalian PLD. Rho, Cdc42, and Rac1 are binding activators of PLD1, and stimulate substrate binding affinity (1/Km).125 Arf and Rho family GTPases synergize to significantly increase PLD1 activity beyond an additive response. PLD1 does not have a putative CRIB (Cdc42 and Rac-interactive binding) motif, but using truncation deletions, the Rho family-PLD1 binding site was mapped to a region in conserved domain IV in the carboxy-terminus of PLD1.379 In a GTP-dependent mechanism, the Rho family GTPases bind PLD through the switch I region.304 However, binding occurs independently from activation. Geranylgeranylation of Cdc42 is not required for PLD binding, but is required for PLD activation.304 Cdc42 activation of PLD1 is mediated through the Rho-insert region, an alpha helix conserved in all Rho GTPases. However, this insert is not necessary for RhoA or Rac activation of PLD1.380 Rho, Cdc42, and Rac1 selectively activate PLD1. However, a recent report suggests that Rac2 may activate PLD2 via previously uncharacterized mechanism.381 This report identifies two poorly conserved CRIB motifs (CRIB1 aa 255–270, and CRIB2 aa 306–326) in or near the PH domain of PLD2. Rac2 co-immunoprecipitates with PLD2, and mutation within these regions disrupts this interaction.381
Two other Ras GTPases have been proposed to directly modulate PLD. RalA, a Ras-like GTPase implicated in cancer cell transformation, co-immunoprecipitated with PLD1 but not PLD2.382 In another study, identification of the RalA binding site on PLD1 was attempted.377 This study suggested that RalA binds at a site independent of Arf, allowing Arf and RalA to synergistically activate PLD1.377 In vitro, RalA enhanced PLD1 activity in a GTP-dependent mechanism.377,382 Rheb, a member of the Ras GTPase family, has also been reported to directly activate PLD1 in vitro.383
10.3.4.2 Kinases
As mentioned in section 10.3.2.2, PLD is phosphorylated post-translationally as a regulatory mechanism. Therefore, it is not surprising that kinases directly interact with PLD to regulate activity. Protein kinase C (PKC) isoforms are the most well studied kinases that directly interact with PLD. Classic PKC isoforms α, β, and γ are stimulated by calcium and DAG, and are therefore responsive to PMA-stimulation. In cells, these classic isoforms stimulate PLD1 and PLD2 activity downstream of PLC activation. PKCα phosphorylates PLD1357 and PLD2384 at serine and threonine residues, but activation is not phosphorylation-dependent. In timecourse studies, PMA-induced PLD activity occurs immediately, and phosphorylation only occurs later with a concomitant decrease in lipase activity, suggesting phosphorylation decreases PLD activity.358,384 The PKC binding domain was mapped to the amino-terminus of PLD1,385 however, PKC is able to activate N-terminally truncated PLD1 in a phosphorylation-independent mechanism.125 PKC modulates PLD activity in a bimodal fashion. PKC enhances kcat as well as substrate binding (Km), and therefore synergistically activates PLD1 in combination with catalytic activator Arf GTPase.125 However, amino-terminally truncated PLD1 constructs only show enhanced Km in response to PKC.
10.3.4.3 Other regulatory proteins
Numerous proteins have been reported to modulate PLD activity in response to signaling pathway activation, and a number of them have been demonstrated to do so directly. PED/PEA-15 (phosphoprotein enriched in diabetes/phosphoprotein enriched in astrocytes) is overexpressed in many tissues in type II diabetes patients. This protein directly binds CR IV of PLD and enhances PKC-activation of PLD.386 This interaction impairs insulin regulation of the glucose transporter and insulin secretion, whereas competing for the PED/PEA-15 protein interaction with expression of the PLD1 CRIV domain restores insulin secretion.386 This interaction is suggested by the authors to be a novel therapeutic target for type II diabetes. Grb2 is another protein that positively regulates PLD activity. Grb2 serves as a scaffolding protein to recruit signaling proteins including Sos, the Ras GEF, to the plasma membrane. As discussed in section 10.3.2.2. the Grb2 SH2 domain binds PLD2 through phospho-tyrosine residues.359 The SH3 domains that flank the SH2 domain have been suggested to stimulate PLD activity.
Direct protein interactions that curb lipase activity have also been described. The heterotrimeric Gβγ subunits, dissociated from the Gα subunit upon GPCR stimulation, directly interact with the catalytic domain of PLD1 and PLD2 to inhibit activity.387 PLD has been implicated in synaptic vesicle trafficking. Two synaptic vesicle-associated proteins, amphiphysin I and AP3 (also called AP180) directly bind PLD and inhibit lipase activity. Amphiphysin I heterodimerizes with Amphiphysin II in order to associate with clathrin coated vesicles. The N-terminus of Amphiphysin I directly binds PLD1 and PLD2 with affinities of roughly 15 nM, inhibiting catalytic activity. Assembly protein 3 (AP3, also called AP180) binds clathrin-coated vesicles and the C-terminus of PLD1 to inhibit lipase activity.
Cytoskeletal components directly modulate PLD activity. Monomeric G actin inhibits PLD activity. Conversely, PLD activity triggers actin polymerization, and polymerized F-actin stimulates PLD activity. This divergent signaling mechanism may enhance cytoskeletal reorganization in localized subdomains of the cell. PLD2 has also been shown to directly bind microtubules, again suggesting that these interactions sequester the protein as a means of ensuring phospholipase activity is limited to the correct locations within the cell. Other proteins originally thought to directly interact with PLD and inhibit activity include α-synuclein, which has subsequently been shown to not inhibit PLD activity in vitro or in cells overexpressing this protein.388
10.4 Recombinant protein expression and purification
A limiting factor in studying the biochemical and structural character of mammalian PLD enzymes is that, to date, the enzymes have proven tremendously difficult to express and purify recombinantly. In contrast to plant and fungal enzymes, which are readily expressed and isolated from bacterial expression systems, mammalian PLD enzymes have not, to date, been expressed as catalytically active proteins in prokaryotic expression systems. Even plant and yeast enzymes with highly conserved regulatory and catalytic domains, such as PLDζ and Spo14, are catalytically active when expressed and purified from bacteria, whereas catalytically competent mammalian PLD enzymes have not been expressed or isolated from bacteria.389 In Escherichia coli, mammalian PLD protein is highly proteolyzed and localizes to inclusion bodies, where insoluble, unfolded, aggregate protein is collected. Attempts to purify and refold mammalian PLD from inclusion bodies have not been reported.
However, there are multiple instances of recombinant mammalian PLD expression in eukaryotic systems, including insect cells, Spodoptera frugiperda, 129,205 yeast, and Schizosaccharomyces pombe.390 Catalytically active mammalian PLD1 and PLD2 can be expressed and partially purified from these eukaryotic recombinant systems. Since post-translational modifications, including lipid modification and phosphorylation, are not necessary for catalysis (see section 10.3.2), and refolding from inclusion bodies has never been successful, this suggests eukaryotic protein chaperones may be integral for proper folding of mammalian PLD enzymes. Intriguing studies from John Exton’s group support this by demonstrating that the amino and carboxy terminal domains can be expressed on separate plasmids and co-purified as catalytically active complex.344 However, mixing of amino and carboxy termini that were expressed and purified in isolation does not yield catalytically active protein.
When expressed in insect cells (monolayer cultures of Sf21 or Sf9 cells), the bulk of mammalian PLD1 protein is soluble or loosely membrane-associate and is easily extracted with mid ionic strength buffers and can be purified in the absence of detergent. Mammalian PLD2, however, is mostly membrane-associated, and efficient protein extraction requires high salt and detergent. Throughout purification, this enzyme is not stable without detergent, which can be used at concentrations below the critical micelle concentration (cmc).
Purification of mammalian PLD1 and PLD2 using classic chromatographic methods, such as ion exchange, heparin, and size-exclusion, yields partially pure fractions. Purity is further enhanced when mammalian PLD is expressed with affinity-tags, the best results are obtained through the use of multiple tandem affinity purification steps coupled with classic chromatographic methods. However, placement of the affinity tag at the amino-terminus is critical. Modification to the carboxy terminus significantly decreases catalytic activity, as would be expected based on PLD2 splice variants with truncated carboxy termini that yield proteins with 8–12 % of the activity of full length PLD2 enzyme.328
Despite the increased purity afforded by tandem affinity tags, mammalian PLD, particularly PLD1, is poorly expressed in insect cells. Low expression levels may be due to the fact that expression of catalytically active PLD enzymes is deleterious to insect cell viability. Supporting this is evidence that expression is significantly increased for catalytically-inactive mutants or amino-terminally truncated constructs that do not exhibit proper localization or catalytic activity in cells. Recent studies demonstrate that truncation of the amino terminus of PLD1 coupled with use of a large affinity tag (bacterial maltose binding protein, commonly used to enhance solubility of recombinant proteins) significantly increase expression and enable one-step affinity purification of homogenous PLD.125,129
10.5 Signaling pathways
More than 15 years after the cloning of the first mammalian PLD, this enzyme, its activity, and products continue to be implicated in a wide range of signaling pathways and cellular functions. These pathways include receptor-mediated responses, growth and survival pathways, and vesicular trafficking. PLD-mediated cytoskeletal reorganization in response to chemoattractants, and pathogenic infection are critical immunologic functions. Only recently have potent and isoenzyme selective small molecule inhibitors of mammalian PLD isoforms become available. Many studies continue to utilize primary alcohols to implicate PLD in different signaling pathways. As described in section 2.2, in the presence of low concentrations (<3 %) of primary alcohol, mammalian PLD will perform transphosphatidylation and generate a metabolically-stable phosphatidylalcohol instead of phosphatidic acid. Discrepancies are now emerging between functions of PLD previously reported using alcohols, and those demonstrated using RNAi knockdown, small molecule inhibitors, or those observed in knockout animals.391 Signaling roles for PLD mentioned here include those determined using primary alcohols as well as knockdown or pharmacological inhibition. However, further characterization of PLD activity using these newer methods is necessary to clarify and validate previously-defined roles of mammalian PLD.
10.5.1 Receptor-mediated signaling
Extracellular stimuli trigger intracellular responses via cell receptors present at the plasma membrane. These include GPCRs, receptor tyrosine kinases (RTKs), and integrins, all of which mediate signaling through PLD activation. The specific mechanisms for receptor-mediated PLD activation differ between cell types, but the canonical pathways are described here.
10.5.1.1 GPCR signaling
G protein-coupled receptors (GPCR) trigger dissociation of Gα and Gβγ heterotrimeric G proteins upon agonist stimulation. Uncoupled heterotrimer subunits elicit signaling cascades through downstream effector proteins. Many of these pathways elicit functional responses through signaling to PLD in multiple ways (Figure 9). In the canonical pathway, upon agonist stimulation, GTP-Gαq stimulates PLCβ hydrolysis of PI(4,5)P2, producing DAG and IP3 (see excellent reviews by Rhee 2001 on PLC subtype activation,392 Hubbard and Hepler 2006 for review on Gq family,393 and Harden review in this issue). IP3 triggers calcium release from the ER, and this coupled with DAG synergistically activates PKCα, which in turn bimodally activates PLD (discussed in section 10.3.4.2). Litosch and colleagues recently showed that this PLCβ signaling is potentiated by PLD-produced PA.394,395 Dissociated Gβγ also activates PLCβ, to indirectly activate PLD in a PKC-dependent manner. Additionally, Preninger et al. demonstrated that the Gβγ subunit of the heterotrimer can directly inhibit PLD activity via interactions through the PLD catalytic domain.387 Gβγ interaction disrupts both basal and Arf-stimulated activity.387,396 As illustrated in Figure 9, levels of PLD activation are intricately titrated in response to specific agonist-mediated or intracellular circumstances.
Figure 9.

The G12/13 class of heterotrimers activates PLD in a small GTPase dependent manner. Gα12 activates RhoA via Pyk2, a focal adhesion tyrosine kinase, which directly stimulates PLD1 activity. As shown in Figure 9C, Gα13 activates the γ subtype of PI3K to generate PIP3. Upon PIP3 binding, ARNO and Rho GEF trigger GDP for GTP exchange on Arf and RhoA, respectively.397 These activated small GTPases then directly activate PLD (as discussed in section 10.3.4.1). Gq and G12 also stimulate Src, which tyrosine-phosphorylates both PLD at the PH domain, and the receptor tyrosine kinase, EGFR (Figure 10; see section 10.5.1.2 for signaling details). PLD phosphorylation does not affect cellular phospholipase activity, but the direct interaction does enhance Src kinase activity.355 EGFR phosphorylation results in homodimerization, autophosphorylation, and GPCR-EGFR transactivation in the absence of EGFR agonist.398,399
Figure 10.

Roles for PLD in pathogenic response have been reported, many of which are GPCR-mediated and result in changes in reactive oxygen species formation, vesicular trafficking or transcription. In leukocytes, PLD1 expression is induced in response to pathogenic and pro-inflammatory stimuli through activation of membrane receptors including the Gi-coupled f-Met-Leu-Phe receptor (fMLPR). PLD activity in macrophages and neutrophils is implicated in respiratory burst,400 engulfment of bacteria, and reorganization of cytoskeletal elements. Recently, PLD was shown to be involved in HIV replication via CCR5, an MIP-1 chemokine receptor that interacts with an HIV glycoprotein.401 In response to CCR5 agonist stimulation, PLD is activated in an ERK1/2-dependent manner to activate transcription factors, including NFκB, that facilitate replication of the latent HIV genome integrated into the host genome.
PLD is a major source of PA generated by cell surface receptor-mediated signaling pathways. Its primary substrate in mammalian cells is PC, but consistent with its catalytic mechanism it can also utilize other amine containing glycerophospholipids as substrates (e.g., PE and PS). The molecular species of PA generated by PLD are predominantly mono- and di-unsaturated species, particularly 16:0/18:1 containing fatty acyl species. Work from Michael Wakelam’s laboratory provided an insightful comparison of DAG and PA species generated from PLC and PLD sources, respectively.402,403 The authors reported differences in cellular targets modulated by these distinct signaling pathways, such as the lack of PKC activation by molecular species of DAGs generated downstream of PLD. Activated in parallel by many of the same cell surface receptors, PLC isoenzymes generate two second messengers from the hydrolysis of PI(4,5)P2, namely DAG and IP3. The DAG generated via the PLC pathway is typically polyunsaturated (e.g., 38:4 DAG) reflecting the major species of the PIP2 substrate available in mammalian cells.404 The polyunsaturated DAG generated from PLC provides a second and distinct signaling source of cellular PA via the transfer of a phosphate from ATP to DAG through the action of a DAG kinase. An excellent review of DAG kinase isoenyzmes types and regulation is provided in this thematic issue by Richard Epand and coauthors.405 Different isoenzymes of DAG kinases have distinct substrate specificities. Recent advances in electrospray ionization mass spectrometry have identified a surprising diversity of DAG molecular species that can now be resolved and quantitated using a linear regression algorithm.406 This type of analysis has revealed that DAK kinase isoenzymes have extremely diverse functionalities and substrate preferences leading to differences in the array and relative concentrations of acyl species of DAGs in cells following perturbations, such as overexpression or genetic knockouts.407,408 For example, the DAG kinase epsilon shows the ability to select acyl chains on both the sn-1 and sn-2 positions of the glycerol backbone of the DAG substrate as well as on its product, PA, which modulates a feedback inhibition of this isoenzyme.409 This PLC-DAG kinase pathway provides a distinct phase of PA that appears later in the temporal sequence of receptor-mediated PA generation. By contrast the PA molecular species generated by PLD appear rapidly after receptor activation, but are also rapidly metabolized into DAG via the actions of lipid phosphatases. The ultimate metabolic fates and functional distinctions of these two sources of signaling PA species are not as yet fully defined, but recent development of new types of lipid probes that utilize alkyne-cobalt chemistry132 provides opportunities to track and identify lipid metabolites even after multiple biotransformations. This will facilitate identification of distant metabolites and allow the functional consequences of different sources of PA production to be unambiguously determined.
10.5.1.2 Canonical RTK signaling via EGFR
The EGFR, is highly conserved in eukaryotic organisms, and is a representative member of the ERBb family of growth factor receptors with intrinsic tyrosine kinase activity. EGFR activates downstream signaling pathways including those responsible for growth, survival, and cytoskeletal reorganization. Aberrant EGFR signaling has been implicated in tumorigenesis.
Upon GPCR transactivation or binding epidermal growth factor (EGF), EGFR homodimerizes and tyrosine phosphorylates the adjacent receptor in the cytosolic region to generate an active receptor complex (activation mechanism reviewed410). These phosphotyrosine residues serve as docking sites for downstream effector proteins, including PLCγ1, Grb2, and PI3K. Even prior to cloning the mammalian PLD isoforms, PLD activity was shown to be activated by EGFR stimulation. Critical characterization of the multiple, and sometimes overlapping, mechanisms in which EGFR signaling activates PLD activity has been performed. For simplification, these are illustrated and described in separate schematics.
PLD2 can be localized to EGFR via its PX domain. In Figure 10A, the PX domain of PLD2 binds the SH3 domain of PLCγ1, which directly localizes to the EGFR. PLCγ1 hydrolyzes PI(4,5)P2 to generate DAG ad IP3. Similar to GPCR-activation of PLCβ, PLC-derived products induce PKCα activation of PLD. Changes in actin polymerization can occur in response to GPCR or RTK signaling. PLD has been shown to directly bind actin, resulting in mutual regulatory interactions (see section 10.3.4.3).
In a separate mechanism of PLD activation PLD2 directly interacts with the EGFR. At the receptor, phosphotyrosine residues in the PLD2 PH domain bind the Grb2 SH2 domain.411 This interaction enhances phospholipase activity, via Grb2 SH3 interaction, to generate PA (see section 10.3.4 for details on regulatory interaction). Recently Zhao et al., demonstrated that the Ras GEF, Sos localizes the PLD2-produced PA, where it is activated by Grb2.412 Subsequent Ras activation elicits a host of signaling cascades. Ras activates PI3K, which generates PIP3 and induces Akt translocation and activation. Ral GEF is also a Ras effector protein, which results in GTP-Ral activation of PLD.382,413 Finally, Ras activates Raf, which localizes to the plasma membrane via PLD-produced PA interactions. Ras signaling through Raf triggers activation of the MAPK pathway and via NFκB, subsequently upregulates transcription of genes involved in survival, proliferation, and differentiation.
Somewhat more controversial is the role of PLD in EGFR-stimulated mTOR signaling (reviewed414,415) illustrated in Figure 10C. Several reports suggest PLD generated PA competes for rapamycin and FKBP binding in the FRB domain of mTOR.5,416 These studies were performed using primary alcohols to show mTORC1 kinase activity was significantly decreased upon diverting PLD activity to generation of transphosphatidylation product. A follow up study used NMR to map the PA binding site within the FRB domain.417 The small GTPase Rheb was recently suggested to stimulate PLD1 as a feed forward mechanism of mTORC1 activation.383 Again these studies relied heavily on the use of primary alcohols, RNAi knockdowns, and a somewhat incomplete biochemical analysis. Subsequent use of PLD-selective small molecule inhibitors and genetic knockouts may illuminate that the role of PLD in mTOR regulation is considerably more complex with both feedforward and feedback modulation.
10.5.1.3 Integrin signaling
Integrins support cell adhesion as well as growth and survival by functioning as both an anchor to the extracellular matrix (ECM) as well as a signaling receptor. Although integrins do not possess intrinsic enzymatic activity, upon ligand binding, these receptors elicit similar signaling pathways to those of growth factor receptors by heterodimerizing and binding various effector proteins at their cytosolic face. Integrins heterodimers can signal independently or complexed with growth factor receptors to trigger chemotaxis, cell differentiation, proliferation, and survival (reviewed418). As in EGFR signaling pathways, PLD is activated downstream of integrin receptors via multiple mechanisms.
Focal adhesion kinase (FAK) directly binds the integrin receptor to induce Ras-mediated signaling and MAPK activation. Ras activates PI3K to generate PIP3. In response, the Rho GTPase, Rac, undergoes guanine nucleotide exchange thereby triggering PLD activation.419 Canonically, PLD1 is directly stimulated by C-terminal interaction with Rac1. However, Pang et al. have shown that Rac2 directly interacts with PLD2 via CRIB domains in the PLD N-terminal regulatory domain.381 In vitro the C-terminus of Rac selectively binds PA. In cells, PLD-produced PA triggers Rac translocation to membrane ruffles and lamellipodia.419 Treatment with n-butanol results in cytosolic localization of GTP-bound Rac, supporting the role of PLD in Rac translocation. At regions of membrane protrusion and lamellipodiae formation, Rac facilitates cytoskeletal reorganization. PLD colocalizes at these membrane microdomains and induces actin polymerization (see section 10.3.4.3 for regulatory mechanisms).
Integrin signaling also mediates Arf activation of PLD. Integrin effector proteins elicit Arf GAP, ASAP, localization to the leading edge of migrating cells to attenuate Arf signaling (reviewed420) and perturb Arf-activation of PLD (reviewed).421 This bimodal mechanism of small GTPase regulation titrates levels of phospholipase activity during integrin-mediated membrane ruffling, cell migration, and invasion.
Similar to the role of PLD in Dictyostelium migration (see section 6.3), mammalian PLD isoforms have been implicated in chemotaxis. These enzymes, stimulated by Rho GTPases downstream of integrin, chemokine, and growth factor receptors, trigger cytoskeletal rearrangement and membrane ruffling. Primary butanol and PLD-selective inhibitors disrupt these pathways, suggesting PA formation as well as protein-protein interactions participate in these signaling responses.
As discussed in section 10.5.1.2, PLD-produced PA has been suggested to directly activate mTOR and facilitate mTOR complex formation and signaling, including mTORC2 and subsequent Akt phosphorylation. Akt and mTORC2 signaling not only support pro-survival signaling via MDM2 stabilization, and BAD and Bcl-XI activation, but also induce cytoskeletal reorganization. mTORC2 induces actin polymerization and triggers myosin II assembly and cell migration via PAK and myosin phosphorylation. PLD activity also induces secretion of proteolytic matrix metalloproteases that degrade surrounding ECM to facilitate cellular movement.
10.5.2 Vesicular Trafficking
Mammalian PLD enzymes differentially localize to cellular membranes to directly and indirectly induce changes in membrane curvature and fusion that facilitate endocytosis/exocytosis and vesicular trafficking. As discussed in section 10.2.1, PLD1 primarily localizes to intracellular membranes including TGN and endosomal membranes and has constitutively low basal activity. Upon cell stimulation, PLD1 translocates to plasma membrane and is activate. PLD2 is generally constitutively localized to the plasma membrane and has high basal activity.
Arf GTPases activate the otherwise low basal activity of PLD1. Arf1 stimulates Golgi-localized PLD,422 while Arf6 stimulates PLD1 at the plasma membrane.423 In an independent mechanism, Arf present at either membrane cooperates with Arf-stimulated PA to facilitate vesicles formation.424,425 In contrast to the Sec14 bypass mechanism in yeast, PA accumulation, rather than DAG, facilitates vesicle budding. This may be due to several PA-related mechanisms. PA is a cone shaped lipid, and induces changes in membrane curvature. Arf and PA also trigger recruitment of coatomer proteins, including COPI.426,427 PA activates PI4P5K, which generates PI(4,5)P2 and induces translocation of coatomer proteins and proteins involved in vesicle budding, including dynamin (a GTPase involved in endocytosis and membrane scission) and AP180 (a clathrin assembly protein) (Figure 11). Following recruitment, AP180 directly inhibits PLD activity.428,429 Recently, PLD was also reported to directly interact with dynamin. This interaction occurred in a GTP-dependent manner, and it was suggested that the PX domain of PLD2 might serve as a GAP for dynamin.430
Figure 11.

PLD and PA-dependent mechanisms function in vesicle formation to facilitate receptor internalization and recycling (Figure 11), SNARE-mediated synaptic vesicle fusion (similar to that observed in Spo14-mediated prospore membrane formation, see section 6.1.2.1.), and exocytic mechanisms including respiratory burst431 and degranulation.432
11. PLD Inhibitors
Until recently, there were few chemical tools to study PLD function, and no small molecules existed to dissect the individual roles of PLD1 and PLD2. Historically, the field relied on the overexpression of catalytically active or inactive forms of either PLD1 or PLD2 in vivo, or employed RNAi for the individual isoforms in an effort to discern discrete roles for PLD1 and PLD2. In order to assess the therapeutic potential of the inhibition of PLD1, PLD2 and/or dual inhibition of both isoforms, the genetic and biological data must be verified with a small molecule inhibitor. Untily recently, direct small molecule PLD inhibitors were not available, and none of the early inhibitors afforded isoform selectivity. Moreover, the most utilized class of molecule to study PLD function over the past 20 years has been primary alcohols, (e.g., n-butanol). Alcohols are often, inaccurately described in the literature as “PLD inhibitors”. It is important to emphasize that alcohols are not PLD inhibitors, rather n-butanol (as well as other primary alcohols) block PLD-catalyzed PA production by competing with water as a nucleophile, thereby causing the formation of phosphatidylbutanol in a transphosphatidylation reaction. Additionally, there are concerns that n-butanol may not fully block PA production and it may also be promiscuous in cell-based assays affecting multiple pathways in addition to transphosphatidylation. Thus, conclusions reached in the literature from studies employing n-butanol alone should be viewed with caution, and the data generated require further confirmation with isoform-selective small molecule PLD inhibitors, RNA interference (RNAi) knockdowns, and genetic knockouts.
Over the past twenty years, a diverse range of chemotypes 1–20 have been reported as inhibitors of either PLD or PLD signaling (Figures 12 and 13) based on activity in an equally diverse array of PLD assays. Thus, quantitative, and in some instances qualitative, comparisons with regards to PLD activity are not possible. As a result, early PLD inhibitors fall into two categories, direct and indirect inhibitors. As many of these inhibitors have not been thoroughly studied, these divisions by mechanism of action must be interpreted with caution.
Figure 12.

Figure 13.

11.1 Indirect Inhibitors of PLD Activity
Several compounds, 1–10, have been identified that inhibit PLD enzymatic activity in cells and/or decrease PLD protein expression in cells, but do not directly inhibit PLD enzymatic activity in vitro (Figure 12). These compounds are not ideal chemical probes, because many of them are not potent and/or have a large number of known molecular targets in a variety of different signaling pathways. Resveratrol (1), a polyphenol found in the skin of red grapes, inhibits the production of PA by human neutrophils with an IC50 of approximately 50 μM. Additionally, in experiments where cells were treated with 1 % ethanol, resveratrol blocked the formation of phosphatidylethanol, which suggests that resveratrol decreases PLD enzymatic activity.433 Honokiol (2), a natural product that was isolated from the seed cones of Magnolia grandiflora, has been shown to have antimicrobial,434 antiangiogenic435 and proapoptotic436 properties. Honokiol (20 μM) was shown to block the formation of phosphatidylbutanol in MDA-MB-231 cells treated with 0.8 % n-butanol indicating that honokiol decreases PLD activity in cells. However, honokiol (concentrations up to 50 μM) had no effect on PLD enzymatic activity in vitro.437 Trans-diethylstilbestrol (3), a synthetic compound that is structurally similar to resveratrol, inhibits the formation of both PA and phosphatidylethanol (in cells treated with 1 % ethanol) slightly more potently than resveratrol.438 Triptolide (4), a diterpene triepoxide isolated from Triptergium wilfordii that has been used in traditional Chinese medicine for centuries, and is currently entering clinical trials439 (semisynthetic derivative), was a hit in a screen designed to identify compounds that decrease PLD expression.440 However, triptolide was also a hit in an earlier screen designed to identify compounds that suppress the human heat shock response441 and more recently Titov et al. identified XPB, a subunit of the transcription factor TFIIH, as a molecular target of triptolide.442 Triptolide’s indirect mechanism of action and other known molecular targets render the compound inadequate as a chemical probe for studying the cellular functions of PLD.
In the mid 1990s, Schering-Plough reported on the isolation of a series of polycyclic ketoepoxide metabolites from fungal cultures. SCH49211 (5) and SCH49212 (6), isolated from cultures of Nattrassia mangiferae, were shown to inhibit PLD activation with IC50s of 11 μM and 12 μM, respectively, in HL60 cells treated with fMLP (formyl-Met-Leu-Phe).443 Shortly after this first report, the same group disclosed SCH53823 (7), isolated from the dead leaves of Ruercus virginiana, and then prepared the corresponding acylated derivative, SCH53827 (8) to enable structure determination. Interestingly, the unnatural product 8 inhibited PLD activation, with an IC50 of 17 μM in HL60 cells employing the fMLP PLD assay.444 Around the same time, Hedge and co-workers described the isolation and characterization of saponin 1 (9) and saponin 2 (10) from the extract of the leaves of Myrsine australis.445 Both natural products were shown to inhibit fMLP stimulated PLD with IC50s of 8 μM and 24 μM, respectively. It has previously been observed that certain ceramide lipids and the aminoglycoside antibiotic neomycin also inhibit PLD activity.
11.2 First generation direct inhibitors of PLD activity
Over the past 10 years several compounds that inhibit PLD directly have been identified. These compounds decrease PLD enzymatic activity measured by transphosphatidylation in cells and measured by the hydrolysis of [3H]-PC in an in vitro reconstitution assay (Figure 13). These direct-acting compounds can be categorized into three classes: (1) phosphate mimetics, (2) natural products and (3) synthetic, drug-like small molecules. The identification and subsequent optimization of some of these compounds was a major advance in the field of lipid cell signaling. Indeed, the lack of small molecule ligands to use as tools to probe both the cellular and in vivo roles of each PLD isoform has arguably hindered the validation of PLD as a potential therapeutic target, and studies with n-butanol (11) have clearly provided some erroneous data.
Crystal structures have not been determined for either human PLD1 or PLD2, but a crystal structure of a bacterial PLD, Streptomyces sp. strain PMF, was published in 2000 and this structure contains a phosphate molecule bound in the enzyme’s active site.105 In 2002 Davies et al. reported that tungstate (12) and vanadate (13) inhibit a PLD superfamily member, tyrosyl-DNA phosphodiesterase, as evidenced by both an in vitro enzyme activity assay and multiple crystal structures.104,446 Subsequently, tungstate and vanadate, both phosphate mimetics, were identified as PLD inhibitors via the in vitro reconstitution assay of PLD. Gomez-Cambronero reported that during purification of PLD from human granulocytes a standard protease cocktail inhibited PLD activity.447 Deconvolution of the six inhibitor cocktail identified the serine protease inhibitor 4-(2-aminoethyl)benzene sulfonyl fluoride, AEBSF (14), as the chemical blocker of PLD activity, with no affect on PLA2 or PLC. AEBSF inhibits both basal and stimulated PLD activity with an IC50 of 75 μM. Interestingly, AEBSF is an electrophilic compound with the capacity to covalently modify proteins, limiting its potential as a PLD inhibitor. Moreover, the S-F bond may be hydrolyzed to the corresponding sulfonic acid by water to generate a phosphate mimetic. SCH420789 (15), a fungal metabolite, was isolated and shown to inhibit PLD in vitro with an IC50 value of approximately 10 μM.448 Calphostin-c (16), a perylenequinone compound from the fungus Cladosporium caldosporoides, was identified as a direct-acting inhibitor of PLD and previously shown to inhibit protein kinase C directly in vitro.449 Protein kinase C activates PLD in cells and directly in vitro129 so it could be reasonably inferred that the most plausible explanation for calphostin-c’s ability to inhibit PLD activity in cells would be its ability to block PKC activation of PLD. However, calphostin-c inhibits both PLD1 and PLD2 directly with reported IC50 values of 100–200 nM for both isoforms.450 Presqualene diphosphate (17), a constituent of human leukocyte membranes, was shown to inhibit both Streptomyces chromofucscus PLD (IC50 = 100 nM) and human PLD1b (IC50 > 1 μM) in vitro.451 Curcumin (18), the predominant yellow pigment in turmeric (Curcuma longa), is a polyphenolic compound that has been used in Ayurvedic medicine for thousands of years and currently is the subject of a large number of basic and clinical research studies.452 Yamamoto et al. showed that curcumin inhibits the PLD activity present in a membrane preparation with an IC50 of 10 μM.453 Furthermore, we have observed that curcumin inhibits recombinant, purified PLD1 and PLD2 in vitro (Scott, Armstrong, and Brown, unpublished observations).
Two selective estrogen receptor modulators (SERMs), raloxifene (19) and 4-OH tamoxifen (20), were identified as direct modulators of human PLD1 and PLD2.454 Their identification as modulators of PLD activity is consistent with an interesting, continuing trend that SERMs appear to have a myriad of estrogen receptor-independent effects. SERMs have hydroxyl groups positioned so as to mimic the structure of estradiol; this allows SERMs to bind to the estrogen receptor and block activation of the receptor by its endogenous ligand. Therefore, SERMs are typically used to treat estrogen receptor-positive breast cancer. However, an interesting observation is that tamoxifen decreases tumor growth in about 10–15 % of estrogen receptor-negative tumors.455 Additionally, tamoxifen inhibits the growth of estrogen receptor-negative cancer cell lines and induces apoptosis in these cells.456 PLD activity and/or expression is frequently increased in breast cancer457 so it is plausible that one of the estrogen receptor-independent effects of SERMs could be PLD inhibition. Indeed, raloxifene inhibits PLD1 (IC50 = 4.3 μM) and PLD2 (IC50 = 3.4 μM) directly in vitro and in several different cell lines (IC50 = 5–10 μM).454,458
The actions of tamoxifen on PLD both in vitro and in cells are more complicated. Tamoxifen is a prodrug; the actions of tamoxifen are realized primarily through its active metabolites, including 4-OH tamoxifen.459 4-OH tamoxifen is 100-fold more potent than tamoxifen at suppressing estrogen receptor-dependent cell proliferation and 4-OH tamoxifen binds to the estrogen receptor with 20 to 30-fold higher affinity than tamoxifen.460,461 Tamoxifen actually stimulates PLD1 and PLD2 activity in vitro and in some cell lines (during a 30 minute treatment); however, the active metabolite of tamoxifen, 4-OH tamoxifen, stimulates PLD1 in vitro yet inhibits PLD2 in vitro, albeit with poor potency (IC50 > 20 μM). 4-OH tamoxifen inhibits PLD1 and PLD2 in cells with an IC50 of about 5 μM on each isoform.454 Interestingly, tamoxifen blocked phorbol ester stimulated PLD activity in an estrogen receptor-negative human breast cancer cell line (MCF-7) at concentrations of 2–5 μM only during longer (24 h) treatments and did not block phorbol ester stimulated PLD activity during a short (0.5 h) treatment.462
11.3 Second generation direct inhibitors of PLD activity: The identification of halopemide as a PLD Inhibitor
A renaissance in the PLD inhibitor field began in 2007 with a brief report from a group at Novartis on a high throughput screen to identify PLD2 inhibitors for use as inflammatory mediators. This effort identified halopemide (21), a psychotropic agent originally reported by Janssen in the late 1970s and early 1980s for numerous neuroscience indications (Figure 14) as a PLD2 inhibitor with an IC50 value of 1.5 μM.463 This short report was limited to a succinct description of the synthesis of fourteen halopemide analogs where alternative amide moieties were surveyed, resulting in the discovery of 22, later coined FIPI, with an IC50 of 200 nM and good rat pharmacokinetics. However, there was no mention of PLD1 inhibition in this initial paper, but it was subsequently found that halopemide (21) potently inhibits both PLD1 (cellular IC50 = 21 nM, in vitro IC50 = 220 nM) and PLD2 (cellular IC50 = 300 nM, in vitro IC50 = 310 nM) as does 22 (PLD1 cellular IC50 = 1 nM, in vitro IC50 = 9.5 nM; PLD2 cellular IC50 = 44 nM, in vitro IC50 = 17 nM).458 Thus, halopemide (21) and all the halopemide analogs presented in this report are more accurately described as dual PLD1/2 inhibitors, and even show a slight preference for PLD1 inhibition. Despite these issues, the halopemide (21) scaffold is an excellent starting point for a PLD inhibitor development campaign due to the potent PLD inhibition, favorable preclinical drug metabolism and pharmacokinetic profile, and most importantly, extensive history in multiple clinical trials.464
Figure 14.

Halopemide (21), also known as R 34301, is related to the butyrphenone-based neuroleptics such as spirerone and haloperidol, and was originally developed as an anti-emetic drug, but later found to possess unique psychotropic effects as a dopamine antagonist.464 21 was found to be a ‘psychic energizer’ having effects on the negative symptoms, as well as the positive symptoms of schizophrenia without the extrapyramidal side effects common to standard atypical antipsychotic agents.465 As eluded above, halopemide was evaluated in five separate clinical trials with over 100 schizophrenic, oligophrenic and autistic patients receiving the drug.464 Efficacy was observed in the majority of patients, and importantly, no adverse side effects or toxicities were noted, despite achieving plasma exposures of 100 ng/mL to 360 ng/mL from the 20 mg/kg and 60 mg/kg doses of 21, respectively.466 At these plasma concentrations, PLD1 was clearly inhibited, suggesting inhibition of PLD by this chemotype is safe in humans and a therapeutically viable mechanism.
11.4 Optimization of halopemide for isoform-selective PLD inhibition
Human PLD1 and PLD2 respond to different stimuli both in vitro and in vivo.129 Additionally, in some cancer types only one PLD isoform is upregulated at the protein expression and/or enzymatic activity level.457,467 More recently, studies in PLD knockout animals have clearly defined, non-overlapping roles and therapeutic potential for both PLD1 and PLD2. For these reasons the development of isoform-selective PLD inhibitors is a desirable goal not only from a discovery science perspective, but also from the vantage point of a drug discovery effort. After the initial report on halopemide synthesis and inhibitor properties, numerous analogs have been synthesized and assayed in an effort to develop isoform-specific PLD inhibitors.458,468,469,470
Since the group from Novartis reported that halopemide is a PLD inhibitor, the Brown and Lindsley groups at Vanderbilt have synthesized and assayed hundreds of halopemide analogs in an effort to develop isoform-specific PLD inhibitors.458,468,469,470 The first phase of isoform-specific PLD inhibitor development was reported in early 2009.458 As shown in Figure 15, a matrix library approach was employed to survey three regions of 21 simultaneously to afford a 3 × 3 × 30 library of ~270 halopemide analogs employing standard solution phase parallel synthesis techniques combined with mass-directed preparative LC-MS. Rigorous pharmacological characterization of a representative subset of the ~270 compounds was performed; IC50 values were reported in cell systems engineered to give only a PLD1 or a PLD2 response as wells as IC50 values that were determined on recombinant PLD1 and PLD2 enzymes purified from insect cells. Data from both an in vitro enzyme activity assay and a cellular activity assay show that the compounds inhibit PLD1/2 directly and that the compounds effectively permeate the cell membrane. Many of the compounds display low nanomolar potency values, and this library produced a number of dual PLD1/2 inhibitors and a number of moderately preferring PLD1 analogs. This first generation effort did afford the first PLD1-selective inhibitor, VU0155069 (23), where the chiral (S)-methyl group significantly enhanced PLD1 preference to ~163-fold over PLD2 in a cell-based assay. Subsequent iterations of lead optimization found the chiral (S)-methyl group as a general moiety that increased PLD1 inhibition. While the piperidinyl benzimidazolone-containing analogs failed to display any preference for PLD2 inhibition, a triazaspirone congener uniformly increased PLD2 inhibition to provide the first PLD2 (10-fold PLD2 preferring) selective inhibitor, VU0155072 (24). Additionally, some of the compounds decreased the ability of several breast cancer cell lines to invade through a Matrigel™ membrane in a transwell migration assay, which is consistent with earlier studies showing this enzyme’s role in regulating cell migration.458,471,472,473
Figure 15.

11.5 Development of highly selective PLD1 and PLD2 inhibitors
While the first generation libraries were diversity-oriented in an effort to explore chemical space and identify molecular entities that would engender PLD isoform-selective inhibition, subsequent optimization strategies were more focused and driven from a medicinal chemistry perspective (Figure 16) to improve PLD1 and PLD2 potency and selectivity within 25 and 26, respectively. Several important pieces of information were discovered or confirmed in this round of analog synthesis based on 25: (1) homologation of the ethyl diamine linker of an analog by just one carbon to a propyl chain eliminated all activity; (2) heteroaromatic and aromatic amides on the right side of analogs confer excellent potency; (3) a racemic trans-cyclopropyl phenyl amide dramatically increased PLD1 selectivity; (4) a 5-bromo substituted benzimidazolone increased potency and PLD1 selectivity.
Figure 16.

The confluence of several fortuitous discoveries, primarily the fact that the (S)-methyl group adjacent to the amide nitrogen and the racemic trans-cyclopropyl phenyl amide both dramatically enhance PLD1 selectivity, led to the discovery (Figures 17 and 18) of a potent (cellular PLD1 IC50 = 3.7 nM), 1,700-fold PLD1-selective inhibitor, VU0359595 (28).
Figure 17.

Figure 18.

With a highly PLD1-selective inhibitor in hand, the focus of the synthesis became the development of a PLD2-selective inhibitor based on 26. Developing a PLD2-selective inhibitor proved to be significantly more challenging for at least 3 reasons: (1) halopemide itself is a slightly PLD1-preferring compound; (2) no structural features in the ethyl diamine linker or amide cap had been identified that improved PLD2 selectivity and (3) PLD2-preferring inhibitors were sparse until the triazaspirone moiety was identified, and require multi-step syntheses to access functionalized congeners. Once again, parallel synthesis afforded quick identification of molecular features that engendered PLD2 inhibition, identifying VU0364739 (36), a PLD2 inhibitor displaying over 75-fold selectivity versus PLD1 inhibition in both biochemical and cell-based assays (Figures 19 and 20).
Figure 19.

Figure 20.

Interestingly, the chiral (S)-methyl group proved to be a ‘molecular switch’ in this scaffold provoking PLD1 preference, converting a 75-fold PLD2-selective inhibitor 36 (cellular PLD1 IC50 = 1,500 nM, PLD2 IC50 = 20 nM), into a 6-fold PLD1-selective inhibitor (PLD1 IC50 = 10 nM, PLD2 IC50 = 60 nM), by virtue of increasing potency for PLD1 by 150-fold.
In vitro anticancer activity data and rat pharmacokinetic data were also reported for the PLD2 inhibitor VU0364739 (36) and the 1,700-fold PLD1-selective inhibitor VU0359595 (28). In a triple negative breast cancer cell line (MDA-MB-231) the PLD2-selective inhibitor VU0364739 decreased cell proliferation significantly more than the PLD1-selective inhibitor VU0359595 and caused a much larger increase in caspase 3/7 activity than the PLD1-selective inhibitor VU0359595. Additionally, the PLD2-selective inhibitor VU0364739 caused a much larger increase in caspase 3/7 activity than the PLD1-selective inhibitor VU0359595 in MDA-MB-231 cells. While the pharmacokinetic properties of both VU0364739 and VU0359595 would need to be optimized for robust in vivo proof-of-concept experiments, the compounds are certainly acceptable starting points for an optimization campaign focused on drug metabolism. Both compounds are highly cleared (approximately Cl = 60 ml/min/kg for both compounds), yet display reasonable half-lives (VU0359595 = 0.78 h and VU0364739 = 1.2 h) due to the relatively large volume of distribution of each compound (VU0359595 = 4.7 L/kg and VU0364739 = 8.1 L/kg). The most interesting drug disposition data obtained is that while VU0359595 was below the level of quantification in the rat brain when administered orally at 10 mg/kg, VU0364739 partitions almost equally between brain and plasma (brain/plasma = 0.73) at the same dose.470 Given recent reports that have suggested the therapeutic potential of PLD inhibition in Alzheimer’s disease7,474,475 and stroke476 the discovery of this centrally penetrant PLD inhibitor should facilitate target validation in animal models of disease.
A patent application has been published claiming composition of matter for novel, small molecule PLD inhibitors.477 The markush structure 44 is shown in Figure 21, and the compound is based on a diazaspirone core, a des-aza analog of 36, where the chirality at the benzylic carbon is reported to impact PLD isoform specificity. This is the first example where a common core can provide potent and selective PLD1 and PLD2 inhibitors as well as dual PLD1/2 inhibitors.
Figure 21.

12. Functional consequences of PLD inhibition or overexpression
Prior to the use of small molecules various groups studied the cell signaling roles of PLD using traditional methods such as dominant-negative mutations, overexpression and, more recently, RNAi. Additionally, it has been known since the mid 1960s that short, primary alcohols can compete with water as a nucleophile in the PLD-catalyzed transphosphatidylation reaction, thereby causing the formation of a phosphatidylalcohol species, and preventing the formation of phosphatidic acid.112 Although clearly an imperfect tool and not a viable starting point for a medicinal chemistry strategy, n-butanol has long been used to study the effects of blocking the production of PA produced by PLD.
There is a growing appreciation that the “classic differences” in effects between n-butanol and t-butanol are not simply accounted for by their effects on PLD activity. This traditional tool has likely led to spurious conclusions as to the functional roles of PLD. While most of the information about PLD’s role in cell signaling networks has been produced by a combination of biochemical approaches (including the use of n-butanol) and RNAi-mediated knockdowns, reports using small molecule inhibitors and genetic knockouts are now being added to look at systematic effects in whole organisms.
12.1 Respiratory burst
Nicotinamide adenine dinucleotide phosphate-oxidase (NADPH oxidase) is a membrane bound enzyme complex that sits quiescently in neutrophils, eosinophils and mononuclear phagocytes until activated during respiratory burst. Upon activation, NADPH oxidase generates superoxide by transferring electrons from NADPH inside the cell across the cell membrane and then coupling the electrons to molecular oxygen. The superoxide radical is further transformed into hydrogen peroxide and hypohalous acids (e.g., hypochlorous acid), which are used as a form of ‘chemical warfare’ by human cells to attack human pathogens.478
A host of experiments done in cells have indicated a role for PLD in NADPH oxidase activation. When PA production in cultured human neutrophils is blocked via the use of n-butanol, the respiratory burst as measured by O2 production, was almost completely blocked.479 There is additional evidence that PLD is involved in regulating NADPH oxidase activity both in cells and in vitro: when the leukotriene B4 receptor is activated, levels of presqualene diphosphate rapidly decline thereby removing a negative regulatory element from inhibiting PLD’s capacity to stimulate NADPH oxidase activity.480
Agwu and colleagues gathered the first evidence suggesting that didecanoyl-PA activates NADPH oxidase in vitro by combining subcellular fractions in order to reconstitute NADPH oxidase activity and showing that didecanoyl-PA activated this combination of subcellular fractions.481 Almost a decade later McPhail et al. showed that, in vitro, PA activates a protein kinase that phosphorylates and activates a component of the NADPH oxidase complex, p47-phox.482 In 2011 Norton et al. utilized small molecule PLD inhibitors and PLD2 knockout mice to show that PLD1 (and not PLD2) regulates the production of reactive oxygen species in neutrophils.131 An excellent review of PLD function in respiratory physiology was provided by Cummings and colleagues.483
12.2 Transport and endocytosis
Vesicles are the primary means by which cells store, move and dispose of a multitude of cellular components. Eukaryotic cells are composed of various organelles that effectively share information and cargo via vesicular trafficking, which involves three steps: budding from the donor compartment; transport and/or targeting to a specific acceptor compartment, and fusion of the vesicle with the acceptor compartment. Vesicular trafficking is one of the most fundamental processes in cell biology and an excellent review is available from Bonifacino and Glick.484 For a concise review of the role of PLD in membrane trafficking the reader is referred to a review by Roth.485 Previous efforts to elucidate the role of PLD in vesicular trafficking relied heavily on primary alcohols to block PLD-mediated PA formation, but more recently drug-like, small molecules have been utilized to study the role of PLD in vesicular trafficking.
The use of primary alcohols to block PLD-mediated PA formation or monitor product formation in cells by several groups yielded the first evidence supporting a role for PLD in exocytosis. Xie et al. measured significant increases in the amount of phosphatidylethanol formed by HL60 granulocytes treated with ethanol during primary granule secretion. Additionally, ethanol dose-dependently decreased the release of myeloperoxidase from the HL60 granulocytes486 and ethanol also blocked IgE-receptor-mediated mast cell degranulation.487 Chen et al. showed that 1 % n-butanol was sufficient to decrease the release of nascent secretory vesicles from the trans-golgi network, which they independently confirmed in parallel by treating permeabilized cells with a catalytically inactive PLD mutant (K898R). PLD also increases the release of nascent secretory vesicles in permabilized cells.425 Additional evidence that PLD plays an important role in exocytosis was acquired via genetic manipulations in Saccharomyces cerevisiae. Ella et al. discovered a PLD gene in yeast and generated a genetic knockout, noting that diploid yeast lacking the PLD gene were unable to sporulate.268 Therefore, these results suggest a broader role for PLD in regulating cell growth and division.
A role for PLD in endocytosis has been described and supported by the use of n-butanol, catalytically inactive mutants, and RNAi. PLD activity is required for agonist mediated-epidermal growth factor receptor internalization: (1) n-butanol decreases the agonist-stimulated internalization of the epidermal growth factor receptor; (2) overexpression of PLD1 or PLD2 increases the agonist-stimulated internalization of the epidermal growth factor receptor and (3) overexpression of catalytically inactive PLD1 or PLD2 decreases the agonist-stimulated internalization of the epidermal growth factor receptor.488 PLD activity is also required for μ-opioid receptor internalization: (1) n-butanol decreases the internalization of the μ-opioid receptor, and (2) overexpression of catalytically inactive PLD2 decreases the internalization of the μ-opioid receptor.489,490 Recently, studies have taken advantage of RNAi as a means to examine the effects of PLD inhibition on endocytosis. Bhattacharya et al. showed that n-butanol, overexpression of a catalytically inactive PLD2, or PLD2 RNAi treatment all decrease the internalization of the mGluR1a metabotropic glutamate receptor.491 Du et al. also utilized RNAi to show that overexpression of a dominant-negative PLD2 (K758R) or transfection with PLD2 RNAi decreased the internalization of the angiotensin II type 1 receptor.492
A role for PLD1 in macroautophagy has been established by a rigorous set of experiments employing n-butanol, a pharmacological inhibitor, RNAi treatment and knockout of PLD1 in a mouse. All of these treatments decreased autophagy as measured by a variety of different readouts.493 Inhibition of autophagy via PLD1 inhibition may be desirable in some disease states and not others; the recent development of a 1700-fold PLD1-selective inhibitor468 should facilitate answering this question. PLD2 ablation via gene targeting in mice rescues memory deficits in a mouse model of Alzheimer’s disease.7 The recent development of a centrally penetrant PLD2-selective inhibitor470 should facilitate the rigorous target validation of PLD2 for Alzheimer’s disease.
12.3 Platelet aggregation
A role for PLD in platelet activation has been previously suggested.494 Due to the lack of small molecule inhibitors, previous studies had to utilize imprecise tools that could not address how inhibiting PLD activity affected platelet aggregation.495 However, in 2009 Disse et al. determined that PLD1 regulates the secretion of von Willebrand factor from endothelial cells utilizing either n-butanol or RNAi.496 Von Willebrand factor is one of the major procoagulant and proinflammatory proteins required for hemostasis and a deficiency of von Willebrand factor (von Willebrand disease) is the most common inherited bleeding disorder. Disse et al. first suggested that PLD might be involved in regulating the secretion of von Willebrand factor from vascular endothelial cells by showing that n-butanol decreased its histamine-induced secretion from vascular endothelial cells. Using an independent method of decreasing PLD activity, RNAi, Sadler et al. showed that PLD1 knockdown dramatically decreased the histamine-induced secretion of von Willebrand factor, whereas PLD2 RNAi had no effect on its histamine-induced secretion.497
12.4 Neuronal physiology
There has long been an association between PLD and neuronal physiology and pathology, but some truly provocative animal model data facilitated by gene targeting have recently emerged. A comprehensive review of the role of PLD in brain function is provided by Oliveira.7 In the 1970s PLD activity was reported in mammalian brain tissue.324,498 Reports have implicated PLD in the process of neurite outgrowth499 and functional roles for PLD in receptor trafficking, specifically the internalization of opioid receptors and metabotropic glutamate receptors have also been reported.490,491
A possible pathophysiological role for PLD in Alzheimer’s disease has been suggested. Two groups independently reported increased PLD activity in brain tissue homogenates from Alzheimer’s patients as compared to controls. 475,500 Overexpressing amyloid precursor protein causes an increase in PLD activity in P19 mouse embryonic carcinoma cells.501 The amyloid β (1–40)-stimulated increase in PLD activity was correlated with the release of lactate dehydrogenase, which makes it reasonable to speculate that some of the neurotoxic actions of amyloid β (1–40) are mediated by PLD.502
The neurotoxic peptide α-synuclein has been implicated in the pathophysiology of both Parkinson’s disease and Alzheimer’s disease.503 Additionally, two point mutations in α-synuclein are genetically linked to familial Parkinson’s disease.504,505 All three naturally occurring synuclein isoforms α, β, and γ-synuclein were reported to inhibit PLD2 in vitro.506 Ahn et al. reported that PLD1 and PLD2 co-immunoprecipitate with α-synuclein, but also showed that PLD does not appear to impact the physiological lesions caused by α-synuclein.507 By contrast, a collaborative investigation by the Selkoe and Brown laboratories found that under numerous experimental conditions α-synuclein does not inhibit PLD in cells or in vitro.388
12.5 Cell invasion and metastasis
Cancer cell invasion and metastasis are distinct, but not unrelated processes. Invasion refers to the ability of cancer cells to invade adjacent normal tissue, whereas metastasis refers to the ability of cancer cells to gain access to a circulatory system (blood or the lymphatic system) and colonize distinct and spatially distant physiological environments.508 One of the critical early steps in metastasis is the invasion of surrounding tissue in order to gain access to either the blood or lymphatic system.509 Some of the hallmarks of this invasion process include rearrangement of the actin cytoskeleton, increased cell motility and secretion of matrix metalloproteinases.509 PLD has been shown to play a role in regulating all of these processes.6
Experiments utilizing inactivating mutations of PLD suggest that inhibiting PLD enzymatic activity decreases cancer cell invasion472 and cells transfected with a dominant-negative PLD1 were unable to form actin stress fibers when stimulated with either phorbol myristate acetate (PMA) or lysophosphatidic acid (LPA), whereas wildtype cells were able to form actin stress fibers.350 A number of studies have shown that increased PLD activity leads to an increase in the invasion of cancer cells as measured by transwell migration assays and decreased PLD activity leads to a decrease in the invasion of cancer cells.458,471,473,510,511 PLD activity also regulates the expression of MMP-2 and MMP-9, the 72 kDa and 92 kDa gelatinases, respectively.471,473,512,513
The signaling pathways that connect PLD enzymatic activity to MMP expression need to be more clearly elucidated in cancer cells. A more complete understanding of how PLD drives MMP expression in specific cancers may lead to a better understanding of how to tailor antimetastatic therapies. To date, the vast majority of evidence implicating PLD in cell invasion comes from experiments utilizing n-butanol, overexpression of wildtype or dominant-negative PLD1/2 and/or RNAi. Furthermore, pharmacological inhibition of PLD1 and PLD2 with a dual-isoform inhibitor, VU0155056, decreases the invasion of several cancer cell lines in a Matrigel™ transwell invasion assay.458 Another group used the halopemide analog (FIPI), which was originally reported by Monovich et al.,463 and showed that PLD inhibition blocked actin cytoskeleton reorganization, cell spreading and chemotaxis.130
12.6 Cell proliferation and apoptosis
Increased PLD expression and enzymatic activity have been observed in a variety of human cancers including breast,457 renal,514 brain471 and colorectal.467 Overexpression of PLD is able to promote cell growth and proliferation despite the presence of a variety of apoptotic stimuli.515,516 Furthermore, PLD activity is required for mutant Ras driven tumorigenesis in mice.517 Experiments utilizing inactivating mutations of PLD suggest that inhibiting PLD enzymatic activity increases cancer cell apoptosis.518 On a molecular level PLD has been implicated in oncogenic signaling pathways involving the epidermal growth factor receptor,519 matrix metalloproteinase (MMP) expression,471,473 p53,520,521 the mammalian target of rapamycin (mTOR)522 and Ras.523 The signaling network interactions between PLD and the various Ras signaling pathways constitute a series of complex interactions. One such example is the observation that the recruitment of Raf-1 kinase (which is activated by Ras) to the plasma membrane is dependent upon a direct interaction with PA.4,524 PLD activity contributes to key events in the oncogenic process including growth signaling, gatekeeper override, suppression of apoptosis and metastasis.6
A wide variety of extracellular factors that stimulate cell proliferation have been shown to increase PLD activity. Platelet derived growth factor (PDGF),525 fibroblast growth factor (FGF)526 and EGF527 are able to significantly increase PLD activity in a variety of different cell lines under physiological conditions. Additionally, cells that are transformed by mutations in several robustly validated oncogenes also display increased PLD activity. Notably, cells transformed by v-Ras517 or v-Raf528 display PLD activity that is several-fold higher than untransformed cells. PLD also facilitates the activation of the mitogen-activated protein kinase (MAPK) cascade.6 Treatment of cells with PA (the enzymatic product of PLD) suppresses p53 expression.365 PLD also acts to suppress the expression of p53 by stabilizing the MDM2-p53 complex.520 There are studies suggesting that PLD regulates the activity of mTOR, but the exact molecular mechanism of the PLD-mTOR interaction is being interrogated.415
The laboratory of David Foster and others have reported that PLD activity regulates hypoxia inducible factor (HIF) expression. Under normoxic conditions the von Hippel-Lindau (VHL) tumor suppressor gene is expressed and encodes part of an E3 ubiquitin ligase that targets the α subunits of HIF for degradation by the proteasome.529 There are two known conditions in which overexpression of HIF occurs and provides a survival advantage to cancer cells: (1) von Hippel-Lindau disease and (2) in the hypoxic tumor microenvironment of an individual with cancer who does not have von Hippel-Lindau disease. The result is that unregulated overexpression of HIF leads to angiogenesis, increased red blood cell production, and a shift to anaerobic metabolism.530 Toschi et al. utilized two VHL-deficient renal cancer cell lines (786-0 and RCC4) to show that HIF2α expression is dependent on PLD. The authors provide evidence for this conclusion by using three independent approaches: (1) treating cells with n-butanol, (2) the expression of dominant-negative PLD constructs and (3) the use of RNAi targeted to PLD1 and/or PLD2.531
It has been shown that decreasing PLD activity via genetic or biochemical approaches can increase cancer cell apoptosis.6,516 There are also previous reports linking PLD to changes in caspase activity.329,365 However, few accounts exist of how pharmacological inhibition of PLD affects cancer cell apoptosis. In 2010, Lavieri et al. showed that a PLD2-selective inhibitor, VU0364739, decreased the proliferation of MDA-MB-231 cells in a dose and time-dependent manner. VU0364739 also caused a several-fold increase in caspase 3/7-activity indicating that VU0364739 likely causes a decrease in cell proliferation by inducing apoptosis.470
The Wnt signaling pathway has emerged as a central regulator of cell proliferation and mutations in this pathway are clearly linked to oncogenesis. Briefly, there are several known Wnt signaling pathways (for reviews of the other Wnt pathways see532,533) and the canonical Wnt signaling pathway leads to the stabilization of β-catenin, which in turn activates T-cell factor-dependent transcription of a variety of target genes.534 Kang et al. utilized n-butanol, RNAi and pharmacological inhibitors to provide good evidence of a relationship between PLD and the Wnt signaling pathway. Their principal findings were that are: (1) Wnt3a increases PLD1 expression and activity in cultured cells, and β-catenin and TCF4 were required for this effect; (2) decreasing PLD activity decreases the ability of β-catenin to increase the transcription of PLD1 and other Wnt target genes; (3) PLD1 is necessary for Wnt-driven anchorage-independent growth and β-catenin/TCF4 are necessary for PLD1-driven anchorage-independent growth; and (4) the expression levels of PLD1 and PLD2 were substantially increased in the colon, liver and stomach tissues of mice after injection with LiCl (a known Wnt pathway agonist).535,536,537
The mitogen activated protein kinase (MAPK) pathway is a well-studied mechanism by which cells transmit extracellular signals from the cell surface to the nucleus and ultimately alter gene transcription. Several components of the MAPK pathway are frequently mutated, overexpressed and/or hyperactivated in human cancers.538 The three known mammalian Raf isforms, A-Raf, B-Raf and C-Raf, are serine/threonine kinases that lie in the middle of the MAPK pathway and have normal physiological roles as well as roles as oncogenes. All three Raf isoforms have been studied extensively in vitro and in cells. Additionally, all three Raf isoforms have been knocked out in mice.539 A-Raf knockout mice die within days of birth,540 while both B-Raf541 and C-Raf542 knockouts are lethal in utero. Raf kinase signaling has also been exploited into FDA approved drugs. At the time of this review sorafenib, a small molecule C-Raf inhibitor developed by Bayer and Onyx is approved for the treatment of advanced renal cell carcinoma543 and advanced hepatocellular carcinoma.544 Additionally, several other companies have Raf inhibitors at various stages of development in their pipelines.545
Recent work done by several different groups has provided evidence for a strong link between PLD and C-Raf kinase. In 1996 Ghosh et al. reported several findings that have been confirmed and expanded by other groups. They found that: (1) C-Raf binds to PA; (2) The PA binding site of C-Raf is between residues 389 and 423; (3) C-Raf does not bind phosphatidylalcohols and (4) treatment of Madin-darby canine kidney cells (MDCK) with 1 % ethanol reduced the translocation of C-Raf from the cytosol to the plasma membrane following treatment with 12-O-tetradecanoylphorbol-13-acetate.3 In agreement with earlier findings, Rizzo et al. showed that C-Raf binds PA and found that mutating arginine 398 to an alanine substantially reduced C-Raf’s ability to bind PA. Phosphatidic acid does not activate C-Raf kinase either in vitro or in vivo.524 Mutations that disrupt the C-Raf-PA interaction prevent the recruitment of C-Raf to membranes, but disruption of the Ras-Raf interaction does not prevent the recruitment of C-Raf to membranes. Expression of a dominant-negative Ras mutant did not prevent insulin-dependent C-Raf translocation to the plasma membrane, but did inhibit the phosphorylation of MAPK and the PA binding region of C-Raf was sufficient to target green fluoresecent protein to membranes. Taken together these results suggest a model whereby PA is both necessary and sufficient to target C-Raf to membranes, whereas Ras is not required to target C-Raf to membranes. However, in order for C-Raf to be activated, Ras must be present. Therefore, PA is required to bring C-Raf into proximity of Ras, then Ras activates C-Raf.4 Much of the data in support of this paradigm were gathered via dominant-negative, overexpression and mutation experiments, because RNAi and pharmacological inhibitors were not widely available at the time. Two reports have also shown that overexpression of C-Raf can either stimulate or inhibit PLD activity depending on the level of C-Raf activity. Low intensity C-Raf activity stimulates PLD activity,528 whereas high intensity C-Raf activity inhibits PLD activity.515
The mammalian target of rapamycin (mTOR) is a serine/threonine kinase that belongs to the phosphoinositide 3-kinase (PI3K)-related kinase family and serves as one of the master regulators of cell growth and division.546 The ability of PI3K to modulate mTOR activity is well established,547 but a detailed, well-substantiated understanding of the interaction between PLD and mTOR is being explored.415 In 2001 Fang et al. showed that the treatment of cells with n-butanol decreases mTOR downstream signaling, as measured by the activity of p70S6K. In the same studies n-butanol blocked a serum-induced increase in p70S6K activity suggesting that PLD might be involved in mTOR signaling. It should be noted that the PA produced by PLD appears to be necessary, but not sufficient, for mTOR signaling. In experiments where cells were deprived of amino acids, PA was not able to stimulate mTOR signaling.5 This original report linking PLD to mTOR provides relatively modest evidence in favor of the conclusion that PLD regulates mTOR signaling, because n-butanol was the only tool used to block PLD-mediated PA production.
In 2003 Chen et al. noted that PLD appears to confer resistance to rapamycin-induced cell death. They showed that the IC50 value for rapamycin-induced cell death in a cell line with relatively low PLD activity was about 10 nM, whereas the IC50 in rapamycin-induced cell death in a cell line with relatively high PLD activity was about 10 μM. Additionally, when a dominant-negative PLD2 was transfected into cells with high PLD activity, the cells showed increased sensitivity to rapamycin.522 A different group showed that PLD1 RNAi decreases the amount of phosphorylated p70S6K in B16 melanoma cells.548 Interestingly, Veverka et al. published a solution NMR structure of phosphatidic acid bound to the FKBP12-rapamycin binding domain of mTOR.248 This is compelling evidence that PA binds to mTOR, but in and of itself does not provide definitive information as to whether or not the PA that binds mTOR in vivo is made by PLD. Data from two different reports suggest the role PLD may play in mTOR signaling. Sun et al. showed that the suppression of TSC2 (via the transfection of a dominant-negative TSC2) strongly activates PLD in cells. They subsequently showed that recombinant Rheb purified from bacteria activates PLD1-immunocomplexes pulled-down from CHO cells. This suggests a model where the small GTPase Rheb (known to be regulated by TSC2549) either activates PLD directly or activates a protein pulled-down with PLD, which then in turn activates PLD.383 Toschi et al., through the use of n-butanol and dominant-negative PLD constructs, showed that PA produced by PLD is required for the formation of mTORC1 and mTORC2 complexes in 786-0 cells.416 Recently, Xu et al. showed that when T24 cells are treated with a combination of both a PLD1 (VU0379595) inhibitor and a PLD2 inhibitor (VU0364739), there is a decrease in mTOR activation as measured by phosphorylated p70S6K.550 Interestingly, Lehman et al. observed that PA produced by PLD can directly activate p70S6K independently of mTOR signaling.551 In 2011 Arous et al. reported that oleaic acid activated mTOR in cultured cells suggesting a possible mechanistic explanation for the increase in liver cancer seen in the obese population. Arous et al. claim that the oleic acid activation of mTOR is dependent on PLD, but provided only indirect evidence for this claim by using n-butanol as a “PLD inhibitor.” Furthermore, the effect of n-butanol on well-validated readouts of mTOR activation, such as p70S6K phosphorylation, is relatively small.552
A preponderance of evidence collected by independent groups indicates that PLD provides a survival signal in human cancer cells. Most of this evidence is from experiments in cultured cells that utilized n-butanol. Some groups have used primary cells isolated from humans and more recently groups have begun using RNAi, dominant-negatives, overexpression and small molecule inhibitors. Additionally, animal model experiments have only recently been published.322,517 Many of the cell signaling details about both PLD1 and PLD2 can now be more rigorously investigated through the use of molecular genetic techniques and small molecule inhibitors in both cell culture and animal models of disease.
13. PLD as a potential therapeutic target
Although the biochemistry, enzymology and pharmacology of PLD have been studied for more than half a century, the systematic investigation of PLD as a therapeutic target began only within the last few years. With the advent and commercialization of RNAi technology a generally better, more direct and more specific method to inhibit PLD has become available. The report of small molecule PLD inhibitors463 and the extensive effort that resulted in the development of drug-like, isoform-selective PLD inhibitors present a new opportunity for research in the field of lipid signaling.458,468,469,470
While the exact mechanism of action of these small molecule PLD inhibitors is still under investigation, the traditional view of PLD signaling is that PLD signals through production of PA, but there are still protein-protein and protein-lipid interactions to be taken into account. In 2011 Doti et al. showed that amino acids 762–801 of PLD1 interact with phosphoprotein enriched in diabetes/phosphoprotein enriched in astrocytes (PED/PEA15).553 This PED/PEA15 protein is overexpressed in several tissues in individuals with type 2 diabetes and its overexpression in cultured cells and transgenic animals impairs insulin regulation of glucose transport by a mechanism that is dependent on its physical interaction with PLD.386 It is interesting to consider the possibility of pharmacological agents that would act not on PLD catalytic activity, but rather block interactions between PLD and PED/PEA15.
In 2010 PLD1−/− and PLD2−/− mice were reported for the first time.391,474,493 The recent publication of viable PLD1 and PLD2 knockout mice and the report of isoform-selective, small molecule PLD inhibitors made PLD a target of interest in several diseases.
13.1 Cancer
Buchanan et al. reported a provocative set of experiments utilizing xenograft tumor models in mice. In order to explore how decreasing PLD activity would affect the ability of oncogenic Ras to transform cells, Buchanan generated rat fibroblasts that stably overexpress a dominant-negative PLD, referred to as Rat-2V25 cells.350 They showed that PLD activity is necessary for the H-Ras induced transformation of Rat-2 fibroblasts. Wildtype Rat-2 fibroblasts transfected with H-RasV12 grow in soft agar and form tumors in nude mice, but Rat-2V25 cells (that overexpress a dominant-negative PLD) do not form colonies in soft agar and do not form tumors in nude mice when transfected with H-RasV12. Additionally, when exogenous PA was added to the Rat-2V25 cells these cells were able to grow in soft agar and form tumors in nude mice.517 This study provided some of the first in vivo validation of PLD as a viable cancer target situated downstream of one of the most commonly mutated genes in human cancer.
Using a zebrafish vertebrate model organism, Zeng et al. showed that zPld1 is required for angiogenesis.322 Zebrafish treated with morpholino oligonucleotides targeted to zPld1 showed impaired intersegmental blood vessel development. While clearly a less specific approach, zebrafish embryos incubated with n-butanol also showed impaired intersegmental blood vessel development. Although intended to investigate the role of PLD in vertebrate development, the major finding of these studies is certainly additional evidence that inhibiting PLD may be a useful therapeutic approach in the treatment of cancer. The identification of PLD as a possible cancer drug target is based on observations of increased PLD activity or expression in tissue samples obtained from cancer patients.457,467,514,517,554,555 Furthermore, PLD1−/− and PLD2−/− mice are viable, develop normally, are fertile and exhibit behavior indistinguishable from wildtype littermates,391,474,493 suggesting that prolonged inhibition of one PLD isoform would be therapeutically viable.
13.2 Alzheimer’s disease
The recent reports of PLD1 and PLD2 knockout mice described viable animals with protection of disease states.391,474,493 The PLD2−/− mice (generated via gene targeting) facilitated research on a possible role of PLD in Alzheimer’s disease.474 Oligomeric amyloid β stimulates PLD activity in cultured neurons and ablation of PLD2 via gene targeting blocks this effect. In vivo PLD activity is increased in the brain of a mouse model of Alzheimer’s disease and PLD2 ablation via gene targeting rescues memory deficits and confers neuronal protection in a mouse model of Alzheimer’s disease despite a significant amyloid β load. Mass spectrometry-based analysis of lipids in the brains of animals with PLD2 knocked out in the background of a wildtype or Alzheimer’s mouse model show striking acyl chain specificity and compensatory mechanisms in PA metabolism. Interestingly, the total amount of PA present in either the mouse model of Alzheimer’s disease or the mouse model of Alzheimer’s disease crossed with a PLD2 knockout mouse (ADxPLD2KO) is essentially the same, but specific PA molecular species change by as much as 50 % in the ADxPLD2KO.474 The preparation of a PLD2 knockout mouse and the cross between a PLD2 knockout mouse and a mouse model of Alzheimer’s disease yielded excellent in vivo data on the role of PLD in a neurodegenerative disease. Additionally, the recent report of a centrally penetrant PLD2 inhibitor sets the stage for potential, preclinical target validation.470
13.3 Thrombotic disease
In 2010 Elvers et al. reported the generation of PLD1 homozygous knockout mice. The PLD1−/− mice display impaired αIIbβ3 intergrin activation in response to major agonists and show defective glycoprotein 1b-dependent aggregate formation under “high shear” conditions. These molecular alterations resulted in protection from thrombosis and ischemic brain injury without increasing bleeding time. Blood flow was monitored in two arterial thrombosis models triggered by chemical or mechanical perturbations and showed decreased occlusion in the PLD1−/− mice compared to wildtype mice, thus showing protection against thrombosis. This impressive study also reported no difference in bleeding time between wildtype mice and PLD1−/− mice. The implications of this work are exciting as the current pharmacological approaches used to prevent stroke and other thrombotic events (e.g., aspirin, clopidogrel and warfarin) increase bleeding times, which can be problematic. In summary, Elvers et al. showed that PLD1 is not required for normal hemostasis, but PLD1 is required for occlusive thrombus formation.391 Clearly, mouse model data must be extrapolated to human physiology with caution, but this study provides exceptionally strong evidence that PLD1 should be interrogated as a therapeutic target in thrombotic disease. The development of a drug that protects against thrombosis and ischemic brain injury without affecting a patient’s ability to form clots in the case of trauma would be a major advancement.
Acknowledgments
We thank Pavlina Ivanova for insightful discussions and Michelle Armstrong for skillful preparation of figures and artwork. We gratefully acknowledge the support of the National Institutes of Health (NIH Training Grant # GM 07628 for PES and RRL, U54 GM069338, and P01 ESO13125), the Pharmaceutical Research and Manufacturers of America Foundation (PhRMA) Predoctoral Fellowship in Pharmacology/Toxicology to RRL, the McDonnell Foundation for Brain Cancer Research, the Department of Pharmacology and the Vanderbilt Institute of Chemical Biology.
Biographies
 H. Alex Brown is Professor of Pharmacology and Chemistry at Vanderbilt University School of Medicine. Alex received his Ph.D. degree in 1992 from the University of North Carolina at Chapel Hill working in the laboratory of T. Kendall Harden and then pursued postdoctoral training in the Department of Pharmacology at the University of Texas-Southwestern Medical Center with Paul Sternweis. Alex joined the faculty at Cornell University in 1996 with appointments in Pharmacology and Biochemistry Molecular & Cell Biology. Alex received the Sidney Kimmel Foundation for Cancer Research Scholar award in 1997. Working with Fred McLafferty at Cornell, Alex developed the field of computational lipidomics in his laboratory utilizing electrospray ionization mass spectrometry. Al Gilman, then the director of the Alliance for Cellular Signaling (AfCS), invited Alex to use this emerging technology to contribute to the AfCS research program. In 2002, Alex was recruited to Vanderbilt University School of Medicine as the Ingram Professor of Cancer Research in Pharmacology. Alex has been the director of the glycerophospholipid core for the LIPID Metabolites and Pathways Strategy (LIPID MAPS) consortium since 2003. He has served on the editorial board for the Journal of Biological Chemistry, NIH-NIGMS & NIHLB study section, ASBMB publications committee, and as associate editor of Molecular Pharmacology from 2006 until 2009. In addition, Alex was editor of a three volume series on Lipidomics and Bioactive Lipids for Methods in Enzymology, co-editor with Lawrence Marnett of a thematic issue on lipid biochemistry for Chemical Reviews in 2011, and has organized multiple international conferences on lipid metabolism & signaling. Alex received the Vanderbilt Ingram Cancer Center (VICC) “High Impact Publications Award” in 2010, as well as the Vanderbilt University Medical Center “Leadership of a Multi-Investigator Team Award” together with Craig Lindsley in 2011. Alex is currently Metabolomics co-director for the Scripps-Vanderbilt Human Chemical Sciences Institute and the associate director of System Analysis for the Vanderbilt Institute of Chemical Biology (VICB). His current research is focused on understanding the roles of lipid molecular species and phospholipases in cellular functions and human disease.
H. Alex Brown is Professor of Pharmacology and Chemistry at Vanderbilt University School of Medicine. Alex received his Ph.D. degree in 1992 from the University of North Carolina at Chapel Hill working in the laboratory of T. Kendall Harden and then pursued postdoctoral training in the Department of Pharmacology at the University of Texas-Southwestern Medical Center with Paul Sternweis. Alex joined the faculty at Cornell University in 1996 with appointments in Pharmacology and Biochemistry Molecular & Cell Biology. Alex received the Sidney Kimmel Foundation for Cancer Research Scholar award in 1997. Working with Fred McLafferty at Cornell, Alex developed the field of computational lipidomics in his laboratory utilizing electrospray ionization mass spectrometry. Al Gilman, then the director of the Alliance for Cellular Signaling (AfCS), invited Alex to use this emerging technology to contribute to the AfCS research program. In 2002, Alex was recruited to Vanderbilt University School of Medicine as the Ingram Professor of Cancer Research in Pharmacology. Alex has been the director of the glycerophospholipid core for the LIPID Metabolites and Pathways Strategy (LIPID MAPS) consortium since 2003. He has served on the editorial board for the Journal of Biological Chemistry, NIH-NIGMS & NIHLB study section, ASBMB publications committee, and as associate editor of Molecular Pharmacology from 2006 until 2009. In addition, Alex was editor of a three volume series on Lipidomics and Bioactive Lipids for Methods in Enzymology, co-editor with Lawrence Marnett of a thematic issue on lipid biochemistry for Chemical Reviews in 2011, and has organized multiple international conferences on lipid metabolism & signaling. Alex received the Vanderbilt Ingram Cancer Center (VICC) “High Impact Publications Award” in 2010, as well as the Vanderbilt University Medical Center “Leadership of a Multi-Investigator Team Award” together with Craig Lindsley in 2011. Alex is currently Metabolomics co-director for the Scripps-Vanderbilt Human Chemical Sciences Institute and the associate director of System Analysis for the Vanderbilt Institute of Chemical Biology (VICB). His current research is focused on understanding the roles of lipid molecular species and phospholipases in cellular functions and human disease.
 Craig W. Lindsley is Professor of Pharmacology and Chemistry, Center Co-Director and Director of Medicinal Chemistry for the Vanderbilt Center for Neuroscience Drug Discovery as well as PI of the Vanderbilt Specialized Chemistry Center (one of two in the MLPCN) at Vanderbilt Medical Center. Craig received the Ph.D. degree in Chemistry from the University of California, Santa Barbara in 1996 and pursued postdoctoral studies in the Department of Chemistry at Harvard University. After brief stints at Parke-Davis and Eli Lilly, Craig joined the Medicinal Chemistry Department at Merck, West Point and established and led the Technology Enabled Synthesis (TES) Group. At Merck, Craig developed a streamlined approach for lead optimization which resulted in the accelerated delivery of six preclinical candidates (cancer, schizophrenia, cognition/Alzheimer’s disease, atrial fibrillation, insomnia) with reduced staffing. Several of these have now provided proof-of-concept in Phase II clinical trials. Importantly, Craig also designed, synthesized and provided preclinical proof-of-concept for the first isoenzyme selective, allosteric AKT kinase inhibitors and the first positive allosteric modulators (PAMs) of mGlu5 (CPPHA, CDPPB) and M1 (BQCA). After 6 years at Merck, Craig joined the faculty of the Department of Pharmacology at Vanderbilt University in late 2006 as an Associate Professor of Pharmacology and Chemistry with a primary mission of facilitating translation of recent advances in basic science to novel therapeutics. In 2010, he was promoted to Professor of Pharmacology and Chemistry with tenure and established himself as a leader in the development of allosteric ligands for the study of GPCRs, kinases and phospholipases (PLD) in psychiatric and neurological disorders. Craig is Editor-in-Chief of ACS Chemical Neuroscience, former Associate Editor of Current Topics in Medicinal Chemistry and serves on the editorial boards of 5 other international journals. Craig is a medicinal chemistry consultant for numerous pharmaceutical companies and foundations. Craig’s current research is focused on development of novel treatment strategies for schizophrenia, Alzheimer’s Disease, Fragile X Syndrome, Parkinson’s disease, brain tumors, as well as other brain disorders.
Craig W. Lindsley is Professor of Pharmacology and Chemistry, Center Co-Director and Director of Medicinal Chemistry for the Vanderbilt Center for Neuroscience Drug Discovery as well as PI of the Vanderbilt Specialized Chemistry Center (one of two in the MLPCN) at Vanderbilt Medical Center. Craig received the Ph.D. degree in Chemistry from the University of California, Santa Barbara in 1996 and pursued postdoctoral studies in the Department of Chemistry at Harvard University. After brief stints at Parke-Davis and Eli Lilly, Craig joined the Medicinal Chemistry Department at Merck, West Point and established and led the Technology Enabled Synthesis (TES) Group. At Merck, Craig developed a streamlined approach for lead optimization which resulted in the accelerated delivery of six preclinical candidates (cancer, schizophrenia, cognition/Alzheimer’s disease, atrial fibrillation, insomnia) with reduced staffing. Several of these have now provided proof-of-concept in Phase II clinical trials. Importantly, Craig also designed, synthesized and provided preclinical proof-of-concept for the first isoenzyme selective, allosteric AKT kinase inhibitors and the first positive allosteric modulators (PAMs) of mGlu5 (CPPHA, CDPPB) and M1 (BQCA). After 6 years at Merck, Craig joined the faculty of the Department of Pharmacology at Vanderbilt University in late 2006 as an Associate Professor of Pharmacology and Chemistry with a primary mission of facilitating translation of recent advances in basic science to novel therapeutics. In 2010, he was promoted to Professor of Pharmacology and Chemistry with tenure and established himself as a leader in the development of allosteric ligands for the study of GPCRs, kinases and phospholipases (PLD) in psychiatric and neurological disorders. Craig is Editor-in-Chief of ACS Chemical Neuroscience, former Associate Editor of Current Topics in Medicinal Chemistry and serves on the editorial boards of 5 other international journals. Craig is a medicinal chemistry consultant for numerous pharmaceutical companies and foundations. Craig’s current research is focused on development of novel treatment strategies for schizophrenia, Alzheimer’s Disease, Fragile X Syndrome, Parkinson’s disease, brain tumors, as well as other brain disorders.
 Paige E. Selvy obtained her Bachelor’s degree in Biochemistry from Mount Holyoke College and is currently a doctoral candidate in the Department of Pharmacology at Vanderbilt University in the laboratory of Dr. H. Alex Brown. In Dr. Brown’s lab, she completed biochemical structure-activity relationship characterization for the first description of isoform-selective small molecule inhibitors of mammalian PLD. Her dissertation research went on to characterize the molecular mechanism of action and small molecule binding site for these novel compounds. Paige received the 2011 Excellence in Chemical Biology student research award at Vanderbilt University for her collaborative and multidisciplinary research. Paige will soon pursue postdoctoral studies in the area of lipid signaling targets in host-pathogen interactions.
Paige E. Selvy obtained her Bachelor’s degree in Biochemistry from Mount Holyoke College and is currently a doctoral candidate in the Department of Pharmacology at Vanderbilt University in the laboratory of Dr. H. Alex Brown. In Dr. Brown’s lab, she completed biochemical structure-activity relationship characterization for the first description of isoform-selective small molecule inhibitors of mammalian PLD. Her dissertation research went on to characterize the molecular mechanism of action and small molecule binding site for these novel compounds. Paige received the 2011 Excellence in Chemical Biology student research award at Vanderbilt University for her collaborative and multidisciplinary research. Paige will soon pursue postdoctoral studies in the area of lipid signaling targets in host-pathogen interactions.
 Robert R. Lavieri received his B.A. in biochemistry and philosophy from DePauw University and is currently a Ph.D. candidate in the Department of Pharmacology at Vanderbilt University, where he works under the joint mentorship of both Craig W. Lindsley and H. Alex Brown. During the first portion of Rob’s dissertation research he utilized medicinal chemistry, including technology enabled synthesis, under the direction of Dr. Lindsley in order to determine structure activity relationships and ultimately to synthesize isoform-selective PLD inhibitors. More recently he has been utilizing these compounds to study cell signaling networks under the direction of Dr. Brown. Rob was awarded a Pharmaceutical Research and Manufacturers Association of America (PhRMA) Predoctoral fellowship in 2010.
Robert R. Lavieri received his B.A. in biochemistry and philosophy from DePauw University and is currently a Ph.D. candidate in the Department of Pharmacology at Vanderbilt University, where he works under the joint mentorship of both Craig W. Lindsley and H. Alex Brown. During the first portion of Rob’s dissertation research he utilized medicinal chemistry, including technology enabled synthesis, under the direction of Dr. Lindsley in order to determine structure activity relationships and ultimately to synthesize isoform-selective PLD inhibitors. More recently he has been utilizing these compounds to study cell signaling networks under the direction of Dr. Brown. Rob was awarded a Pharmaceutical Research and Manufacturers Association of America (PhRMA) Predoctoral fellowship in 2010.
References
- 1.Voelker DR. Microbiol Rev. 1991;55:543. doi: 10.1128/mr.55.4.543-560.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stace CL, Ktistakis NT. Biochim Biophys Acta. 2006;1761:913. doi: 10.1016/j.bbalip.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 3.Ghosh S, Strum JC, Sciorra VA, Daniel L, Bell RM. J Biol Chem. 1996;271:8472. doi: 10.1074/jbc.271.14.8472. [DOI] [PubMed] [Google Scholar]
- 4.Rizzo MA, Shome K, Watkins SC, Romero G. J Biol Chem. 2000;275:23911. doi: 10.1074/jbc.M001553200. [DOI] [PubMed] [Google Scholar]
- 5.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Science. 2001;294:1942. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 6.Foster DA, Xu L. Mol Cancer Res. 2003;1:789. [PubMed] [Google Scholar]
- 7.Oliveira TG, Di Paolo G. Biochim Biophys Acta. 2010;1801:799. doi: 10.1016/j.bbalip.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Siess W, Tigyi G. J Cell Biochem. 2004;92:1086. doi: 10.1002/jcb.20108. [DOI] [PubMed] [Google Scholar]
- 9.Athenstaedt K, Daum G. Eur J Biochem. 1999;266:1. doi: 10.1046/j.1432-1327.1999.00822.x. [DOI] [PubMed] [Google Scholar]
- 10.Athenstaedt K, Weys S, Paltauf F, Daum G. J Bacteriol. 1999;181:1458. doi: 10.1128/jb.181.5.1458-1463.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gibellini F, Smith TK. IUBMB Life. 2010;62:414. doi: 10.1002/iub.337. [DOI] [PubMed] [Google Scholar]
- 12.Kennedy EP. Am J Clin Nutr. 1958;6:216. doi: 10.1093/ajcn/6.3.216. [DOI] [PubMed] [Google Scholar]
- 13.Hanahan DJ, Chaikoff IL. J Biol Chem. 1947;168:233. [PubMed] [Google Scholar]
- 14.Hanahan DJ, Chaikoff IL. J Biol Chem. 1947;169:699. [PubMed] [Google Scholar]
- 15.Hanahan DJ, Chaikoff IL. J Biol Chem. 1948;172:191. [PubMed] [Google Scholar]
- 16.Heller M. Adv Lipid Res. 1978;16:267. doi: 10.1016/b978-0-12-024916-9.50011-1. [DOI] [PubMed] [Google Scholar]
- 17.Liebecq C. Biochemical nomenclature and related documents. 2. Portland Press; London: 1992. [Google Scholar]
- 18.Ponting CP, Kerr ID. Protein Sci. 1996;5:914. doi: 10.1002/pro.5560050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koonin EV. Trends Biochem Sci. 1996;21:242. [PubMed] [Google Scholar]
- 20.Imamura S, Horiuti Y. J Biochem. 1979;85:79. doi: 10.1093/oxfordjournals.jbchem.a132334. [DOI] [PubMed] [Google Scholar]
- 21.Yoshioka K, Mizoguchi M, Takahara M, Iwanamura S, Beppu T, Horinouchi S. Japanese Patent. 1991;H3-187382
- 22.Zambonelli C, Roberts MF. Prog Nucleic Acid Res Mol Biol. 2005;79:133. doi: 10.1016/S0079-6603(04)79003-0. [DOI] [PubMed] [Google Scholar]
- 23.Zambonelli C, Casali M, Roberts MF. J Biol Chem. 2003;278:52282. doi: 10.1074/jbc.M310252200. [DOI] [PubMed] [Google Scholar]
- 24.Yang H, Roberts MF. Protein Sci. 2003;12:2087. doi: 10.1110/ps.03192503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stieglitz K, Seaton B, Roberts MF. J Biol Chem. 1999;274:35367. doi: 10.1074/jbc.274.50.35367. [DOI] [PubMed] [Google Scholar]
- 26.Yang H, Roberts MF. Protein Sci. 2002;11:2958. doi: 10.1110/ps.0225302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geng D, Chura J, Roberts MF. J Biol Chem. 1998;273:12195. doi: 10.1074/jbc.273.20.12195. [DOI] [PubMed] [Google Scholar]
- 28.Geng D, Baker DP, Foley SF, Zhou C, Stieglitz K, Roberts MF. Biochim Biophys Acta. 1999;1430:234. doi: 10.1016/s0167-4838(99)00005-9. [DOI] [PubMed] [Google Scholar]
- 29.McNamara PJ, Cuevas WA, Songer JG. Gene. 1995;156:113. doi: 10.1016/0378-1119(95)00002-n. [DOI] [PubMed] [Google Scholar]
- 30.Cuevas WA, Songer JG. Infect Immun. 1993;61:4310. doi: 10.1128/iai.61.10.4310-4316.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yabu T, Imamura S, Yamashita M, Okazaki T. J Biol Chem. 2008;283:29971. doi: 10.1074/jbc.M805402200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hodgson AL, Krywult J, Corner LA, Rothel JS, Radford AJ. Infect Immun. 1992;60:2900. doi: 10.1128/iai.60.7.2900-2905.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McNamara PJ, Bradley GA, Songer JG. Mol Microbiol. 1994;12:921. doi: 10.1111/j.1365-2958.1994.tb01080.x. [DOI] [PubMed] [Google Scholar]
- 34.Soucek A, Souckova A. J Hyg Epidemiol Microbiol Immunol. 1974;18:327. [PubMed] [Google Scholar]
- 35.Hodgson AL, Bird P, Nisbet IT. J Bacteriol. 1990;172:1256. doi: 10.1128/jb.172.3.1256-1261.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lucas EA, Billington SJ, Carlson P, McGee DJ, Jost BH. BMC Microbiol. 2010;10:270. doi: 10.1186/1471-2180-10-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Talay SR, editor. Gram-positive adhesins. Vol. 12. Karger: Basel; 2005. [DOI] [PubMed] [Google Scholar]
- 38.van Meeteren LA, Frederiks F, Giepmans BN, Pedrosa MF, Billington SJ, Jost BH, Tambourgi DV, Moolenaar WH. J Biol Chem. 2004;279:10833. doi: 10.1074/jbc.C300563200. [DOI] [PubMed] [Google Scholar]
- 39.Eggleton DG, Middleton HD, Doidge CV, Minty DW. Aust Vet J. 1991;68:317. doi: 10.1111/j.1751-0813.1991.tb03085.x. [DOI] [PubMed] [Google Scholar]
- 40.Murakami MT, Fernandes-Pedrosa MF, Tambourgi DV, Arni RK. J Biol Chem. 2005;280:13658. doi: 10.1074/jbc.M412437200. [DOI] [PubMed] [Google Scholar]
- 41.Futrell JM. Am J Med Sci. 1992;304:261. doi: 10.1097/00000441-199210000-00008. [DOI] [PubMed] [Google Scholar]
- 42.Tambourgi DV, Magnoli FC, van den Berg CW, Morgan BP, de Araujo PS, Alves EW, Da Silva WD. Biochem Biophys Res Commun. 1998;251:366. doi: 10.1006/bbrc.1998.9474. [DOI] [PubMed] [Google Scholar]
- 43.Tambourgi DV, Petricevich VL, Magnoli FC, Assaf SL, Jancar S, Dias Da Silva W. Toxicon. 1998;36:391. doi: 10.1016/s0041-0101(97)00063-9. [DOI] [PubMed] [Google Scholar]
- 44.Murakami MT, Fernandes-Pedrosa MF, de Andrade SA, Gabdoulkhakov A, Betzel C, Tambourgi DV, Arni RK. Biochem Biophys Res Commun. 2006;342:323. doi: 10.1016/j.bbrc.2006.01.123. [DOI] [PubMed] [Google Scholar]
- 45.Lee S, Lynch KR. Biochem J. 2005;391:317. doi: 10.1042/BJ20050043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moolenaar WH, van Meeteren LA, Giepmans BN. Bioessays. 2004;26:870. doi: 10.1002/bies.20081. [DOI] [PubMed] [Google Scholar]
- 47.de Giuseppe PO, Ullah A, Silva DT, Gremski LH, Wille AC, Chaves Moreira D, Ribeiro AS, Chaim OM, Murakami MT, Veiga SS, Arni RK. Biochem Biophys Res Commun. 2011;409:622. doi: 10.1016/j.bbrc.2011.05.053. [DOI] [PubMed] [Google Scholar]
- 48.Jones DR, Avila MA, Sanz C, Varela-Nieto I. Biochem Biophys Res Commun. 1997;233:432. doi: 10.1006/bbrc.1997.6475. [DOI] [PubMed] [Google Scholar]
- 49.Raikwar NS, Bowen RF, Deeg MA. Biochem J. 2005;391:285. doi: 10.1042/BJ20050656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li JY, Hollfelder K, Huang KS, Low MG. J Biol Chem. 1994;269:28963. [PubMed] [Google Scholar]
- 51.Deeg MA, Verchere CB. Endocrinology. 1997;138:819. doi: 10.1210/endo.138.2.4940. [DOI] [PubMed] [Google Scholar]
- 52.Deeg MA, Raikwar NS, Johnson C, Williams CD. Transl Res. 2007;150:153. doi: 10.1016/j.trsl.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 53.Naghibalhossaini F, Ebadi P. Cancer Lett. 2006;234:158. doi: 10.1016/j.canlet.2005.03.028. [DOI] [PubMed] [Google Scholar]
- 54.Raymond FD, Fortunato G, Moss DW, Castaldo G, Salvatore F, Impallomeni M. Clin Sci (Lond) 1994;86:447. doi: 10.1042/cs0860447. [DOI] [PubMed] [Google Scholar]
- 55.Chalasani N, Vuppalanchi R, Raikwar NS, Deeg MA. J Clin Endocrinol Metab. 2006;91:2279. doi: 10.1210/jc.2006-0075. [DOI] [PubMed] [Google Scholar]
- 56.Deeg MA, Bowen RF, Williams MD, Olson LK, Kirk EA, LeBoeuf RC. Am J Physiol Endocrinol Metab. 2001;281:E147. doi: 10.1152/ajpendo.2001.281.1.E147. [DOI] [PubMed] [Google Scholar]
- 57.Schofield JN, Stephens JW, Hurel SJ, Bell KM, de Souza JB, Rademacher TW. Mol Genet Metab. 2002;75:154. doi: 10.1006/mgme.2001.3287. [DOI] [PubMed] [Google Scholar]
- 58.Tang JH, He WJ, Huang H, Tan CC, Duan Q, Wang KJ, Yuan XY, Zhu XJ. Clin Biochem. 2009;42:400. doi: 10.1016/j.clinbiochem.2008.11.019. [DOI] [PubMed] [Google Scholar]
- 59.Okamoto Y, Morishita J, Tsuboi K, Tonai T, Ueda N. J Biol Chem. 2004;279:5298. doi: 10.1074/jbc.M306642200. [DOI] [PubMed] [Google Scholar]
- 60.Wang J, Okamoto Y, Morishita J, Tsuboi K, Miyatake A, Ueda N. J Biol Chem. 2006;281:12325. doi: 10.1074/jbc.M512359200. [DOI] [PubMed] [Google Scholar]
- 61.Schmid PC, Reddy PV, Natarajan V, Schmid HH. J Biol Chem. 1983;258:9302. [PubMed] [Google Scholar]
- 62.Petersen G, Hansen HS. FEBS Lett. 1999;455:41. doi: 10.1016/s0014-5793(99)00861-3. [DOI] [PubMed] [Google Scholar]
- 63.Wang J, Ueda N. Prostaglandins Other Lipid Mediat. 2009;89:112. doi: 10.1016/j.prostaglandins.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 64.Okamoto Y, Tsuboi K, Ueda N. Vitam Horm. 2009;81:1. doi: 10.1016/S0083-6729(09)81001-7. [DOI] [PubMed] [Google Scholar]
- 65.Guo Y, Wang H, Okamoto Y, Ueda N, Kingsley PJ, Marnett LJ, Schmid HH, Das SK, Dey SK. J Biol Chem. 2005;280:23429. doi: 10.1074/jbc.C500168200. [DOI] [PubMed] [Google Scholar]
- 66.Wang H, Xie H, Sun X, Kingsley PJ, Marnett LJ, Cravatt BF, Dey SK. Prostaglandins Other Lipid Mediat. 2007;83:62. doi: 10.1016/j.prostaglandins.2006.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhu C, Solorzano C, Sahar S, Realini N, Fung E, Sassone-Corsi P, Piomelli D. Mol Pharmacol. 2011;79:786. doi: 10.1124/mol.110.070201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leung D, Saghatelian A, Simon GM, Cravatt BF. Biochemistry. 2006;45:4720. doi: 10.1021/bi060163l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ueda N, Okamoto Y, Tsuboi K. Curr Med Chem. 2005;12:1413. doi: 10.2174/0929867054020918. [DOI] [PubMed] [Google Scholar]
- 70.Burch J, McKenna C, Palmer S, Norman G, Glanville J, Sculpher M, Woolacott N. Health Technol Assess. 2009;13(Suppl 3):13. doi: 10.3310/hta13suppl3/03. [DOI] [PubMed] [Google Scholar]
- 71.Tyagi P, Jain VK, Nadler E. Support Cancer Ther. 2005;3:11. doi: 10.1016/s1543-2912(13)60113-7. [DOI] [PubMed] [Google Scholar]
- 72.Zhang H, Hilton DA, Hanemann CO, Zajicek J. Brain Pathol. 2011;21:544. doi: 10.1111/j.1750-3639.2011.00477.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zajicek J, Fox P, Sanders H, Wright D, Vickery J, Nunn A, Thompson A. Lancet. 2003;362:1517. doi: 10.1016/S0140-6736(03)14738-1. [DOI] [PubMed] [Google Scholar]
- 74.Teare L, Zajicek J. Expert Opin Investig Drugs. 2005;14:859. doi: 10.1517/13543784.14.7.859. [DOI] [PubMed] [Google Scholar]
- 75.Yun CH, Ahn T, Guengerich FP, Yamazaki H, Shimada T. Arch Biochem Biophys. 1999;367:81. doi: 10.1006/abbi.1999.1254. [DOI] [PubMed] [Google Scholar]
- 76.Guengerich FP. Chem Res Toxicol. 2001;14:611. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- 77.Yoo JS, Ishizaki H, Yang CS. Carcinogenesis. 1990;11:2239. doi: 10.1093/carcin/11.12.2239. [DOI] [PubMed] [Google Scholar]
- 78.Zhou SF, Yang LP, Zhou ZW, Liu YH, Chan E. AAPS J. 2009;11:481. doi: 10.1208/s12248-009-9127-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cho EY, Yun CH, Chae HZ, Chae HJ, Ahn T. Biochem Biophys Res Commun. 2008;376:584. doi: 10.1016/j.bbrc.2008.09.023. [DOI] [PubMed] [Google Scholar]
- 80.Omura T. J Biochem. 2010;147:297. doi: 10.1093/jb/mvq001. [DOI] [PubMed] [Google Scholar]
- 81.Tokumura A, Majima E, Kariya Y, Tominaga K, Kogure K, Yasuda K, Fukuzawa K. J Biol Chem. 2002;277:39436. doi: 10.1074/jbc.M205623200. [DOI] [PubMed] [Google Scholar]
- 82.Tsuda S, Okudaira S, Moriya-Ito K, Shimamoto C, Tanaka M, Aoki J, Arai H, Murakami-Murofushi K, Kobayashi T. J Biol Chem. 2006;281:26081. doi: 10.1074/jbc.M602925200. [DOI] [PubMed] [Google Scholar]
- 83.Umezu-Goto M, Kishi Y, Taira A, Hama K, Dohmae N, Takio K, Yamori T, Mills GB, Inoue K, Aoki J, Arai H. J Cell Biol. 2002;158:227. doi: 10.1083/jcb.200204026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Aoki J, Taira A, Takanezawa Y, Kishi Y, Hama K, Kishimoto T, Mizuno K, Saku K, Taguchi R, Arai H. J Biol Chem. 2002;277:48737. doi: 10.1074/jbc.M206812200. [DOI] [PubMed] [Google Scholar]
- 85.Clair T, Aoki J, Koh E, Bandle RW, Nam SW, Ptaszynska MM, Mills GB, Schiffmann E, Liotta LA, Stracke ML. Cancer Res. 2003;63:5446. [PubMed] [Google Scholar]
- 86.van Meeteren LA, Ruurs P, Christodoulou E, Goding JW, Takakusa H, Kikuchi K, Perrakis A, Nagano T, Moolenaar WH. J Biol Chem. 2005;280:21155. doi: 10.1074/jbc.M413183200. [DOI] [PubMed] [Google Scholar]
- 87.Tania M, Khan A, Zhang H, Li J, Song Y. Biochem Biophys Res Commun. 2010;401:493. doi: 10.1016/j.bbrc.2010.09.114. [DOI] [PubMed] [Google Scholar]
- 88.Nakanaga K, Hama K, Aoki J. J Biochem. 2010;148:13. doi: 10.1093/jb/mvq052. [DOI] [PubMed] [Google Scholar]
- 89.Hausmann J, Kamtekar S, Christodoulou E, Day JE, Wu T, Fulkerson Z, Albers HM, van Meeteren LA, Houben AJ, van Zeijl L, Jansen S, Andries M, Hall T, Pegg LE, Benson TE, Kasiem M, Harlos K, Kooi CW, Smyth SS, Ovaa H, Bollen M, Morris AJ, Moolenaar WH, Perrakis A. Nat Struct Mol Biol. 2011;18:198. doi: 10.1038/nsmb.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nishimasu H, Okudaira S, Hama K, Mihara E, Dohmae N, Inoue A, Ishitani R, Takagi J, Aoki J, Nureki O. Nat Struct Mol Biol. 2011;18:205. doi: 10.1038/nsmb.1998. [DOI] [PubMed] [Google Scholar]
- 91.Tanaka M, Okudaira S, Kishi Y, Ohkawa R, Iseki S, Ota M, Noji S, Yatomi Y, Aoki J, Arai H. J Biol Chem. 2006;281:25822. doi: 10.1074/jbc.M605142200. [DOI] [PubMed] [Google Scholar]
- 92.Fotopoulou S, Oikonomou N, Grigorieva E, Nikitopoulou I, Paparountas T, Thanassopoulou A, Zhao Z, Xu Y, Kontoyiannis DL, Remboutsika E, Aidinis V. Dev Biol. 2010;339:451. doi: 10.1016/j.ydbio.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 93.Dennis J, White MA, Forrest AD, Yuelling LM, Nogaroli L, Afshari FS, Fox MA, Fuss B. Mol Cell Neurosci. 2008;37:412. doi: 10.1016/j.mcn.2007.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yuelling LM, Fuss B. Biochim Biophys Acta. 2008;1781:525. doi: 10.1016/j.bbalip.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kanda H, Newton R, Klein R, Morita Y, Gunn MD, Rosen SD. Nat Immunol. 2008;9:415. doi: 10.1038/ni1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nakasaki T, Tanaka T, Okudaira S, Hirosawa M, Umemoto E, Otani K, Jin S, Bai Z, Hayasaka H, Fukui Y, Aozasa K, Fujita N, Tsuruo T, Ozono K, Aoki J, Miyasaka M. Am J Pathol. 2008;173:1566. doi: 10.2353/ajpath.2008.071153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhou Z, Subramanian P, Sevilmis G, Globke B, Soehnlein O, Karshovska E, Megens R, Heyll K, Chun J, Saulnier-Blache JS, Reinholz M, van Zandvoort M, Weber C, Schober A. Cell Metab. 2011;13:592. doi: 10.1016/j.cmet.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 98.Liscovitch M, Czarny M, Fiucci G, Tang X. Biochem J. 2000;345(Pt 3):401. [PMC free article] [PubMed] [Google Scholar]
- 99.Rudolph AE, Stuckey JA, Zhao Y, Matthews HR, Patton WA, Moss J, Dixon JE. J Biol Chem. 1999;274:11824. doi: 10.1074/jbc.274.17.11824. [DOI] [PubMed] [Google Scholar]
- 100.Abergel C, Abousalham A, Chenivesse S, Riviere M, Moustacas-Gardies AM, Verger R. Acta Crystallogr D Biol Crystallogr. 2001;57:320. doi: 10.1107/s0907444900020825. [DOI] [PubMed] [Google Scholar]
- 101.Leiros I, Hough E, D’Arrigo P, Carrea G, Pedrocchi-Fantoni G, Secundo F, Servi S. Acta Crystallogr D Biol Crystallogr. 2000;56:466. doi: 10.1107/s0907444900000925. [DOI] [PubMed] [Google Scholar]
- 102.Stuckey JA, Dixon JE. Nat Struct Biol. 1999;6:278. doi: 10.1038/6716. [DOI] [PubMed] [Google Scholar]
- 103.Davies DR, Interthal H, Champoux JJ, Hol WG. Structure. 2002;10:237. doi: 10.1016/s0969-2126(02)00707-4. [DOI] [PubMed] [Google Scholar]
- 104.Davies DR, Interthal H, Champoux JJ, Hol WG. J Mol Biol. 2002;324:917. doi: 10.1016/s0022-2836(02)01154-3. [DOI] [PubMed] [Google Scholar]
- 105.Leiros I, Secundo F, Zambonelli C, Servi S, Hough E. Structure. 2000;8:655. doi: 10.1016/s0969-2126(00)00150-7. [DOI] [PubMed] [Google Scholar]
- 106.Uesugi Y, Hatanaka T. Biochim Biophys Acta. 2009;1791:962. doi: 10.1016/j.bbalip.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 107.Uesugi Y, Arima J, Iwabuchi M, Hatanaka T. FEBS J. 2007;274:2672. doi: 10.1111/j.1742-4658.2007.05802.x. [DOI] [PubMed] [Google Scholar]
- 108.Uesugi Y, Arima J, Iwabuchi M, Hatanaka T. Protein Sci. 2007;16:197. doi: 10.1110/ps.062537907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ogino C, Daido H, Ohmura Y, Takada N, Itou Y, Kondo A, Fukuda H, Shimizu N. Biochim Biophys Acta. 2007;1774:671. doi: 10.1016/j.bbapap.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 110.Orth ES, Brandao TA, Souza BS, Pliego JR, Vaz BG, Eberlin MN, Kirby AJ, Nome F. J Am Chem Soc. 2010;132:8513. doi: 10.1021/ja1034733. [DOI] [PubMed] [Google Scholar]
- 111.Leiros I, McSweeney S, Hough E. J Mol Biol. 2004;339:805. doi: 10.1016/j.jmb.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 112.Yang SF, Freer S, Benson AA. J Biol Chem. 1967;242:477. [PubMed] [Google Scholar]
- 113.Stanacev NZ, Stuhne-Sekalec L. Biochim Biophys Acta. 1970;210:350. doi: 10.1016/0005-2760(70)90183-9. [DOI] [PubMed] [Google Scholar]
- 114.Sung TC, Roper RL, Zhang Y, Rudge SA, Temel R, Hammond SM, Morris AJ, Moss B, Engebrecht J, Frohman MA. EMBO J. 1997;16:4519. doi: 10.1093/emboj/16.15.4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gottlin EB, Rudolph AE, Zhao Y, Matthews HR, Dixon JE. Proc Natl Acad Sci USA. 1998;95:9202. doi: 10.1073/pnas.95.16.9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dawson RM. Biochem J. 1967;102:205. doi: 10.1042/bj1020205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ella KM, Dolan JW, Meier KE. Biochem J. 1995;307(Pt 3):799. doi: 10.1042/bj3070799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Iwasaki Y, Horiike S, Matsushima K, Yamane T. Eur J Biochem. 1999;264:577. doi: 10.1046/j.1432-1327.1999.00669.x. [DOI] [PubMed] [Google Scholar]
- 119.Berg OG, Gelb MH, Tsai MD, Jain MK. Chem Rev. 2001;101:2613. doi: 10.1021/cr990139w. [DOI] [PubMed] [Google Scholar]
- 120.Deems RA. Anal Biochem. 2000;287:1. doi: 10.1006/abio.2000.4766. [DOI] [PubMed] [Google Scholar]
- 121.Jain MK, Berg OG. Biochim Biophys Acta. 1989;1002:127. doi: 10.1016/0005-2760(89)90281-6. [DOI] [PubMed] [Google Scholar]
- 122.Carman GM, Deems RA, Dennis EA. J Biol Chem. 1995;270:18711. doi: 10.1074/jbc.270.32.18711. [DOI] [PubMed] [Google Scholar]
- 123.Buser CA, McLaughlin S. Methods Mol Biol. 1998;84:267. doi: 10.1385/0-89603-488-7:267. [DOI] [PubMed] [Google Scholar]
- 124.Qin C, Wang C, Wang X. J Biol Chem. 2002;277:49685. doi: 10.1074/jbc.M209598200. [DOI] [PubMed] [Google Scholar]
- 125.Henage LG, Exton JH, Brown HA. J Biol Chem. 2006;281:3408. doi: 10.1074/jbc.M508800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.James SR, Paterson A, Harden TK, Downes CP. J Biol Chem. 1995;270:11872. doi: 10.1074/jbc.270.20.11872. [DOI] [PubMed] [Google Scholar]
- 127.Kung C. Nature. 2005;436:647. doi: 10.1038/nature03896. [DOI] [PubMed] [Google Scholar]
- 128.Brown HA, Gutowski S, Moomaw CR, Slaughter C, Sternweis PC. Cell. 1993;75:1137. doi: 10.1016/0092-8674(93)90323-i. [DOI] [PubMed] [Google Scholar]
- 129.Brown HA, Henage LG, Preininger AM, Xiang Y, Exton JH. Methods Enzymol. 2007;434:49. doi: 10.1016/S0076-6879(07)34004-4. [DOI] [PubMed] [Google Scholar]
- 130.Su W, Yeku O, Olepu S, Genna A, Park JS, Ren H, Du G, Gelb MH, Morris AJ, Frohman MA. Mol Pharmacol. 2009;75:437. doi: 10.1124/mol.108.053298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Norton LJ, Zhang Q, Saqib KM, Schrewe H, Macura K, Anderson KE, Lindsley CW, Brown HA, Rudge SA, Wakelam MJ. J Cell Sci. 2011;124:1973. doi: 10.1242/jcs.082008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Milne SB, Tallman KA, Serwa R, Rouzer CA, Armstrong MD, Marnett LJ, Lukehart CM, Porter NA, Brown HA. Nat Chem Biol. 2010;6:205. doi: 10.1038/nchembio.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Smith GL, Vanderplasschen A, Law M. J Gen Virol. 2002;83:2915. doi: 10.1099/0022-1317-83-12-2915. [DOI] [PubMed] [Google Scholar]
- 134.Hirt P, Hiller G, Wittek R. J Virol. 1986;58:757. doi: 10.1128/jvi.58.3.757-764.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Eckert D, Williams O, Meseda CA, Merchlinsky M. J Virol. 2005;79:15084. doi: 10.1128/JVI.79.24.15084-15090.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hiller G, Eibl H, Weber K. J Virol. 1981;39:903. doi: 10.1128/jvi.39.3.903-913.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hiller G, Weber K. J Virol. 1985;55:651. doi: 10.1128/jvi.55.3.651-659.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Grosenbach DW, Hruby DE. J Virol. 1998;72:5108. doi: 10.1128/jvi.72.6.5108-5120.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Husain M, Moss B. J Virol. 2003;77:9008. doi: 10.1128/JVI.77.16.9008-9019.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Blasco R, Moss B. J Virol. 1991;65:5910. doi: 10.1128/jvi.65.11.5910-5920.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Husain M, Weisberg A, Moss B. Virology. 2003;308:233. doi: 10.1016/s0042-6822(03)00063-1. [DOI] [PubMed] [Google Scholar]
- 142.Baek SH, Kwak JY, Lee SH, Lee T, Ryu SH, Uhlinger DJ, Lambeth JD. J Biol Chem. 1997;272:32042. doi: 10.1074/jbc.272.51.32042. [DOI] [PubMed] [Google Scholar]
- 143.Husain M, Moss B. J Virol. 2002;76:7777. doi: 10.1128/JVI.76.15.7777-7789.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Roper RL, Moss B. J Virol. 1999;73:1108. doi: 10.1128/jvi.73.2.1108-1117.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Honeychurch KM, Yang G, Jordan R, Hruby DE. J Virol. 2007;81:7310. doi: 10.1128/JVI.00034-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chen Y, Honeychurch KM, Yang G, Byrd CM, Harver C, Hruby DE, Jordan R. Virol J. 2009;6:44. doi: 10.1186/1743-422X-6-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Baker RO, Bray M, Huggins JW. Antiviral Res. 2003;57:13. doi: 10.1016/S0166-3542(02)00196-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kato N, Eggers HJ, Rolly H. J Exp Med. 1969;129:795. doi: 10.1084/jem.129.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schmutz C, Payne LG, Gubser J, Wittek R. J Virol. 1991;65:3435. doi: 10.1128/jvi.65.7.3435-3442.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yang G, Pevear DC, Davies MH, Collett MS, Bailey T, Rippen S, Barone L, Burns C, Rhodes G, Tohan S, Huggins JW, Baker RO, Buller RL, Touchette E, Waller K, Schriewer J, Neyts J, DeClercq E, Jones K, Hruby D, Jordan R. J Virol. 2005;79:13139. doi: 10.1128/JVI.79.20.13139-13149.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Grosenbach DW, Berhanu A, King DS, Mosier S, Jones KF, Jordan RA, Bolken TC, Hruby DE. Proc Natl Acad Sci USA. 2010;107:838. doi: 10.1073/pnas.0912134107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Reddy MK, Bauer WR. J Biol Chem. 1989;264:443. [PubMed] [Google Scholar]
- 153.DeMasi J, Du S, Lennon D, Traktman P. J Virol. 2001;75:10090. doi: 10.1128/JVI.75.21.10090-10105.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Schmiel DH, Miller VL. Microbes Infect. 1999;1:1103. doi: 10.1016/s1286-4579(99)00205-1. [DOI] [PubMed] [Google Scholar]
- 155.Winans SC, Walker GC. J Bacteriol. 1983;154:1117. doi: 10.1128/jb.154.3.1117-1125.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Winans SC, Walker GC. J Bacteriol. 1985;161:402. doi: 10.1128/jb.161.1.402-410.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lackey D, Walker GC, Keng T, Linn S. J Bacteriol. 1977;131:583. doi: 10.1128/jb.131.2.583-588.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Pohlman RF, Liu F, Wang L, More MI, Winans SC. Nucleic Acids Res. 1993;21:4867. doi: 10.1093/nar/21.21.4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lagunavicius A, Sasnauskas G, Halford SE, Siksnys V. J Mol Biol. 2003;326:1051. doi: 10.1016/s0022-2836(03)00020-2. [DOI] [PubMed] [Google Scholar]
- 160.Zaremba M, Urbanke C, Halford SE, Siksnys V. J Mol Biol. 2004;336:81. doi: 10.1016/j.jmb.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 161.Grazulis S, Manakova E, Roessle M, Bochtler M, Tamulaitiene G, Huber R, Siksnys V. Proc Natl Acad Sci USA. 2005;102:15797. doi: 10.1073/pnas.0507949102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Bartolucci C, Lamba D, Grazulis S, Manakova E, Heumann H. J Mol Biol. 2005;354:940. doi: 10.1016/j.jmb.2005.09.096. [DOI] [PubMed] [Google Scholar]
- 163.Titball RW. Microbiol Rev. 1993;57:347. doi: 10.1128/mr.57.2.347-366.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Titball RW. Symp Ser Soc Appl Microbiol. 1998;27:127S. [PubMed] [Google Scholar]
- 165.Songer JG. Trends Microbiol. 1997;5:156. doi: 10.1016/S0966-842X(97)01005-6. [DOI] [PubMed] [Google Scholar]
- 166.Ochman H, Lawrence JG, Groisman EA. Nature. 2000;405:299. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- 167.Edwards JL, Entz DD, Apicella MA. Infect Immun. 2003;71:6381. doi: 10.1128/IAI.71.11.6381-6391.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Edwards JL, Shao JQ, Ault KA, Apicella MA. Infect Immun. 2000;68:5354. doi: 10.1128/iai.68.9.5354-5363.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Edwards JL, Brown EJ, Ault KA, Apicella MA. Cell Microbiol. 2001;3:611. doi: 10.1046/j.1462-5822.2001.00140.x. [DOI] [PubMed] [Google Scholar]
- 170.Edwards JL, Brown EJ, Uk-Nham S, Cannon JG, Blake MS, Apicella MA. Cell Microbiol. 2002;4:571. doi: 10.1046/j.1462-5822.2002.t01-1-00215.x. [DOI] [PubMed] [Google Scholar]
- 171.Edwards JL, Apicella MA. Cell Microbiol. 2006;8:1253. doi: 10.1111/j.1462-5822.2006.00707.x. [DOI] [PubMed] [Google Scholar]
- 172.Grassme H, Gulbins E, Brenner B, Ferlinz K, Sandhoff K, Harzer K, Lang F, Meyer TF. Cell. 1997;91:605. doi: 10.1016/s0092-8674(00)80448-1. [DOI] [PubMed] [Google Scholar]
- 173.Hinnebusch BJ. Curr Issues Mol Biol. 2005;7:197. [PubMed] [Google Scholar]
- 174.Ajl SJ, Reedal JS, Durrum EL, Warren J. J Bacteriol. 1955;70:158. doi: 10.1128/jb.70.2.158-169.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Walker RV. Curr Top Microbiol Immunol. 1967;41:23. doi: 10.1007/978-3-642-46062-3_2. [DOI] [PubMed] [Google Scholar]
- 176.Schar M, Meyer KF. Schweiz Z Pathol Bakteriol. 1956;19:51. [PubMed] [Google Scholar]
- 177.Hinnebusch BJ, Rudolph AE, Cherepanov P, Dixon JE, Schwan TG, Forsberg A. Science. 2002;296:733. doi: 10.1126/science.1069972. [DOI] [PubMed] [Google Scholar]
- 178.Lindler LE, Plano GV, Burland V, Mayhew GF, Blattner FR. Infect Immun. 1998;66:5731. doi: 10.1128/iai.66.12.5731-5742.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Cocchiaro JL, Valdivia RH. Cell Microbiol. 2009;11:1571. doi: 10.1111/j.1462-5822.2009.01364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kalman S, Mitchell W, Marathe R, Lammel C, Fan J, Hyman RW, Olinger L, Grimwood J, Davis RW, Stephens RS. Nat Genet. 1999;21:385. doi: 10.1038/7716. [DOI] [PubMed] [Google Scholar]
- 181.Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov RL, Zhao Q, Koonin EV, Davis RW. Science. 1998;282:754. doi: 10.1126/science.282.5389.754. [DOI] [PubMed] [Google Scholar]
- 182.Read TD, Myers GS, Brunham RC, Nelson WC, Paulsen IT, Heidelberg J, Holtzapple E, Khouri H, Federova NB, Carty HA, Umayam LA, Haft DH, Peterson J, Beanan MJ, White O, Salzberg SL, Hsia RC, McClarty G, Rank RG, Bavoil PM, Fraser CM. Nucleic Acids Res. 2003;31:2134. doi: 10.1093/nar/gkg321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Nelson DE, Crane DD, Taylor LD, Dorward DW, Goheen MM, Caldwell HD. Infect Immun. 2006;74:73. doi: 10.1128/IAI.74.1.73-80.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Cocchiaro JL, Kumar Y, Fischer ER, Hackstadt T, Valdivia RH. Proc Natl Acad Sci USA. 2008;105:9379. doi: 10.1073/pnas.0712241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Jacobs AC, Hood I, Boyd KL, Olson PD, Morrison JM, Carson S, Sayood K, Iwen PC, Skaar EP, Dunman PM. Infect Immun. 2010;78:1952. doi: 10.1128/IAI.00889-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Wilderman PJ, Vasil AI, Johnson Z, Vasil ML. Mol Microbiol. 2001;39:291. doi: 10.1046/j.1365-2958.2001.02282.x. [DOI] [PubMed] [Google Scholar]
- 187.Chater KF, Biro S, Lee KJ, Palmer T, Schrempf H. FEMS Microbiol Rev. 2010;34:171. doi: 10.1111/j.1574-6976.2009.00206.x. [DOI] [PubMed] [Google Scholar]
- 188.Chater KF. Philos Trans R Soc Lond B Biol Sci. 2006;361:761. doi: 10.1098/rstb.2005.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ulbrich-Hofmann R, Lerchner A, Oblozinsky M, Bezakova L. Biotechnol Lett. 2005;27:535. doi: 10.1007/s10529-005-3251-2. [DOI] [PubMed] [Google Scholar]
- 190.Masayama A, Takahashi T, Tsukada K, Nishikawa S, Takahashi R, Adachi M, Koga K, Suzuki A, Yamane T, Nakano H, Iwasaki Y. ChemBioChem. 2008;9:974. doi: 10.1002/cbic.200700528. [DOI] [PubMed] [Google Scholar]
- 191.Dippe M, Mrestani-Klaus C, Schierhorn A, Ulbrich-Hofmann R. Chem Phys Lipids. 2008;152:71. doi: 10.1016/j.chemphyslip.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 192.Iwasaki Y, Nishiyama T, Kawarasaki Y, Nakano H, Yamane T. J Biosci Bioeng. 2000;89:506. doi: 10.1016/s1389-1723(00)89107-0. [DOI] [PubMed] [Google Scholar]
- 193.Iwasaki Y, Mishima N, Mizumoto K, Nakano H, Yamane T. J Ferment Bioeng. 1995;79:417. [Google Scholar]
- 194.Lee JS, Bat-Ochir M, Demirev AV, Nam DH. J Microbiol. 2009;47:116. doi: 10.1007/s12275-008-0161-8. [DOI] [PubMed] [Google Scholar]
- 195.Hagishita T, Nishikawa M, Hatanaka T. Biotech Lett. 2000;22:1587. [Google Scholar]
- 196.Uesugi Y, Mori K, Arima J, Iwabuchi M, Hatanaka T. J Biol Chem. 2005;280:26143. doi: 10.1074/jbc.M414319200. [DOI] [PubMed] [Google Scholar]
- 197.Mori K, Mukaihara T, Uesugi Y, Iwabuchi M, Hatanaka T. Appl Environ Microbiol. 2005;71:754. doi: 10.1128/AEM.71.2.754-760.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Aikens CL, Laederach A, Reilly PJ. Proteins. 2004;57:27. doi: 10.1002/prot.20180. [DOI] [PubMed] [Google Scholar]
- 199.Wang X. Curr Opin Plant Biol. 2002;5:408. doi: 10.1016/s1369-5266(02)00283-2. [DOI] [PubMed] [Google Scholar]
- 200.Wang X. Plant Physiol. 1999;120:645. doi: 10.1104/pp.120.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Munnik T. Trends Plant Sci. 2001;6:227. doi: 10.1016/s1360-1385(01)01918-5. [DOI] [PubMed] [Google Scholar]
- 202.Wang X. Prog Lipid Res. 2000;39:109. doi: 10.1016/s0163-7827(00)00002-3. [DOI] [PubMed] [Google Scholar]
- 203.Wang X, Xu L, Zheng L. J Biol Chem. 1994;269:20312. [PubMed] [Google Scholar]
- 204.Rose K, Rudge SA, Frohman MA, Morris AJ, Engebrecht J. Proc Natl Acad Sci USA. 1995;92:12151. doi: 10.1073/pnas.92.26.12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hammond SM, Altshuller YM, Sung TC, Rudge SA, Rose K, Engebrecht J, Morris AJ, Frohman MA. J Biol Chem. 1995;270:29640. doi: 10.1074/jbc.270.50.29640. [DOI] [PubMed] [Google Scholar]
- 206.Hong Y, Zhang W, Wang X. Plant, Cell Environ. 2010;33:627. doi: 10.1111/j.1365-3040.2009.02087.x. [DOI] [PubMed] [Google Scholar]
- 207.Qin C, Wang X. Plant Physiol. 2002;128:1057. doi: 10.1104/pp.010928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Elias M, Potocky M, Cvrckova F, Zarsky V. BMC Genomics. 2002;3:2. doi: 10.1186/1471-2164-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Li M, Hong Y, Wang X. Biochim Biophys Acta. 2009;1791:927. doi: 10.1016/j.bbalip.2009.02.017. [DOI] [PubMed] [Google Scholar]
- 210.Hwang IS, Park SJ, Roh T, Choi M, Kim HJ. Rapid Commun Mass Spectrom. 2001;15:110. doi: 10.1002/1097-0231(20010130)15:2<110::AID-RCM200>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 211.Pappan K, Austin-Brown S, Chapman KD, Wang X. Arch Biochem Biophys. 1998;353:131. doi: 10.1006/abbi.1998.0640. [DOI] [PubMed] [Google Scholar]
- 212.Qin W, Pappan K, Wang X. J Biol Chem. 1997;272:28267. doi: 10.1074/jbc.272.45.28267. [DOI] [PubMed] [Google Scholar]
- 213.Rizo J, Sudhof TC. J Biol Chem. 1998;273:15879. doi: 10.1074/jbc.273.26.15879. [DOI] [PubMed] [Google Scholar]
- 214.Sutton RB, Davletov BA, Berghuis AM, Sudhof TC, Sprang SR. Cell. 1995;80:929. doi: 10.1016/0092-8674(95)90296-1. [DOI] [PubMed] [Google Scholar]
- 215.Essen LO, Perisic O, Cheung R, Katan M, Williams RL. Nature. 1996;380:595. doi: 10.1038/380595a0. [DOI] [PubMed] [Google Scholar]
- 216.Grobler JA, Essen LO, Williams RL, Hurley JH. Nat Struct Biol. 1996;3:788. doi: 10.1038/nsb0996-788. [DOI] [PubMed] [Google Scholar]
- 217.Perisic O, Fong S, Lynch DE, Bycroft M, Williams RL. J Biol Chem. 1998;273:1596. doi: 10.1074/jbc.273.3.1596. [DOI] [PubMed] [Google Scholar]
- 218.Zheng L, Krishnamoorthi R, Zolkiewski M, Wang X. J Biol Chem. 2000;275:19700. doi: 10.1074/jbc.M001945200. [DOI] [PubMed] [Google Scholar]
- 219.Zheng L, Shan J, Krishnamoorthi R, Wang X. Biochemistry. 2002;41:4546. doi: 10.1021/bi0158775. [DOI] [PubMed] [Google Scholar]
- 220.Pappan K, Zheng L, Krishnamoorthi R, Wang X. J Biol Chem. 2004;279:47833. doi: 10.1074/jbc.M402789200. [DOI] [PubMed] [Google Scholar]
- 221.Pappan K, Qin W, Dyer JH, Zheng L, Wang X. J Biol Chem. 1997;272:7055. doi: 10.1074/jbc.272.11.7055. [DOI] [PubMed] [Google Scholar]
- 222.Pappan K, Wang X. Arch Biochem Biophys. 1999;368:347. doi: 10.1006/abbi.1999.1325. [DOI] [PubMed] [Google Scholar]
- 223.Austin-Brown SL, Chapman KD. Plant Physiol. 2002;129:1892. doi: 10.1104/pp.001974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Lein W, Saalbach G. Biochim Biophys Acta. 2001;1530:172. doi: 10.1016/s1388-1981(00)00182-7. [DOI] [PubMed] [Google Scholar]
- 225.Bargmann BOR, Laxalt AM, Ter Riet B, Testerink C, Merquiol E, Mosblech A, Leon-Reyes A, Pieterse CMJ, Haring MA, Heilmann I, Bartels D, Munnik T. Plant, Cell Environ. 2009;32:837. doi: 10.1111/j.1365-3040.2009.01962.x. [DOI] [PubMed] [Google Scholar]
- 226.Stumpe S, Konig S, Ulbrich-Hofmann R. FEBS J. 2007;274:2630. doi: 10.1111/j.1742-4658.2007.05798.x. [DOI] [PubMed] [Google Scholar]
- 227.Hong Y, Pan X, Welti R, Wang X. Plant Cell. 2008;20:803. doi: 10.1105/tpc.107.056390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Hong Y, Pan X, Welti R, Wang X. Plant Signal Behav. 2008;3:1099. doi: 10.4161/psb.3.12.7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Novotna Z, Linek J, Hynek R, Martinec J, Potocky M, Valentova O. FEBS Lett. 2003;554:50. doi: 10.1016/s0014-5793(03)01093-7. [DOI] [PubMed] [Google Scholar]
- 230.Cullis PR, Hope MJ, Tilcock CP. Chem Phys Lipids. 1986;40:127. doi: 10.1016/0009-3084(86)90067-8. [DOI] [PubMed] [Google Scholar]
- 231.Chang IF, Curran A, Woolsey R, Quilici D, Cushman JC, Mittler R, Harmon A, Harper JF. Proteomics. 2009;9:2967. doi: 10.1002/pmic.200800445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Huang S, Gao L, Blanchoin L, Staiger CJ. Mol Biol Cell. 2006;17:1946. doi: 10.1091/mbc.E05-09-0840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Lee S, Park J, Lee Y. Mol Cells. 2003;15:313. [PubMed] [Google Scholar]
- 234.Wang C, Wang X. Plant Physiol. 2001;127:1102. [PMC free article] [PubMed] [Google Scholar]
- 235.Katagiri T, Takahashi S, Shinozaki K. Plant J. 2001;26:595. doi: 10.1046/j.1365-313x.2001.01060.x. [DOI] [PubMed] [Google Scholar]
- 236.Gardiner JC, Harper JD, Weerakoon ND, Collings DA, Ritchie S, Gilroy S, Cyr RJ, Marc J. Plant Cell. 2001;13:2143. doi: 10.1105/TPC.010114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Zhang W, Wang C, Qin C, Wood T, Olafsdottir G, Welti R, Wang X. Plant Cell. 2003;15:2285. doi: 10.1105/tpc.013961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Hong Y, Devaiah SP, Bahn SC, Thamasandra BN, Li M, Welti R, Wang X. Plant J. 2009;58:376. doi: 10.1111/j.1365-313X.2009.03788.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Taniguchi YY, Taniguchi M, Tsuge T, Oka A, Aoyama T. Planta. 2010;231:491. doi: 10.1007/s00425-009-1052-x. [DOI] [PubMed] [Google Scholar]
- 240.Li M, Qin C, Welti R, Wang X. Plant Physiol. 2006;140:761. doi: 10.1104/pp.105.070995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Li G, Xue HW. Plant Cell. 2007;19:281. doi: 10.1105/tpc.106.041426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Munnik T, de Vrije T, Irvine RF, Musgrave A. J Biol Chem. 1996;271:15708. doi: 10.1074/jbc.271.26.15708. [DOI] [PubMed] [Google Scholar]
- 243.Tiwari K, Paliyath G. Plant Physiol Biochem. 2011;49:329. doi: 10.1016/j.plaphy.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 244.Munnik T, Arisz SA, De Vrije T, Musgrave A. Plant Cell. 1995;7:2197. doi: 10.1105/tpc.7.12.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Agredano-Moreno LT, Reyes de la Cruz H, Martinez-Castilla LP, Sanchez de Jimenez E. Mol Biosyst. 2007;3:794. doi: 10.1039/b705803a. [DOI] [PubMed] [Google Scholar]
- 246.Menand B, Desnos T, Nussaume L, Berger F, Bouchez D, Meyer C, Robaglia C. Proc Natl Acad Sci USA. 2002;99:6422. doi: 10.1073/pnas.092141899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Mahfouz MM, Kim S, Delauney AJ, Verma DP. Plant Cell. 2006;18:477. doi: 10.1105/tpc.105.035931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Veverka V, Crabbe T, Bird I, Lennie G, Muskett FW, Taylor RJ, Carr MD. Oncogene. 2008;27:585. doi: 10.1038/sj.onc.1210693. [DOI] [PubMed] [Google Scholar]
- 249.Crespo JL, Diaz-Troya S, Florencio FJ. Plant Physiol. 2005;139:1736. doi: 10.1104/pp.105.070847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Sormani R, Yao L, Menand B, Ennar N, Lecampion C, Meyer C, Robaglia C. BMC Plant Biol. 2007;7:26. doi: 10.1186/1471-2229-7-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Reyes de la Cruz H, Aguilar R, Sanchez de Jimenez E. Biochemistry. 2004;43:533. doi: 10.1021/bi035222z. [DOI] [PubMed] [Google Scholar]
- 252.Stepanova AN, Alonso JM. Curr Opin Plant Biol. 2009;12:548. doi: 10.1016/j.pbi.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 253.Jakubowicz M, Galganska H, Nowak W, Sadowski J. J Exp Bot. 2010;61:3475. doi: 10.1093/jxb/erq177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Grill E, Himmelbach A. Curr Opin Plant Biol. 1998;1:412. doi: 10.1016/s1369-5266(98)80265-3. [DOI] [PubMed] [Google Scholar]
- 255.Zhang YY, Zhu HY, Zhang Q, Li MY, Yan M, Wang R, Wang LL, Welti R, Zhang WH, Wang XM. Plant Cell. 2009;21:2357. doi: 10.1105/tpc.108.062992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Mishra G, Zhang W, Deng F, Zhao J, Wang X. Science. 2006;312:264. doi: 10.1126/science.1123769. [DOI] [PubMed] [Google Scholar]
- 257.Ryu SB, Wang X. Biochim Biophys Acta. 1996;1303:243. doi: 10.1016/0005-2760(96)00096-3. [DOI] [PubMed] [Google Scholar]
- 258.Wang C, Zien CA, Afitlhile M, Welti R, Hildebrand DF, Wang X. Plant Cell. 2000;12:2237. doi: 10.1105/tpc.12.11.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Lee S, Hirt H, Lee Y. Plant J. 2001;26:479. doi: 10.1046/j.1365-313x.2001.01037.x. [DOI] [PubMed] [Google Scholar]
- 260.Cornell RB, Arnold RS. Chem Phys Lipids. 1996;81:215. [Google Scholar]
- 261.Gfeller A, Dubugnon L, Liechti R, Farmer EE. Sci Signal. 2010;3:cm3. doi: 10.1126/scisignal.3109cm3. [DOI] [PubMed] [Google Scholar]
- 262.Zhao Y, Thilmony R, Bender CL, Schaller A, He SY, Howe GA. Plant J. 2003;36:485. doi: 10.1046/j.1365-313x.2003.01895.x. [DOI] [PubMed] [Google Scholar]
- 263.Brooks DM, Bender CL, Kunkel BN. Mol Plant Pathol. 2005;6:629. doi: 10.1111/j.1364-3703.2005.00311.x. [DOI] [PubMed] [Google Scholar]
- 264.Dharmalingam K, Jayaraman J. Biochem Biophys Res Commun. 1971;45:1115. doi: 10.1016/0006-291x(71)90134-3. [DOI] [PubMed] [Google Scholar]
- 265.Grossman S, Cobley J, Hogue PK, Kearney EB, Singer TP. Arch Biochem Biophys. 1973;158:744. doi: 10.1016/0003-9861(73)90569-9. [DOI] [PubMed] [Google Scholar]
- 266.Kishida M, Shimoda C. Curr Genet. 1986;10:443. doi: 10.1007/BF00419871. [DOI] [PubMed] [Google Scholar]
- 267.Honigberg SM, Conicella C, Espositio RE. Genetics. 1992;130:703. doi: 10.1093/genetics/130.4.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Ella KM, Dolan JW, Qi C, Meier KE. Biochem J. 1996;314(Pt 1):15. doi: 10.1042/bj3140015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Harkins AL, Yuan G, London SD, Dolan JW. FEMS Yeast Res. 2010;10:717. doi: 10.1111/j.1567-1364.2010.00646.x. [DOI] [PubMed] [Google Scholar]
- 270.Kanoh H, Nakashima S, Zhao Y, Sugiyama Y, Kitajima Y, Nozawa Y. Biochim Biophys Acta. 1998;1398:359. doi: 10.1016/s0167-4781(98)00067-0. [DOI] [PubMed] [Google Scholar]
- 271.Rudge SA, Engebrecht J. Biochim Biophys Acta. 1999;1439:167. doi: 10.1016/s1388-1981(99)00092-x. [DOI] [PubMed] [Google Scholar]
- 272.Tang X, Waksman M, Ely Y, Liscovitch M. Eur J Biochem. 2002;269:3821. doi: 10.1046/j.1432-1033.2002.03073.x. [DOI] [PubMed] [Google Scholar]
- 273.Neiman AM. J Cell Biol. 1998;140:29. doi: 10.1083/jcb.140.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Harkins AL, London SD, Dolan JW. FEMS Yeast Res. 2008;8:237. doi: 10.1111/j.1567-1364.2007.00336.x. [DOI] [PubMed] [Google Scholar]
- 275.Hube B, Hess D, Baker CA, Schaller M, Schafer W, Dolan JW. Microbiology. 2001;147:879. doi: 10.1099/00221287-147-4-879. [DOI] [PubMed] [Google Scholar]
- 276.Dolan JW, Bell AC, Hube B, Schaller M, Warner TF, Balish E. Med Mycol. 2004;42:439. doi: 10.1080/13693780410001657162. [DOI] [PubMed] [Google Scholar]
- 277.Honigberg SM, Esposito RE. Proc Natl Acad Sci USA. 1994;91:6559. doi: 10.1073/pnas.91.14.6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Waksman M, Eli Y, Liscovitch M, Gerst JE. J Biol Chem. 1996;271:2361. doi: 10.1074/jbc.271.5.2361. [DOI] [PubMed] [Google Scholar]
- 279.Sciorra VA, Rudge SA, Prestwich GD, Frohman MA, Engebrecht J, Morris AJ. EMBO J. 1999;18:5911. doi: 10.1093/emboj/18.21.5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Rudge SA, Zhou C, Engebrecht J. Genetics. 2002;160:1353. doi: 10.1093/genetics/160.4.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Rudge SA, Morris AJ, Engebrecht J. J Cell Biol. 1998;140:81. doi: 10.1083/jcb.140.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Xie Z, Fang M, Rivas MP, Faulkner AJ, Sternweis PC, Engebrecht JA, Bankaitis VA. Proc Natl Acad Sci USA. 1998;95:12346. doi: 10.1073/pnas.95.21.12346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Sciorra VA, Rudge SA, Wang J, McLaughlin S, Engebrecht J, Morris AJ. J Cell Biol. 2002;159:1039. doi: 10.1083/jcb.200205056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Rudge SA, Cavenagh MM, Kamath R, Sciorra VA, Morris AJ, Kahn RA, Engebrecht J. Mol Biol Cell. 1998;9:2025. doi: 10.1091/mbc.9.8.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Connolly JE, Engebrecht J. Eukaryot Cell. 2006;5:112. doi: 10.1128/EC.5.1.112-124.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Mayr JA, Kohlwein SD, Paltauf F. FEBS Lett. 1996;393:236. doi: 10.1016/0014-5793(96)00893-9. [DOI] [PubMed] [Google Scholar]
- 287.Waksman M, Tang X, Eli Y, Gerst JE, Liscovitch M. J Biol Chem. 1997;272:36. doi: 10.1074/jbc.272.1.36. [DOI] [PubMed] [Google Scholar]
- 288.Sreenivas A, Patton-Vogt JL, Bruno V, Griac P, Henry SA. J Biol Chem. 1998;273:16635. doi: 10.1074/jbc.273.27.16635. [DOI] [PubMed] [Google Scholar]
- 289.Kupiec M, Byers B, Esposito RE, Mitchell AP. The Molecular and Cellular Biology of the Yeast Saccharomyces, Cell Cycle and Cell Biology. Vol. 3 Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1997. [Google Scholar]
- 290.Nakanishi H, de los Santos P, Neiman AM. Mol Biol Cell. 2004;15:1802. doi: 10.1091/mbc.E03-11-0798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Davis L, Barbera M, McDonnell A, McIntyre K, Sternglanz R, Jin Q, Loidl J, Engebrecht J. Genetics. 2001;157:1179. doi: 10.1093/genetics/157.3.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, Remor M, Hofert C, Schelder M, Brajenovic M, Ruffner H, Merino A, Klein K, Hudak M, Dickson D, Rudi T, Gnau V, Bauch A, Bastuck S, Huhse B, Leutwein C, Heurtier MA, Copley RR, Edelmann A, Querfurth E, Rybin V, Drewes G, Raida M, Bouwmeester T, Bork P, Seraphin B, Kuster B, Neubauer G, Superti-Furga G. Nature. 2002;415:141. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 293.Riedel CG, Mazza M, Maier P, Korner R, Knop M. J Biol Chem. 2005;280:37846. doi: 10.1074/jbc.M504244200. [DOI] [PubMed] [Google Scholar]
- 294.Mendonsa R, Engebrecht J. Biochim Biophys Acta. 2009;1791:970. doi: 10.1016/j.bbalip.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 295.Loewen CJ, Gaspar ML, Jesch SA, Delon C, Ktistakis NT, Henry SA, Levine TP. Science. 2004;304:1644. doi: 10.1126/science.1096083. [DOI] [PubMed] [Google Scholar]
- 296.Cleves AE, McGee TP, Whitters EA, Champion KM, Aitken JR, Dowhan W, Goebl M, Bankaitis VA. Cell. 1991;64:789. doi: 10.1016/0092-8674(91)90508-v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Phillips SE, Vincent P, Rizzieri KE, Schaaf G, Bankaitis VA, Gaucher EA. Crit Rev Biochem Mol Biol. 2006;41:21. doi: 10.1080/10409230500519573. [DOI] [PubMed] [Google Scholar]
- 298.Bankaitis VA, Malehorn DE, Emr SD, Greene R. J Cell Biol. 1989;108:1271. doi: 10.1083/jcb.108.4.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Bankaitis VA, Aitken JR, Cleves AE, Dowhan W. Nature. 1990;347:561. doi: 10.1038/347561a0. [DOI] [PubMed] [Google Scholar]
- 300.Bankaitis VA, Mousley CJ, Schaaf G. Trends Biochem Sci. 2010;35:150. doi: 10.1016/j.tibs.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Li X, Routt SM, Xie Z, Cui X, Fang M, Kearns MA, Bard M, Kirsch DR, Bankaitis VA. Mol Biol Cell. 2000;11:1989. doi: 10.1091/mbc.11.6.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Hairfield ML, Ayers AB, Dolan JW. FEMS Yeast Res. 2001;1:225. doi: 10.1111/j.1567-1364.2001.tb00038.x. [DOI] [PubMed] [Google Scholar]
- 303.Waltermann C, Klipp E. Biochem Soc Trans. 2010;38:1257. doi: 10.1042/BST0381257. [DOI] [PubMed] [Google Scholar]
- 304.Walker SJ, Wu WJ, Cerione RA, Brown HA. J Biol Chem. 2000;275:15665. doi: 10.1074/jbc.M000076200. [DOI] [PubMed] [Google Scholar]
- 305.Whiteway M, Bachewich C. Annu Rev Microbiol. 2007;61:529. doi: 10.1146/annurev.micro.61.080706.093341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.McLain N, Dolan JW. Microbiology. 1997;143(Pt 11):3521. doi: 10.1099/00221287-143-11-3521. [DOI] [PubMed] [Google Scholar]
- 307.Li SI, Purugganan MD. Trends Genet. 2011;27:48. doi: 10.1016/j.tig.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 308.Devreotes P. Science. 1989;245:1054. doi: 10.1126/science.2672337. [DOI] [PubMed] [Google Scholar]
- 309.Chen Y, Rodrick V, Yan Y, Brazill D. Eukaryot Cell. 2005;4:694. doi: 10.1128/EC.4.4.694-702.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Raghu P, Manifava M, Coadwell J, Ktistakis NT. Biochim Biophys Acta. 2009;1791:889. doi: 10.1016/j.bbalip.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 311.Zouwail S, Pettitt TR, Dove SK, Chibalina MV, Powner DJ, Haynes L, Wakelam MJ, Insall RH. Biochem J. 2005;389:207. doi: 10.1042/BJ20050085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Ray S, Chen Y, Ayoung J, Hanna R, Brazill D. Cell Signal. 2011;23:335. doi: 10.1016/j.cellsig.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Nagasaki A, Uyeda TQ. Exp Cell Res. 2008;314:1136. doi: 10.1016/j.yexcr.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 314.Nakashima S, Hisamoto N, Banno Y, Matsumoto K, Nozawa Y. Worm Breeder’s Gazette. 2000;16:1. [Google Scholar]
- 315.Liu LX, Spoerke JM, Mulligan EL, Chen J, Reardon B, Westlund B, Sun L, Abel K, Armstrong B, Hardiman G, King J, McCague L, Basson M, Clover R, Johnson CD. Genome Res. 1999;9:859. doi: 10.1101/gr.9.9.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Matthies DS, Fleming PA, Wilkes DM, Blakely RD. J Neurosci. 2006;26:6200. doi: 10.1523/JNEUROSCI.5036-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Kinchen JM, Doukoumetzidis K, Almendinger J, Stergiou L, Tosello-Trampont A, Sifri CD, Hengartner MO, Ravichandran KS. Nat Cell Biol. 2008;10:556. doi: 10.1038/ncb1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.LaLonde M, Janssens H, Yun S, Crosby J, Redina O, Olive V, Altshuller YM, Choi SY, Du G, Gergen JP, Frohman MA. BMC Dev Biol. 2006;6:60. doi: 10.1186/1471-213X-6-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Raghu P, Coessens E, Manifava M, Georgiev P, Pettitt T, Wood E, Garcia-Murillas I, Okkenhaug H, Trivedi D, Zhang Q, Razzaq A, Zaid O, Wakelam M, O’Kane CJ, Ktistakis N. J Cell Biol. 2009;185:129. doi: 10.1083/jcb.200807027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.LaLonde MM, Janssens H, Rosenbaum E, Choi SY, Gergen JP, Colley NJ, Stark WS, Frohman MA. J Cell Biol. 2005;169:471. doi: 10.1083/jcb.200502122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Ghosh S, Moore S, Bell RM, Dush M. J Biol Chem. 2003;278:45690. doi: 10.1074/jbc.M302933200. [DOI] [PubMed] [Google Scholar]
- 322.Zeng XX, Zheng X, Xiang Y, Cho HP, Jessen JR, Zhong TP, Solnica-Krezel L, Brown HA. Dev Biol. 2009;328:363. doi: 10.1016/j.ydbio.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Liu HY, Lee N, Tsai TY, Ho SY. Biochim Biophys Acta. 2010;1801:1330. doi: 10.1016/j.bbalip.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 324.Saito M, Kanfer J. Biochem Biophys Res Commun. 1973;53:391. doi: 10.1016/0006-291x(73)90674-8. [DOI] [PubMed] [Google Scholar]
- 325.Park SK, Provost JJ, Bae CD, Ho WT, Exton JH. J Biol Chem. 1997;272:29263. doi: 10.1074/jbc.272.46.29263. [DOI] [PubMed] [Google Scholar]
- 326.Colley WC, Sung TC, Roll R, Jenco J, Hammond SM, Altshuller Y, Bar-Sagi D, Morris AJ, Frohman MA. Curr Biol. 1997;7:191. doi: 10.1016/s0960-9822(97)70090-3. [DOI] [PubMed] [Google Scholar]
- 327.Lopez I, Arnold RS, Lambeth JD. J Biol Chem. 1998;273:12846. doi: 10.1074/jbc.273.21.12846. [DOI] [PubMed] [Google Scholar]
- 328.Steed PM, Clark KL, Boyar WC, Lasala DJ. FASEB J. 1998;12:1309. doi: 10.1096/fasebj.12.13.1309. [DOI] [PubMed] [Google Scholar]
- 329.Jang YH, Ahn BH, Namkoong S, Kim YM, Jin JK, Kim YS, Min do S. Cell Signal. 2008;20:2198. doi: 10.1016/j.cellsig.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 330.Hammond SM, Jenco JM, Nakashima S, Cadwallader K, Gu Q, Cook S, Nozawa Y, Prestwich GD, Frohman MA, Morris AJ. J Biol Chem. 1997;272:3860. doi: 10.1074/jbc.272.6.3860. [DOI] [PubMed] [Google Scholar]
- 331.Stahelin RV, Ananthanarayanan B, Blatner NR, Singh S, Bruzik KS, Murray D, Cho W. J Biol Chem. 2004;279:54918. doi: 10.1074/jbc.M407798200. [DOI] [PubMed] [Google Scholar]
- 332.Sugars JM, Cellek S, Manifava M, Coadwell J, Ktistakis NT. J Biol Chem. 1999;274:30023. doi: 10.1074/jbc.274.42.30023. [DOI] [PubMed] [Google Scholar]
- 333.Liu MY, Gutowski S, Sternweis PC. J Biol Chem. 2001;276:5556. doi: 10.1074/jbc.M006404200. [DOI] [PubMed] [Google Scholar]
- 334.Tsukahara T, Tsukahara R, Fujiwara Y, Yue J, Cheng Y, Guo H, Bolen A, Zhang C, Balazs L, Re F, Du G, Frohman MA, Baker DL, Parrill AL, Uchiyama A, Kobayashi T, Murakami-Murofushi K, Tigyi G. Mol Cell. 2010;39:421. doi: 10.1016/j.molcel.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Cao JX, Koop BF, Upton C. Virus Res. 1997;48:11. doi: 10.1016/s0168-1702(96)01422-0. [DOI] [PubMed] [Google Scholar]
- 336.Munck A, Bohm C, Seibel NM, Hashemol Hosseini Z, Hampe W. FEBS J. 2005;272:1718. doi: 10.1111/j.1742-4658.2005.04601.x. [DOI] [PubMed] [Google Scholar]
- 337.Pedersen KM, Finsen B, Celis JE, Jensen NA. J Biol Chem. 1998;273:31494. doi: 10.1074/jbc.273.47.31494. [DOI] [PubMed] [Google Scholar]
- 338.Cole R, Proulx P. J Bacteriol. 1975;124:1148. doi: 10.1128/jb.124.3.1148-1152.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Madesh M, Balasubramanian KA. Lipids. 1997;32:471. doi: 10.1007/s11745-997-0061-9. [DOI] [PubMed] [Google Scholar]
- 340.Choi SY, Huang P, Jenkins GM, Chan DC, Schiller J, Frohman MA. Nat Cell Biol. 2006;8:1255. doi: 10.1038/ncb1487. [DOI] [PubMed] [Google Scholar]
- 341.Hughes WE, Parker PJ. Biochem J. 2001;356:727. doi: 10.1042/0264-6021:3560727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Sugars JM, Cellek S, Manifava M, Coadwell J, Ktistakis NT. J Biol Chem. 2002;277:29152. doi: 10.1074/jbc.M112169200. [DOI] [PubMed] [Google Scholar]
- 343.Du G, Altshuller YM, Vitale N, Huang P, Chasserot-Golaz S, Morris AJ, Bader MF, Frohman MA. J Cell Biol. 2003;162:305. doi: 10.1083/jcb.200302033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Xie Z, Ho WT, Exton JH. J Biol Chem. 1998;273:34679. doi: 10.1074/jbc.273.52.34679. [DOI] [PubMed] [Google Scholar]
- 345.Balboa MA, Insel PA. J Biol Chem. 1995;270:29843. doi: 10.1074/jbc.270.50.29843. [DOI] [PubMed] [Google Scholar]
- 346.Jang YH, Min do S. J Biol Chem. 2011;286:4680. doi: 10.1074/jbc.M110.162602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Lee S, Park JB, Kim JH, Kim Y, Shin KJ, Lee JS, Ha SH, Suh PG, Ryu SH. J Biol Chem. 2001;276:28252. doi: 10.1074/jbc.M008521200. [DOI] [PubMed] [Google Scholar]
- 348.Honda A, Nogami M, Yokozeki T, Yamazaki M, Nakamura H, Watanabe H, Kawamoto K, Nakayama K, Morris AJ, Frohman MA, Kanaho Y. Cell. 1999;99:521. doi: 10.1016/s0092-8674(00)81540-8. [DOI] [PubMed] [Google Scholar]
- 349.Madesh M, Ibrahim SA, Balasubramanian KA. Free Radic Biol Med. 1997;23:271. doi: 10.1016/s0891-5849(97)00093-2. [DOI] [PubMed] [Google Scholar]
- 350.Kam Y, Exton JH. Mol Cell Biol. 2001;21:4055. doi: 10.1128/MCB.21.12.4055-4066.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Dennis EA, Deems RA, Harkewicz R, Quehenberger O, Brown HA, Milne SB, Myers DS, Glass CK, Hardiman G, Reichart D, Merrill AH, Jr, Sullards MC, Wang E, Murphy RC, Raetz CR, Garrett TA, Guan Z, Ryan AC, Russell DW, McDonald JG, Thompson BM, Shaw WA, Sud M, Zhao Y, Gupta S, Maurya MR, Fahy E, Subramaniam S. J Biol Chem. 2010;285:39976. doi: 10.1074/jbc.M110.182915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Xie Z, Ho WT, Exton JH. Biochim Biophys Acta. 2002;1580:9. doi: 10.1016/s1388-1981(01)00168-8. [DOI] [PubMed] [Google Scholar]
- 353.Gomez-Cambronero J. J Interferon Cytokine Res. 1995;15:877. doi: 10.1089/jir.1995.15.877. [DOI] [PubMed] [Google Scholar]
- 354.Min DS, Ahn BH, Jo YH. Mol Cells. 2001;11:369. [PubMed] [Google Scholar]
- 355.Ahn BH, Kim SY, Kim EH, Choi KS, Kwon TK, Lee YH, Chang JS, Kim MS, Jo YH, Min DS. Mol Cell Biol. 2003;23:3103. doi: 10.1128/MCB.23.9.3103-3115.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Singer WD, Brown HA, Bokoch GM, Sternweis PC. J Biol Chem. 1995;270:14944. doi: 10.1074/jbc.270.25.14944. [DOI] [PubMed] [Google Scholar]
- 357.Kim Y, Han JM, Park JB, Lee SD, Oh YS, Chung C, Lee TG, Kim JH, Park SK, Yoo JS, Suh PG, Ryu SH. Biochemistry. 1999;38:10344. doi: 10.1021/bi990579h. [DOI] [PubMed] [Google Scholar]
- 358.Hu T, Exton JH. J Biol Chem. 2003;278:2348. doi: 10.1074/jbc.M210093200. [DOI] [PubMed] [Google Scholar]
- 359.Di Fulvio M, Lehman N, Lin X, Lopez I, Gomez-Cambronero J. Oncogene. 2006;25:3032. doi: 10.1038/sj.onc.1209340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Di Fulvio M, Frondorf K, Gomez-Cambronero J. Cell Signal. 2008;20:176. doi: 10.1016/j.cellsig.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Henkels KM, Short S, Peng HJ, Di Fulvio M, Gomez-Cambronero J. Biochem Biophys Res Commun. 2009;389:224. doi: 10.1016/j.bbrc.2009.08.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Henkels KM, Peng HJ, Frondorf K, Gomez-Cambronero J. Mol Cell Biol. 2010;30:2251. doi: 10.1128/MCB.01239-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Yin H, Gui Y, Du G, Frohman MA, Zheng XL. J Biol Chem. 2010;285:13580. doi: 10.1074/jbc.M109.046359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Riebeling C, Bourgoin S, Shields D. Biochim Biophys Acta. 2008;1781:376. doi: 10.1016/j.bbalip.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 365.Jang YH, Namkoong S, Kim YM, Lee SJ, Park BJ, Min DS. Cell Death Differ. 2008;15:1782. doi: 10.1038/cdd.2008.111. [DOI] [PubMed] [Google Scholar]
- 366.Hodgkin MN, Masson MR, Powner D, Saqib KM, Ponting CP, Wakelam MJ. Curr Biol. 2000;10:43. doi: 10.1016/s0960-9822(99)00264-x. [DOI] [PubMed] [Google Scholar]
- 367.Nakamura S, Kiyohara Y, Jinnai H, Hitomi T, Ogino C, Yoshida K, Nishizuka Y. Proc Natl Acad Sci USA. 1996;93:4300. doi: 10.1073/pnas.93.9.4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Hoer A, Cetindag C, Oberdisse E. Biochim Biophys Acta. 2000;1481:189. doi: 10.1016/s0167-4838(00)00108-4. [DOI] [PubMed] [Google Scholar]
- 369.Kawabe K, Kodaki T, Katayama K, Okamura S, Mori M, Yamashita S. J Biochem. 1998;123:870. doi: 10.1093/oxfordjournals.jbchem.a022018. [DOI] [PubMed] [Google Scholar]
- 370.Cockcroft S, Thomas GM, Fensome A, Geny B, Cunningham E, Gout I, Hiles I, Totty NF, Truong O, Hsuan JJ. Science. 1994;263:523. doi: 10.1126/science.8290961. [DOI] [PubMed] [Google Scholar]
- 371.Brown HA, Gutowski S, Kahn RA, Sternweis PC. J Biol Chem. 1995;270:14935. doi: 10.1074/jbc.270.25.14935. [DOI] [PubMed] [Google Scholar]
- 372.Brown HA, Sternweis PC. Methods Enzymol. 1995;257:313. doi: 10.1016/s0076-6879(95)57035-7. [DOI] [PubMed] [Google Scholar]
- 373.Sung TC, Altshuller YM, Morris AJ, Frohman MA. J Biol Chem. 1999;274:494. doi: 10.1074/jbc.274.1.494. [DOI] [PubMed] [Google Scholar]
- 374.Ge M, Cohen JS, Brown HA, Freed JH. Biophys J. 2001;81:994. doi: 10.1016/S0006-3495(01)75757-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Vinggaard AM, Jensen T, Morgan CP, Cockcroft S, Hansen HS. Biochem J. 1996;319(Pt 3):861. doi: 10.1042/bj3190861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Jiang X, Gutowski S, Singer WD, Sternweis PC. Methods Enzymol. 2002;345:328. doi: 10.1016/s0076-6879(02)45026-4. [DOI] [PubMed] [Google Scholar]
- 377.Kim JH, Lee SD, Han JM, Lee TG, Kim Y, Park JB, Lambeth JD, Suh PG, Ryu SH. FEBS Lett. 1998;430:231. doi: 10.1016/s0014-5793(98)00661-9. [DOI] [PubMed] [Google Scholar]
- 378.Jones DH, Bax B, Fensome A, Cockcroft S. Biochem J. 1999;341(Pt 1):185. [PMC free article] [PubMed] [Google Scholar]
- 379.Cai S, Exton JH. Biochem J. 2001;355:779. doi: 10.1042/bj3550779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Walker SJ, Brown HA. J Biol Chem. 2002;277:26260. doi: 10.1074/jbc.M201811200. [DOI] [PubMed] [Google Scholar]
- 381.Peng HJ, Henkels KM, Mahankali M, Dinauer MC, Gomez-Cambronero J. J Biol Chem. 2011;286:16308. doi: 10.1074/jbc.M110.206672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Luo JQ, Liu X, Hammond SM, Colley WC, Feig LA, Frohman MA, Morris AJ, Foster DA. Biochem Biophys Res Commun. 1997;235:854. doi: 10.1006/bbrc.1997.6793. [DOI] [PubMed] [Google Scholar]
- 383.Sun Y, Fang Y, Yoon MS, Zhang C, Roccio M, Zwartkruis FJ, Armstrong M, Brown HA, Chen J. Proc Natl Acad Sci USA. 2008;105:8286. doi: 10.1073/pnas.0712268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Chen JS, Exton JH. J Biol Chem. 2004;279:22076. doi: 10.1074/jbc.M311033200. [DOI] [PubMed] [Google Scholar]
- 385.Kook S, Exton JH. Cell Signal. 2005;17:1423. doi: 10.1016/j.cellsig.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 386.Viparelli F, Cassese A, Doti N, Paturzo F, Marasco D, Dathan NA, Monti SM, Basile G, Ungaro P, Sabatella M, Miele C, Teperino R, Consiglio E, Pedone C, Beguinot F, Formisano P, Ruvo M. J Biol Chem. 2008;283:21769. doi: 10.1074/jbc.M803771200. [DOI] [PubMed] [Google Scholar]
- 387.Preininger AM, Henage LG, Oldham WM, Yoon EJ, Hamm HE, Brown HA. Mol Pharmacol. 2006;70:311. doi: 10.1124/mol.105.021451. [DOI] [PubMed] [Google Scholar]
- 388.Rappley I, Gitler AD, Selvy PE, LaVoie MJ, Levy BD, Brown HA, Lindquist S, Selkoe DJ. Biochemistry. 2009;48:1077. doi: 10.1021/bi801871h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Xie Z, Kim HK, Exton JH. Methods Enzymol. 2002;345:255. doi: 10.1016/s0076-6879(02)45021-5. [DOI] [PubMed] [Google Scholar]
- 390.Katayama K, Kodaki T, Nagamachi Y, Yamashita S. Biochem J. 1998;329(Pt 3):647. doi: 10.1042/bj3290647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Elvers M, Stegner D, Hagedorn I, Kleinschnitz C, Braun A, Kuijpers ME, Boesl M, Chen Q, Heemskerk JW, Stoll G, Frohman MA, Nieswandt B. Sci Signal. 2010;3:ra1. doi: 10.1126/scisignal.2000551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Rhee SG. Annu Rev Biochem. 2001;70:281. doi: 10.1146/annurev.biochem.70.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Hubbard KB, Hepler JR. Cell Signal. 2006;18:135. doi: 10.1016/j.cellsig.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 394.Litosch I. Biochem Biophys Res Commun. 2009;390:603. doi: 10.1016/j.bbrc.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 395.Litosch I, Pujari R, Lee SJ. Cell Signal. 2009;21:1379. doi: 10.1016/j.cellsig.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 396.Le Stunff H, Dokhac L, Bourgoin S, Bader MF, Harbon S. Biochem J. 2000;352(Pt 2):491. doi: 10.1042/0264-6021:3520491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Plonk SG, Park SK, Exton JH. J Biol Chem. 1998;273:4823. doi: 10.1074/jbc.273.9.4823. [DOI] [PubMed] [Google Scholar]
- 398.Biscardi JS, Maa MC, Tice DA, Cox ME, Leu TH, Parsons SJ. J Biol Chem. 1999;274:8335. doi: 10.1074/jbc.274.12.8335. [DOI] [PubMed] [Google Scholar]
- 399.Tice DA, Biscardi JS, Nickles AL, Parsons SJ. Proc Natl Acad Sci USA. 1999;96:1415. doi: 10.1073/pnas.96.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Locati M, Riboldi E, Bonecchi R, Transidico P, Bernasconi S, Haribabu B, Morris AJ, Mantovani A, Sozzani S. Biochem J. 2001;358:119. doi: 10.1042/0264-6021:3580119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Paruch S, Heinis M, Lemay J, Hoeffel G, Maranon C, Hosmalin A, Perianin A. FASEB J. 2007;21:4038. doi: 10.1096/fj.06-7325com. [DOI] [PubMed] [Google Scholar]
- 402.Pettitt TR, Martin A, Horton T, Liossis C, Lord JM, Wakelam MJ. J Biol Chem. 1997;272:17354. doi: 10.1074/jbc.272.28.17354. [DOI] [PubMed] [Google Scholar]
- 403.Pettitt TR, McDermott M, Saqib KM, Shimwell N, Wakelam MJ. Biochem J. 2001;360:707. doi: 10.1042/0264-6021:3600707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Milne SB, Ivanova PT, DeCamp D, Hsueh RC, Brown HA. J Lipid Res. 2005;46:1796. doi: 10.1194/jlr.D500010-JLR200. [DOI] [PubMed] [Google Scholar]
- 405.Shulga YV, Topham MK, Epand RM. Chem Rev. 2011 doi: 10.1021/cr1004106. [DOI] [PubMed] [Google Scholar]
- 406.Callender HL, Forrester JS, Ivanova P, Preininger A, Milne S, Brown HA. Anal Chem. 2007;79:263. doi: 10.1021/ac061083q. [DOI] [PubMed] [Google Scholar]
- 407.Milne SB, Ivanova PT, Armstrong MD, Myers DS, Lubarda J, Shulga YV, Topham MK, Brown HA, Epand RM. Biochemistry. 2008;47:9372. doi: 10.1021/bi800492c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Shulga YV, Myers DS, Ivanova PT, Milne SB, Brown HA, Topham MK, Epand RM. Biochemistry. 2010;49:312. doi: 10.1021/bi901551e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Lung M, Shulga YV, Ivanova PT, Myers DS, Milne SB, Brown HA, Topham MK, Epand RM. J Biol Chem. 2009;284:31062. doi: 10.1074/jbc.M109.050617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Riese DJ, 2nd, Gallo RM, Settleman J. Bioessays. 2007;29:558. doi: 10.1002/bies.20582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Di Fulvio M, Frondorf K, Henkels KM, Lehman N, Gomez-Cambronero J. J Mol Biol. 2007;367:814. doi: 10.1016/j.jmb.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Zhao C, Du G, Skowronek K, Frohman MA, Bar-Sagi D. Nat Cell Biol. 2007;9:706. doi: 10.1038/ncb1594. [DOI] [PubMed] [Google Scholar]
- 413.Voss M, Weernink PA, Haupenthal S, Moller U, Cool RH, Bauer B, Camonis JH, Jakobs KH, Schmidt M. J Biol Chem. 1999;274:34691. doi: 10.1074/jbc.274.49.34691. [DOI] [PubMed] [Google Scholar]
- 414.Sun Y, Chen J. Cell Cycle. 2008;7:3118. doi: 10.4161/cc.7.20.6881. [DOI] [PubMed] [Google Scholar]
- 415.Foster DA. Biochim Biophys Acta. 2009;1791:949. doi: 10.1016/j.bbalip.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Toschi A, Lee E, Xu L, Garcia A, Gadir N, Foster DA. Mol Cell Biol. 2009;29:1411. doi: 10.1128/MCB.00782-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Leone M, Crowell KJ, Chen J, Jung D, Chiang GG, Sareth S, Abraham RT, Pellecchia M. Biochemistry. 2006;45:10294. doi: 10.1021/bi060976+. [DOI] [PubMed] [Google Scholar]
- 418.Ivaska J, Heino J. Cell Tissue Res. 2010;339:111. doi: 10.1007/s00441-009-0857-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Chae YC, Kim JH, Kim KL, Kim HW, Lee HY, Heo WD, Meyer T, Suh PG, Ryu SH. Mol Biol Cell. 2008;19:3111. doi: 10.1091/mbc.E07-04-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Sabe H, Onodera Y, Mazaki Y, Hashimoto S. Curr Opin Cell Biol. 2006;18:558. doi: 10.1016/j.ceb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 421.Hiroi T. Front Biosci. 2009;14:717. doi: 10.2741/3275. [DOI] [PubMed] [Google Scholar]
- 422.Ktistakis NT, Brown HA, Waters MG, Sternweis PC, Roth MG. J Cell Biol. 1996;134:295. doi: 10.1083/jcb.134.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Caumont AS, Galas MC, Vitale N, Aunis D, Bader MF. J Biol Chem. 1998;273:1373. doi: 10.1074/jbc.273.3.1373. [DOI] [PubMed] [Google Scholar]
- 424.Siddhanta A, Shields D. J Biol Chem. 1998;273:17995. doi: 10.1074/jbc.273.29.17995. [DOI] [PubMed] [Google Scholar]
- 425.Chen YG, Siddhanta A, Austin CD, Hammond SM, Sung TC, Frohman MA, Morris AJ, Shields D. J Cell Biol. 1997;138:495. doi: 10.1083/jcb.138.3.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Kuai J, Boman AL, Arnold RS, Zhu X, Kahn RA. J Biol Chem. 2000;275:4022. doi: 10.1074/jbc.275.6.4022. [DOI] [PubMed] [Google Scholar]
- 427.Yang JS, Gad H, Lee SY, Mironov A, Zhang L, Beznoussenko GV, Valente C, Turacchio G, Bonsra AN, Du G, Baldanzi G, Graziani A, Bourgoin S, Frohman MA, Luini A, Hsu VW. Nat Cell Biol. 2008;10:1146. doi: 10.1038/ncb1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Lee C, Kang HS, Chung JK, Sekiya F, Kim JR, Han JS, Kim SR, Bae YS, Morris AJ, Rhee SG. J Biol Chem. 1997;272:15986. doi: 10.1074/jbc.272.25.15986. [DOI] [PubMed] [Google Scholar]
- 429.Cho JH, Oh DY, Kim HJ, Park SY, Choi HJ, Kwon SJ, Lee KS, Han JS. Cancer Lett. 2011;302:144. doi: 10.1016/j.canlet.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 430.Lee CS, Kim IS, Park JB, Lee MN, Lee HY, Suh PG, Ryu SH. Nat Cell Biol. 2006;8:477. doi: 10.1038/ncb1401. [DOI] [PubMed] [Google Scholar]
- 431.Hu T, Liu Z, Shen X. J Cell Mol Med. 2010 doi: 10.1111/j.1582-4934.2010.01035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Cohen JS, Brown HA. Biochemistry. 2001;40:6589. doi: 10.1021/bi0103011. [DOI] [PubMed] [Google Scholar]
- 433.Tou J, Urbizo C. Cell Signal. 2001;13:191. doi: 10.1016/s0898-6568(01)00137-1. [DOI] [PubMed] [Google Scholar]
- 434.Clark AM, El-Feraly FS, Li WS. J Pharm Sci. 1981;70:951. doi: 10.1002/jps.2600700833. [DOI] [PubMed] [Google Scholar]
- 435.Bai X, Cerimele F, Ushio-Fukai M, Waqas M, Campbell PM, Govindarajan B, Der CJ, Battle T, Frank DA, Ye K, Murad E, Dubiel W, Soff G, Arbiser JL. J Biol Chem. 2003;278:35501. doi: 10.1074/jbc.M302967200. [DOI] [PubMed] [Google Scholar]
- 436.Shigemura K, Arbiser JL, Sun SY, Zayzafoon M, Johnstone PA, Fujisawa M, Gotoh A, Weksler B, Zhau HE, Chung LW. Cancer. 2007;109:1279. doi: 10.1002/cncr.22551. [DOI] [PubMed] [Google Scholar]
- 437.Garcia A, Zheng Y, Zhao C, Toschi A, Fan J, Shraibman N, Brown HA, Bar-Sagi D, Foster DA, Arbiser JL. Clin Cancer Res. 2008;14:4267. doi: 10.1158/1078-0432.CCR-08-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Tou JS, Urbizo C. Steroids. 2008;73:216. doi: 10.1016/j.steroids.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 439.Kitzen JJ, de Jonge MJ, Lamers CH, Eskens FA, van der Biessen D, van Doorn L, Ter Steeg J, Brandely M, Puozzo C, Verweij J. Eur J Cancer. 2009;45:1764. doi: 10.1016/j.ejca.2009.01.026. [DOI] [PubMed] [Google Scholar]
- 440.Kang DW, Lee JY, Oh DH, Park SY, Woo TM, Kim MK, Park MH, Jang YH, Min do S. Exp Mol Med. 2009;41:678. doi: 10.3858/emm.2009.41.9.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Westerheide SD, Kawahara TL, Orton K, Morimoto RI. J Biol Chem. 2006;281:9616. doi: 10.1074/jbc.M512044200. [DOI] [PubMed] [Google Scholar]
- 442.Titov DV, Gilman B, He QL, Bhat S, Low WK, Dang Y, Smeaton M, Demain AL, Miller PS, Kugel JF, Goodrich JA, Liu JO. Nat Chem Biol. 2011 doi: 10.1038/nchembio.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Chu M, Truumees I, Patel MG, Gullo VP, Pai J-K, Das PR, Puar MS. Bioorg, Med Chem Lett. 1994;4:1539. [Google Scholar]
- 444.Chu M, Patel MG, Pai J-K, Das PR, Puar MS. Bioorg Med Chem Lett. 1996;6:579. [Google Scholar]
- 445.Hegde VR, Silver J, Patel MG, Bryant R, Pai J, Das PR, Puar MS, Cox PA. J Nat Prod. 1995;58:1492. doi: 10.1021/np50124a003. [DOI] [PubMed] [Google Scholar]
- 446.Davies DR, Interthal H, Champoux JJ, Hol WG. Chem Biol. 2003;10:139. doi: 10.1016/s1074-5521(03)00021-8. [DOI] [PubMed] [Google Scholar]
- 447.Andrews B, Bond K, Lehman JA, Horn JM, Dugan A, Gomez-Cambronero J. Biochem Biophys Res Commun. 2000;273:302. doi: 10.1006/bbrc.2000.2938. [DOI] [PubMed] [Google Scholar]
- 448.McDonald LA, Barbieri LR, Bernan VS, Janso J, Lassota P, Carter GT. J Nat Prod. 2004;67:1565. doi: 10.1021/np049924g. [DOI] [PubMed] [Google Scholar]
- 449.Diwu Z, Zimmermann J, Meyer T, Lown JW. Biochem Pharmacol. 1994;47:373. doi: 10.1016/0006-2952(94)90029-9. [DOI] [PubMed] [Google Scholar]
- 450.Sciorra VA, Hammond SM, Morris AJ. Biochemistry. 2001;40:2640. doi: 10.1021/bi002528m. [DOI] [PubMed] [Google Scholar]
- 451.Levy BD. Br J Pharmacol. 2005;146:344. doi: 10.1038/sj.bjp.0706338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Hatcher H, Planalp R, Cho J, Torti FM, Torti SV. Cell Mol Life Sci. 2008;65:1631. doi: 10.1007/s00018-008-7452-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Yamamoto H, Hanada K, Kawasaki K, Nishijima M. FEBS Lett. 1997;417:196. doi: 10.1016/s0014-5793(97)01280-5. [DOI] [PubMed] [Google Scholar]
- 454.Eisen SF, Brown HA. Mol Pharmacol. 2002;62:911. doi: 10.1124/mol.62.4.911. [DOI] [PubMed] [Google Scholar]
- 455.Jaiyesimi IA, Buzdar AU, Decker DA, Hortobagyi GN. J Clin Oncol. 1995;13:513. doi: 10.1200/JCO.1995.13.2.513. [DOI] [PubMed] [Google Scholar]
- 456.Perry RR, Kang Y, Greaves B. Annals of Surgical Oncology. 1995;2:238. doi: 10.1007/BF02307030. [DOI] [PubMed] [Google Scholar]
- 457.Noh DY, Ahn SJ, Lee RA, Park IA, Kim JH, Suh PG, Ryu SH, Lee KH, Han JS. Cancer Lett. 2000;161:207. doi: 10.1016/s0304-3835(00)00612-1. [DOI] [PubMed] [Google Scholar]
- 458.Scott SA, Selvy PE, Buck JR, Cho HP, Criswell TL, Thomas AL, Armstrong MD, Arteaga CL, Lindsley CW, Brown HA. Nat Chem Biol. 2009;5:108. doi: 10.1038/nchembio.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Borgna JL, Rochefort H. J Biol Chem. 1981;256:859. [PubMed] [Google Scholar]
- 460.Jordan VC, Collins MM, Rowsby L, Prestwich G. J Endocrinol. 1977;75:305. doi: 10.1677/joe.0.0750305. [DOI] [PubMed] [Google Scholar]
- 461.Coezy E, Borgna JL, Rochefort H. Cancer Res. 1982;42:317. [PubMed] [Google Scholar]
- 462.Kiss Z, Anderson WH. FEBS Lett. 1997;400:145. doi: 10.1016/s0014-5793(96)01377-4. [DOI] [PubMed] [Google Scholar]
- 463.Monovich L. Bioorg Med Chem Lett. 2007;17:2310. doi: 10.1016/j.bmcl.2007.01.059. [DOI] [PubMed] [Google Scholar]
- 464.Loonen AJM, Soudijn W. Pharm Weekbl, Sci Ed. 1985;7:1. doi: 10.1007/BF01962862. [DOI] [PubMed] [Google Scholar]
- 465.Loonen AJ, Soe-Agnie CJ, Soudijn W. Brain Res. 1981;210:485. doi: 10.1016/0006-8993(81)90932-x. [DOI] [PubMed] [Google Scholar]
- 466.van Rooij HH, Waterman RL, Kraak JC. J Chromatogr. 1979;164:177. doi: 10.1016/s0378-4347(00)81186-x. [DOI] [PubMed] [Google Scholar]
- 467.Yamada Y, Hamajima N, Kato T, Iwata H, Yamamura Y, Shinoda M, Suyama M, Mitsudomi T, Tajima K, Kusakabe S, Yoshida H, Banno Y, Akao Y, Tanaka M, Nozawa Y. J Mol Med. 2003;81:126. doi: 10.1007/s00109-002-0411-x. [DOI] [PubMed] [Google Scholar]
- 468.Lewis JA, Scott SA, Lavieri R, Buck JR, Selvy PE, Stoops SL, Armstrong MD, Brown HA, Lindsley CW. Bioorg Med Chem Lett. 2009;19:1916. doi: 10.1016/j.bmcl.2009.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Lavieri R, Scott SA, Lewis JA, Selvy PE, Armstrong MD, Alex Brown H, Lindsley CW. Bioorg Med Chem Lett. 2009;19:2240. doi: 10.1016/j.bmcl.2009.02.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Lavieri RR, Scott SA, Selvy PE, Kim K, Jadhav S, Morrison RD, Daniels JS, Brown HA, Lindsley CW. J Med Chem. 2010;53:6706. doi: 10.1021/jm100814g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Park MH, Ahn BH, Hong YK, Min DS. Carcinogenesis. 2009;30:356. doi: 10.1093/carcin/bgn287. [DOI] [PubMed] [Google Scholar]
- 472.Zheng Y, Rodrik V, Toschi A, Shi M, Hui L, Shen Y, Foster DA. J Biol Chem. 2006;281:15862. doi: 10.1074/jbc.M600660200. [DOI] [PubMed] [Google Scholar]
- 473.Williger BT, Ho WT, Exton JH. J Biol Chem. 1999;274:735. doi: 10.1074/jbc.274.2.735. [DOI] [PubMed] [Google Scholar]
- 474.Oliveira TG, Chan RB, Tian H, Laredo M, Shui G, Staniszewski A, Zhang H, Wang L, Kim TW, Duff KE, Wenk MR, Arancio O, Di Paolo G. J Neurosci. 2010;30:16419. doi: 10.1523/JNEUROSCI.3317-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Jin JK, Ahn BH, Na YJ, Kim JI, Kim YS, Choi EK, Ko YG, Chung KC, Kozlowski PB, Min do S. Neurobiol Aging. 2007;28:1015. doi: 10.1016/j.neurobiolaging.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 476.Elvers M, Pozgaj R, Pleines I, May F, Kuijpers MJ, Heemskerk JM, Yu P, Nieswandt B. J Thromb Haemostasis. 2010;8:1353. doi: 10.1111/j.1538-7836.2010.03838.x. [DOI] [PubMed] [Google Scholar]
- 477.Brown HA, Lindsley CW, Waterson AG, Scott SA. 2010-US43045. US Patent application WO. 2010 July 23;
- 478.Babior BM, Lambeth JD, Nauseef W. Arch Biochem Biophys. 2002;397:342. doi: 10.1006/abbi.2001.2642. [DOI] [PubMed] [Google Scholar]
- 479.Rossi F, Grzeskowiak M, Della Bianca V, Calzetti F, Gandini G. Biochem Biophys Res Commun. 1990;168:320. doi: 10.1016/0006-291x(90)91711-z. [DOI] [PubMed] [Google Scholar]
- 480.Levy BD, Clark JM, Wakelam MJO, Serhan CN. Am J Resp Crit Care. 1999;159:A188. [Google Scholar]
- 481.Agwu DE, McPhail LC, Sozzani S, Bass DA, McCall CE. J Clin Invest. 1991;88:531. doi: 10.1172/JCI115336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.McPhail LC, Waite KA, Regier DS, Nixon JB, Qualliotine-Mann D, Zhang WX, Wallin R, Sergeant S. Biochim Biophys. 1999;1439:277. doi: 10.1016/s1388-1981(99)00100-6. [DOI] [PubMed] [Google Scholar]
- 483.Cummings R, Parinandi N, Wang L, Usatyuk P, Natarajan V. Mol Cell Biochem. 2002;234–235:99. [PubMed] [Google Scholar]
- 484.Bonifacino JS, Glick BS. Cell. 2004;116:153. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- 485.Roth MG. Traffic. 2008;9:1233. doi: 10.1111/j.1600-0854.2008.00742.x. [DOI] [PubMed] [Google Scholar]
- 486.Xie MS, Jacobs LS, Dubyak GR. J Clin Invest. 1991;88:45. doi: 10.1172/JCI115303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Gruchalla RS, Dinh TT, Kennerly DA. J Immunol. 1990;144:2334. [PubMed] [Google Scholar]
- 488.Shen Y, Xu L, Foster DA. Mol Cell Biol. 2001;21:595. doi: 10.1128/MCB.21.2.595-602.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Koch T, Brandenburg LO, Liang Y, Schulz S, Beyer A, Schroder H, Hollt V. J Neurochem. 2004;88:680. doi: 10.1046/j.1471-4159.2003.02189.x. [DOI] [PubMed] [Google Scholar]
- 490.Koch T, Brandenburg LO, Schulz S, Liang Y, Klein J, Hollt V. J Biol Chem. 2003;278:9979. doi: 10.1074/jbc.M206709200. [DOI] [PubMed] [Google Scholar]
- 491.Bhattacharya M, Babwah AV, Godin C, Anborgh PH, Dale LB, Poulter MO, Ferguson SS. J Neurosci. 2004;24:8752. doi: 10.1523/JNEUROSCI.3155-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Du G, Huang P, Liang BT, Frohman MA. Mol Biol Cell. 2004;15:1024. doi: 10.1091/mbc.E03-09-0673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Dall’armi C, Hurtado-Lorenzo A, Tian H, Morel E, Nezu A, Chan RB, Yu WH, Robinson KS, Yeku O, Small SA, Duff K, Frohman MA, Wenk MR, Yamamoto A, Di Paolo G. Nat Commun. 2010;1:142. doi: 10.1038/ncomms1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Vorland M, Holmsen H. Platelets. 2008;19:300. doi: 10.1080/09537100801910838. [DOI] [PubMed] [Google Scholar]
- 495.Haslam RJ, Coorssen JR. Programmed Cell Death in Cancer Progression and Therapy. 1993;344:149. [Google Scholar]
- 496.Disse J, Vitale N, Bader M-F, Gerke V. Blood. 2009;113:973. doi: 10.1182/blood-2008-06-165282. [DOI] [PubMed] [Google Scholar]
- 497.Sadler JE. Annu Rev Biochem. 1998;67:395. doi: 10.1146/annurev.biochem.67.1.395. [DOI] [PubMed] [Google Scholar]
- 498.Kobayashi M, Kanfer JN. J Neurochem. 1987;48:1597. doi: 10.1111/j.1471-4159.1987.tb05707.x. [DOI] [PubMed] [Google Scholar]
- 499.Zhang Y, Huang P, Du G, Kanaho Y, Frohman MA, Tsirka SE. Glia. 2004;46:74. doi: 10.1002/glia.10322. [DOI] [PubMed] [Google Scholar]
- 500.Kanfer JN, Singh IN, Pettegrew JW, McCartney DG, Sorrentino G. J Lipid Mediat Cell Signal. 1996;14:361. doi: 10.1016/0929-7855(96)00545-7. [DOI] [PubMed] [Google Scholar]
- 501.Lee MJ, Oh JY, Park HT, Uhlinger DJ, Kwak JY. Neurosci Lett. 2001;315:159. doi: 10.1016/s0304-3940(01)02339-4. [DOI] [PubMed] [Google Scholar]
- 502.Cox DA, Cohen ML. Neurosci Lett. 1997;229:37. doi: 10.1016/s0304-3940(97)00407-2. [DOI] [PubMed] [Google Scholar]
- 503.Leong SL, Cappai R, Barnham KJ, Pham CL. Neurochem Res. 2009;34:1838. doi: 10.1007/s11064-009-9986-8. [DOI] [PubMed] [Google Scholar]
- 504.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Nat Genet. 1998;18:106. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 505.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Science. 1997;276:2045. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 506.Jenco JM, Rawlingson A, Daniels B, Morris AJ. Biochemistry. 1998;37:4901. doi: 10.1021/bi972776r. [DOI] [PubMed] [Google Scholar]
- 507.Ahn BH, Rhim H, Kim SY, Sung YM, Lee MY, Choi JY, Wolozin B, Chang JS, Lee YH, Kwon TK, Chung KC, Yoon SH, Hahn SJ, Kim MS, Jo YH, Min DS. J Biol Chem. 2002;277:12334. doi: 10.1074/jbc.M110414200. [DOI] [PubMed] [Google Scholar]
- 508.Poste G, Fidler IJ. Nature. 1980;283:139. doi: 10.1038/283139a0. [DOI] [PubMed] [Google Scholar]
- 509.Bacac M, Stamenkovic I. Annu Rev Pathol. 2008;3:221. doi: 10.1146/annurev.pathmechdis.3.121806.151523. [DOI] [PubMed] [Google Scholar]
- 510.Knoepp SM, Chahal MS, Xie YH, Zhang ZH, Brauner DJ, Hallman MA, Robinson SA, Han SJ, Imai M, Tomlinson S, Meier KE. Mol Pharmacol. 2008;74:574. doi: 10.1124/mol.107.040105. [DOI] [PubMed] [Google Scholar]
- 511.Imamura F. Biochem Biophys Res Commun. 1993;193:497. doi: 10.1006/bbrc.1993.1651. [DOI] [PubMed] [Google Scholar]
- 512.Kato Y. J Biol Chem. 2005;280:10938. doi: 10.1074/jbc.M411313200. [DOI] [PubMed] [Google Scholar]
- 513.Reich R, Blumenthal M, Liscovitch M. Clin Exp Metastasis. 1995;13:134. doi: 10.1007/BF00133618. [DOI] [PubMed] [Google Scholar]
- 514.Zhao Y, Ehara H, Akao Y, Shamoto M, Nakagawa Y, Banno Y, Deguchi T, Ohishi N, Yagi K, Nozawa Y. Biochem Biophys Res Commun. 2000;278:140. doi: 10.1006/bbrc.2000.3719. [DOI] [PubMed] [Google Scholar]
- 515.Joseph T, Bryant A, Frankel P, Wooden R, Kerkhoff E, Rapp UR, Foster DA. Oncogene. 2002;21:3651. doi: 10.1038/sj.onc.1205380. [DOI] [PubMed] [Google Scholar]
- 516.Zhong M, Shen Y, Zheng Y, Joseph T, Jackson D, Foster DA. Biochem Biophys Res Commun. 2003;302:615. doi: 10.1016/s0006-291x(03)00229-8. [DOI] [PubMed] [Google Scholar]
- 517.Buchanan FG, McReynolds M, Couvillon A, Kam Y, Holla VR, Dubois RN, Exton JH. Proc Natl Acad Sci USA. 2005;102:1638. doi: 10.1073/pnas.0406698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Shi M, Zheng Y, Garcia A, Xu L, Foster DA. Cancer Lett. 2007;258:268. doi: 10.1016/j.canlet.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Snider AJ, Zhang Z, Xie Y, Meier KE. Am J Physiol Cell Physiol. 2010;298:C163. doi: 10.1152/ajpcell.00001.2009. [DOI] [PubMed] [Google Scholar]
- 520.Hui L, Abbas T, Pielak RM, Joseph T, Bargonetti J, Foster DA. Mol Cell Biol. 2004;24:5677. doi: 10.1128/MCB.24.13.5677-5686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Hui L, Zheng Y, Yan Y, Bargonetti J, Foster DA. Oncogene. 2006;25:7305. doi: 10.1038/sj.onc.1209735. [DOI] [PubMed] [Google Scholar]
- 522.Chen Y, Rodrik V, Foster DA. Oncogene. 2005;24:672. doi: 10.1038/sj.onc.1208099. [DOI] [PubMed] [Google Scholar]
- 523.Zhao C. Nat Cell Biol. 2007;9:706. doi: 10.1038/ncb1594. [DOI] [PubMed] [Google Scholar]
- 524.Rizzo MA. J Biol Chem. 1999;274:1131. doi: 10.1074/jbc.274.2.1131. [DOI] [PubMed] [Google Scholar]
- 525.Plevin R, Cook SJ, Palmer S, Wakelam MJ. Biochem J. 1991;279(Pt 2):559. doi: 10.1042/bj2790559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Motoike T, Bieger S, Wiegandt H, Unsicker K. FEBS Lett. 1993;332:164. doi: 10.1016/0014-5793(93)80505-o. [DOI] [PubMed] [Google Scholar]
- 527.Song J, Jiang YW, Foster DA. Cell Growth Differ. 1994;5:79. [PubMed] [Google Scholar]
- 528.Frankel P, Ramos M, Flom J, Bychenok S, Joseph T, Kerkhoff E, Rapp UR, Feig LA, Foster DA. Biochem Biophys Res Commun. 1999;255:502. doi: 10.1006/bbrc.1999.0234. [DOI] [PubMed] [Google Scholar]
- 529.Kaelin WG., Jr Clin Cancer Res. 2007;13:680s. doi: 10.1158/1078-0432.CCR-06-1865. [DOI] [PubMed] [Google Scholar]
- 530.Ohh M. Neoplasia. 2006;8:623. doi: 10.1593/neo.06442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Toschi A, Edelstein J, Rockwell P, Ohh M, Foster DA. Oncogene. 2008;27:2746. doi: 10.1038/sj.onc.1210927. [DOI] [PubMed] [Google Scholar]
- 532.Katoh M. Oncol Rep. 2005;14:1583. [PubMed] [Google Scholar]
- 533.Kohn AD, Moon RT. Cell Calcium. 2005;38:439. doi: 10.1016/j.ceca.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 534.Clevers H. Cell. 2006;127:469. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 535.Kang DW, Choi KY, Min DS. Cancer Res. 2011;71:293. doi: 10.1158/0008-5472.CAN-10-2463. [DOI] [PubMed] [Google Scholar]
- 536.Kang DW, Lee SH, Yoon JW, Park WS, Choi KY, Min do S. Cancer Res. 2010;70:4233. doi: 10.1158/0008-5472.CAN-09-3470. [DOI] [PubMed] [Google Scholar]
- 537.Kang DW, Min do S. PLoS One. 2010;5:e12109. doi: 10.1371/journal.pone.0012109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, Stivala F, Libra M, Basecke J, Evangelisti C, Martelli AM, Franklin RA. Biochim Biophys Acta. 2007;1773:1263. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Leicht DT, Balan V, Kaplun A, Singh-Gupta V, Kaplun L, Dobson M, Tzivion G. Biochim Biophys Acta. 2007;1773:1196. doi: 10.1016/j.bbamcr.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Pritchard CA, Bolin L, Slattery R, Murray R, McMahon M. Curr Biol. 1996;6:614. doi: 10.1016/s0960-9822(02)00548-1. [DOI] [PubMed] [Google Scholar]
- 541.Wojnowski L, Zimmer AM, Beck TW, Hahn H, Bernal R, Rapp UR, Zimmer A. Nat Genet. 1997;16:293. doi: 10.1038/ng0797-293. [DOI] [PubMed] [Google Scholar]
- 542.Wojnowski L, Stancato LF, Zimmer AM, Hahn H, Beck TW, Larner AC, Rapp UR, Zimmer A. Mech Dev. 1998;76:141. doi: 10.1016/s0925-4773(98)00111-7. [DOI] [PubMed] [Google Scholar]
- 543.http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2005/021923ltr.pdf
- 544.http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2007/021923s004,s005,s006,s007.pdf
- 545.Schreck R, Rapp UR. Int J Cancer. 2006;119:2261. doi: 10.1002/ijc.22144. [DOI] [PubMed] [Google Scholar]
- 546.Sabatini DM. Nat Rev Cancer. 2006;6:729. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 547.Vivanco I, Sawyers CL. Nat Rev Cancer. 2002;2:489. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 548.Ohguchi K, Banno Y, Nakagawa Y, Akao Y, Nozawa Y. J Cell Physiol. 2005;205:444. doi: 10.1002/jcp.20421. [DOI] [PubMed] [Google Scholar]
- 549.Zhang Y. Nat Cell Biol. 2003;5:578. doi: 10.1038/ncb999. [DOI] [PubMed] [Google Scholar]
- 550.Xu L, Salloum D, Medlin PS, Saqcena M, Yellen P, Perrella B, Foster DA. J Biol Chem. 2011;286:25477. doi: 10.1074/jbc.M111.249631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551.Lehman N, Ledford B, Di Fulvio M, Frondorf K, McPhail LC, Gomez-Cambronero J. FASEB J. 2007;21:1075. doi: 10.1096/fj.06-6652com. [DOI] [PubMed] [Google Scholar]
- 552.Arous C, Naimi M, Van Obberghen E. Diabetologia. 2011;54:954. doi: 10.1007/s00125-010-2032-1. [DOI] [PubMed] [Google Scholar]
- 553.Doti N, Cassese A, Marasco D, Paturzo F, Sabatella M, Viparelli F, Dathan N, Monti SM, Miele C, Formisano P, Beguinot F, Ruvo M. Mol Biosyst. 2010;6:2039. doi: 10.1039/c005272h. [DOI] [PubMed] [Google Scholar]
- 554.Uchida N, Okamura S, Nagamachi Y, Yamashita S. J Cancer Res Clin Oncol. 1997;123:280. doi: 10.1007/BF01208639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555.Shen Q, Stanton ML, Feng W, Rodriguez ME, Ramondetta L, Chen L, Brown RE, Duan X. Int J Clin Exp Pathol. 2010;4:13. [PMC free article] [PubMed] [Google Scholar]