

Summary
Ras-driven tumors are often refractory to conventional therapies. Here we identify a promising targeted therapeutic strategy for two Ras-driven cancers: Nf1-deficient malignancies and KRas/p53-mutant lung cancer. We show that agents that enhance proteotoxic stress, including the HSP90 inhibitor IPI-504, induce tumor regression in aggressive mouse models, but only when combined with rapamycin. These agents synergize by promoting irresolvable ER stress, resulting in catastrophic ER and mitochondrial damage. This process is fueled by oxidative stress, which is caused by IPI-504-dependent production of reactive oxygen species, and the rapamycin-dependent suppression of glutathione, an important endogenous antioxidant. Notably, the mechanism by which these agents cooperate reveals a therapeutic paradigm that can be expanded to develop additional combinations.
Introduction
While significant advances have been made in developing targeted therapies, identifying treatments for tumors driven by mutations that do not affect a targetable protein represents a major challenge in cancer research. Ras-driven cancers are a classic example of this challenge and despite the fact that Ras signaling has been studied for >25 years there are still no effective targeted therapies (Young et al., 2009). Small-molecule inhibitors that target Ras effectors are being evaluated; however, studies suggest that the therapeutic efficacy of single targeted agents may be limited, underscoring the need to identify additional targets and/or more effective drug combinations (Engelman et al., 2008; Young et al., 2009).
To develop new therapies we initially focused on a distinct subset of Ras-driven tumors: those that possess mutations in the NF1 tumor suppressor. NF1 encodes a Ras GTPase-activating protein (RasGAP), which negatively regulates Ras by catalyzing the hydrolysis of Ras-GTP (Martin et al., 1990; Cawthon et al., 1990). Accordingly, NF1-deficient tumors are driven by aberrant Ras activation (DeClue et al., 1992; Johannessen et al., 2005). NF1 mutations underlie neurofibromatosis type 1 (NF1) (Martin et al., 1990;Cawthon et al., 1990) and NF1 is mutated or suppressed in sporadic glioblasotoma (TCGA Consortium, 2008; Parsons et al., 2008; McGillicuddy et al., 2009), NSCLC (Ding et al., 2008) and neuroblastoma (Holzel et al., 2010), demonstrating a broader role for NF1-loss in cancer. The most common malignancy associated with NF1 are malignant peripheral nerve sheath tumors (MPNSTs), which are highly aggressive and frequently metastasize. Despite radiation and in some cases chemotherapy, inoperable tumors rapidly progress and can be lethal within months. As such, identifying an effective treatment for these tumors is critical.
We and others have reported that mTOR is hyperactivated in NF1-deficient tumors as a consequence of aberrant Ras signaling (Johannessen et al., 2005, Dasgupta et al., 2005). Using a Nf1/p53-mutant MPNST model we further demonstrated that the mTOR inhibitor rapamycin suppressed tumor growth (Johannessen et al., 2008). However while the response to rapamycin was potent, effects were cytostatic. Therefore, we have been using this model to develop more effective mTOR-inhibitor based combination therapies, with the expectation that successful combinations might also be effective in other Ras and/or mTOR-driven tumors.
To identify additional therapeutic agents we considered drugs that might exploit specific cellular vulnerabilities of cancer cells. In addition to the pro-tumorigenic hallmarks of cancer (Hanahan and Weinberg, 2000), tumor cells often exhibit specific stress-related phenotypes caused by insults such as excessive DNA damage, as well as replicative, metabolic, and proteotoxic stress (Luo et al., 2009). Accordingly, it has been suggested that agents that further enhance or sensitize cancer cells to these stresses could be developed as potential therapies (Luo et al., 2009;Taipale et al, 2010.). In this study we investigated agents that augment proteotoxic or ER (endoplasmic reticulum) stress. ER stress is induced when unfolded proteins accumulate in the endoplasmic reticulum (Ron and Walter, 2007). Cancer cells frequently exhibit high levels of ER stress, caused by factors such as high mutational load, copy number variation, oxidative stress, hypoxia, and nutrient deprivation (Luo et al., 2009; Taipale et al, 2010). Aneuploidy in particular, has recently been shown to induce proteotoxic stress in both normal and cancer cells (Tang et al., 2011). Oncogenic RAS also causes ER stress (Denoyelle et al., 2006). Once triggered, ER stress activates a signal transduction pathway known as the unfolded protein response (UPR) (Ron and Walter, 2007). The UPR is initially engaged as a protective mechanism to reduce protein accumulation; however, when ER stress levels become insurmountable cell death ensues (Ron and Walter, 2007). This observation has led to the speculation that agents that further enhance ER stress in vulnerable cancer cells could be developed as anti-cancer therapies (Luo et al., 2009; Tang et al., 2011). In this study we evaluated the therapeutic effects of compounds that augment proteotoxic stress in cancer cells, alone and in combination with mTOR inhibitors, in two Ras-driven mouse tumor models.
Results
MPNSTs are sensitive to agents that enhance ER stress
To determine whether MPNSTs might be sensitive to agents that induce ER stress, we first evaluated basal stress levels. MPNSTs are highly aneuploid and are driven by constitutive activation of Ras, and therefore might be subject to substantial ER stress. Indeed, ER stress levels were much higher in tumors as compared to normal peripheral nerve, as confirmed by three independent markers of UPR activation: BiP upregulation, phosphorylation of eukaryotic translational initiation factor 2α (eIF2α), and accumulation of the spliced active form of XBP-1 (sXBP-1) (Figure 1A) (Ron and Walter, 2007). Next, we assessed the sensitivity of human and mouse MPNSTs to classic ER stress-inducing agents: thapsigargin (an ER calcium ATPase inhibitor) and tunicamycin (a glycosylation inhibitor). Both agents enhanced ER stress (Figure 1B), and triggered cell death at concentrations that did not impact the viability of normal cells (Figure 1C, D), indicating that MPNSTs are hypersensitive to these ER stress-inducing agents.
Figure 1. Therapeutic effects of rapamycin and ER stress inducing agents on MPNSTs.
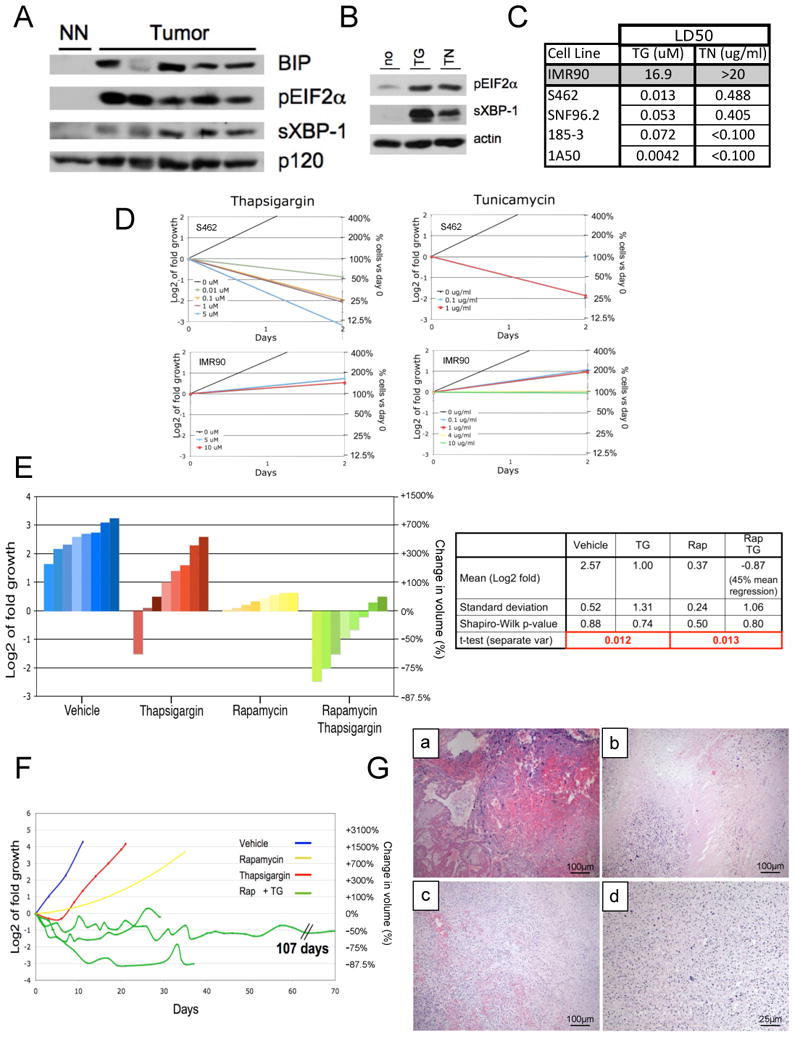
(A) Immunoblots of tumor tissue derived from Nf1/p53 mutant murine MPNSTs and normal peripheral nerve (NN). BIP, phospho-EIF2a (pEIF2α) and the spliced form of XBP-1 (sXBP-1) indicate UPR activation. (B) Immunoblots of pEIF2α and sXBP-1 in MPNST cells after 4 hours of 100nM thapsigargin (TG) or 0.5ug/ml tunicamycin (TN). Actin is a loading control. (C) LD50 values in response to TG or TN (48 hours) in normal cells (IMR90), human MPNST cell lines (S462, SNF96.2) and mouse MPNST cell lines (185-3, 1A50). (D) Growth curves comparing the effects of different doses of thapsigargin and tunicamycin in S462 human MPNSTs and IMR90s. (E) Waterfall plot depicting tumor growth after 10 days of treatment with vehicle (blue), thapsigargin (red), rapamycin (yellow) and rapamycin/thapsigargin (green). The left y-axis indicates the log2 of tumor fold growth vs. day 0 and the right y-axis shows the change in fold volume. The table shown reports mean and standard deviation for each treatment arm (n=8) and mean tumor shrinkage. (F) Graph depicting the change in tumor size over time. Three animals on the rapamycin/thapsigargin combination are shown (green). For simplicity the yellow line is an average volume of rapamycin treated tumors (n=8). Blue and red lines represent vehicle and thapsigargin treated animals respectively. (G) H&E stained tumor remnants from animals treated with rapamycin/thapsigargin. Sections from tumors after (a) 107 days of treatment, (b) 35 days (c), 21 days (d) 4 days, showing pyknotic nuclei throughout the tumor. All images were taken using 10× objective, except (d) which has been magnified to 40×. (See also Figure S1)
ER stress inducing agents promote tumor regression in vivo but only when combined with rapamycin
Based on the hypersensitivity of MPNST cells to these agents in vitro, we hypothesized that they might promote tumor regression. In the Nf1/p53 tumor model animals develop MPNSTs in ∼5 months (Cichowski et al., 1999) and survive an average of 10.7 days after tumor detection (Johannessen et al., 2008). Tumor bearing animals were treated with vehicle, thapsigargin, or rapamycin (Figure 1E). Thapsigargin exhibited minimal efficacy (red bars) and was less potent than rapamycin (yellow bars). This finding was unexpected given the cytotoxic versus cytostatic effects of thapsigargin and rapamycin observed in vitro (Figure 1D and Johannessen et al., 2008). However, combined rapamycin/thapsigargin treatment triggered rapid tumor regression (green bars; p=0.013). On average tumors shrank 45%; however, some tumors regressed >75% (Figure 1F) and remaining masses were largely comprised of hemorrhage and cellular debris (Figure 1G). Maximal effects were observed within 10 days, although significant tumor regression was detected in 3 days (Figure 1F, G). Extensive long-term survival studies were not performed because mice often scratched or bit these rapidly shrinking lesions, resulting in ulceration that necessitated euthanasia. Nevertheless, when animals were successfully treated for longer duration tumors did not re-grow (Figure 1F). One animal survived 107 days after tumor development with no evidence of relapse, surviving > 10 times as long as control animals (Figure 1F, G). Tunicamycin also induced tumor regression when co-administered with rapamycin, consistent with the conclusion that excessive ER stress was a critical driver of this response (Figure S1).
The HSP90 inhibitor IPI-504 cooperates with rapamycin to promote tumor regression
While these observations were striking, thapsigargin and tunicamycin do not represent clinically viable agents. HSP90 inhibitors are another class of drugs known to induce ER stress and are currently being investigated in the clinic (clinicaltrials.gov). HSP90 maintains protein homeostasis by folding newly synthesized and misfolded proteins, assembling and dissembling protein complexes, and resolving protein aggregates (Whitesell and Lindquist, 2005). HSP90 also directly stabilizes two key stress-sensing components of the UPR: IRE1 and pPERK/PERK (Marcu et al., 2002). Therefore, HSP90 inhibitors would be expected to promote ER stress in cancer cells via two cooperating mechanisms: first, by directly impairing global protein folding in these already compromised tumor cells and second, by inactivating subsequent adaptive responses provided by two arms of the UPR. Accordingly, we assessed the therapeutic effects of IPI-504, a hydroquinone hydrochloride salt of the geldanamycin-derivative 17-AAG (Sydor et al., 2006).
As predicted, IPI-504 rapidly induced ER stress and activates the UPR, demonstrated by the upregulation of BiP, eIF2α, sXBP-1, IRE1 and phosphorylated PERK within 2-4 hours (Figure 2A) (Healy et al., 2009). However, prolonged exposure to IPI-504 resulted in a destabilization of IRE1 and pPERK/PERK (Figure 2A). Consequently, downstream UPR signals including sXBP-1 and pEIF2α were inactivated by 8 hours, as expected (Marcu et al., 2002)(Figure 2A). Notably, BiP levels, which are not dependent on IRE1 and PERK, was elevated further by 16 hours, demonstrating that ER stress was enhanced in two phases in response to IPI-504 (Figure 2A) (Marcu et al., 2002). Similar to thapsigargin and tunicamycin, MPNST cells were sensitive to low doses of IPI-504 in vitro (Figure 2B, C).
Figure 2. Rapamycin and IPI-504 promote MPNST regression.
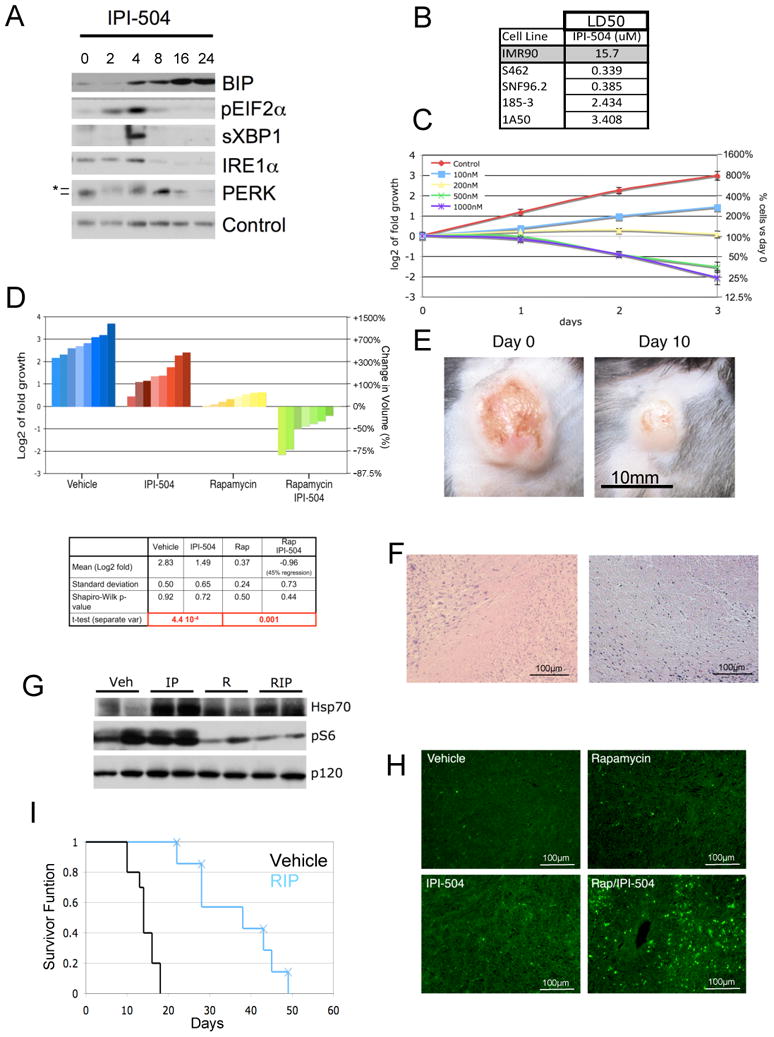
(A) Immunoblots of BIP, pEIF2α, sXBP-1, IRE1α and PERK in human MPNSTs treated with IPI-504 over time (hours). Note that the activation of pEIF2α, sXBP1, IRE1 and PERK (*denotes activated phosphorylated PERK) and initial upregualtion of BIP occurs within 2-4 hours. A second wave of BIP upregulation occurs between 8-16 hours, as pEIF2α, sXBP-1, IRE1α and PERK become suppressed. Actin serves as a loading control. (B) LD50 values in response to IPI-504 (72 hours) for normal cells (IMR90), human MPNST cell lines (S462, SNF96.2) and mouse MPNST cell lines (185-3, 1A50). (C) Growth curves of the S462 cell line treated with different concentrations of IPI-504. (D) Waterfall plot depicting tumor growth after 10 days of treatment with vehicle (blue), IPI-504 (red), rapamycin (yellow) and rapamycin/IPI-504(green). The left y-axis indicates the log2 of tumor fold growth vs. day 0 and the right y-axis shows the change in fold volume. The table reports mean and standard deviation for each treatment arm (n=8) and mean tumor shrinkage. The Shapiro-Wilk test shows that all data sets have a normal distribution. (E) A photograph of an MPNST is shown at day 0 and after 10 days of treatment with Rapamycin/IPI-504. (F) H&E stained tumors from animals treated with rapamycin/IPI-504. (G) Pharmacodynamic analysis of lung tissue after 16h of treatment as shown by an Hsp70 and phosphoS6 immunoblots. p120 serves as a loading control. (H) TUNEL staining of tumors treated for 16h. (I) Kaplain-Meier curve of tumor bearing Nf1/p53 mutant mice treated with vehicle (black) or rapamycin (blue) as described. X's indicate and animal that was euthanized due to skin ulceration. All error bars show +/− SD. (See also Figure S2)
Using a previously established dosing schedule of IPI-504 (Douglas et al., 2009), we assessed the effects of this agent alone and in combination with rapamycin in vivo. Like thapsigargin, IPI-504 was unable to promote tumor regression as a single agent but did so when combined with rapamycin (Figure 2D) (p=0.001). On average tumors shrank 49% (Figure 2D, green bars). Tumor regression was visually apparent (Figure 2E) and histological analysis revealed massive cell death and accumulating debris (Figure 2F).
The pharmacodynamic response to IPI-504 in clinical trials is assessed by measuring HSP70 levels, which increase when HSP90 is effectively inhibited (Ramanathan et al., 2007). Target inhibition was confirmed in vivo using this readout (Figure 2G). Rapamycin also effectively suppressed the mTOR pathway (Figure 2G). Maximal tumor regression in response to rapamycin/IPI-504 treatment occurred within 3-5 days and no toxicity was observed in the course of this study as determined by weight, grooming, or body score (Figure S2). TUNEL staining was apparent within 16 hours, which was not observed in tumors from animals exposed to rapamycin or IPI-504 alone (Figure 2H). To mimic the dose of IPI-504 used in clinical trials, IPI-504 was administered once rather than twice per week at 100mg/kg. This treatment schedule also promoted tumor shrinkage and significantly prolonged survival (Figure 2I) (p=8.9 × 10−5). This Kaplan-Meier curve likely underestimates survival, as most animals were euthanized due to self-inflicted damage at the site of residual lesions (denoted by Xs) (Figure 2I). No long-term toxicity was observed as determined after 50 days of treatment.
IPI-504 mediates its therapeutic effects by suppressing HSP90 and promoting ER stress
HSP90 is encoded by more than one gene, is extremely abundant, and interacts with more than 20 co-chaperones (Taipale et al., 2010). Therefore it is not possible to completely inactivate HSP90 activity by genetically suppressing a single gene. However, two additional structurally distinct HSP90 inhibitors, BEP800 and AUY-922 (Massey et al., 2010), as well as 17-AAG itself, killed MPNSTs, induced ER stress, and impacted the UPR with the same kinetics as IPI-504 (Figure 3A, B, C and Figure S3A), confirming that these agents all function by suppressing HSP90.
Figure 3. Cell death induced by IPI-504 is due to inhibition of HSP90 and subsequent effects on the UPR and ER stress.
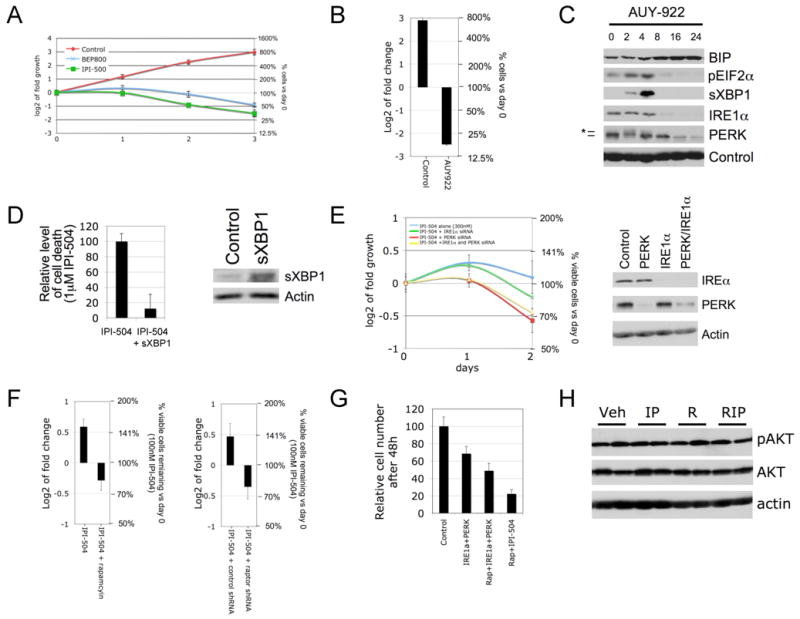
(A) Growth curves of the MPNST cell line S462 treated with two different HSP90 inhibitors (500nM IPI-504 and 500nM BEP800). (B) S462 cells were treated with the HSP90 inhibitor AUY-922 (100nM), for 72 hours. The left y-axis indicates the log2 of tumor fold change vs. day 0 and the right y-axis shows the relative change in cell number as compared to day 0. (C) Immunoblots showing the effects of AUY-922 on BIP, pEIF2α, sXBP-1, IRE1α and PERK in human MPNST cells over time (hours). Actin serves as a loading control. (D) Relative level of cell death in the presence of 1μM IPI-504 with and without over-expression of sXBP1 (the activated spliced form of XBP1). The right hand panel confirms expression of sXBP1. (E) Growth curves in response to low doses of IPI-504 in cells where IRE1α and/or PERK are knocked down by siRNA. The immunoblot confirms knock down. (F) S462 cells were treated with 100nM IPI-504 for 72 hours, with and without rapamycin pretreatment (100nM) (left) or Raptor shRNA (right). The left y-axis indicates the log2 of tumor fold change vs. day 0 and the right y-axis shows the relative change in cell number as compared to day 0. (G) Relative number of S462 cells after combined knock down of IRE1α and PERK with or without rapamycin (100nM) as compared to rapamycin plus IPI-504 (300nM). (F) pAKT/AKT mmunoblots of tumor tissue from animals treated for 16 hours in mice exposed to vehicle (Veh), IPI-504 (IP), rapamycin (R) and rapamycin/IPI-504 (RIP). All error bars show +/− SD. (See also Figure S3)
The observation that HSP90 inhibitors enhance ER stress, and that three distinct ER stress-inducing agents (IPI-504, thapsigargin, tunicamycin) induce the same therapeutic response, supports the hypothesis that IPI-504 mediates its effects by triggering excessive ER stress. To formally address this possibility we assessed whether ectopic expression of sXBP1, a downstream UPR component that can reduce ER stress (Ozcan et al., 2008), might attenuate the therapeutic effects of IPI-504. Notably, sXBP1 expression reduced and delayed cell death in response to IPI-504 (Figure 3D). Conversely, siRNAs that recognize PERK and IRE1 sensitized MPNSTs to sub-threshold doses of IPI-504 (Figure 3E). Together these data indicate that excessive ER stress plays a causal role in driving the therapeutic response.
Rapamycin sensitized MPNSTs to IPI-504 in vitro as it does in vivo (Figure 3F) and genetic ablation of raptor, a critical component of TORC1, did so as well (Figure 3F). Rapamycin also enhanced the suppressive effects of PERK and IRE1 siRNAs (Figure 3G). However, this combination was not as potent as rapamycin and IPI-504, consistent with the notion that PERK and IRE1 destabilization contribute to the therapeutic response, but do not entirely mediate the effects of HSP90 suppression, which has a more global effect on protein homeostasis in these impaired cancer cells. Thus, both genetic and chemical studies demonstrate that IPI-504 and rapamycin function through their intended targets (HSP90 and TORC1) and that ER stress is an important mediator of the therapeutic response. Interestingly, while the proteasome inhibitor bortezomib can induce proteotoxic stress in professional secretory cells (e.g. multiple myeloma cells), bortezomib did not substantially induce ER stress in MPNSTs and did not promote tumor regression when combined with rapamycin (Figure S3B), further underscoring the importance of the ER stress response in mediating the observed therapeutic effects.
The limited clinical efficacy of mTOR inhibitors has been proposed to result from AKT activation that can occur via the suppression of negative feedback pathways (Dancey et al., 2009). However, as we have previously reported, rapamycin did not induce AKT activation in MPNSTs in vivo (Figure 3H) (Johannessen et al., 2008). Moreover, combined rapamcyin/IPI-504 treatment did not suppress AKT phosphorylation or expression levels, indicating that this combination is not more effective because it inhibits AKT (Figure 3H). However, it is still possible that an mTOR kinase inhibitor or dual PI3K/mTOR inhibitor may synergize even more potently with HSP90 inhibitors, by concomitantly suppressing this well-established survival pathway.
Rapamycin and IPI-504 trigger a catastrophic destruction of the ER and mitochondria in MPNSTs in vivo
To elucidate the biological consequences of combined rapamycin/IPI-504 treatment we performed transmission electron microscopy on MPNSTs in vivo. Within 7 hours rapamycin/IPI-504 induced a massive accumulation of double-membraned vesicles (Figure 4A) (n=5). These structures exhibited cellular hallmarks of autophagosomes and contained visible cargo (Figure 4B) (Klionsky et al., 2008). The ER and mitochondria can both act as a source of membranes for autophagosomes (Hayashi-Nishino et al., 2009; Yla-Anttila et al., 2009; Hailey et al.). We detected autophagosomes emerging from both organelles in response to rapamycin/IPI-504, the significance of which is discussed below (Figure 4C). The appearance of autophagic vessicles can be caused by autophagy induction or can occur when productive autophagy is blocked (Klionsky et al., 2008). However rapamycin/IPI-504 induced the degradation of p62/SQSTM1 in these tumors, which is degraded as a consequence of productive autophagy (Figure 4D) (Klionsky et al., 2008). In addition, rapamycin/IPI-504 triggered a rapid increase in LC3-expressing punctae in MPNSTs, which fused with lysozomes shortly thereafter, indicating that autophagy was induced rather than blocked (Figure 4E, F) (N'Diaye et al., 2009; Pankiv et al., 2007). Notably, excessive ER stress actively triggers autophagy, which is engaged as a protective mechanism to degrade unfolded protein aggregates (Hotamisligil, 2010). However while IPI-504 and rapamycin can both induce signals that promote autophagy, individually each agent was unable to elicit a potent autophagic response in vivo (Figure 4A), suggesting that these agents were somehow synergizing.
Figure 4. IPI-504 and rapamycin trigger a catastrophic destruction of the ER and mitochondria in MPNSTs.
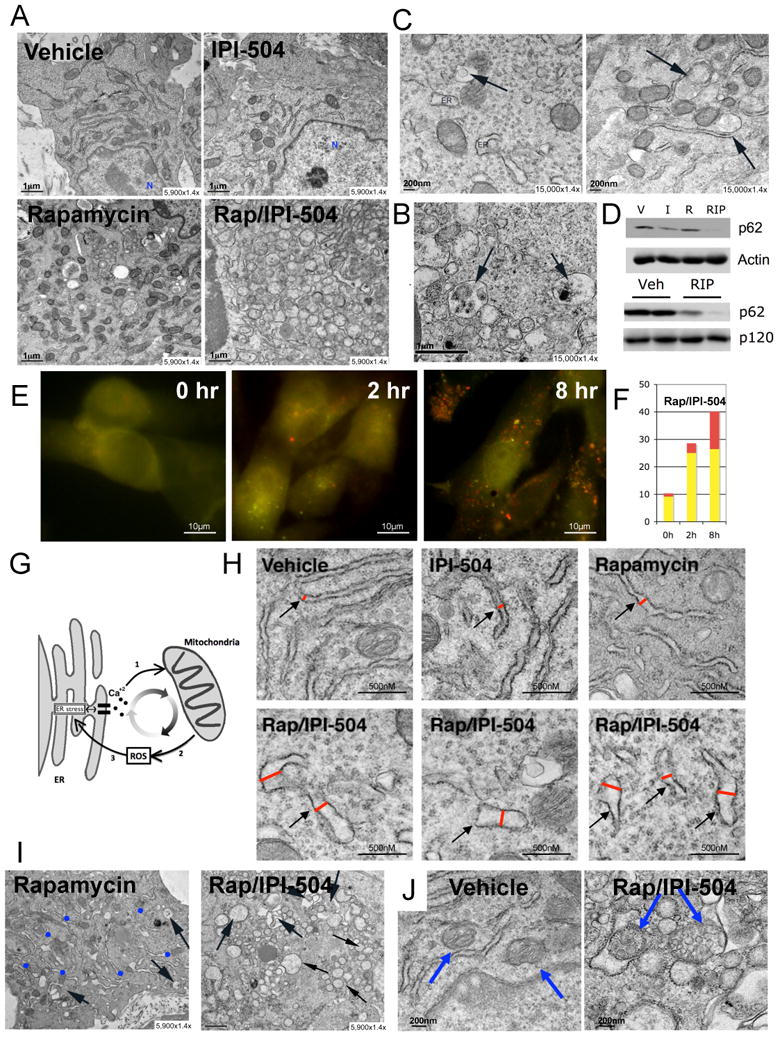
(A) Transmission Electron Microscopy (TEM) images (5,900×1.4×) of tumors after 7h of treatment with vehicle, IPI-504, rapamycin or rapamycin/IPI-504. (B) TEM images (15000×1.4×) of tumors treated for 7 hours with rapamcyin/IPI-504. Black arrows indicate double-membraned autophagosomes with cargo. (C) TEM images of a tumor treated for 7 hours with rapamycin/IPI-504. The black arrows show autophagosomes emanating from a mitochondrion (left panel) and ER (right panel). (D) (Top) Immunoblot for p62 (SQSTM1) from MPNST tissue in animals treated with vehicle (V), IPI-504 (I), rapmycin (R) or rapamycin and IPI-504 (RIP). (Bottom) A second immunoblot demonstrating a decrease in p62 levels in additional RIP treated tumors. (E) S462 cells were infected with mCherry-EGFP-LC3B and treated for 2 and 8 hours with 100nM Rapamycin and 4μM IPI-504. Yellow/green spots punctae represent autophagosomes. Red spots represent autophagolysosomes. (F) Bargraph representing the average number of autophagosomes (yellow bar) and autophagolysosomes (red bar) after 0, 2 and 8 hours of treatment of rapamycin and IPI-504. (G) Model illustrating cross-talk between ER stress, mitochondria and ROS production. (H) TEM depicting the relative size of the ER in tumors exposed to vehicle, IPI-504, rapamycin or rapamycin/IPI-504. (I) TEM showing numerous ER membranes in rapamycin treated tumors (blue dots) in contrast to tumors exposed to rapamycin/IPI-504 for 16 hours, where they are not visible. Black arrows show a few autophagosomes in rapamycin treated tumors and many in rapamycin/IPI-504 treated tumors. (J) TEM showing normal mitochondria in vehicle treated tumors (blue arrows, left) versus swollen vesicularized mitochondria in tumors treated with rapamyin/IPI-504 for 16 hours (blue arrows, right). The mitochondria on the left in this panel is being engulfed by an autophagosome. (See also Figure S4)
To investigate this synergy we examined ER and mitochondria in MPNSTs. Notably, there is a complex interdependent relationship between the ER and mitochondria in response to ER stress (Malhotra and Kaufman, 2007) (Figure 4G). ER stress triggers intra-luminal calcium release, which promotes mitochondrial membrane depolarization and ROS production (Malhotra and Kaufman, 2007;Kim et al., 2008). ROS further promotes protein misfolding, thereby enhancing ER stress. In response to low levels of ER stress, adaptive responses are engaged; however, when ER stress levels become insurmountable a vicious cycle ensues, resulting in catastrophic damage to the ER, mitochondria, and cell death (Malhotra and Kaufman, 2007). Consistent with notion that rapamycin and IPI-504 synergize to induce irresolvable ER stress, we observed severe ER swelling within 7 hours (Figure 4H). In addition, we detected a dramatic accumulation of polyubiquitinated protein aggregates, which occurs when unfolded proteins accumulate (Figure S4). Interestingly, after 16 hours ER membranes were nearly undetectable in rapamycin/IPI-504 treated tumors (Figure 4I), suggesting that ER membranes may have been depleted by excessive and continuous autophagy emanating from these membranes. Finally, as excessive ER stress ultimately triggers severe mitochondrial damage, after 16 hours mitochondria became swollen, highly vesicularized, and were engulfed by autophagosomes (mitophagy) (Figure 4J). The dramatic effects of combined rapamycin/IPI-504 treatment on autophagy, ER swelling and destruction, and mitochondrial damage were observed in all tumors examined (≥ 5/5 for each condition) and were not detected in tumors from animals exposed to single agents.
Oxidative stress plays a critical role in mediating the therapeutic response to rapamycin and IPI-504
These observations suggest that rapamycin and IPI-504 promote tumor regression by inducing irresolvable ER stress, continuous autophagy, and progressive damage to ER and mitochondria (Figure 4G). Because ROS are thought to play a critical role in fueling this vicious cycle, we assessed the requirement for ROS in the therapeutic response in vitro and in vivo. Importantly, IPI-504 triggered ROS production (Figure 5A) and the antioxidant Vitamin C suppressed MPNST cell death by 73% (Figure 5B). More strikingly, when mice were pre-treated with Vitamin C, rapamycin/IPI-504 was no longer capable of inducing tumor regression (Figure 5C). Because geldanamcyin-derivatives may induce ROS via mechanisms in addition to effects on HSP90 (Sreedhar et al., 2003), we evaluated a structurally unrelated HSP90 inhibitor. Notably, BEP800 also induced an increase in ROS production (Figure S5A). Vitamin C also suppressed the therapeutic effects of this agent by 78 % (Figure 5D), providing additional evidence that ROS is a general mediator of cell death in response to this class of drugs.
Figure 5. Oxidative stress plays a critical role in mediating the therapeutic response to IPI-504 and rapamycin.
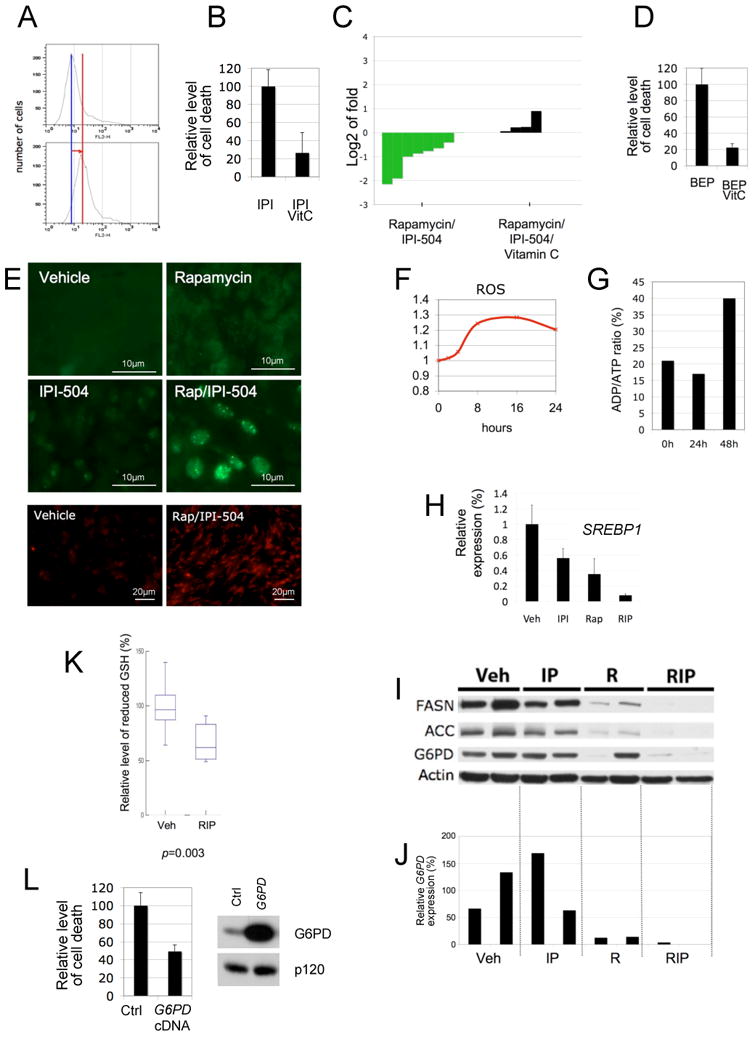
(A) Levels of ROS induced by IPI-504 in MPNST cell lines in vitro. The red line depicts the shift in fluorescence intensity, reflecting ROS production. (B) Relative levels of cell death in the presence of 500nM IPI-504 +/− 100uM Vitamin C. (C) Waterfall plot depicting tumor growth after 10 days of treatment with rapamycin/IPI-504 as shown in Figure 2 (green) versus rapamycin/IPI-504 and Vitamin C (black). The left y-axis indicates the log2 of tumor fold growth vs. day 0. (D) Relative levels of cell death in response to 500nM BEP800 +/− 100uM Vitamin C. (E) (Top 4 panels) Immunocytochemistry using a PML antibody (green) on tumors treated for 7 hours as indicated. (Bottom 2 panels) Dihydroethidium staining (red) of frozen MPNST tumor tissue depicting an increase in ROS levels in response to Rap/IPI-504 after 7 hours of treatment. (F) Kinetics of ROS induction in S462 cells in response to IPI-504/Rap. (G) Kinetics of the ratio of ADP/ATP in S462 cells in response to IPI-504/Rap. Note that ROS production precedes the increase in ADP/ATP levels (decrease in ATP). (H) SREBP1 mRNA levels in tumors from animals that were treated for 7 hours as indicated. (I) Immunoblots showing expression of FASN, ACC and G6PD in tumors from animals treated for 7h as indicated. Actin serves as a loading control. (J) G6PD mRNA levels in individual tumors treated as described in i. (K) Relative levels of reduced glutathione (GSH) in tumors treated with vehicle or rapamycin/IPI-504 (n-6). (L) Relative levels of cell death caused by IPI-504 in cells ectopically expressing G6PD or a GFP control plasmid. Immunblot demonstrating G6PD protein levels in MPNSTs used in the left panel. (See also Figure S5)
Given the demonstrated importance of ROS we investigated whether IPI-504 and rapamycin might be synergizing in these tumors by enhancing oxidative stress. PML has been proposed to be an in vivo sensor of oxidative stress, as it becomes associated with nuclear bodies in a ROS-dependent manner (Jeanne et al., 2010). Interestingly, only rapamycin/IPI-504 treatment induced the formation of PML-containing nuclear bodies in MPNSTs (Figure 5E), suggesting that rapamycin and IPI-504 together were required to achieve maximal levels of oxidative stress. These results were confirmed by measuring ROS levels in tumor tissue using dihydroethidium (DHE), where ROS was elevated within 7 hours of treatment (Figure 5E). Moreover, Vitamin C suppressed the accumulation of polyubiquitin aggregates (Figure S5B), the formation of nuclear PML bodies (Figure S5C), the robust and sustained autophagic response (Figure S5D), ER swelling and destruction and mitochondrial damage (Figure S5E). Collectively, these results demonstrate that oxidative stress is required for the therapeutic response in vivo. The finding that ER swelling, protein aggregation and ROS production all occur within 7 hours of treatment (see Figures 4H, S4, 5E, 5F) and precede ATP depletion and mitochondrial destruction (Figures 5G and 4J), demonstrates that these effects are triggered by ER stress and are not a secondary consequence of a mitochondrial metabolic collapse. This conclusion is further supported by the observation that sXBP1 suppress cell death (Figure 3D).
Rapamycin and IPI-504 promote excessive oxidative stress by inducing ROS and simultaneously suppressing the G6PD/glutathione antioxidant pathway
Oxidative stress is caused by an imbalance between ROS production and ROS clearance pathways. Given that IPI-504 stimulates ROS production, we investigated whether rapamycin might be enhancing the effects of IPI-504 by suppressing endogenous antioxidants. Because of its high concentration and central role in maintaining redox state, the reduced form of glutathione (GSH) is one of the most important endogenous cellular antioxidants (Meister and Anderson, 1983). Glutathione reduction is dependent on NADPH, which is primarily produced by the Pentose Phosphate Pathway (PPP). The first and rate-limiting enzyme of the PPP is Glucose 6-phosphate dehydrogenase (G6PD). Accordingly, G6PD plays a well-established role in protecting cells from oxidative stress via its effects on GSH production (Pandolfi et al., 1995; Xu et al; Efferth et al., 2006). The importance of G6PD in this pathway is highlighted by the observation that hypomorphic mutations in G6PD underlie favism, which causes acute hemolytic anemia in affected individuals exposed to fava beans and other oxidative stressors (Belsey, 1973). Interestingly, a direct connection between G6PD and the mTOR pathway has recently been established. Specifically, G6PD expression can be suppressed by mTOR inhibitors in vitro, through inhibitory effects on the transcription factor SREBP1 (Duvel et al., 2010). Therefore, we examined the components of this pathway (SREBP1, G6PD, GSH) in MPNSTs.
Consistent with cellular studies showing that SREPB1 is regulated by mTOR (Duvel et al., 2010; Porstmann et al., 2008), rapamycin significantly decreased the expression of known SREBP targets including SREBP1 itself, ACC and FASN, in vivo within 7 hours (Figure 5H, I). IPI-504 exerted a slightly suppressive effect on SREBP1, but together both agents reduced SREBP1 expression by 92% and effectively suppressed ACC and FASN (Figure 5H, I). Rapamycin also potently suppressed G6PD mRNA levels in MPNST tumor tissue (Figure 5J). However, rapamycin alone had inconsistent effects on G6PD protein levels (Figure 5I), perhaps reflecting a slower turnover of G6PD protein within this short time frame. Nevertheless, rapamycin and IPI-504 together dramatically suppressed both G6PD mRNA and protein expression in MPNSTs in vivo (Figure 5I, J). Accordingly, rapamycin/IPI-504 caused a 34% decrease in reduced glutathione levels in these tumors (Figure 5K, p=0.003). The magnitude of this decrease in GSH is particularly significant given that individuals affected by favism similarly exhibit a 34% mean reduction of GSH in red blood cells, which sensitizes these cells to oxidative stressors, resulting in severe protein misfolding and protein aggregate formation (Szeinberg et al., 1958). Finally, to genetically confirm that G6PD can play a functional role in protecting tumor cells from IPI-504-induced oxidative stress, we ectopically expressed G6PD in MPNSTs. Importantly, G6PD suppressed IPI-504-induced MPNST cell death by 50% (Figure 5L). Taken together these data suggest that rapamycin and IPI-504 synergize by promoting excessive oxidative stress, which is a consequence of both IPI-504-induced ROS production and rapamycin-dependent suppression of G6PD and GSH.
Rapamycin/IPI-504 promotes tumor regression in a model of Kras/p53 mutant NSCLC
To determine whether the efficacy of this combination might extend to other KRAS-mutant tumors we performed a similar study in a mouse model of NSCLC (Jackson et al., 2005). Notably, NSCLCs are also highly aneuploid, illustrating an additional similarity between these two tumor types. In this model lung adenocarcinomas are induced by intranasal administration of adenoviral Cre, which causes the concomitant expression of a single KrasG12D allele and loss of p53 (termed LSL-KrasG12D/+; p53fl/fl). 8.5-9 weeks after infection, tumor burden was assessed by MRI. Animals were re-imaged 1 week later to assess the rate of tumor growth and treatment commenced thereafter. In this mixed genetic background 50-80% of the tumors were confirmed to be adenocarcinomas within 10 weeks after Cre exposure (DuPage et al., 2009)(Figure S6). Neither rapamycin nor IPI-504 induced tumor regression alone; however, combined rapamycin/IPI-504 treatment resulted in dramatic tumor shrinkage (Figure 6A). 6/8 mice exhibited this potent response and individual responding masses shrunk up to 82% as determined by MRI analysis (Figure 6B). The overall reduction in total tumor volume, which is the sum of numerous independent tumors per mouse, is shown for each animal (Figure 6C). Histological analysis of tumors two weeks after treatment confirmed substantial tumor regression (Figure 6D). However, while tumor regression in response to rapamycin/IPI-504 was robust, three types of tumor remnants were detected. Minimal tumor remnants comprised of a few cells surrounding alveolar space were observed (Figure 6E,a,b). Slightly larger remnants surrounding alveoli were also detected (Figure 6E c,d). Finally some lesions, albeit vastly smaller than vehicle, rapamycin or IPI-504 treated tumors, were found (Figure 6E e,f). However, even in these cases significant gaps between tumor cells were observed, resulting in increased alveolar space (Figure 6E, f) in contrast to the dense, high-grade lesions observed in control animals. Moreover, there was a rapid and qualitatively obvious improvement in breathing in rapamycin/IPI-504 treated animals. The observation that not all tumors exhibit the identical therapeutic response is consistent with the fact that each individual lung tumor in this model represents an independent genetic event. Notably, while combined MEK and PI3K inhibitors have been shown to promote tumor regression in murine NSCLCs harboring the KrasG12D mutation alone (Engelman et al., 2008), no targeted therapy has been shown to promote the regression of the more aggressive KrasG12D, p53-deficient tumors, underscoring the significance of this finding and its potential impact on therapeutic development in KRAS-mutant NSCLC.
Figure 6. Rapamcyin/IPI-504 promotes regression of KrasG12D, p53-deficient NSCLC.
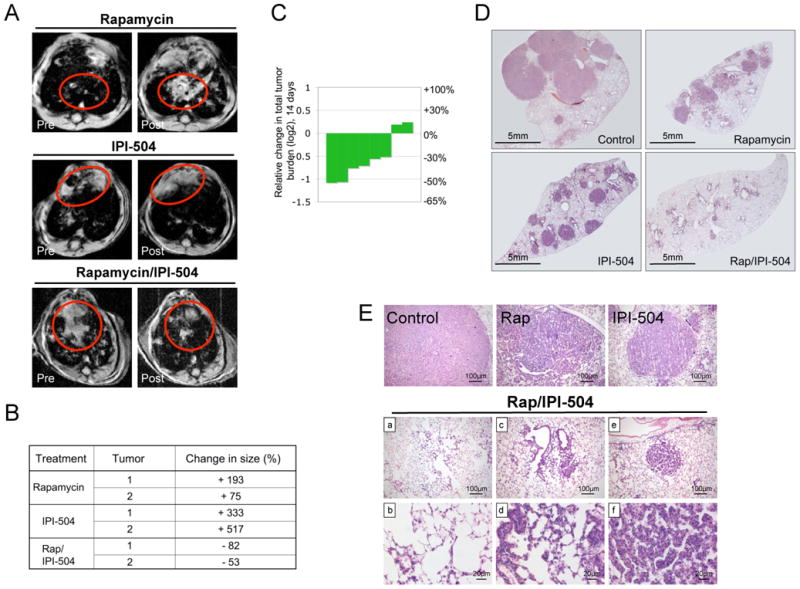
(A) MRI images of animals pre and post treatment as specified. The red circles highlight tumor masses. (B) Table listing the volumetric change of individual tumor masses as determined by MRI. (C) Waterfall plot depicting the reduction of total tumor volume in individual animals treated with rapamycin and IPI-504. (D) H&E stain of histological sections of the lung after 14 days of treatment (2×). (E) H&E stain of lesions from animals treated with vehicle, rapamycin, IPI-504 or rapamyin/IPI-504 for 2 weeks. Images b, d and f are enlargements of images a, c and e respectively. (See also Figure S6)
Discussion
Currently, there are no effective targeted therapies for Ras-driven cancers. Moreover, in some cancers KRAS mutations are used to exclude patients from being treated with specific targeted agents (Karapetis et al., 2008). As such, identifying a targeted therapy for Ras-driven tumors is an important endeavor. In this study we took an orthogonal therapeutic approach: combining an agent that targets an important downstream oncogenic pathway (mTOR) with agents that capitalize on a cancer-associated cellular vulnerability, specifically the enhanced sensitivity to proteotoxic stress (Luo et al., 2009). Importantly, we found that several agents that induce ER stress, including the HSP90 inhibitor IPI-504, cooperated with rapamycin to promote dramatic tumor regression in two distinct Ras driven-cancers. To date no targeted agents have been shown to be capable of causing tumor regression in either of these highly aggressive genetically-engineered models or, more importantly, in cognate human tumors. Given that these human cancers are generally refractory to standard therapies there is an urgent need to develop improved treatments. Thus, these studies have identified a promising therapeutic strategy for these two aggressive malignancies.
However, while we found that this combination was effective in two specific Ras-driven cancers, it will be important to determine whether its efficacy will extend to other Ras driven-tumors, other mTOR-driven tumors, and/or other tumors that exhibit high levels of ER stress. Our data suggest that a combination of these factors will be involved and that responsive tumors will require some dependence on mTOR and will also exhibit high levels of ER stress. On a molecular level, mutations in RAS, NF1 and possibly other genes that affect the mTOR pathway, may promote sensitivity to these combined agents. Although other variables, such as the extent of aneuploidy or copy number variation will likely impact the therapeutic response due to direct effects on basal ER stress levels. The recent observation that aneuploidy confers sensitivity to proteotoxic agents in normal and cancer cells in some settings, supports this hypothesis (Tang et al., 2011).
Finally, while these studies provide compelling data to support the clinical investigation of rapamycin and IPI-504, they also serve as a foundation for developing combinations using other related agents. For example, mTOR kinase or dual mTOR/PI3K inhibitors may enhance the efficacy of this combination. Similarly, there are several structurally unrelated HSP90 inhibitors in clinical development, which should provide an array of compounds that may differ in efficacy and/or toxicity. Moreover, the mechanism by which IPI-504 and rapamycin cooperate reveals an even broader range of drug options. For example, other agents that enhance proteotoxic stress and/or alter the heat shock response could be combined with agents that either suppress antioxidant pathways or further stimulate ROS production. In this respect it is noteworthy that ROS production is thought to play a functional role in mediating the cytoxic effects of many conventional chemotherapies. However, targeted agents may prove to be more effective if they are better tolerated and consequently confer a greater therapeutic index. Regardless, the potential utility of these agents may be overlooked if they are assessed exclusively as mono-therapies in genetically heterogeneous tumors, where individually they may exhibit minimal activity. Indeed, none of the single agents investigated in this study exerted a cytotoxic response when administered individually. Moreover, our studies suggest that potential drug combinations need to be empirically tested in rigorous models in vivo. For example, while the proteasome inhibitor bortezomib can induce proteotoxic stress in professional secretory cells (e.g. multiple myeloma cells), bortezomib did not substantially induce ER stress in MPNSTs and therefore did not promote tumor regression when combined with rapamycin. These observations highlight the challenge of developing effective combination therapies and underscore the utility of using robust animal to rapidly identify the most effective drug combinations among numerous possibilities.
Experimental Procedures
Cell Lines and Reagents
S462, SNF96.2 and IMR90s (ATCC). 1A50 and 237-1 are mouse Nf1/p53-deficient MPNST cell lines (Johannessen et al., 2008). Antibodies were obtained from the following sources: Cell Signaling Technologies: pAKT (4060), AKT (9272), pEIF2α (3557), pS6 (2211), total S6 (2317), BIP (3183), FASN (3180), ACC (3676) and IREα (3294), PERK (3192); Anti-p120 (G12920) (Trans. Labs); Hsp70 (Sc24) and p62 (sc-10117) (Santa Cruz Biotechnology); actin (A2066) (Sigma); XBP-1 (619502) (Biolegend); poly-Ub (FK1)(Biolegend Int); PML (05-718)(Millipore); G6PD (A300-404A)(Bethyl Labs). IPI-504 and IPI-504 vehicle were supplied by Infinity Pharmaceuticals; Thapsigargin, tunicamycin 17-AAG and ascorbic acid (Vitamin C) (Sigma); BEP800 (Selleck Chemicals); AUY922 (Chemietek); Rapamycin (LC Labs).
Real Time PCR
Tissues were crushed using an in liquid nitrogen cooled Bessman Tissue Pulverizer and dissolved in Trizol reagent (Invitrogen). RNA was treated with DNaseI (Roche) and reverse transcribed using the qScript Reverse transcriptase kit (Quanta). Real time PCR analysis was performed using the PerfeCTa SYBR Green kit (Quanta) for the following genes: mouse G6PDx (5′-cctaccatctggtggctgtt-3′ 5′-tggctttaaagaagggctca-3′); human G6PD (5′-aagaacgtgaagctccctga-3′ 5′-aatataggggatgggcttgg-3′); mouse SREBP1 (5′-gatcaaagaggagccagtgc-3′ 5′-tagatggtggctgctgagtg-3′); human SREBP1 (5′-tgcattttctgacacgcttc-3′ 5′- ccaagctgtacaggctctcc-3′).
RNAi
Non-targeting siRNAs and siRNAs against PERK were purchased from Dharmacon (D-001810-10-05 and L-004883-00 respectively); siRNA targeting IREα (Qiagen, S100605248). siRNAs were transfected using lipofectamine RNAiMAX from Invitrogen. A lentiviral pLKO vector containing the following shRNA (5′-CGACTACTACATCTCCGTGTA-3′) was used to target Raptor.
Immunofluorescence
Tissues were fixed in formalin and embedded in paraffin. A standard Immunofluorescence protocol was followed. Antigen unmasking was performed by boiling the slides in 10mM Citrate (pH 6) for 10 min followed by 30 min cooling. Blocking and hybridization were performed in 1×PBS with 5% serum and 0.3% Triton X-100. Antibodies were diluted 1:250.
TUNEL Staining, ROS detection, GSH assay and ADP/ATP ratio
TUNEL staining was performed with the ApopTag Fluorescein In Situ Apoptosis Detection Kit (Millipore). Reactive Oxygen species were evaluated by MitoSOX Red (M36008) in vitro and by dihydroethidium staining in snap frozen tumor sections (D11347) (both Invitrogen). GSH was measured using the GSH-glo Glutathione Assay Kit (Promega, V6911. The ADP/ATP ratio was determined using the ApoSENSOR ADP/ATP Ratio Assay Kit (K255-200) (Biovision).
Constructs
Human G6PD (Open Biosystems) and mouse sXBP-1 were cloned into a pLenti CMV/TO Puro vector. The pBabe-puro mCherry-EGFP-LC3B construct was obtained from Addgene. Lentiviral and retroviral infections were performed as previously described (Johannessen et al., 2005).
Drug treatment and dosing schedule
Animal procedures were approved by the Center for Animal and Comparative Medicine in Harvard Medical School in accordance with the NIH Guild for the Care and Use of Laboratory Animals and the Animal Welfare Act. IPI-504 (100mg/kg) was administered once or twice weekly. Thapsigargin (0.2mg/kg) and tunicamycin (0.2mg/kg) were administered twice a week. Rapamycin was administered daily at 5mg/kg (Johannessen et al., 2008). Compounds given in combination were administered sequentially. Mice were treated daily with 40mg/kg Vitamin C by oral gavage prior to IP injections of Rapamycin and IPI-504.
Tumor volume measurements
MPNST model
Mice were enrolled in the study when tumor size reached 300- 700mm3. Tumor size was measured every 2-3 days by Vernier calipers. Tumor volume was calculated using the standard formula L × W2 × 52. Tumor volume and log2 of fold growth versus day 0 was calculated and graphed.
Lung cancer model
Mice were infected with Adenoviral Cre (University of Iowa) by nasal instillation (Jackson et al., 2005). Tumor burden was determined by MRI eight weeks after inhalation and again one week later (Engelman et al., 2008). Nine weeks after inhalation tumor bearing mice were divided into cohorts and were treated with single or double agents for 2 weeks. Lung tissues were harvested for histopathology after the last MRI. Fixed lung tissues were stained by H&E, tumor burden was subsequently analyzed using the ImageJ software (NIH).
EM
Tumor tissues were fixed for EM after 7 and 16h of a single dose and further processed for standard EM techniques (Barth et al.).
Statistics
All statistical analysis was performed using SYSTAT 12 software. For each data set basic statistical values (mean and standard deviation) were calculated and normality determined (Shapiro-Wilk normality test); all data sets were normally distributed. Thapsigargin and IPI-504 were compared to vehicle treated tumors; rapamycin/thapsigargin and rapamycin/IPI-504 were compared to Rapamycin only treated tumors all by Student t-test. Survival analysis was analyzed using the Mandel method. For the comparison of lung tumor burden in the lung cancer model we performed an ANOVA test, followed by Student t-test.
Supplementary Material
Highlights.
We describe a promising combination therapy for two aggressive Ras-driven cancers
mTOR and HSP90 inhibitors cooperate to exert potent activity in mouse models of MPNST and NSCLC
These agents function by promoting irresolvable ER and oxidative stress
Combinatorial therapy can capitalize on cellular vulnerabilities of cancer cells
Significance.
Ras-driven tumors are often refractory to conventional therapies and a clinically effective targeted therapy has not yet been developed. Moreover, in some cancers KRAS mutations are used to exclude patients from being treated with specific targeted agents. As such, developing an effective targeted therapy for Ras-driven tumors is an important endeavor. We have identified a promising therapy for two distinct Ras-driven cancers: Nf1-deficient nervous system malignancies and KRas/p53-mutant non-small cell lung cancer (NSCLC). Importantly, these studies have defined a specific drug combination that can now be assessed in patients with these largely untreatable cancers. Moreover, these studies establish a therapeutic paradigm that can be expanded to develop additional drug combinations.
Acknowledgments
We thank Wade Harper, Steve Elledge, Nathanel Gray, Heather Harding and Hugues de Thé for helpful discussions. This work was supported by the NCI KC: (CA129814) and the Ludwig Center at DF/HCC; KWW: (CA122794, CA140594). TD is a recipient of the Young Investigator Award of the Children's Tumor Foundation. OM is a Postdoctoral Fellow of the Research Foundation Flanders.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth S, Glick D, Macleod KF. Autophagy: assays artifacts. J Pathol. 221:117–124. doi: 10.1002/path.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsey MA. The epidemiology of favism. Bull World Health Organ. 1973;48:1–13. [PMC free article] [PubMed] [Google Scholar]
- Cawthon RM, Weiss R, Xu GF, Viskochil D, Culver M, Stevens J, Robertson M, Dunn D, Gesteland R, O'Connell P, et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell. 1990;62:193–201. doi: 10.1016/0092-8674(90)90253-b. [DOI] [PubMed] [Google Scholar]
- Cichowski K, Shih TS, Schmitt E, Santiago S, Reilly K, McLaughlin ME, Bronson RT, Jacks T. Mouse models of tumor development in neurofibromatosis type 1. Science. 1999;286:2172–2176. doi: 10.1126/science.286.5447.2172. [DOI] [PubMed] [Google Scholar]
- Dancey JE, Curiel R, Purvis J. Evaluating temsirolimus activity in multiple tumors: a review of clinical trials. Semin Oncol. 2009;36 3:S46–58. doi: 10.1053/j.seminoncol.2009.10.010. [DOI] [PubMed] [Google Scholar]
- Dasgupta B, Yi Y, Chen DY, Weber JD, Gutmann DH. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res. 2005;65:2755–2760. doi: 10.1158/0008-5472.CAN-04-4058. [DOI] [PubMed] [Google Scholar]
- DeClue JE, Papageorge AG, Fletcher JA, Diehl SR, Ratner N, Vass WC, Lowy DR. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell. 1992;69:265–273. doi: 10.1016/0092-8674(92)90407-4. [DOI] [PubMed] [Google Scholar]
- Denoyelle C, Abou-Rjaily G, Bezrookove V, Verhaegen M, Johnson TM, Fullen DR, Pointer JN, Gruber SB, Su LD, Nikiforov MA, et al. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat Cell Biol. 2006;8:1053–1063. doi: 10.1038/ncb1471. [DOI] [PubMed] [Google Scholar]
- Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas M, Lim AR, Porter JR, West K, Pink MM, Ge J, Wylie AA, Tibbits TT, Biggs K, Curtis M, et al. The antiproliferative activity of the heat shock protein 90 inhibitor IPI-504 is not dependent on NAD(P)H:quinone oxidoreductase 1 activity in vivo. Mol Cancer Ther. 2009;8:3369–3378. doi: 10.1158/1535-7163.MCT-09-0568. [DOI] [PubMed] [Google Scholar]
- DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc. 2009;4:1064–1072. doi: 10.1038/nprot.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efferth T, Schwarzl SM, Smith J, Osieka R. Role of glucose-6-phosphate dehydrogenase for oxidative stress and apoptosis. Cell Death Differ. 2006;13:527–528. doi: 10.1038/sj.cdd.4401807. author reply 529-530. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 141:656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11:1433–1437. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- Healy SJ, Gorman AM, Mousavi-Shafaei P, Gupta S, Samali A. Targeting the endoplasmic reticulum-stress response as an anticancer strategy. Eur J Pharmacol. 2009;625:234–246. doi: 10.1016/j.ejphar.2009.06.064. [DOI] [PubMed] [Google Scholar]
- Holzel M, Huang S, Koster J, Ora I, Lakeman A, Caron H, Nijkamp W, Xie J, Callens T, Asgharzadeh S, et al. NF1 is a tumor suppressor in neuroblastoma that determines retinoic acid response and disease outcome. Cell. 142:218–229. doi: 10.1016/j.cell.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, Jacks T. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005;65:10280–10288. doi: 10.1158/0008-5472.CAN-05-2193. [DOI] [PubMed] [Google Scholar]
- Jeanne M, Lallemand-Breitenbach V, Ferhi O, Koken M, Le Bras M, Duffort S, Peres L, Berthier C, Soilihi H, Raught B, de The H. PML/RARA oxidation and arsenic binding initiate the antileukemia response of As2O3. Cancer Cell. 2010;18:88–98. doi: 10.1016/j.ccr.2010.06.003. [DOI] [PubMed] [Google Scholar]
- Johannessen CM, Johnson BW, Williams SM, Chan AW, Reczek EE, Lynch RC, Rioth MJ, McClatchey A, Ryeom S, Cichowski K. TORC1 is essential for NF1-associated malignancies. Curr Biol. 2008;18:56–62. doi: 10.1016/j.cub.2007.11.066. [DOI] [PubMed] [Google Scholar]
- Johannessen CM, Reczek EE, James MF, Brems H, Legius E, Cichowski K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci U S A. 2005;102:8573–8578. doi: 10.1073/pnas.0503224102. Epub 2005 Jun 8573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- Marcu MG, Doyle M, Bertolotti A, Ron D, Hendershot L, Neckers L. Heat shock protein 90 modulates the unfolded protein response by stabilizing IRE1alpha. Mol Cell Biol. 2002;22:8506–8513. doi: 10.1128/MCB.22.24.8506-8513.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GA, Viskochil D, Bollag G, McCabe PC, Crosier WJ, Haubruck H, Conroy L, Clark R, O'Connell P, Cawthon RM, et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell. 1990;63:843–849. doi: 10.1016/0092-8674(90)90150-d. [DOI] [PubMed] [Google Scholar]
- Massey AJ, Schoepfer J, Brough PA, Brueggen J, Chene P, Drysdale MJ, Pfaar U, Radimerski T, Ruetz S, Schweitzer A, et al. Preclinical antitumor activity of the orally available heat shock protein 90 inhibitor NVP-BEP800. Mol Cancer Ther. 2010;9:906–919. doi: 10.1158/1535-7163.MCT-10-0055. [DOI] [PubMed] [Google Scholar]
- McGillicuddy LT, Fromm JA, Hollstein PE, Kubek S, Beroukhim R, De Raedt T, Johnson BW, Williams SM, Nghiemphu P, Liau LM, et al. Proteasomal and genetic inactivation of the NF1 tumor suppressor in gliomagenesis. Cancer Cell. 2009;16:44–54. doi: 10.1016/j.ccr.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister A, Anderson ME. Glutathione. Annu Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- N'Diaye EN, Kajihara KK, Hsieh I, Morisaki H, Debnath J, Brown EJ. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep. 2009;10:173–179. doi: 10.1038/embor.2008.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan U, Ozcan L, Yilmaz E, Duvel K, Sahin M, Manning BD, Hotamisligil GS. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol Cell. 2008;29:541–551. doi: 10.1016/j.molcel.2007.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandolfi PP, Sonati F, Rivi R, Mason P, Grosveld F, Luzzatto L. Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defense against oxidative stress. Embo J. 1995;14:5209–5215. doi: 10.1002/j.1460-2075.1995.tb00205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung YL, Schulze A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8:224–236. doi: 10.1016/j.cmet.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan RK, Egorin MJ, Eiseman JL, Ramalingam S, Friedland D, Agarwala SS, Ivy SP, Potter DM, Chatta G, Zuhowski EG, et al. Phase I and pharmacodynamic study of 17-(allylamino)-17-demethoxygeldanamycin in adult patients with refractory advanced cancers. Clin Cancer Res. 2007;13:1769–1774. doi: 10.1158/1078-0432.CCR-06-2233. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Sreedhar AS, Mihaly K, Pato B, Schnaider T, Stetak A, Kis-Petik K, Fidy J, Simonics T, Maraz A, Csermely P. Hsp90 inhibition accelerates cell lysis. Anti-Hsp90 ribozyme reveals a complex mechanism of Hsp90 inhibitors involving both superoxide- and Hsp90-dependent events. J Biol Chem. 2003;278:35231–35240. doi: 10.1074/jbc.M301371200. [DOI] [PubMed] [Google Scholar]
- Sydor JR, Normant E, Pien CS, Porter JR, Ge J, Grenier L, Pak RH, Ali JA, Dembski MS, Hudak J, et al. Development of 17-allylamino-17-demethoxygeldanamycin hydroquinone hydrochloride (IPI-504), an anti-cancer agent directed against Hsp90. Proc Natl Acad Sci U S A. 2006;103:17408–17413. doi: 10.1073/pnas.0608372103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeinberg A, Asher Y, Sheba C. Studies on glutathione stability in erythrocytes of cases with past history of favism or sulfa-drug-induced hemolysis. Blood. 1958;13:348–358. [PubMed] [Google Scholar]
- Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11:515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- Tang YC, Williams BR, Siegel JJ, Amon A. Identification of aneuploidy-selective antiproliferation compounds. Cell. 2011;144:499–512. doi: 10.1016/j.cell.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- Xu Y, Zhang Z, Hu J, Stillman IE, Leopold JA, Handy DE, Loscalzo J, Stanton RC. Glucose-6-phosphate dehydrogenase-deficient mice have increased renal oxidative stress and increased albuminuria. FASEB J. 2009;24:609–616. doi: 10.1096/fj.09-135731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yla-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy. 2009;5:1180–1185. doi: 10.4161/auto.5.8.10274. [DOI] [PubMed] [Google Scholar]
- Young A, Lyons J, Miller AL, Phan VT, Alarcon IR, McCormick F. Ras signaling and therapies. Adv Cancer Res. 2009;102:1–17. doi: 10.1016/S0065-230X(09)02001-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.