

Abstract
The pan Bcl-2 family antagonist Obatoclax (GX15-070), currently in clinical trials, was shown to sensitize TRAIL-resistant tumors to TRAIL-mediated apoptosis via the release of Bak and Bim from Mcl-1 or Bcl-2/Bcl-XL complexes or by the activation of Bax, though other mechanisms were not examined. Herein, we hypothesize that Obatoclax-mediated sensitization to TRAIL apoptosis may also result from alterations of the apoptotic pathways. The TRAIL-resistant B-cell line Ramos was used as a model for investigation. Treatment of Ramos cells with obatoclax significantly inhibited the expression of several members of the Bcl-2 family, dissociated Bak from Mcl-1 and inhibited the NFκB activity. Cells treated with Mcl-1 siRNA were sensitized to TRAIL apoptosis. We examined whether the sensitization of Ramos to TRAIL by Obatoclax resulted from signaling of the DR4 and/or DR5. Transfection with DR5 siRNA, but not with DR4 siRNA, sensitized the cells to apoptosis following treatment with Obatoclax and TRAIL. The signaling via DR5 correlated with Obatoclax-induced inhibition of the DR5 repressor Yin Yang 1 (YY1). Transfection with YY1 siRNA sensitized the cells to TRAIL apoptosis following treatment with Obatoclax and TRAIL. Overall, the present findings reveal a new mechanism of Obatoclax-induced sensitization to TRAIL apoptosis and the involvement of the inhibition of NFκB activity and downstream Mcl-1 and YY1 expressions and activities.
Key words: Obatoclax, TRAIL, YY1, DR5, lymphoma, immunosensitization
Introduction
Current cancer treatment modalities with clinical response consist of chemotherapy, hormonal therapy, radiation and/or immunotherapy (antibodies, vaccines, gene therapy). However, several of these therapies are toxic and not curative. Many patients relapse, and tumor resistance gradually develops.1 Therefore, there is an urgent need to develop novel, non-toxic therapies to overcome resistance. One of the currently clinically examined treatments of malignancies is the use of TRAIL/agonist and/or anti-DR4 and DR5 monoclonal antibodies.2,3 Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a type II transmembrane protein expressed in cytotoxic cells (e.g., CTL, NK, macrophages) and a few selected tissues.4 TRAIL binds to death receptors DR4 and DR5 as well as to decoy receptors DcR1 and DcR25 or osteoprotegerin (OPG).6 TRAIL is selectively cytotoxic to sensitive transformed cells, with minimal toxicity to normal tissues and stem cells.7,8 TRAIL and agonist anti-DR4/DR5 monoclonal antibodies are now in phase I or II clinical trials against a variety of tumors.9,10 It should be noted, however, that many hematological malignancies are resistant to TRAIL,11 limiting its therapeutic effectiveness as monotherapy. Since most tumors are not sensitive to TRAIL-induced apoptosis, there is the need to be sensitized by other agents to reverse resistance.7 The mechanisms by which malignant cells develop resistance to TRAIL have been poorly explored.12,13 Notably, a common mechanism in TRAIL resistance is overexpression of Bcl-2 and its anti-apoptotic family members, which function by blocking the mitochondrial pathway.14 Members of the Bcl-2 family overexpression protect chronic lymphocytic leukemia, prostate, breast, colon cancers, melanoma and lymphoma from TRAIL-induced apoptosis.11,13,15 Therefore, strategies to modulate the Bcl-2 pathway may represent an important therapeutic target that may increase the efficacy of TRAIL for the treatment of human cancers.
Obatoclax is a small-molecule Bcl-2 antagonist that binds the hydrophobic groove in Bcl-2, Bcl-XL and Mcl-116 and mimics pro-apoptotic BH3-only proteins, such as Bad, Bim, Noxa and Puma, from sequestering the multidomain pro-apoptotic proteins (Bak and Bax). The Bcl-2 anti-apoptotic family members exert their activity by different mechanisms. For instance, Bcl-2 controls the permeability of the mitochondrial outer membrane and permits the release of factors, such as cytochrome C and Smac/DIABLO.16 These activate caspase 9 and other caspases of the core apoptotic pathway. It has been proposed to antagonize the pro-survivor members by BH3 mimetics and its potential therapeutic intervention.14
Nguyen et al. reported the development of a small molecular inhibitor Obatoclax (GX15-070) of pro-survival Bcl-2 members including Mcl-1, and demonstrated that it overcomes the resistance conferred by Bcl-XL.17 These findings provide the rationale of developing Obatoclax for therapeutic use in combination with other targeted cancer treatments.
Recent studies reported the sensitization of several solid tumors by Obatoclax to TRAIL-mediated apoptosis. Various mechanisms were reported using different tumors, such as the release of Bim from the anti-apoptotic proteins Mcl-1 and Bcl-2 or direct activation of Bax with no alteration of gene products related to the Bcl-2 family members or the TRAIL receptors DR4 and/or DR5.18–20 However, the effect of Obatoclax on the regulation of pro-survival pathways that might be involved in the sensitization to TRAIL has not been considered. We hypothesized that the Obatoclax-mediated effect as a BH3 mimetic may also antagonize Bcl-2 anti-apoptotic members through inhibition of their expression by interfering upstream with the activation of anti-survival or deactivation of pro-survival pathways. The TRAIL-resistant B-Non Hodgkin Lymphoma (B-NHL) cell line, Ramos, was used as a model for investigation. The above hypothesis was examined as follows: (1) Does Obatoclax sensitize Ramos cells to TRAIL apoptosis, and does it activate the type II mitochondrial apoptotic pathway? (2) Does Obatoclax inhibit the constitutively activated NFκB pathway and downstream anti-apoptotic gene products? (3) Does Obatoclaxmediated sensitization to TRAIL result via signaling of the DR4 and/or DR5 pathway? and (4) Does the DR5 transcription repressor YY1 play a role in Obatoclax-induced sensitization to TRAIL? The findings show that Obatoclax inhibits NFκB activity and demonstrate the involvement of Mcl-1 and YY1 inhibition by Obatoclax and upregulation of DR5 in the sensitization of TRAIL apoptosis.
Results
Treatment of Ramos cells with Obatoclax inhibits the expression of anti-apoptotic gene products.
Obatoclax has been reported to antagonize and inhibit the activity of anti-apoptotic members of the Bcl-2 family including Mcl-1 in solid tumors.21–24 To examine the effects of Obatoclax in lymphoid tumors, Ramos cells were treated with two optimal concentrations of Obatoclax (14 and 28 nM) for 24 h, and cell lysates were prepared for analysis of various gene products. The findings in Figure 1A demonstrate that following treatment of Ramos cells with Obatoclax (14 and 28 nM) resulted in significant inhibition of Mcl-1, Bcl-XL, XIAP and cIAP 1/2 protein expression, and that the effect was more pronounced with 28 nM of Obatoclax. Similar results were observed in the Daudi cell line (data not shown). In addition, Ramos cells treated with Obatoclax for 6, 12 and 48 h were also analyzed for expression for Mcl-1, Bcl-XL and XIAP, and no changes were observed at 6 and 12 h. The inhibition observed at 24 h was similar to 48 h (data not shown). Analyses of various pro-apoptotic gene products revealed that there were inhibitions of Bad, Bid and Bax and no significant induction of Bim (Fig. 1B). Reports by Nguyen et al. and Mott et al. demonstrated that Obatoclax inhibits the association between Mcl-1 and Bak in intact cells.17,19 These findings were confirmed here in Ramos cells treated with Obatoclax for 24 h, and the lysates were first immunoprecipitated with Mcl-1 antibody and developed for western blotting with both anti-Bak and anti-Mcl-1 antibodies. The findings shown in Figure 1C demonstrate that there were significantly higher levels of Bak and Mcl-1 in the untreated cells when compared with cells treated with Obatoclax, suggesting that there were losses of association of Mcl-1 with Bak, and these findings demonstrate that Obatoclax dissociates the Bak/Mcl-1 complexes.
Figure 1.

Obatoclax modifies anti-apoptotic gene products expression. (A and B) Ramos cells were treated with the indicated concentrations of Obatoclax for 24 h. Mcl-1, Bcl-XL, XIAp, c-IAP 1/2, Bad, Bim, Bid, Bax and Bak protein expressions were analyzed by western blot. The β-actin protein expression was used as a loading control. Densitometric analysis is shown in the right. (C) Ramos cells were treated or not with Obatoclax (28 nM) for 24 h. Immunoprecipitation was done with anti-Mcl-1 antibody, and western blotting was developed with an anti-Bak antibody. Whole-cell lysates were used as control.
Overall, the above findings demonstrated that Obatoclax treatment of Ramos cells resulted in significant inhibition of the expression of several gene products of the Bcl-2 family. These findings suggested that Obatoclax, originally developed as a BH3-mimetic inhibitor of the anti-apoptotic gene products of the Bcl-2 family, exerts other activities by regulating gene expression as well. The gene products modified by Obatoclax may have resulted, in part, from Obatoclax-induced regulation upstream of survival pathways that regulate such gene products, such as the NFκB survival pathway.25–28
Obatoclax inhibits the constitutive NFκB pathway.
The findings above shown in Figure 1 suggested that inhibition of the anti-apoptotic gene products by Obatoclax may have resulted from its upstream inhibition of the constitutively activated NFκB pathway, which regulates, in part, the transcription of these gene products.25–28 This hypothesis was tested by examining the effect of Obatoclax treatment of Ramos cells on the activity of NFκB. Ramos cells were treated with two concentrations of Obatoclax (14 and 28 nM), and the lysates were prepared for analyses for gene products by western blortting and densitometry. The data in Figure 2A demonstrate that Obatoclax inhibited the phosphorylation of p65 (Ser 536) and IKK expression. The inhibition of NFκB activity by Obatoclax was corroborated using the NFκB luciferase reporter assay. Treatment with various Obatoclax concentrations resulted in significant inhibition of luciferase activity in a concentration-dependent fashion. The NFκB inhibitor DHMEQ was used as a positive control (Fig. 2B). In addition to the above, Obatoclax inhibited NFκB DNA-binding activity as determined by EMSA (Fig. 2C). The inhibition by Obatoclax downstream of anti-apoptotic gene products regulated by NFκB was also observed following treatment with DHMEQ.29–32 The data in Figure 2D show that DHMEQ sensitized Ramos cells to TRAIL apoptosis in a concentration-dependent manner. Similar results were observed in Daudi and DHL-4 cell lines (data not shown).
Figure 2.

Obatoclax-mediated inhibitions of NFκB expression and DNA-binding activity. (A) Ramos cells were treated with Obatoclax (14 or 28 nM) for 24 h. Whole cell extracts were analyzed by western blot using specific antibodies. Densitometric analysis is shown (right). The β-actin protein expression was used as a loading control. (B) PC3 cells were transfected with the NFκB-Luc reporter plasmid and, 24 h after, the cells were treated with DMSO or Obatoclax. twenty four hours after, the luciferase activity was evaluated as described in The Materials and Methods. Cells treated with 10 µg/ml DHMEQ, a chemical inhibitor of NFκB, served as a positive control for inhibition of NFκB promoter activity. (C) Ramos cells were treated for 24 h with Obatoclax, and nuclear extracts were tested by EMSA for NFκB DNA-binding activity. (D) Ramos cells were left untreated or treated with DHMEQ (1, 2.5, 5 and 10 µg/ml) and apoptosis was analyzed. The data represent the mean ± SD of the three independent experiments performed in duplicate *p < 0.05.
Altogether, the above findings suggested that Obatoclax inhibits the activity of the NFκB pathway and its inhibition is responsible, in part, for sensitization of Ramos cells to TRAIL-induced apoptosis.
Sensitization of TRAIL-resistant Ramos cells to TRAIL-induced apoptosis by Obatoclax and underlying molecular mechanism of sensitization.
(A) Sensitization to TRAIL apoptosis. NFκB inhibitors such as DHMEQ have been shown to sensitize TRAIL-resistant tumor cells to apoptosis by TRAIL as confirmed here (Fig. 2D). These findings imply that Obatoclax-mediated inhibition of NFκB is involved in the sensitization to TRAIL.
We examined the sensitizing effect of Obatoclax treatment on the TRAIL-resistant Ramos cells to TRAIL apoptosis. The cells were treated with various concentrations of TRAIL (2.5–10 ng/ml) and Obatoclax (7, 14, 21 and 28 nM). The cells were pre-treated with Obatoclax for 24 h and treated with TRAIL for an additional 18 h, and the cells were tested for viability (Fig. 3A) and for apoptosis (Fig. 3B). In addition, the same experiments were performed at 24 or 48 h (Fig. 3C). The findings demonstrated that significant apoptosis was shown with the combination-treatment of Obatoclax and TRAIL, and it was dependent on the concentration used. The significant potentiation of apoptosis was synergistic as determined by an isobologram analysis (Fig. 3B, right part). Similar results were observed with Daudi and Raji NHL cell lines (data not shown). The effect of Obatoclax, TRAIL and combination on toxicity of normal peripheral blood cells obtained from four normal individuals did not show any significant loss in cell viability by any of the treatments above (Fig. 3D).
Figure 3.

Obatoclax-induced sensitization of lymphoma cell lines to TRAIL-mediated apoptosis. (A) Ramos cells were treated with Obatoclax at the indicated concentrations in the presence or not of various concentrations of TRAIL (2.5, 5 or 10 ng/ml) for 18 h. Cell viability was examined microscopically by trypan blue dye exclusion and by the Xtt assay. Mean percentage relative to control untreated cells ± SD, (n = 3). (B) Left, Ramos cell were pretreated for 24 h with increasing concentrations of Obatoclax followed by treatment with TRAIL (2.5, 5, 10 or 20 ng/ml) for 18 h. Apoptosis was determined by activation of pro-caspase 3 as described in Materials and Methods. The data represent the mean ± SD from three independent experiments *p < 0.05, **p < 0.01, ***p < 0.001. Right, synergy between Obatoclax and TRAIL was achieved, as indicated by the isobologram analysis. F.I.C., fractional inhibitory concentration. (C) Ramos cells were treated as above for 24 and 48 h, and apoptosis was determined as indicated in (B). (D) PBLC from four healthy donors were treated with several concentrations of Obatoclax (14, 28 and 35 nM) for 24 h in combination or not with different concentrations of TRAIL (2.5, 5, 10 and 20 ng/ml), and percent of viability was determined. D1 through D4 represent normal donors. (E) Ramos cells were pre-treated or not with Obatoclax (28 nM) for 24 h and then with TRAIL (10 ng/ml) for an additional 18 h. The depolarization of the mitochondrial membrane was assessed by staining with DiOC6 and then analyzed by flow cytometry. Left, percentage of depolarization is shown in bar form. The data represent the values expressed as % of depolarization of the MM relative to the control and as 100% and represent the mean ± SEM of at least three independent experiments *p < 0.05: treated vs. untreated cells. Right, representative histogram of decreased polarization of MM. The upper part shown the MFI of each of the conditions used and the values of the % of decreased polarization of MM relative to the control. (F) Ramos cells were pretreated with DMSO or Obatoclax and then treated or not with TRAIL (10 ng/ml). protein lysates were screened by western blot for caspase 3, 8 and 9.
Altogether, the above findings demonstrate that Obatoclax sensitized TRAIL-resistant tumor cells to TRAIL-induced apoptosis in a synergistic manner.
(B) Obatoclax sensitizes Ramos cells to TRAIL-induced apoptosis by activation of the type II mitochondrial pathway. We examined the effect of treatment of Ramos cells with Obatoclax, TRAIL or combination on the mitochondrial membrane potential. The data in Figure 3E demonstrate that Obatoclax treatment had a modest effect on the depolarization of the mitochondrial membrane potential, and TRAIL treatment alone had no effect. However, treatment with the combination of Obatoclax and TRAIL resulted in significant depolarization of the mitochondrial membrane potential (46%) (see Table above Fig. 3E), (Fig. 3E, left part). A representative histogram is shown in Figure 3E (right part). The above data suggested that the combination treatment might have acted through the activation of mitochondrial type II apoptotic pathway, leading to the activation of caspases 9 and3 (Fig. 3E) and resulting in apoptosis. Analyses of lysates from cells treated with a single agent or the combination show clearly that TRAIL treatment had no effect on the activation of caspases 9 and 3. Treatment with Obatoclax showed activation of caspase 3 and no effect on caspase 9 and 8. The combination treatment, however, resulted in the activation of caspases 3 and9 (Fig. 3E). Caspase 8 was activated by the low concentration of Obatoclax (14 nM) but not with the higher concentration (28 nM). Recent reports in reference 33 suggested that high concentrations of Obatoclax can induce caspase-independent activation of apoptosis.
Overall, the above findings demonstrate that Obatoclaxinduced sensitization to TRAIL results in the activation of the mitochondrial type II apoptotic pathway. These results are concordant with the inhibition of anti-apoptotic gene products shown above in Figure 1, which regulate the mitochondrial membrane potential and activate caspases 9 and 3.34
(C) Role of Obatoclax-induced inhibition of Mcl-1 in the sensitization to TRAIL apoptosis. Previous reports in references 35 and 36 and our present findings have demonstrated that Obatoclax antagonizes Mcl-1 association with Bak, and it has also been shown here to inhibit Mcl-1 expression (Fig. 1A). The possible role of Mcl-1 inhibition by Obatoclax in the sensitization to TRAIL was examined. Ramos cells were transfected with Mcl-1 siRNA and control siRNA for 48 h and then treated with TRAIL (2.5–20 ng/ml) for an additional 24 h. Thereafter, the cells were tested for apoptosis by the activation of caspase 3. The findings in Figure 4A demonstrate that treatment with Mcl-1 siRNA significantly inhibited Mcl-1 expression compared with cells treated with control siRNA. The cells transfected with Mcl-1 siRNA but not with control siRNA were sensitized to TRAIL-mediated apoptosis. The extent of apoptosis was a function of the TRAIL concentration used (Fig. 4B). These findings suggest the possible involvement of the Mcl-1 inhibition in Obatoclax-mediated sensitization of Ramos cells TRAIL apoptosis.
Figure 4.
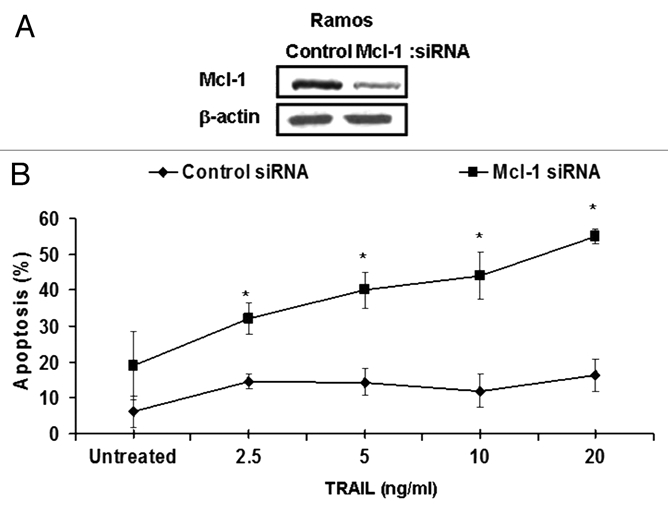
Mcl-1 plays a roll in the Obatoclax-induced sensitization of Ramos cells to TRAIL-mediated apoptosis. Ramos cells were grown in media without antibiotic for 24 h and then transfected with Mcl-1 siRNA or control siRNA. After 24 h, the cells were treated with the indicated TRAIL concentrations, and 18 h after, the cells were analyzed for apoptosis by activation of caspase 3 by flow (B) or analyzed by western blot (A). The data represent the mean ± SD from three independent experiments *p < 0.01.
Direct role of Obatoclax-induced inhibition of YY1 and induction of DR5 in the sensitization to TRAIL apoptosis.
We have previously reported that the inhibition of NFκB activity and downstream DR5 transcriptional repressor YY1 are responsible, in large part, for tumor cell sensitization to TRAIL apoptosis.37–40 We have shown here that Obatoclax inhibits NFκB activity and results in tumor cell sensitization to TRAIL-mediated apoptosis (Fig. 2). Hence, we examined whether the sensitization by Obatoclax resulted from the inhibition of YY1 and upregulation of DR5.
(A) Obatoclax inhibits YY1 expression and activity. Ramos cells were treated with Obatoclax (14 and 28 nM) for 24 h, and the lysates were examined for YY1 expression by western blot. The findings demonstrate that Obatoclax treatment significantly inhibited the YY1 expression by both Obatoclax concentrations used (Fig. 5A, top part). These findings are also shown by densitometry analysis (Fig. 5A, bottom part). The inhibition of YY1 activity by Obatoclax was examined both by EMSA and by an YY1 luciferase reporter assay. In EMSA, treatment with Obatoclax significantly inhibited YY1 DNA-binding activity (Fig. 5B). PC3 cells transfected with an YY1 luciferase reporter and treated with Obatoclax resulted in significant inhibition of the luciferase reporter activity. The NFκB inhibitor DHMEQ was used as an internal control (Fig. 5C).
Figure 5.
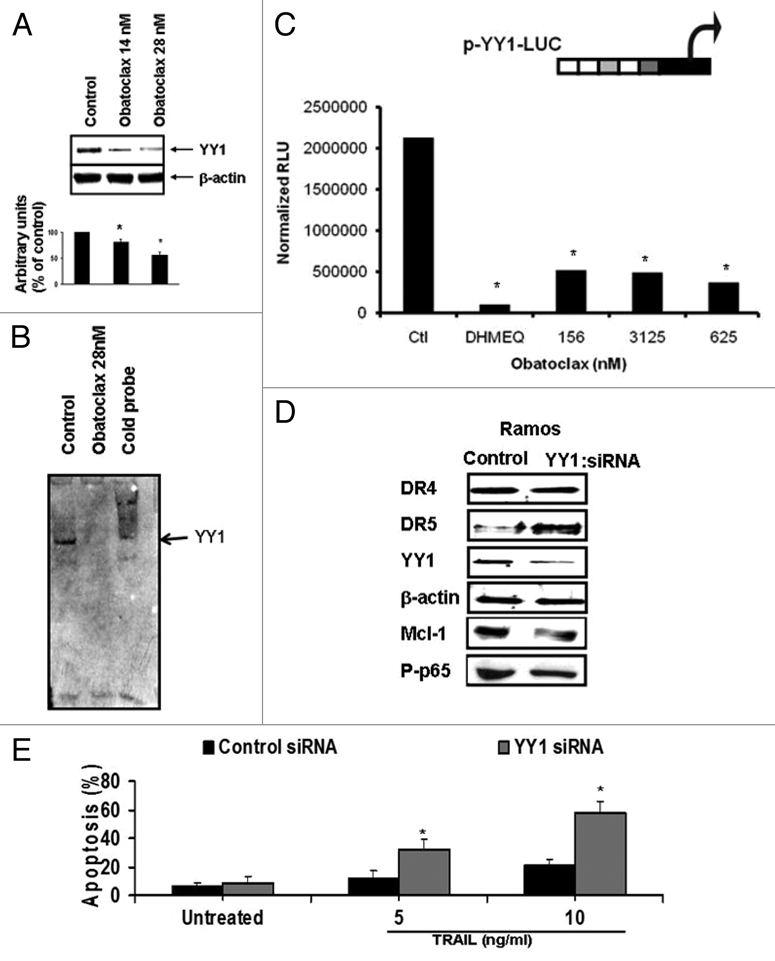
Downregulation of YY1 by Obatoclax can be in part responsible to TRAIL sensitization through DR5 upregulation. (A) Ramos cells were treated with Obatoclax (14 and 28 nM) and cell lysates were analyzed by western blot for YY1 expression. The low part shows the densitometry analysis (mean ± SD, n = 3 *p < 0.05). (B) Ramos cells were treated with Obatoclax (28 nM) or DMSO for 24 h and the nuclear extracts were tested for YY1 DNA-binding activity by EMSA. (C) PC3 cells were transfected with the pYY1-Luc reporter construct and then treated with DMSO or Obatoclax for 24 h (mean ± SD, n = 3 *p < 0.05). The YY1 reporter luciferase activity was determined and normalized as described in experimental procedures. (D) Ramos cells were transfected with either YY1 siRNA or control siRNA and 24 h after transfection the cells were lyzed and analyzed by western blot for DR4, DR5, YY1, Mcl-1 and p-p65. The β-actin protein expression was used as a loading control. (e) Ramos cells were transfected with either YY1 siRNA or control siRNA and after 24 h the cells were treated with the indicated TRAIL concentrations for 18 h and apoptosis was determined by cleavage caspase-3 by flow (mean ± SD, n = 3 *p < 0.05).
(B) Role of YY1-induced inhibition by Obatoclax in the sensitization to Ramos cell of TRAIL apoptosis. We examined the direct role of Obatoclax-induced inhibition of YY1 activity in the sensitization to TRAIL by treating the cells with YY1 siRNA. Ramos cells were transfected with YY1 siRNA or control siRNA for 48 h. Treatment with YY1 siRNA resulted in the upregulation of DR5, and there was no noticeable change on DR4 (Fig. 5D, left part). In addition, YY1 siRNA treatment resulted in the inhibition of Mcl-1 and Phospho-p65 (Fig. 5D, left part). The cells transfected with YY1 siRNA and treated with various TRAIL concentrations for 18 h were examined for apoptosis. The findings demonstrate that transfection with YY1 siRNA, but not with control siRNA, resulted in significant sensitization to TRAIL apoptosis, and the level of apoptosis was a function of the TRAIL concentration used (Fig. 5E).
(C) The important role of DR5 upregulation by Obatoclax, but not DR4, in Obatoclax-mediated sensitization to TRAIL mediated apoptosis. (1) Upregulation of DR5 by Obatoclax via YY1 inhibition. We have reported that YY1 is a transcriptional repressor of the TRAIL death receptor DR5, and its inhibition results in the upregulation of DR5 expression.38,39,41 Here, we examined whether Obatoclax-induced inhibition of YY1 results in the upregulation of DR5 expression. Ramos cells were treated with Obatoclax, and cell lysates were examined for expression of both DR4 and DR5 by western blot. The data in Figure 6A demonstrate that treatment with Obatoclax results in significant upregulation of DR5 expression, both with 14 and 28 nM Obatoclax concentrations. In contrast, there was no upregulation of DR4 expression (Fig. 6A, top part). The data were also analyzed by densitometry (Fig. 6A, bottom part). The upregulation of DR5 was also analyzed by flow cytometry, and a representative histogram is shown in Figure 6B. Clearly, a significant upregulation of surface DR5 obtained following Obatoclax treatment and the expression of DR5 was higher with 28 nM as compared with 14 nM. In addition to Ramos cells, other cancer cell lines that included lymphoma and solid tumors treated with Obatoclax resulted in upregulation of DR5 (Fig. 6C), although the upregulation in DHL4 cells was modest.
Figure 6.

Obatoclax upregulates DR5 expression. (A) Ramos cells were treated with Obatoclax at the indicated concentrations for 24 h and then analyzed by western blot with specific antibodies. The densitometry analysis is shown (lower part). (B) Ramos cells were treated with DMSO or Obatoclax (14 or 20 nM) for 24 h, and the expression of cell surface DR5 was determined by flow. Anti-DR5 antibody conjugated with pe was used, and control IgG-PE was used. (C) 2F7, DHL4, PC3, SW620 and Jurkat cells were treated with Obatoclax at the indicated concentrations for 24 h and the expressions of DR4 and DR5 were determined by western blot. (D) Ramos cells were transfected with siRNA for DR4 and DR5 for 24 h and analyzed by western blot or were treated with DMSO or with the indicated Obatoclax concentrations and, 24 h after, the cells were treated with TRAIL (10 ng/ml) and then assessed for apoptosis by cleavage caspase-3 by flow (mean ± SD, n = 3 *p < 0.05). Upper part, cells were analyzed by western blot.
(2) Sensitization by Obatoclax to TRAIL apoptosis in Ramos cells is mediated by activation of DR5 but not by DR4. The above findings with Obatoclax-induced DR5 upregulation via YY1 inhibition, did not address whether, in the absence of upregulation of DR4, signaling of DR4 by TRAIL might have been involved in TRAIL-induced apoptosis. To determine the direct roles each for DR4 and DR5 in Obatoclax-induced TRAIL sensitization, Ramos cells were treated with either DR4 or DR5 siRNA. The findings in Figure 6D show that treatment with DR4 siRNA or DR5 siRNA resulted in the specific inhibition of the corresponding proteins as expected. DR5 silencing antagonizes Obatoclaxdependent sensitization to TRAIL. However, cells treated with DR4 siRNA in the presence of Obatoclax and TRAIL showed significant apoptosis. These findings suggested that Obatoclaxmediated sensitization to TRAIL in Ramos cells is through the activation of the DR5 signaling pathway in the absence of triggering the DR4 pathway.
Overall, the above findings suggest that Obatoclax-induced YY1 inhibition and DR5 upregulation are responsible, in large part, for the sensitization to TRAIL apoptosis. TRAIL sensitization is mediated by the induction of DR5 but not DR4.
Discussion
The present findings provide evidence for a novel mechanism of Obatoclax-induced gene regulation in B-NHL tumor cells that is involved in the reversal of resistance to TRAIL-induced apoptosis. Obatoclax has been designed as a BH3-mimetic; however, the present findings suggested novel activities mediated by Obatoclax, namely, inhibition of the constitutively activated NFκB survival/anti-apoptotic pathway and the inhibition of several gene products involved in apoptosis. We suggested the involvement of both Mcl-1 and the DR5-repressor YY1, inhibited by Obatoclax, in the sensitization to TRAIL apoptosis. Obatoclax-induced sensitization to TRAIL apoptosis occurs primarily via activation of the death receptor DR5 but not DR4. Thus, Obatoclax exerts several activities, namely, directly as a BH3 mimetic, as previously reported, and indirectly, as reported here, by inhibiting the survival/anti-apoptotic NFκB pathway and downstream anti-apoptotic target genes.
Obatoclax-mediated sensitization to TRAIL apoptosis correlated with Obatoclax-induced inhibition of YY1, upregulation of DR5 and with inhibition of various anti-apoptotic gene products, namely, Mcl-1. Reported studies with non-lymphoid tumor cells also showed their sensitization by Obatoclax to TRAIL apoptosis, although through a different mechanism, namely, by the release of Bak and Bim from Mcl-1.19,24,35,36 There were no alterations of cellular expression of other gene products, such as Mcl-1, in contrast to our findings here.19 Another study reported by Huang et al. showed that Obatoclax sensitized human pancreatic cancer cells to TRAIL apoptosis.18 The mechanism of sensitization was due to the dissociation of Bak and Bim from Bcl-2, Bcl-XL or Mcl-1. The direct role of Bax upregulation by Obatoclax was reported by Smoot et al., in which treatment of cholangiocarcinoma cells with Obatoclax resulted in the translocation of Bax into the mitochondria, which contributed directly to cell death.20 The discrepancy between these reported two studies and ours herein is not clear but may be due to the type of tumor cells studied.
Inhibition of NFκB by Obatoclax resulted in the inhibition downstream of several anti-apoptotic gene products, such as Mcl1, Bcl-XL, cIAP and XIAP (as expected) as well as the pro-apoptotic gene product Bax at a high Obatoclax concentration. Of interest, Obatoclax modestly inhibited Bax expression, possibly as a consequence of NFκB inhibition or the modification of other transcription factors that may lead to p53-mediated inhibition of Bax.42 Recently, Brem et al.33 demonstrated that Obatoclax induces downregulation of p53 in B-NHL cell lines, and it is known that p53 regulates the expression of Bax.43 The Obatoclax-mediated inhibition of Bax is not sufficient to regulate Obatoclax-mediated sensitization to TRAIL apoptosis. In comparison to B-NHL cells, Obatoclax does not affect the expression of Bax and Bak in normal cells. Brem et al.33 studied the effect of Obatoclax on Rituximab-resistant cell lines using a combination of Obatoclax and chemotherapeutic drugs. They reported that Obatoclax enhanced the activity of Rituximab, and the combination was synergistic. The Obatoclax concentration used by them was higher than the concentration used by us here (0.1–1 µM vs. 14–28 nM). Their findings were interpreted by Obatoclaxinduced inhibition of p53 and upregulation of pro-apoptotic genes, such as Noxa and Puma. Further, they proposed additional pathways of cell death in a caspase-independent manner, such as autophagy. However, the concentration that they used to inhibit p53 and upregulate Puma and Noxa was over 10-fold higher than the concentration used for analyses of cell death. Our findings herein show that the induction of apoptosis with the combination of low concentrations of Obatoclax and TRAIL was caspase-dependent and synergistic. In comparison to B-NHL cells, Obatoclax does not affect the expression of Bax and Bak in normal cells (unpublished).
The present findings show that Obatoclax treatment of Ramos cells inhibited NFκB activity through an unknown mechanism. It may be postulated to be the direct inhibition by Obatoclax of transcription factors or promoters resulting in the regulation of transcription. In addition, previous reports indicated that Bcl-2 family members, such as Bcl-2 and Bcl-XL, can form a complex with NFκB, and this complex inhibits the translocation of NFκB and its function in the nucleus.44 The modification of the flexible loop regulatory domain (FLD) of Bcl-2 that lies between the BH4 and BH3 regions as a dephosphorylated form can be implicated in the interaction with transcriptional factors such as p53 and can also be implicated in the interaction with NFκB and its subsequent inhibition.42,45,46 These various possibilities warrant further investigation in the future. In addition, we have shown here that Obatoclax induced the inhibition of different BH3-only proteins, such as Bid and Bad. Each one belongs to two subclasses of BH3 peptides: one called activators, including the BH3 domain of Bid, that can directly activate Bak, and the other subclass, called sensitizers, represented by Bad and others, which can't activate Bak directly but, instead, bind to an antiapoptotic Bcl-2 to displace Bid-like peptides.47 It is not clear how Obatoclax induces the inhibition of these proteins, and previous data about this family of BH-3 protein have been reported to be regulated also by p53.48,49 Our findings suggest that regulation of Bcl-2 family members through Obatoclax can affect the regulation of transcription factors that are implicated in the regulation of pro-apoptotic signaling events.
The findings here demonstrate that Obatoclax inhibits several anti-apoptotic gene products, such as Mcl-1, Bcl-XL, cIAP and XIAP, and these inhibitory effects are consistent with the inhibition upstream of NFκB.50–53 The findings here are distinct from the original description of Obatoclax as a BH3-mimetic.54 Reported studies demonstrated the role of Obatoclax in the translocation of Bax and Bim from Bcl-XL/Mcl-1 to the mitochondria, leading to its activation. We confirm here in B-NHL cells that treatment of Ramos cells with Obatoclax dissociated Bak from Mcl-1. Nguyen et al. reported that Obatoclax antagonized Mcl-1 resistance to apoptosis.17 In the present findings, we have shown that Obatoclax inhibited further the expression of Mcl-1 in addition to its release from Bak. We also show the involvement of Mcl-1 inhibition by Obatoclax in TRAIL sensitization in Ramos cells through the treatment of tumor cells with Mcl-1 siRNA. However, the direct role may be examined in cells treated with Obatoclax, in which Mcl-1 was overexpressed and then treated with TRAIL.
While the direct role of YY1 in the reversal of TRAIL apoptosis has been demonstrated by us previously and here,39,41 the direct role of DR5 regulation in the sensitization has not been reported. In the present study, we show that Obatoclax-induced inhibition of YY1 and upregulation of DR5 in sensitization took place via activation of the DR5 signaling pathway and not the DR4 pathway, hence suggesting that the induction of DR5 by Obatoclax via the inhibition of YY1 may be involved in the reversal of TRAIL resistance. The above findings demonstrated that both YY1 and Mcl-1 play important roles in the reversal of TRAIL resistance following treatment with Obatoclax. The contribution of each of these modifications in the reversal of resistance is not clear, although we show that Mcl-1 may be regulated by YY1 (Fig. 5D). It has been shown that Mcl-1 can be regulated by several transcriptional factors in cancer as well as in normal cells.55 Those transcriptional factors include NFκB,56 which, in turn, can regulate Mcl-1 transcription by binding to the promoter region. As we observed in Figure 5D, inhibition of YY1 dowregulates the NFκB activation (p65 phosphorylation). This data suggested that a loop probably exists between the regulation of NFκB and YY1 and the target genes for each of these transcriptional factors that are affected in the regulation of the loop NFκB/YY1. This can explain, in part, our results, whereby the regulation of Mcl-1 occurs via the inhibition of YY1 through the NFκB loop. Additional experiments to establish the mechanism linking NFκB and YY1 are the subject of another investigation.
The treatment of normal peripheral blood leukocytes with high concentrations of Obatoclax, either alone or in combination with TRAIL, did not show any detectable cytotoxic activity. These findings are important in the potential therapeutic application of Obatoclax alone or in combination with TRAIL. The findings also suggest that Obatoclax may sensitize in vivo tumor cells to TRAIL-expressing host cytotoxic cells.41 The clinical application of Obatoclax in combination with therapeutics agents was reported recently in a phase II study in lung cancer in combination with Topotecan.57 Obatoclax is safe and tolerant when given in conjunction with Topotecan in patients with advanced solid tumor malignances.58 Studies testing the efficacy of Obatoclax in hematological malignances in combination of therapeutics drugs have not been reported. Thus, the results in this study offer the possibility of exploring the efficacy of Obatoclax in combination with chemo-immunotherapeutic drugs in the treatment of hematological malignances. Like TRAIL, the above findings imply that Obatoclax may also sensitize chemotherapy-resistant tumor cells to chemotherapy-induced apoptosis by the same underlying mechanisms, since the inhibition of NFκB also reverses resistance to chemotherapeutic drugs.30,31 Overall, we propose a dysregulated resistant circuit, namely, the NFκB/YY1/Mcl-1/DR5 that is modified by Obatoclax, leading to reversal of resistance (see diagram in Fig. 7).
Figure 7.
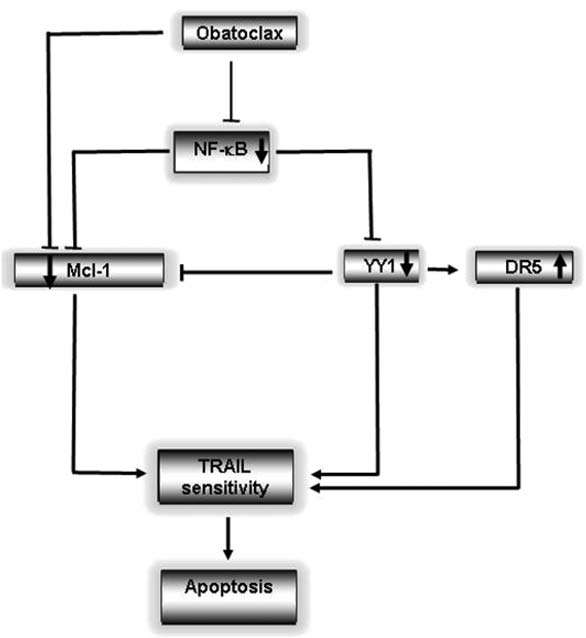
Schematic diagram of the mechanism by which Obatoclax sensitizes B-NHL to apoptosis by TRAIL. B-NHL cells maintain their resistance to TRAIL-induced apoptosis through the constitutive activation of NFκB and YY1, which negatively regulates DR5 expression. Cell treatment with Obatoclax inhibits NFκB and YY1 activities, leading to increased expression of DR5. In parallel, Obatoclax targets Mcl-1 to unsequester pro-apoptotic binding partners and also inhibits expression of Mcl-1. The inhibition by Obatoclax of NFκB and downstream YY1 and Mcl-1 all participate in the sensitization of B-NHL cells to apoptosis by TRAIL.
Materials and Methods
Cell lines and reagents.
Ramos, Daudi, Raji, DHL4, PC3, SW620 and Jurkat cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA). 2F7 cells were kindly provided by Dr. Otoniel Martinez-Maza (Jonsson Comprehensive Cancer Center, Los Angeles, CA). The cell lines were cultured as described previously in references 59 and 60. The following antibodies were obtained from these sources: Bcl-XL, Mcl-1, Bad, Bid, Bim, Bax, Bak (pan antibodies), p50, p65, Phospho-p65 (phosphorelation at Ser 536), IκBα, PhosphoIκBα, IKK, YY1, DR4, caspase 3, caspase 9, caspase 8 and cIAP1/2 (Santa Cruz Biotechnology, Santa Cruz, CA); XIAP (Cell Signaling, Beverly, MA); DR5 (Ψ Scientific, Poway, CA) and β-actin (Chemicon, Temecula, CA); PE-labeled anti-DR5, anti-DR4 and FITC-labeled anti-active caspase-3 antibodies as well as the corresponding IgG isotype controls were obtained from BD PharMingen (San Diego, CA). Soluble recombinant human TRAIL was purchased from Pepro-Tech Inc., (Rocky Hills, NJ). The NFκB inhibitor dehydroxymethylepoxyquinomicin (DHMEQ) was provided by Dr. K. Umezawa (Keio University, Tokyo, Japan)61 and diluted in DMSO. Obatoclax (GX15-070) was obtained from Gemin X Pharmaceuticals (Montreal, QC Canada) and dissolved in DMSO at a stock concentration of 5 mmol/L that was aliquoted, stored at −20°C and then diluted as needed in cell culture medium.
Viability assay.
Cell viability was assessed by either trypan blue exclusion assay by microscopy or by XTT dye absorbance according to the manufacturer's instruction (Roche Diagnostic GmbH, Nonnenwald Germany) and as previously described in reference 59. The viability of the untreated cells was set at 100%. Each experimental condition was performed in triplicate, and the SD was calculated.
Western blot analysis for protein expression.
Cell lines were incubated at 37°C for 24 h. The cells treated with Obatoclax or left untreated, were lysed on ice with 100 µl of ice-cold RIPA buffer (1% Nonidet P-40, 0.1% SDS, 0.5% deoxycholic acid and complete protease inhibitor mixture tablet from Roche). Lysates were prepared as described in reference 41.
Determination of apoptosis.
Apoptosis was assessed in Ramos cells pretreated for 24 h or 48 h with different Obatoclax concentrations, followed by an 18 h treatment with various TRAIL concentrations. Apoptosis was determined by cleavage of procaspase 3 using flow cytometry.39,60 For DHMEQ treatment, the cells were pretreated with different DHMEQ concentrations and then treated for additional 18 h with the indicated TRAIL concentrations, and apoptosis was determined as above.
Flow cytometry.
Ramos cells treated for 24 h with different concentrations of Obatoclax were subjected to flow cytometry for evaluation of DR4 or DR5 surface expression. Cells were stained with PE-conjugated specific antibody for 1 h at 4°C according to the manufacturer's instruction. A flow cytometric analysis was done using an excitation wavelength of 488 on a Flow Epics XL-MCL (Coulter) using System II Software, and both the mean fluorescence intensity and the percentage of apoptotic cells was recorded.
Transfection with siRNA.
Scrambled RNA, Mcl-1, DR4, DR5 and YY1 small interfering RNAs were obtained from Santa Cruz Biotechnology. Ramos cells were cultured at a density of 2.5 x 105/ml in RPMI 1640 devoid of antibiotics for 24 h. Cells were then transfected with 50 nM/liter siRNA in a final volume of 100 µl of medium in the presence of 10 µl of lipofectamine 2000 (Invitrogen, Invitrogen Life Technology, Carlsbad, CA) in opti-MEM. After 24 h of transfection, the cells were treated with TRAIL at the indicated concentrations for 18 h. Ramos cells were collected and analyzed for the relevant protein by western blot or for apoptosis by flow as indicated above.
Reported assay.
Transient transfections with the reporter plasmids pYY1-Luc or pNFκB-Luc were performed in 6-well dishes containing exponentially grown PC3 cells.39 The luciferase pYY1 constructs carrying the full length of the relevant wild-type promoter sequence were synthesized as previously described in reference 60. The pNFκB-Luc plasmid was purchased from Invitrogen (Carlsbad, CA). Transfections were performed using the Transfectol Transfection Kit (Gene Choice) as described in reference 40. Data were normalized to protein concentration levels using the Bio-Rad protein assay. Prostate cell lines were treated with standardized concentration of Obatoclax (156 and 132 nM) to induce upregulation of DR5.
Immunoprecipitation.
Ramos cells were treated with Obatoclax or DMSO for 24 h, and then the cells were lysed on ice with 100 µl of ice-cold RIPA buffer [1% Nonidet P-40, 0.1% SDS, 0.5% deoxycholic acid and complete protease inhibitor mixture tablet (Roche, Diagnostic GmbH, Nonnenwald Germany)]. Lysates were added with 10 µl of anti-Mcl-1 antibody (Santa Cruz Biotechnology) and incubated overnight at 4°C, constantly and gently mixed on a shaker. 100 µl of agarose-conjugate suspension (Roche, Diagnostic GmbH, Nonnenwald Germany) were added and incubated for 1 h at 4°C and constantly and gently mixed on a shaker. Immunoprecipitated complexes were collected by centrifugation at 3,000x g for 2 min at 4°C. The supernatant was discarded, and the pellet was washed three times with 1 ml of cold Dulbeco (Invitrogene Life technology, Carlsbad, CA) and centrifugated at 30,000x g for 2 min at 4°C. Laemmli sample buffer (Bio-Rad) was added to each sample, and then the samples were boiled for 5 min, separated on 12% SDS-polyacrylamide minigels and transferred to nitrocellulose membrane Hybond ECL (Amersham Biosciences). Primary Abs for Bak and Mcl-1 (Santa Cruz Biotechnology) were used at predetermined optimal concentrations overnight. The blots were developed by Luminol Reagent and peroxide (Santa Cruz Biotechnology).
Mitochondrial membrane depolarization.
The mitochondria-specific dye 3,3′-dihexyloxacarbocyanine (DiOC6; Molecular Probes Inc., Eugene, OR) was used to measure the mitochondrial membrane potential37 in Ramos cells after 24 h of treatment with different Obatoclax concentrations. Cells were washed with PBS and resuspended in 900 µl of PBS containing 10 µl of DiOC6 (final concentration 40 nM). Cells were mixed, incubated for 30 min at 37°C in the dark and kept on ice until analyzed by flow. Cells with loss DiOC6 fluorescence represent cells with permeabilized mitochondria.
EMSA.
Ramos cells were harvested after treatment with Obatoclax (28 nM) and washed twice with ice-cold PBS. After washing, the cells were lysed in 1 ml of Nonidet P-40 lysis buffer to purify the nuclear proteins and sonicated for 10 sec at 4°C. The protein concentration was determined using the Bio-Rad protein assay kit. Ten µl of nuclear proteins were mixed with the biotin probe for the transcriptional factors NFκB and YY1, using the EMSA Kit from PanomicsTM (Fremont, CA) following the manufacturer's instructions and as previously described in reference 62. Ten µl of the samples were separated on 5% polyacrylamide minigels and transferred to Nylon Hybond-N+ membrane (Amersham Biosciences). Membranes were exposed to UV light for 3 min. The membranes were exposed to film blots and were developed by Luminol Reagent and peroxide (Santa Cruz Biotechnology).
Isobologram analysis for synergy determination.
The isobologram analysis was used to evaluate the effect of the Obatoclax/TRAIL combination.63 Traditional isobolograms were constructed from treatments combining Obatoclax (7, 14, 21 and 28 nM) with TRAIL (2.5, 5, 10 and 20 ng/ml). Analysis was done according to the dose-response curves using cleavage caspase 3 analysis for Ramos cells treated with TRAIL alone or Obatoclax alone and the combination for 18 h, and these analyses generated dose-response curves (isoboles) of 25 ± 5% cytotoxicity to evaluate the effect of the agents in combination.
Statistical analysis.
All results were expressed as the mean ± SD of three independent experiments in triplicate or duplicate. The statistical analyses were performed using Graph Pad Prism-4. The statistical significance of differences between group means was determined using one-way ANOVA to compare variance. Significant differences were considered for probabilities < 5% (p < 0.05).
Acknowledgments
The authors acknowledge the assistance of Kerry Choy, Daphne Liang and Melissa Cao in the preparation of the manuscript and Dr. Kazueo Umezawa for the NFκB inhibitor and Dr. Otoniel Martinez-Maza for valuable input and support.
Abbreviations
- TRAIL
tumor necrosis factor (TNF)-related apoptosis-inducing ligand
- DR5
death receptor 5
- siRNA
small interfering RNA
- YY1
Yin Yang 1
- DHMEQ
dehydroxymethylepoxyquinomicin
- Mcl-1
myeloid cell leukemia sequence 1 (BCL2-related)
Financial Support
This study was supported in part by academic support from the CONACYT, Mexico (M.M.P. 165639) and Grant FIS/IMSS/PROT/G10/819 from the IMSS (M.M.P.), Jonsson Comprehensive Cancer Center (M.I.V.), UCLA AIDS Institute (M.I.V.) and Fogarty International Center Fellowship (D43 TW00013-14) (M.I.V. and S.H.Y.).
References
- 1.Mimeault M, Batra SK. New advances on critical implications of tumor- and metastasis-initiating cells in cancer progression, treatment resistance and disease recurrence. Histol Histopathol. 2010;25:1057–1073. doi: 10.14670/hh-25.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sung ES, Park KJ, Lee SH, Jang YS, Park SK, Park YH, et al. A novel agonistic antibody to human death receptor 4 induces apoptotic cell death in various tumor cells without cytotoxicity in hepatocytes. Mol Cancer Ther. 2009;8:2276–2285. doi: 10.1158/1535-7163.MCT-09-0235. [DOI] [PubMed] [Google Scholar]
- 3.Zinonos I, Labrinidis A, Lee M, Liapis V, Hay S, Ponomarev V, et al. Apomab, a fully human agonistic antibody to DR5, exhibits potent antitumor activity against primary and metastatic breast cancer. Mol Cancer Ther. 2009;8:2969–2980. doi: 10.1158/1535-7163.MCT-09-0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wiezorek J, Holland P, Graves J. Death receptor agonists as a targeted therapy for cancer. Clin Cancer Res. 2010;16:1701–1708. doi: 10.1158/10780432.CCR-09-1692. [DOI] [PubMed] [Google Scholar]
- 5.Sheikh MS, Fornace AJ., Jr Death and decoy receptors and p53-mediated apoptosis. Leukemia. 2000;14:150913. doi: 10.1038/sj.leu.2401865. [DOI] [PubMed] [Google Scholar]
- 6.Reid P, Holen I. Pathophysiological roles of osteoprotegerin (OPG) Eur J Cell Biol. 2009;88:1–17. doi: 10.1016/j.ejcb.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 7.Kruyt FA. TRAIL and cancer therapy. Cancer Lett. 2008;263:14–25. doi: 10.1016/j.canlet.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 8.Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem. 1996;271:12687–12690. doi: 10.1074/jbc.271.22.12687. [DOI] [PubMed] [Google Scholar]
- 9.Camidge DR, Herbst RS, Gordon MS, Eckhardt SG, Kurzrock R, Durbin B, et al. A phase I safety and pharmacokinetic study of the death receptor 5 agonistic antibody PRO95780 in patients with advanced malignancies. Clin Cancer Res. 2010;16:1256–1263. doi: 10.1158/1078-0432.CCR-091267. [DOI] [PubMed] [Google Scholar]
- 10.Trarbach T, Moehler M, Heinemann V, Kohne CH, Przyborek M, Schulz C, et al. Phase II trial of mapatumumab, a fully human agonistic monoclonal antibody that targets and activates the tumour necrosis factor apoptosis-inducing ligand receptor-1 (TRAIL-R1), in patients with refractory colorectal cancer. Br J Cancer. 2010;102:506–512. doi: 10.1038/sj.bjc.6605507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Testa U. TRAIL/TRAIL-R in hematologic malignancies. J Cell Biochem. 2010;110:21–34. doi: 10.1002/jcb.22549. [DOI] [PubMed] [Google Scholar]
- 12.Mahalingam D, Szegezdi E, Keane M de JS, Samali A. TRAIL receptor signalling and modulation: Are we on the right TRAIL? Cancer Treat Rev. 2009;35:280–288. doi: 10.1016/j.ctrv.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 13.Zhang L, Fang B. Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther. 2005;12:228–237. doi: 10.1038/sj.cgt.7700792. [DOI] [PubMed] [Google Scholar]
- 14.Vogler M, Dinsdale D, Dyer MJ, Cohen GM. Bcl-2 inhibitors: small molecules with a big impact on cancer therapy. Cell Death Differ. 2009;16:360–367. doi: 10.1038/cdd.2008.137. [DOI] [PubMed] [Google Scholar]
- 15.Ray S, Bucur O, Almasan A. Sensitization of prostate carcinoma cells to Apo2L/TRAIL by a Bcl-2 family protein inhibitor. Apoptosis. 2005;10:1411–1418. doi: 10.1007/s10495-005-2490-y. [DOI] [PubMed] [Google Scholar]
- 16.Kang MH, Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res. 2009;15:1126–1132. doi: 10.1158/1078-0432.CCR-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy M, et al. Small molecule Obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci USA. 2007;104:19512–19517. doi: 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang S, Okumura K, Sinicrope FA. BH3 mimetic Obatoclax enhances TRAIL-mediated apoptosis in human pancreatic cancer cells. Clin Cancer Res. 2009;15:150–159. doi: 10.1158/10780432.CCR-08-1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mott JL, Bronk SF, Mesa RA, Kaufmann SH, Gores GJ. BH3-only protein mimetic Obatoclax sensitizes cholangiocarcinoma cells to Apo2L/TRAIL-induced apoptosis. Mol Cancer Ther. 2008;7:2339–2347. doi: 10.1158/1535-7163.MCT08-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smoot RL, Blechacz BR, Werneburg NW, Bronk SF, Sinicrope FA, Sirica AE, et al. A Bax-mediated mechanism for Obatoclax-induced apoptosis of cholangiocarcinoma cells. Cancer Res. 2010;70:1960–1969. doi: 10.1158/0008-5472.CAN09-3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang CC, Wroblewski D, Yang F, Hersey P, Zhang XD. Human melanoma cells under endoplasmic reticulum stress are more susceptible to apoptosis induced by the BH3 mimetic Obatoclax. Neoplasia. 2009;11:945–955. doi: 10.1593/neo.09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Konopleva M, Watt J, Contractor R, Tsao T, Harris D, Estrov Z, et al. Mechanisms of antileukemic activity of the novel Bcl-2 homology domain-3 mimetic GX15-070 (Obatoclax) Cancer Res. 2008;68:3413–3420. doi: 10.1158/0008-5472.CAN07-1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J, Viallet J, Haura EB. A small molecule pan-Bcl-2 family inhibitor, GX15-070, induces apoptosis and enhances cisplatin-induced apoptosis in non-small cell lung cancer cells. Cancer Chemother Pharmacol. 2008;61:525–534. doi: 10.1007/s00280007-0499-3. [DOI] [PubMed] [Google Scholar]
- 24.Pérez-Galán P, Roue G, Villamor N, Campo E, Colomer D. The BH3-mimetic GX15-070 synergizes with bortezomib in mantle cell lymphoma by enhancing Noxa-mediated activation of Bak. Blood. 2007;109:4441–4449. doi: 10.1182/blood2006-07-034173. [DOI] [PubMed] [Google Scholar]
- 25.Dixit V, Mak TW. NFkappaB signaling. Many roads lead to madrid. Cell. 2002;111:615–619. doi: 10.1016/S0092-8674(02)01166-2. [DOI] [PubMed] [Google Scholar]
- 26.Gasparian AV, Yao YJ, Kowalczyk D, Lyakh LA, Karseladze A, Slaga TJ, et al. The role of IKK in constitutive activation of NFkappaB transcription factor in prostate carcinoma cells. J Cell Sci. 2002;115:141–151. doi: 10.1242/jcs.115.1.141. [DOI] [PubMed] [Google Scholar]
- 27.Ghosh S, Karin M. Missing pieces in the NFkappaB puzzle. Cell. 2002;109:81–96. doi: 10.1016/S0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 28.Lee HH, Dadgostar H, Cheng Q, Shu J, Cheng G. NFkappaB-mediated upregulation of Bcl-x and Bfl-1/A1 is required for CD40 survival signaling in B lymphocytes. Proc Natl Acad Sci USA. 1999;96:9136–9141. doi: 10.1073/pnas.96.16.9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jazirehi AR, Vega MI, Bonavida B. Development of rituximab-resistant lymphoma clones with altered cell signaling and cross-resistance to chemotherapy. Cancer Res. 2007;67:1270–1281. doi: 10.1158/0008-5472.CAN-06-2184. [DOI] [PubMed] [Google Scholar]
- 30.Kimura N, Miyakawa Y, Kohmura K, Umezawa K, Ikeda Y, Kizaki M. Targeting NFkappaB and induction of apoptosis by novel NFkappaB inhibitor dehydroxymethylepoxyquinomicin (DHMEQ) in Burkitt lymphoma cells. Leuk Res. 2007;31:1529–1535. doi: 10.1016/j.leukres.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 31.Vega MI, Martinez-Paniagua M, Jazirehi AR, Huerta-Yepez S, Umezawa K, Martinez-Maza O, et al. The NFkappaB inhibitors (bortezomib and DHMEQ) sensitise rituximab-resistant AIDS-B-non-Hodgkin lymphoma to apoptosis by various chemotherapeutic drugs. Leuk Lymphoma. 2008;49:1982–1994. doi: 10.1080/10428190802357071. [DOI] [PubMed] [Google Scholar]
- 32.Watanabe M, Ohsugi T, Shoda M, Ishida T, Aizawa S, Maruyama-Nagai M, et al. Dual targeting of transformed and untransformed HTLV-1-infected T cells by DHMEQ, a potent and selective inhibitor of NFkappaB, as a strategy for chemoprevention and therapy of adult T-cell leukemia. Blood. 2005;106:246–271. doi: 10.1182/blood-2004-093646. [DOI] [PubMed] [Google Scholar]
- 33.Brem EA, Thudium K, Khubchandani S, Tsai PC, Olejniczak SH, Bhat S, et al. Distinct cellular and therapeutic effects of Obatoclax in rituximab-sensitive and -resistant lymphomas. Br J Haematol. 2011;153:599–611. doi: 10.1111/j.1365-2141.2011.08669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–192. doi: 10.1016/S15356108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 35.Pérez-Galán P, Roue G, Lopez-Guerra M, Nguyen M, Villamor N, Montserrat E, et al. BCL-2 phosphorylation modulates sensitivity to the BH3 mimetic GX15070 (Obatoclax) and reduces its synergistic interaction with bortezomib in chronic lymphocytic leukemia cells. Leukemia. 2008;22:1712–1720. doi: 10.1038/leu.2008.175. [DOI] [PubMed] [Google Scholar]
- 36.Trudel S, Li ZH, Rauw J, Tiedemann RE, Wen XY, Stewart AK. Preclinical studies of the pan-Bcl inhibitor Obatoclax (GX015-070) in multiple myeloma. Blood. 2007;109:5430–5438. doi: 10.1182/blood-2006-10-047951. [DOI] [PubMed] [Google Scholar]
- 37.Huerta-Yepez S, Vega M, Escoto-Chavez SE, Murdock B, Sakai T, Baritaki S, et al. Nitric oxide sensitizes tumor cells to TRAIL-induced apoptosis via inhibition of the DR5 transcription repressor Yin Yang1. Nitric Oxide. 2009;20:39–52. doi: 10.1016/j.niox.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 38.Baritaki S, Katsman A, Chatterjee D, Yeung KC, Spandidos DA, Bonavida B. Regulation of tumor cell sensitivity to TRAIL-induced apoptosis by the metastatic suppressor Raf kinase inhibitor protein via Yin Yang 1 inhibition and death receptor 5 upregulation. J Immunol. 2007;179:5441–5453. doi: 10.4049/jimmunol.179.8.5441. [DOI] [PubMed] [Google Scholar]
- 39.Baritaki S, Huerta-Yepez S, Sakai T, Spandidos DA, Bonavida B. Chemotherapeutic drugs sensitize cancer cells to TRAIL-mediated apoptosis: upregulation of DR5 and inhibition of Yin Yang 1. Mol Cancer Ther. 2007;6:1387–1399. doi: 10.1158/1535-7163.MCT-06-0521. [DOI] [PubMed] [Google Scholar]
- 40.Baritaki S, Suzuki E, Umezawa K, Spandidos DA, Berenson J, Daniels TR, et al. Inhibition of Yin Yang 1-dependent repressor activity of DR5 transcription and expression by the novel proteasome inhibitor NPI-0052 contributes to its TRAIL-enhanced apoptosis in cancer cells. J Immunol. 2008;180:6199–6210. doi: 10.4049/jimmunol.180.9.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vega MI, Baritaki S, Huerta-Yepez S, Martinez-Paniagua MA, Bonavida B. A potential mechanism of rituximab-induced inhibition of tumor growth through its sensitization to tumor necrosis factor-related apoptosis-inducing ligand-expressing host cytotoxic cells. Leuk Lymphoma. 2011;52:108–121. doi: 10.3109/10428194.2010.531408. [DOI] [PubMed] [Google Scholar]
- 42.Deng X, Gao F, Flagg T, Anderson J, May WS. Bcl2′s flexible loop domain regulates p53 binding and survival. Mol Cell Biol. 2006;26:4421–4434. doi: 10.1128/MCB.01647-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Graupner V, Alexander E, Overkamp T, Rothfuss O, De Laurenzi V, Gillissen BF, et al. Differential regulation of the proapoptotic multidomain protein Bak by p53 and p73 at the promoter level. Cell Death Differ. 2011;18:1130–1139. doi: 10.1038/cdd.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Massaad CA, Portier BP, Taglialatela G. Inhibition of transcription factor activity by nuclear compartment-associated Bcl-2. J Biol Chem. 2004;279:54470–54478. doi: 10.1074/jbc.M407659200. [DOI] [PubMed] [Google Scholar]
- 45.Grimm S, Bauer MK, Baeuerle PA, Schulze-Osthoff K. Bcl-2 downregulates the activity of transcription factor NFkappaB induced upon apoptosis. J Cell Biol. 1996;134:13–23. doi: 10.1083/jcb.134.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hour TC, Chen L, Lin JK. Suppression of transcription factor NFkappaB activity by Bcl-2 protein in NIH3T3 cells: implication of a novel NFkappaB p50-Bcl-2 complex for the anti-apoptotic function of Bcl-2. Eur J Cell Biol. 2000;79:121–129. doi: 10.1078/S0171-9335(04)70014-X. [DOI] [PubMed] [Google Scholar]
- 47.Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, et al. BH3 domains of BH3-only proteins differentially regulate Baxmediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17:525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 48.Jiang P, Du W, Heese K, Wu M. The Bad guy cooperates with good cop p53: Bad is transcriptionally upregulated by p53 and forms a Bad/p53 complex at the mitochondria to induce apoptosis. Mol Cell Biol. 2006;26:9071–9082. doi: 10.1128/MCB.01025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ, El-Deiry WS. BID regulation by p53 contributes to chemosensitivity. Nat Cell Biol. 2002;4:842–849. doi: 10.1038/ncb866. [DOI] [PubMed] [Google Scholar]
- 50.Glasgow JN, Qiu J, Rassin D, Grafe M, Wood T, Perez-Pol JR. Transcriptional regulation of the BCL-X gene by NFkappaB is an element of hypoxic responses in the rat brain. Neurochem Res. 2001;26:647–659. doi: 10.1023/A:1010987220034. [DOI] [PubMed] [Google Scholar]
- 51.Petlickovski A, Laurenti L, Li X, Marietti S, Chiusolo P, Sica S, et al. Sustained signaling through the B-cell receptor induces Mcl-1 and promotes survival of chronic lymphocytic leukemia B cells. Blood. 2005;105:4820–4827. doi: 10.1182/blood2004-07-2669. [DOI] [PubMed] [Google Scholar]
- 52.Stehlik C. de MR, Binder BR, Lipp J. Cytokine induced expression of porcine inhibitor of apoptosis protein (iap) family member is regulated by NFkappaB. Biochem Biophys Res Commun. 1998;243:827–832. doi: 10.1006/bbrc.1998.8185. [DOI] [PubMed] [Google Scholar]
- 53.Wu M, Lee H, Bellas RE, Schauer SL, Arsura M, Katz D, et al. Inhibition of NFkappaB/Rel induces apoptosis of murine B cells. EMBO J. 1996;15:4682–4690. [PMC free article] [PubMed] [Google Scholar]
- 54.Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov. 2010;9:447–464. doi: 10.1038/nrd3137. [DOI] [PubMed] [Google Scholar]
- 55.Akgul C. Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell Mol Life Sci. 2009;66:1326–1336. doi: 10.1007/s00018008-8637-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feng CY, Rise ML. Characterization and expression analyses of anti-apoptotic Bcl-2-like genes NR-13, Mcl1, Bcl-X1 and Bcl-X2 in Atlantic cod (Gadus morhua) Mol Immunol. 2010;47:763–784. doi: 10.1016/j.molimm.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 57.Paik PK, Rudin CM, Pietanza MC, Brown A, Rizvi NA, Takebe N, et al. A phase II study of obatoclax mesylate, a Bcl-2 antagonist, plus topotecan in relapsed small cell lung cancer. Lung Cancer. 2011 doi: 10.1016/j.lungcan.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paik PK, Rudin CM, Brown A, Rizvi NA, Takebe N, Travis W, et al. A phase I study of Obatoclax mesylate, a Bcl-2 antagonist, plus topotecan in solid tumor malignancies. Cancer Chemother Pharmacol. 2010;66:1079–1085. doi: 10.1007/s00280010-1265-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vega MI, Huerta-Yepez S, Martinez-Paniagua M, Martinez-Miguel B, Hernandez-Pando R, Gonzalez-Bonilla CR, et al. Rituximab-mediated cell signaling and chemo/immuno-sensitization of drug-resistant B-NHL is independent of its Fc functions. Clin Cancer Res. 2009;15:6582–6594. doi: 10.1158/1078-0432.CCR-09-1234. [DOI] [PubMed] [Google Scholar]
- 60.Huerta-Yepez S, Vega M, Garban H, Bonavida B. Involvement of the TNFalpha autocrine-paracrine loop, via NFkappaB and YY1, in the regulation of tumor cell resistance to Fas-induced apoptosis. Clin Immunol. 2006;120:297–309. doi: 10.1016/j.clim.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 61.Umezawa K, Chaicharoenpong C. Molecular design and biological activities of NFkappaB inhibitors. Mol Cells. 2002;14:163–167. [PubMed] [Google Scholar]
- 62.Vega MI, Huerta-Yepaz S, Garban H, Jazirehi A, Emmanouilides C, Bonavida B. Rituximab inhibits p38 MAPK activity in 2F7 B NHL and decreases IL-10 transcription: pivotal role of p38 MAPK in drug resistance. Oncogene. 2004;23:3530–3540. doi: 10.1038/sj.onc.1207336. [DOI] [PubMed] [Google Scholar]
- 63.Berenbaum MC. A method for testing for synergy with any number of agents. J Infect Dis. 1978;137:122–130. doi: 10.1093/infdis/137.2.122. [DOI] [PubMed] [Google Scholar]