

Abstract
Hematopoietic stem cells provide an indispensible source for replenishing the blood with all its constituents throughout the organism's lifetime. Mice with a compromised hematopoietic stem cell compartment cannot survive. p53, a major tumor suppressor gene, has been implicated in regulation of hematopoiesis. In particular, p53 plays a role in homeostasis by regulating HSC quiescence and self renewal. We recently utilized a hypomorphic p53515C allele in conjunction with Mdm2, a negative regulator of p53, to gain insights into the role of p53 in hematopoietic regulation. Our analyses revealed that p53515C/515CMdm2−/− double mutant mice die soon after birth due to hematopoietic failure. Further mechanistic studies revealed that in the absence of Mdm2, ROS-induced postnatal p53 activity depletes hematopoietic stem cells, progenitors and differentiated cells.
Key words: HSC, reactive oxygen species, ROS, p53, Mdm2
The p53 Tumor Suppressor in Hematopoiesis
The p53 tumor suppressor and transcription factor is activated in response to DNA damage signals including ionizing radiation (IR), hypoxia, nucleotide tri-phosphate (NTP) depletion and reactive oxygen species (ROS).1 Active p53 impacts multiple pathways as it transactivates genes involved in apoptosis, cell cycle arrest, senescence, metabolism, fertility and differentiation. Not surprisingly then, p53 levels are constantly regulated in a cell. Mdm2 and Mdm4 play major roles in this regulation as genetic ablation of either gene in mice results in embryonic lethal phenotypes that are rescued by deletion of both p53 alleles.2–4
Hematopoietic defects are observed in mice with Mdm2 hypomorphic and null alleles that express approximately 30% the total levels of Mdm2.5 Mdm2+/− Mdm4+/− mice also succumb to p53-dependent hematopoietic defects and die shortly after birth.6 Additionally, while Mdm2+/− and Mdm4+/− mice are normal and viable, they are highly sensitive to sublethal irradiation and succumb to bone marrow ablation within days after radiation.6 Mice with enhanced p53 activity due to a mutant p53 allele (p53m) display deficiency in hematopoietic engraftment.7 On the other hand, p53-null mice and mice treated with pifithrin-α, a chemical inhibitor of p53 activity, do not succumb to radiation induced hematopoietic syndrome.8 Similarly, mice with homozygous deletion of Puma or Bax, two major p53 target genes that induce an apoptotic response, are overtly radio-resistant.9,10 These genetically engineered mice do not undergo typical degeneration of the hematopoietic organs after radiation exposure. Thus, these data support an essential role of p53 in development and maintenance of the hematopoietic system.
Dissecting the Hematopoietic Hierarchy
The hematopoietic system is one of the most well-studied systems in normal physiology.11 Hematopoietic stem cells (HSCs) are at the apex of the hematopoietic system. HSCs give rise to shortterm progenitors that produce more committed progenitors, namely common myeloid progenitors (CMP) and common lymphoid progenitors (CLP).12,13 CMP and CLP produce the committed precursors which subsequently differentiate into mature hematopoietic cells that are relatively short-lived. HSCs repeat this cycle throughout their lifetime in order to replenish the blood and bone marrow with appropriate cell types, while still maintaining their ability to self-renew.11,14 HSCs are routinely identified using Linc−Kit+Sca1+ (LKS) markers. These HSCs when transplanted in lethally irradiated mice reconstitute the bone marrow and allow survival of the organism.
The developmental origins and the processes of hematopoiesis are generally conserved among vertebrates.11 Most of these processes have been delineated from studies in mice. Hematopoiesis progresses at different anatomical sites during embryogenesis and adulthood.14 Hematopoiesis initially starts in the yolk sac at around embryonic day (E) 8 in mice, shifts to the fetal liver at E12.5, and finally the bone marrow at E16.5.15,16 The bone marrow remains the major hematopoietic organ after birth and in adult mice. The molecular and signaling pathways that regulate HSC function and activities remain unidentified.
p53 in Stem Cell Regulation
Recent studies have emphasized the role of p53 in regulation of stem cells. Expression of Nanog, a gene regulating self-renewal of embryonic stem (ES) cells, is regulated by p53.17 Also, loss of p53 diminishes spontaneous apoptosis and differentiation of ES cells.18 More recently, several laboratories have shown that loss of p53 improves the generation of induced pluripotent stem cells (iPSC) from adult cells.19–23
In HSCs, p53 is preferentially expressed in LKS cells where it negatively regulates self-renewal and maintains quiescence.24 Mice deficient in p53 show enhanced HSC self-renewal and have an increased HSC pool size.24 Recent studies have provided further evidence of p53 function in HSC regulation. Loss of MEF, a positive regulator of the p53 inhibitor Mdm2 in mice results in increased p53 activity and enhanced stem cell quiescence.25 Similarly, overexpression of miR-33, a microRNA which negatively regulates p53, enhances the transplantation efficiency of wild-type HSCs.26,27 Conversely, mice harboring an extra genomic copy of p53 (super-p53) have lower levels of miR-33 and HSCs compared with wild-type mice.26,27 Lastly, p53 also negatively regulates neural stem cell proliferation and self renewal.28 Thus, p53 is an important regulator of stem cell behavior.
Mdm2−/− p53515C/515C as a Model for p53's Role in Hematopoiesis
Numerous p53 mutant mice have been generated.29 One in particular contains a p53 allele (p53515C) encoding the p53R172P protein that retains the ability to induce cell cycle arrest and senescence, but not apoptosis.30,31 To delineate the role of p53 cell cycle arrest and senescence activities in the absence of Mdm2, p53515C/C and Mdm2+/− mice were intercrossed.32,33 Interestingly, Mdm2−/−p53515C/515C mice were born at normal Mendelian ratios but died before weaning. Histological examination of hematopoietic organs revealed a normal cellularity of fetal liver at E14.5 but a progressively acellular bone marrow after birth. Additionally, immunohistochemical analyses revealed that low p53R172P levels were present in fetal livers of Mdm2−/−p53515C/515C and the control Mdm2+/−p53515C/515C littermates.33 However, soon after birth, p53R172P levels became elevated in bone marrows of Mdm2−/−p53515C/515C mice, and this increase was coupled with severe depletion of hematopoietic cells. Since p53 is a stress-response gene, these data suggest that a stress signal instigated the increase in p53R172P levels and activity.
ROS as a p53 Activating Signal
Oxygen tension is a major difference between pre- and post-natal mice and we surmised ROS may be different preand post-birth. ROS damages DNA and hence p53 is activated in response to this stress.34 To characterize whether ROS was the stress signal for p53R172P stabilization, we first analyzed ROS levels in embryonic E14.5 fetal livers and postnatal day (P) 6 bone marrows. As expected, P6 bone marrows from Mdm2+/−p53515C/515C mice had a 3.8-fold higher ROS level compared with the corresponding fetal liver samples.33 Surprisingly, the postnatal Mdm2−/−p53515C/515C bone marrows had 3.3-fold higher ROS levels than the Mdm2+/−p53515C/515C controls. These data suggest that adequate Mdm2 protein levels in Mdm2+/+p53515C/515C and Mdm2+/−p53515C/515C dampen p53 activity. However, in the absence of Mdm2-mediated inhibition, ROS-induced p53R172P activity depleted all HSCs and progenitors cells in Mdm2−/−p53515C/515C bone marrows.33 In agreement with these data, we could revive the hematopoietic potential of Mdm2−/−p53515C/515C hematopoietic cells by culturing them at low oxygen levels.33
Moreover, as p53R172P is defective for inducing apoptosis,30 these data suggest that other modes of cell loss are involved in ablation of hematopoiesis in Mdm2−/−p53515C/515C mice. A significant increase in senescence markers p21, Dcr2 and p15 also implicate senescence as a possible pathway in abrogation of hematopoiesis.33 These data are corroborated by higher SA-βgal activity in freshly isolated postnatal bone marrows from Mdm2−/−p53515C/515C mice as compared with Mdm2+/−p53515C/515C. p21 loss partially rescued the postnatal lethality of Mdm2−/−p53515C/515C mice. Also, p53 activated ROS inducing genes, dubbed p53-induced genes (PIG), which are involved in inducing cell death.33,35 Finally, we could rescue the bone marrow cellularity of postnatal Mdm2−/−p53515C/515C pups by injection of N-Acetyl-Cysteine (NAC), an antioxidant that antagonizes ROS. These data confirmed that ROS contributes to the hematopoietic defect in Mdm2−/−p53515C/515C mice through activation of p53R172P. Since Mdm2+/−Mdm4+/− mice with wild-type p53 have a similar hematopoietic phenotype,6 these data may not necessarily be limited to the p53R172P protein but are also likely a characteristic of wild-type p53 activity.
Other Models of HSC Defects Due to ROS
Elevated ROS in HSCs is correlated with defects in other pathways. For instance, HSCs of Atm−/− mice accumulate ROS which limits the self-renewal capacity of HSCs in serial transplantation experiments.36 Inappropriate activation of mTOR due to targeted mutation of its negative regulator Tsc1 stimulates mitochondrial ROS biogenesis, concomitant with increased cycling of HSCs and ultimately loss of HSC “stemness.”37,38 Also, Bmi1 deletion abrogates mitochondrial function and instigates higher ROS levels, thus damaging HSC function.24 Deletion of Chk2, a DNA damage response gene and activator of p53, alleviates the hematopoietic defect of Bmi1-null mice.24 Based on our model, it is plausible that the Bmi1 effect is mediated through Chk2 activation of p53. This is further supported by the partial rescue of Bmi1-loss on hematopoiesis by deletion of p53.39 Our model suggests that p53 activation eventually leads to increased transcription of a set of ROS-inducing genes which results in depletion of HSCs and their progenitors, subsequently causing cell death.
Possible Mechanisms for ROS-Dependent p53 Activation in HSCs
Evidently, elevated ROS levels negatively influence HSC and progenitor activities. But how exactly is this achieved? Recent studies in Drosophila demonstrate that elevated ROS levels in myeloid progenitors induce their differentiation via downregulation of Polycomb complexes and upregulation of Jnk and Foxo signaling pathways.40 Since p53 is clearly implicated in differentiation,41 this remains a plausible effect of p53R172P-dependent ROS production on HSCs, leading to their eventual disappearance in Mdm2−/− p53515C/515C mice. Specifically, rapid differentiation of HSCs into progenitors or differentiation of progenitors into more differentiated cells would result in shortlived cells that die off within days. Testing such a hypothesis would require lineage tracing of Mdm2−/− p53515C/515C HSCs after sorting. However, the low number of HSCs in our mouse model remains an obstacle to addressing this question.
Similarly, p53 is also implicated in mesenchymal lineage differentiation.42,43 To examine possible effects on mesenchymal differentiation, we collected P6 whole bone marrows of Mdm2−/− p53515C/515C mice and control littermates, and plated them at low cell density using mesenchymal differentiation specific media. Typically, cells are plated for a few days until fibroblast-like colonies form which are then induced to differentiate by adding lineage specific factors to the media. Surprisingly, cultures of Mdm2−/− p53515C/515C bone marrows differentiated into osteoblastic lineage as seen by increased alkaline phosphatase staining without addition of lineage specific factors (Fig. 1). These data indicated that activation of p53R172P in the absence of Mdm2 could also alter differentiation capacity leading to defective hematopoiesis.
Figure 1.

Increased osteoblastic differentiation of P6 bone marrows of Mdm2−/− p53515C/515C mice in vitro. P6 whole bone marrows were allowed to grow for MSC colony formation. Mdm2−/− p53515C/515C (right) cells spontaneously differentiated into osteoblastic lineage as seen by red alkaline phosphatase activity while Mdm2+/− p53515C/515C (left) retained undifferentiated state.
Alternatively, as in the Atm−/− mouse model,36 elevated ROS could have abrogated the self-renewal capacity of HSCs in Mdm2−/−p53515C/515C mice. ROS-dependent depletion of bone marrow cellularity could have exhausted the HSC pool and impaired their self-renewal capacity while trying to regenerate bone marrows. This is supported by the failure of HSCs from P6 bone marrows of Mdm2−/− p53515C/515C to rescue lethally irradiated recipients.33 However, more studies are required to address whether the failure of Mdm2−/−p53515C/515C HSCs to reconstitute hematopoiesis is due to intrinsic or extrinsic defects. One approach could be to transplant Mdm2−/−p53515C/515C HSCs from E14.5 fetal livers (when HSCs numbers and ROS levels are normal) into lethally irradiated mice. Also, to test whether the Mdm2−/−p53515C/515C niche is intact, wild-type HSCs can be transplanted into Mdm2−/−p53515C/515C neonates. These experiments would importantly differentiate between defects that could be intrinsic to HSCs, their niche or both. In the light of the role of p53 in altering mesenchymal lineages, it would not be surprising to find that the niche is also defective. In summary, a p53-dependent increase of ROS levels in HSCs obstructs their normal function, although exactly how this happens is still not perfectly clear.
Quiescence Markers in Mdm2−/−p53515C/515C Bone Marrows
Slow cycling hematopoietic stem cells are associated with hypoxic regions of capillaries suggesting that low ROS maintains quiescence and self-renewal.44 Necdin and Gfi-1 are newly identified targets of p53 in HSCs that induce HSC quiescence.24 In Mdm2−/−p53515C/515C mice, the frequency of LKS is quite low impeding the measurement of RNA levels of these target genes by Real Time-quantitative Polymerase Chain Reaction (RT-qPCR). We therefore isolated RNA from whole bone marrows of neonatal mice. We examined Necdin and Gfi-1 levels in whole tissues and did not see any differences in Necdin levels at P6, but Gfi1 levels were lower in Mdm2−/−p53515C/515C cells as compared with control Mdm2+/−p53515C/515C cells (Fig. 2). The significance of lower Gfi1 in Mdm2−/−p53515C/515C mice warrants further investigations.
Figure 2.
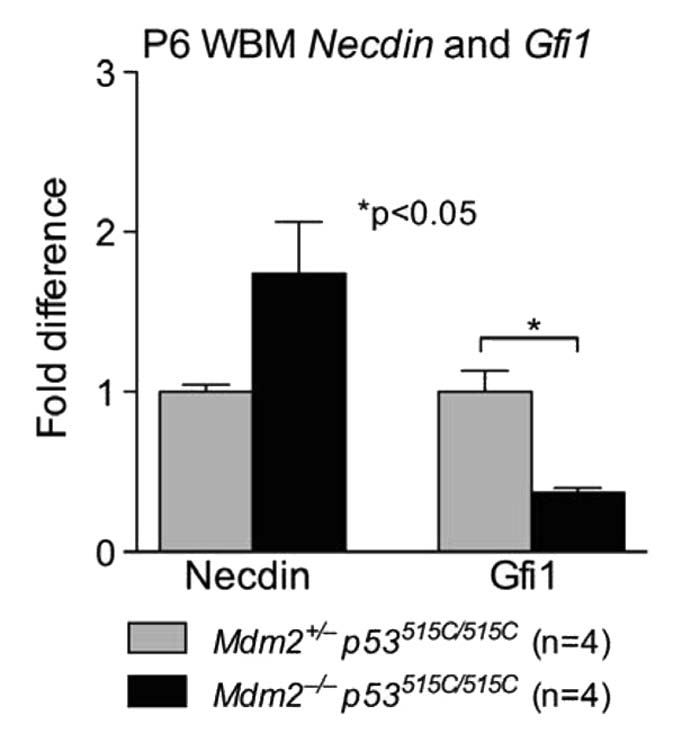
Necdin and Gfi1 RNA levels in Mdm2−/− p53515C/515C mice compared with control Mdm2+/− p53515C/515C littermates. Necdin levels were not different among genotypes, but Gfi1 levels were significantly lower in moribund mice.
p16: At the Crossroads of ROS, p53 and HSCs
p16 overexpression in HSCs inhibits their proliferation purportedly via ROS regulatory pathways that are still poorly understood.36,39,45 Our study further supports the possibility that ROS induces p16 expression and exacerbates the phenotype in Mdm2−/−p53515C/515C mice. p16 is also implicated in senescence.46 Senescent cells generate ROS which could create a positive feedback loop further stabilizing p53R172P and abrogating hematopoiesis.33,46 Since p16 deletion only partially rescues hematopoiesis and the survival of Mdm2−/−p53515C/515C mice, further studies are needed to address the importance of other players affecting HSC function. In a recent study, Cited2 deletion increased p53 levels dampening the HSC numbers which could be rescued by deletion of both p16 and p19ARF.47 It would be interesting to measure ROS levels in Cited2−/− HSCs and bone marrows to delineate the physiological mechanisms for HSC loss.
c-kit Downregulation in Mdm2−/−p53515C/515C Bone Marrows
c-kit is a major tyrosine kinase receptor expressed in hematopoietic stem cells and myeloid progenitors.48 c-kit activation induces self-renewal, differentiation and expansion of hematopoietic stem cells. Loss of c-kit abrogates hematopoiesis, and is detrimental to development; its overexpression is oncogenic.49,50 Specifically, c-kit mutant mice have severe hematopoietic defects and die shortly after birth, reminiscent of Mdm2−/−p53515C/515C mice.51,52 Hence, we measured c-kit RNA levels using RT-PCR in P6 and P10 bone marrows of Mdm2−/−p53515C/515C and found a 5-fold (p = 0.0001) and 7-fold (p = 0.0003) drop in c-kit RNA levels compared with control Mdm2+/−p53515C/515C littermates (Fig. 3A and B).
Figure 3.
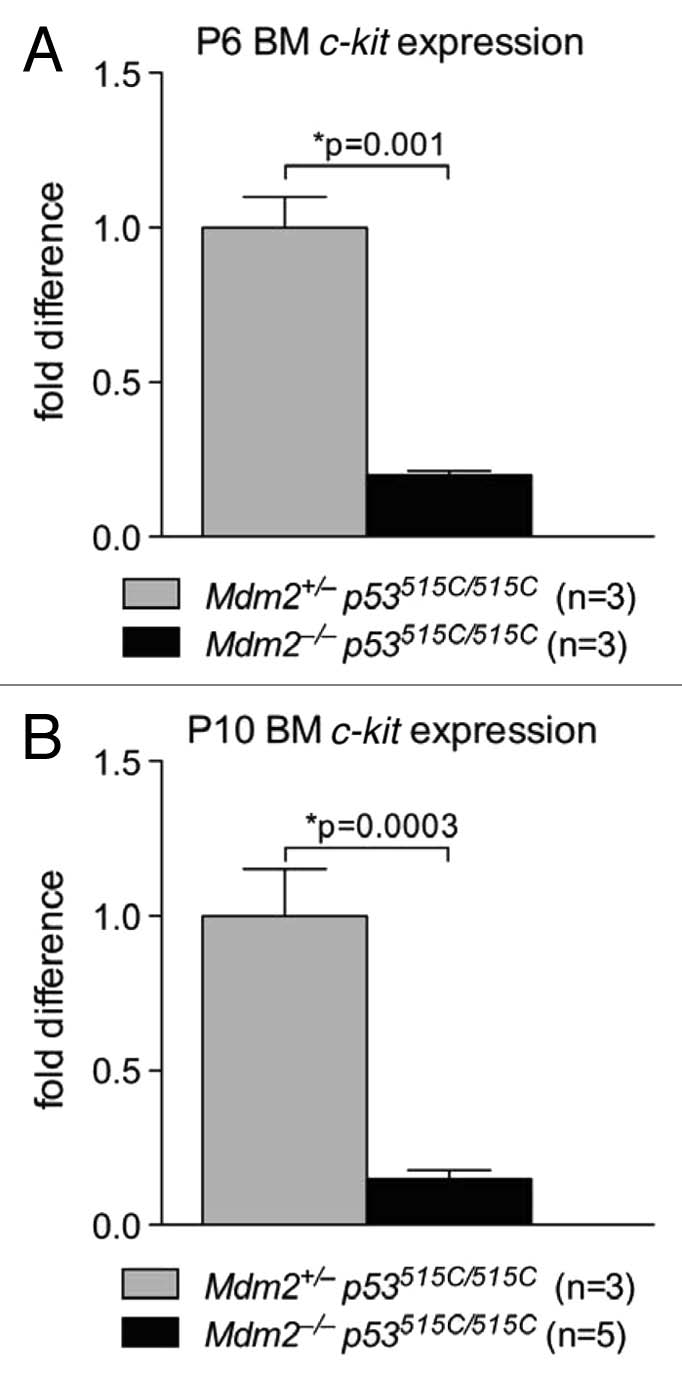
c-kit mRNA levels are significantly lower at P6 and P10 in Mdm2−/− p53515C/515C bone marrows. c-kit mRNA expression measured with RT-qPCR at P6 (A) and P10 (B) of whole bone marrow (BM) cells from Mdm2+/− p53515C/515C and Mdm2−/− p53515C/515C mice. Error bars represent standard error of the mean.
We therefore hypothesized that p53R172P binding to a c-kit response element could be inhibiting its transcription. We screened the promoter region upstream of c-kit transcriptional start site (∼5,000 bases) with 7 different ChIP primer sets (Table 1). However, we could not detect any significant binding of p53 on the c-kit promoter (data not shown). Thus the mechanism by which c-kit is regulated is unknown. Regardless, we believe that loss of c-kit expression is a significant modifier of the Mdm2−/−p53515C/515C hematopoietic phenotype given the consistent and severe decrease in its levels.
Table 1.
List of primers used to span the c-kit promoter for p53 binding
| Primer | Forward primer | Reverse primer |
| ckitRE 1 | CTC CAG GTG CGC TAT GCA | TGG GTG CTT TGC CTG TTT CT |
| ckitRE 2 | TGT AGC GCC AGC ACT TGT G | AGC TGA GGA TGG CTT TGA ACT C |
| ckitRE 3 | CCA ACA GAG CAA CAC AAA GCA | GAA TA G GTT TCC CCC TCC AT C T |
| ckitRE 4 | GCG CAG CGT TCA ACC TGT A | CCT GAG ACA CCC ACC TCA CA |
| ckitRE 5 | CAG GGC TCC CAT CTC AGA TC | AGC GAG GCA CTG TTA GTA GAT GTG |
| ckitRE 8 | TGG AGA AA C TGA GCA TGA AAA ATT | GCA CCC TGA CCT CAG AAA AG |
| ckitRE 9 | CCG GTG GTT GTC CTT TAT TGT C | GCC ACG AGC GCA TTA GGT A |
| Puma | GGA CGG TCG CCT TGC A | CAC CTT AGT CCC AGT GAT GAA A |
| AchR | CC TCC CCC AA C TCC ACT TTT | GGA GGT TGG AGG GAG AA G GA |
Puma and Acetylcholine Receptor (AchR) primers sets were used as positive and negative controls respectively.
Implications to Leukemia Treatment
Aberrant regulation/expression of p53 is common in hematologic malignancies. p53 deletions and mutations have been reported in acute leukemias and in chronic myelogenous leukemia in blast crisis.53 Self-renewing normal HSCs and leukemia stem cells (LSCs) share multiple features.54,55 For instance, both cell types express CD34, lose expression of CD38, are dormant early and can give rise to progeny that are more differentiated.54 Other studies have also indicated that LSCs could very well originate from normal HSCs.56 These data, however, do not rule out the possibility of transformation of a committed progenitor or differentiated cell in the blood lineage into a LSC. Leukemiagenesis proceeds via uncontrolled proliferation and differentiation, two pathways which are regulated by p53. Accordingly, it is plausible that pathways that sensitize normal HSCs could have a similar effect on LSCs. Our results, that Mdm2 acts as a critical regulator of p53 in the hematopoietic system, imply that careful monitoring of hematopoiesis should occur in leukemia patients when treating with drugs that target p53-Mdm2 association or re-activate wild-type p53.
Acknowledgments
This work was supported by NIH grant CA82577 to G.L. and the Cancer Center Support Grant CA16672 to MDACC.
References
- 1.Junttila MR, Evan GI. p53—a Jack of all trades but master of none. Nat Rev Cancer. 2009;9:821–829. doi: 10.1038/nrc2728. [DOI] [PubMed] [Google Scholar]
- 2.Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–208. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- 3.Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–206. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- 4.Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, et al. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet. 2001;29:92–95. doi: 10.1038/ng714. [DOI] [PubMed] [Google Scholar]
- 5.Mendrysa SM, McElwee MK, Michalowski J, O'Leary KA, Young KM, Perry ME. mdm2 Is critical for inhibition of p53 during lymphopoiesis and the response to ionizing irradiation. Mol Cell Biol. 2003;23:462–472. doi: 10.1128/MCB.23.2.462-73.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Terzian T, Wang Y, Van Pelt CS, Box NF, Travis EL, Lozano G. Haploinsufficiency of Mdm2 and Mdm4 in tumorigenesis and development. Mol Cell Biol. 2007;27:5479–5485. doi: 10.1128/MCB.00555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dumble M, Moore L, Chambers SM, Geiger H, Van Zant G, Goodell MA, et al. The impact of altered p53 dosage on hematopoietic stem cell dynamics during aging. Blood. 2007;109:1736–1742. doi: 10.1182/blood-2006-03-010413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leonova KI, Shneyder J, Antoch MP, Toshkov IA, Novototskaya LR, Komarov PG, et al. A small molecule inhibitor of p53 stimulates amplification of hematopoietic stem cells but does not promote tumor development in mice. Cell Cycle. 2010;9:1434–1443. doi: 10.4161/cc.9.7.11508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shao L, Sun Y, Zhang Z, Feng W, Gao Y, Cai Z, et al. Deletion of proapoptotic Puma selectively protects hematopoietic stem and progenitor cells against high-dose radiation. Blood. 2010;115:4707–4714. doi: 10.1182/blood-2009-10-248872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirsch DG, Santiago PM, di Tomaso E, Sullivan JM, Hou WS, Dayton T, et al. p53 controls radiation-induced gastrointestinal syndrome in mice independent of apoptosis. Science. 2010;327:593–596. doi: 10.1126/science.1166202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orkin SH, Zon LI. Hematopoiesis and stem cells: plasticity versus developmental heterogeneity. Nat Immunol. 2002;3:323–328. doi: 10.1038/ni0402-323. [DOI] [PubMed] [Google Scholar]
- 13.Orkin SH, Zon LI. SnapShot: hematopoiesis. Cell. 2008;132:712. doi: 10.1016/j.cell.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 14.Mikkola HK, Orkin SH. The journey of developing hematopoietic stem cells. Development. 2006;133:3733–3744. doi: 10.1242/dev.02568. [DOI] [PubMed] [Google Scholar]
- 15.Morrison SJ, Hemmati HD, Wandycz AM, Weissman IL. The purification and characterization of fetal liver hematopoietic stem cells. Proc Natl Acad Sci USA. 1995;92:10302–10306. doi: 10.1073/pnas.92.22.10302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ema H, Nakauchi H. Expansion of hematopoietic stem cells in the developing liver of a mouse embryo. Blood. 2000;95:2284–2288. [PubMed] [Google Scholar]
- 17.Lin T, Chao C, Saito S, Mazur SJ, Murphy ME, Appella E, et al. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol. 2005;7:165–171. doi: 10.1038/ncb1211. [DOI] [PubMed] [Google Scholar]
- 18.Qin H, Yu T, Qing T, Liu Y, Zhao Y, Cai J, et al. Regulation of apoptosis and differentiation by p53 in human embryonic stem cells. J Biol Chem. 2007;282:5842–5852. doi: 10.1074/jbc.M610464200. [DOI] [PubMed] [Google Scholar]
- 19.Utikal J, Polo JM, Stadtfeld M, Maherali N, Kulalert W, Walsh RM, et al. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature. 2009;460:1145–1148. doi: 10.1038/nature08285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong H, Takahashi K, Ichisaka T, Aoi T, Kanagawa O, Nakagawa M, et al. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature. 2009;460:1132–1135. doi: 10.1038/nature08235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawamura T, Suzuki J, Wang YV, Menendez S, Morera LB, Raya A, et al. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature. 2009;460:1140–1144. doi: 10.1038/nature08311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Collado M, Villasante A, Strati K, Ortega S, Canamero M, et al. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature. 2009;460:1136–1139. doi: 10.1038/nature08290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marión RM, Strati K, Li H, Murga M, Blanco R, Ortega S, et al. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature. 2009;460:1149–1153. doi: 10.1038/nature08287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y, Elf SE, Miyata Y, Sashida G, Huang G, Di Giandomenico S, et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. 2009;4:37–48. doi: 10.1016/j.stem.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, Elf SE, Asai T, Miyata Y, Sashida G, Huang G, et al. The p53 tumor suppressor protein is a critical regulator of hematopoietic stem cell behavior. Cell Cycle. 2009;8:3120–3124. doi: 10.4161/cc.8.19.9627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fuster JJ, Andres V. A role for miR-33 in p53 regulation: New perspectives for hematopoietic stem cell research. Cell Cycle. 2010;9:3397–3398. doi: 10.4161/cc.9.17.13070. [DOI] [PubMed] [Google Scholar]
- 27.Herrera-Merchan A, Cerrato C, Luengo G, Dominguez O, Piris MA, Serrano M, et al. miR-33-mediated downregulation of p53 controls hematopoietic stem cell self-renewal. Cell Cycle. 2010;9:3277–3285. doi: 10.4161/cc.9.16.12598. [DOI] [PubMed] [Google Scholar]
- 28.Meletis K, Barnabe-Heider F, Carlen M, Evergren E, Tomilin N, Shupliakov O, et al. Spinal cord injury reveals multilineage differentiation of ependymal cells. PLoS Biol. 2008;6:182. doi: 10.1371/journal.pbio.0060182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donehower LA, Lozano G. 20 years studying p53 functions in genetically engineered mice. Nat Rev Cancer. 2009;9:831–841. doi: 10.1038/nrc2731. [DOI] [PubMed] [Google Scholar]
- 30.Liu G, Parant JM, Lang G, Chau P, Chavez-Reyes A, El-Naggar AK, et al. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet. 2004;36:63–68. doi: 10.1038/ng1282. [DOI] [PubMed] [Google Scholar]
- 31.Post SM, Quintas-Cardama A, Terzian T, Smith C, Eischen CM, Lozano G. p53-dependent senescence delays Emu-myc-induced B-cell lymphomagenesis. Oncogene. 2010;29:1260–1269. doi: 10.1038/onc.2009.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu G, Terzian T, Xiong S, Van Pelt CS, Audiffred A, Box NF, et al. The p53-Mdm2 network in progenitor cell expansion during mouse postnatal development. J Pathol. 2007;213:360–368. doi: 10.1002/path.2238. [DOI] [PubMed] [Google Scholar]
- 33.Abbas HA, Maccio DR, Coskun S, Jackson JG, Hazen AL, Sills TM, et al. Mdm2 is required for survival of hematopoietic stem cells/progenitors via dampening of ROS-induced p53 activity. Cell Stem Cell. 2010;7:606–617. doi: 10.1016/j.stem.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vigneron A, Vousden KH. p53, ROS and senescence in the control of aging. Aging (Albany NY) 2010;2:471–474. doi: 10.18632/aging.100189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 36.Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431:997–1002. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- 37.Chen C, Liu Y, Liu R, Ikenoue T, Guan KL, Zheng P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med. 2008;205:2397–2408. doi: 10.1084/jem.20081297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen C, Liu Y, Zheng P. The axis of mTOR-mitochondria-ROS and stemness of the hematopoietic stem cells. Cell Cycle. 2009;8:1158–1160. doi: 10.4161/cc.8.8.8139. [DOI] [PubMed] [Google Scholar]
- 39.Akala OO, Park IK, Qian D, Pihalja M, Becker MW, Clarke MF. Long-term haematopoietic reconstitution by Trp53−/−p16Ink4a−/−p19Arf−/− multipotent progenitors. Nature. 2008;453:228–232. doi: 10.1038/nature06869. [DOI] [PubMed] [Google Scholar]
- 40.Owusu-Ansah E, Banerjee U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature. 2009;461:537–541. doi: 10.1038/nature08313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shaulsky G, Goldfinger N, Peled A, Rotter V. Involvement of wild-type p53 in pre-B-cell differentiation in vitro. Proc Natl Acad Sci USA. 1991;88:8982–8986. doi: 10.1073/pnas.88.20.8982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X, Kua HY, Hu Y, Guo K, Zeng Q, Wu Q, et al. p53 functions as a negative regulator of osteoblastogenesis, osteoblast-dependent osteoclastogenesis and bone remodeling. J Cell Biol. 2006;172:115–125. doi: 10.1083/jcb.200507106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Molchadsky A, Shats I, Goldfinger N, Pevsner-Fischer M, Olson M, Rinon A, et al. p53 plays a role in mesenchymal differentiation programs, in a cell fate dependent manner. PLoS ONE. 2008;3:3707. doi: 10.1371/journal.pone.0003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suda T, Arai F, Hirao A. Hematopoietic stem cells and their niche. Trends Immunol. 2005;26:426–433. doi: 10.1016/j.it.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 45.Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302–305. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- 46.Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 47.Kranc KR, Schepers H, Rodrigues NP, Bamforth S, Villadsen E, Ferry H, et al. Cited2 is an essential regulator of adult hematopoietic stem cells. Cell Stem Cell. 2009;5:659–665. doi: 10.1016/j.stem.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Okada S, Nakauchi H, Nagayoshi K, Nishikawa S, Nishikawa S, Miura Y, et al. Enrichment and characterization of murine hematopoietic stem cells that express c-kit molecule. Blood. 1991;78:1706–1712. [PubMed] [Google Scholar]
- 49.Masson K, Ronnstrand L. Oncogenic signaling from the hematopoietic growth factor receptors c-Kit and Flt3. Cell Signal. 2009;21:1717–1726. doi: 10.1016/j.cellsig.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 50.Kent D, Copley M, Benz C, Dykstra B, Bowie M, Eaves C. Regulation of hematopoietic stem cells by the steel factor/KIT signaling pathway. Clin Cancer Res. 2008;14:1926–1930. doi: 10.1158/1078-0432.CCR-07-5134. [DOI] [PubMed] [Google Scholar]
- 51.Waskow C, Paul S, Haller C, Gassmann M, Rodewald HR. Viable c-Kit(W/W) mutants reveal pivotal role for c-kit in the maintenance of lymphopoiesis. Immunity. 2002;17:277–288. doi: 10.1016/S1074-7613(02)00386-2. [DOI] [PubMed] [Google Scholar]
- 52.Trevisan M, Yan XQ, Iscove NN. Cycle initiation and colony formation in culture by murine marrow cells with long-term reconstituting potential in vivo. Blood. 1996;88:4149–4158. [PubMed] [Google Scholar]
- 53.Drexler HG, Fombonne S, Matsuo Y, Hu ZB, Hamaguchi H, Uphoff CC. p53 alterations in human leukemia-lymphoma cell lines: in vitroartifact or prerequisite for cell immortalization? Leukemia. 2000;14:198–206. doi: 10.1038/sj.leu.2401604. [DOI] [PubMed] [Google Scholar]
- 54.Passegué E, Jamieson CH, Ailles LE, Weissman IL. Normal and leukemic hematopoiesis: are leukemias a stem cell disorder or a reacquisition of stem cell characteristics? Proc Natl Acad Sci USA. 2003;100:11842–11849. doi: 10.1073/pnas.2034201100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Passegué E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med. 2005;202:1599–1611. doi: 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miyamoto T, Weissman IL, Akashi K. AML1/ETO-expressing nonleukemic stem cells in acute myelogenous leukemia with 8;21 chromosomal translocation. Proc Natl Acad Sci USA. 2000;97:7521–7526. doi: 10.1073/pnas.97.13.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]