

Abstract

The development of a novel intermolecular oxidative amination reaction, a synthetic transformation that involves the simultaneous functionalization of both an N-H and C-H bond, is described. The process, which is mediated by an I(III) oxidant and contains no metal catalysts, provides a rapid and green method for synthesizing protected anilines from simple arenes and phthalimide. Mechanistic investigations indicate that the reaction proceeds via nucleophilic attack of the phthalimide on an aromatic radical cation, as opposed to the electrophilic aromatic amination that has been reported for other I(III) amination reactions. The application of this new reaction to the synthesis of a variety of substituted aniline derivatives is demonstrated.
1. INTRODUCTION
The formation of carbon-carbon (C-C) bonds via the oxidative cross-coupling of two carbon-hydrogen (C-H) bonds has recently become a field of intense interest and has resulted in the discovery of numerous novel synthetic methods.1 The analogous technology for the oxidative cross-coupling of C-H and nitrogen-hydrogen (N-H) bonds to form carbon-nitrogen (C-N) bonds would also be an important synthetic advance, as amine and amide functional groups are ubiquitous in biologically active molecules (Figure 1). However, a relatively small amount of work has focused on the development of oxidative methods for constructing N-arylamines and amides via tandem C-H/N-H activation. The majority of literature in this field describes the insertion of nitrenoid intermediates into C-H bonds, mediated by transition metal catalysts.2 Cu and Pdcatalyzed reactions, as well as metal-free conditions, have also been recently explored, but these methods are limited to the intramolecular synthesis of carbazoles, oxindoles and diazapenones.3-5 The only reported examples of intermolecular oxidative amination involve azole-type heterocycles and proceed by attack of an amine nucleophile on the substrate's imine moiety.6,7 Herein, we disclose a novel intermolecular reaction that oxidatively constructs the C-N bond of phthalimide-protected anilines via the tandem activation of N-H and C-H bonds.
Figure 1.

Examples of high-value molecules containing anilines
2. DISCUSSION
2.1. Reaction Discovery
We recently discovered that heating a solution of phthalimide (1) and PhI(OAc)2 in benzene provides the phthalimide-protected aniline (2) in an 88% yield (Table 1, entry 4). We initially explored the potential of Cu(I)/(III)-catalysts to perform this oxidative amination, but quickly realized that a metal catalyst was not necessary.5 We are confident that trace metals are not the cause of the transformation, as reactions performed in new, acid washed flasks were similar in both yield and rate to those performed in old flasks. Furthermore, reagents from different commercial sources perform similarly.
Table 1.
Discovery of the intermolecular oxidative aminatio[a]
 | |||||
|---|---|---|---|---|---|
| Oxidant (equiv) | Solvent | Temp. [°C] | Time [h] | Yield [%][c] | |
| 1 | PIDA (1.5) | PhH | 145[b] | 12 | 26 |
| 2 | PIDA (1.5) | PhH | 145 | 3 | 28 |
| 3 | PIDA (2.0) | PhH | 145 | 3 | 45 |
| 4 | PIDA (2.5) | PhH | 145 | 3 | 88 |
| 5 | PIDA (2.5) | PhH | 120 | 3 | no reaction |
| 6 | PIDA (2.5) | TFE | 145 | 3 | 13 |
| 7 | PIDA (2.5) | DMF | 145 | 3 | 4[d] |
| 8 | PIDA (2.5) | DMSO | 145 | 3 | no reaction |
| 9 | PIDA (2.5) | MeCN | 145 | 3 | 51 |
| 10 | NCS (2.5) | MeCN | 145 | 3 | 3[d] |
| 11 | Oxone (2.5) | MeCN | 145 | 3 | no reaction |
| 12 | IBX (2.5) | MeCN | 145 | 3 | no reaction |
| 13 | PIFA (1.0) | PhH | 145 | 3 | 5[d] |
| 14 | PIFA (1.25) | PhH | 145 | 3 | decomposition |
| 15 | PIFA (1.0) | PhH | 100 | 3 | Trace |
| 16 | PIFA (1.0) | PhH | 25[b] | 3 | 0 |
| 17 | PIFA (1.0) | TFE | 145 | 3 | 3.5[d] |
| 18 | PIFA (1.0) | MeCN | 145 | 3 | 3.5[d] |
General reaction conditions: 1 (0.68 mmol), oxidant (1.5 - 2.5 equiv), benzene (1.5 equiv or solvent), solvent (4 mL), microwave heating. PIDA = phenyliodine(III) diacetate, PIFA = phenyliodine(III) bis(trifluoroacetate), NCS = N-chlorosuccinimide, IBX = 2-iodoxybenzoic acid, Oxone = potassium peroxymonosulfate, TFE = 2,2,2-trifluoro-ethanol.
Oil bath.
Yield of isolated product after column chromatography.
GC yield calculated using dodecane as internal standard.
While optimizing the process, we quickly discovered that the best conditions employed microwave heating and 2.5 equivalents of the I(III) oxidant, phenyliodine(III) diacetate (PIDA). Less than two equivalents of PIDA did not allow the reactions to proceed to completion (compare entries 2 and 3).
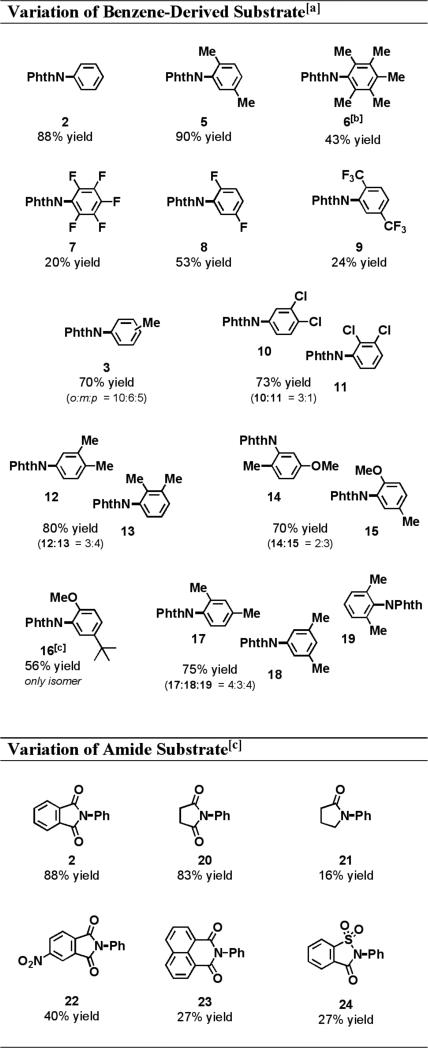
Importantly, we found that the arene substrate does not need to be in large excess (i.e. solvent) for the intermolecular oxidative amination reaction to occur. This is in stark contrast to many of the oxidative arylation reactions that have been previously studied.8 As shown in Table 1, entry 9, acetonitrile was used as the solvent and the concentration of the arene substrate was lowered to a near-stoichiometric level (1.5 equiv). Under these conditions, the yield for the amination of benzene dropped from 88% to 51%, but we hypothesized that this was due to the volatility of the arene. This was verified by the reaction of the less volatile substrate, p-xylene, which provided an 80% yield (Scheme 1A).
Scheme 1.

Oxidative amination of substituted arenes
Other solvent and oxidant combinations proved to be less effective. In particular, 2,2,2-trifluoroethanol (TFE), a solvent that has been shown to be particularly compatible with I(III) mediated arene substitutions provided a minimal yield (entry 6). The fluorinated derivative of PIDA, phenlyiodine(III) bis(trifluoroacetate) (PIFA), proved to be too harsh of an oxidant for these reactions. Any loading of the oxidant in excess of 1 equiv resulted in a complex mixture of products. Lowering the reaction temperature and altering the co-solvent, also failed to provide the desired anilne 2 (entries 13-18).
To determine whether the arene that participated in the oxidative amination originated from the benzene solvent or the oxidant, a crossover experiment was performed (Scheme 1B). When toluene was used as the solvent and PhI(OAc)2 was the oxidant, the reaction did not produce any N-phenylphthalimide (2). Rather, an inseparable mixture of regiomers arising from the oxidative amination of the sp2-hybridized C-H bonds of toluene was observed (3). This confirmed our hypothesis that the source of the aryl group that forms the new C-N bond was the solvent, not the phenyl group that resides on the oxidant.
Cleavage of the phthalimide protecting group allowed for the determination of the regiomeric ratios by comparison to the commercially available toluidine isomers. Interestingly, the major product of the reaction was o-toluidine (4). Amination of the sp3-hybridized C-H bonds was not observed.
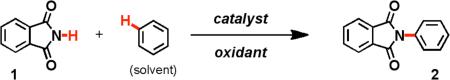
2.2. Substrate Scope
In addition to benzene, toluene and p-xylene, a variety of other simple arenes could be oxidatively coupled to phthalimide (Table 2). Sterically encumbered arenes (6) and those that contain electron-withdrawing groups (7-11) were oxidatively aminated, albeit at reduced yields. Like toluene, monosubstituted and asymmetrically disubstituted benzenes produced mixtures of protected aniline products (10-19). Electron-rich heterocycles, such as furan, decomposed under the reaction conditions, and electron-poor hetero-cycles, such as benzoxazole failed to produce any aminated products.
Table 2.
Substrate scope of the intermolecular oxidative amination
|
The regioselectivity of these amination reactions appears to be slightly directed by electronic factors. For example, p-methylanisole was 50% more likely to be aminated at the position ortho to the larger, but more electron donating, methoxy group (15). Likewise, both the ortho and para-positions of toluene were 67% more likely to be aminated than the meta-position. The only exception to this rule was the amination of p-tert-butylanisole (16), where the size of the tert-butyl group prevented ortho-amination.
Other amine sources, in addition to phthalimide, were also investigated. Succinimide also provided very good yields of oxidative amination products (20), while pyrrolidin-2-one performed the oxidative amination reaction, but resulted in a poor yield (16%, 21). N-Tosylamide and pyrrolidine failed to oxidatively aminate benzene. These data led us to conclude that the requirements for the amine coupling partner were twofold: 1) the N-H bond must be relatively acidic (phthalimide pKa = 8.3) and 2) the amine coupling partner must be secondary, preferably a cyclic imide. In keeping with these requirements, other imides with acidic N-H bonds performed the oxidative amination reaction, albeit in lower yields (22-24). Surprisingly, acyclic imides, such as diacetamide, N-acetylanilines and hetercycles such as indole and benzimidazole produced only trace amounts of products.
2.3. Competition and Kinetic Studies
Competition studies were performed to explore the mechanism of the novel process (Table 3). In all cases, the amination of electron-rich arenes was favored. In particular, the competition between p-xylene and p-difluorobenzene shows a dramatic preference for the amination of the electron rich p-xylene substrate (entry 2).
Table 3.
Competition reactions
 | ||||
|---|---|---|---|---|
 |
 |
|||
| 1 | p-xylene | benzene | 71 | 29 |
| 2 | p-xylene | p-difluoro-benzene | 96 | 4 |
| 3 | benzene | p-difluoro-benzene | 86 | 14 |
[a] Mole fractions determined by GC/MS.
The kinetic isotope effect (KIE) of the reaction was assessed using a competition experiment between equimolar amounts of benzene and benzene-d6. The near-unity KIE of 1.03 implies that C-H bond breaking is not involved in the rate-determining step of the reaction. This is also a stark contrast with the literature describing oxidative arylation processes for forming C-C bonds, where C-H bond breaking is often rate limiting, as indicated by large KIEs.9 The KIE of the reaction involving equimolar amounts of phthalimide and phthalimide-d was observed to be 0.98, which indicates that the cleavage of the N-H bond is also not rate-limiting.
Kinetic analysis of the oxidative amination provided more mechanistic clues. The reaction rate was unaffected by changing the concentration of the arene but was dependent on the concentration of the both oxidant and phthalimide. Both reagents demonstrated first-order dependencies (Figure 2).
Figure 2.

Kinetic analysis of oxidative amination reaction with various amounts of PhI(OAc)2 (oxidant) and phthalimide.
The preference for imides and electron-rich arenes is consistent with two possible mechanisms (Scheme 2). The first involves the in situ formation of a PhI(OAc)(NR2) species (25 and 26), which is highly electrophilic, functioning essentially as an R2N+ equivalent.10 Electrophilic aromatic substitution then forms the desired C-N bond (2) (Scheme 2A).11 Alternatively, a single electron transfer may occur, forming an ion pair with the arene substrate and the I(III) reagent. The radical cation 27 could then undergo a nucleophilic attack by phthalimide (or its anion), giving rise to the desired product (3, Scheme 2B). Kita has previously described such a mechanism for oxidative reactions between arenes and soft nucleophiles such as β-diketones and TMS-N3.12
Scheme 2.

Two possible mechanisms for the oxidative amination
N-centered radicals have also been proposed in I(III) mediated C-N forming reactions,13 but they are not likely to be involved in the reactions shown in Table 1. The weaker C-H bonds of the methyl group of toluene were not aminated, as one would expect for reactions involving N-centered radicals.
2.5 Proposed Mechanism
Based these data, a mechanism involving electrophilic aromatic substitution (EAS) seems unlikely, as the ratio of products obtained from the oxidative amination of toluene (3) are not indicative of the expected EAS regioselectivity (Scheme 2A). While the ortho and para- aminated products were somewhat favored in the reaction of arenes containing electron donating groups, reactions operating via SEAr mechanisms tend to form little, if any, meta-substituted products.
Alternatively, PhI(OAc)2 could oxidize the electron-rich arene substrate to a radical cation (27), and nucleophilic attack on such a radical cation should be relatively non-regioselective (Scheme 2B). The consequent radical intermediate (28) could then be oxidized to form a Wheland-type arenium ion. These two individual oxidation steps may indicate why two equivalents of PhI(OAc)2 are required to achieve complete conversion. The fates of the reduced iodine intermediates are less clear, but the two other by-products that are observed in all of these reactions are iodobenzene and phenyl acetate.
Consequently, we hypothesize that a single electron transfer mechanism is operating in these oxidative amination reactions. This hypothesis is supported by the observation that the reaction was partially inhibited by BHT and completely inhibited by TEMPO, common radical inhibitors. Our hypothesis is supported by the prior work of Kita, who has extensively shown that arene radical cation intermediates such as 27 are formed by the action of I(III) oxidants and can be directly observed by EPR spectroscopy.12
It should be noted that Cho and Chang have recently proposed an electrophilic mechanism for the same reaction described herein.7b This hypothesis was supported by the observation of 25 by mass spectrometry. In comparing our data with those of Kita and both Cho and Chang, it seems likely that several I(III) species are simultaneously present in solution. However, we reason that the observed regioselectivites for aminations of monosubstituted arenes that were observed by both us and Cho and Chang are best explained by the intermediacy of a radical cation intermediate (27).
Furthermore, the unique regioselectivities that were observed in the oxidative amination of toluene (o:m:p = 10:6:5) corroborate the intermediacy of an aromatic radical cation (Scheme 3). The yields of ortho and para-aminated products, which arise from the nucleophilic attack on resonance forms 27b and 27c, are statistically equivalent. This seems plausible as both contain a tertiary radical and a secondary carbocation. Additionally, reactions with p-methylanisole and p-tert-butylanisole (28) not only produced the aforementioned aminated products (14-16), but substitution products, arising from SNAr-type attack on the tertiary cation intermediate (29b) followed by ejection of the methoxide leaving group (30), were also observed (Scheme 4).
Scheme 3.

Resonance structures of the aryl radical cation explain the observed regioselectivities.
Scheme 4.

Formation of substitution products corroborates the intermediacy of arene radical cations.
3. CONCLUSION
In conclusion, the ability to oxidatively couple phthalimide to unfunctionalized arenes is a useful method for synthesizing anilines that is orthogonal to conventional amination techniques, which rely on electrophilic nitration/reduction strategies or metal catalyzed coupling of pre-functionalized arenes. Phthalimide, in particular, is an ideal starting point for the development of the aforementioned oxidative amination technology. It is commercially available, inexpensive, easy to handle and, once coupled, it can be readily converted to a primary amine, which can be further derivatized. Additionally, N-arylphthalimides like those shown in Table 1 have recently been shown to have anti-cancer activity.14 Future work in our laboratory will be dedicated to the mechanistic study and application of this unique method for constructing C-N bonds.
4. EXPERIMENTAL SECTION
Representative Procedure with Arene Solvent
A magnetically stirred solution of the imide substrate (0.68 mmol) and iodobenzene diacetate (1.7 mmol) in 4 mL of the simple arene substrate was microwave heated at 145 °C for 3h. The excess solvent from the mixture was removed at reduced pressure by rotary evaporation, and the crude product was purified by column chromatography.
N-phenylphthalimide 2. Yield = 0.133 g (88 %). The NMR spectra matched the previously published data.17 Rf = 0.31, hexane/ethyl acetate (8:2 v/v). 1H-NMR (400 MHz, CDCl3): δ = 7.39-7.53 (m, 5H), 7.80 (dd, J = 5.4 Hz, 2.8 Hz, 2H), 7.96 (dd, J = 5.6 Hz, 3.2 Hz, 2H). 13C-NMR (100 MHz, CDCl3): δ = 123.7, 126.5, 128.1, 129.1, 131.7, 134.4, 167.30. LRMS EI (m/z): [M+] calculated for C14H9NO2 223.06, observed 223.10 m/z.
3 (o: m: p = 10:6:5) 0.1071 g (70 %). The isomers were identified by comparing with known NMR spectra. 18 Rf =0.2, hexane/ethyl acetate (9:1 v/v) . 1H-NMR (300 MHz, CDCl3): δ = 2.21 (s, 3H), 2.41 (s, 1H), 2.42 (s, 2H), 7.18 – 7.43 (m, 10H), 7.80 (dd, J = 5.4, 3.1 Hz, 5H), 7.92 – 7.99 (m, 4H). 13C NMR (75 MHz, CDCl3): δ = 18.06, 21.23, 21.42, 123.70, 123.72, 123.78, 126.47, 126.89, 127.29, 128.73, 128.95, 129.07, 129.47, 129.80, 130.57, 131.17, 131.48, 131.80, 132.02, 134.33, 134.35, 136.55, 138.20, 139.15, 167.37. LRMS EI (m/z): [M+] calc'd for C14H9NO2 237.08, observed 237.10 m/z.
2-(2, 5-dimethylphenyl) isoindoline-1, 3-dione 5. Yield= 0.1535g (90 %). Rf =0.19, hexane/ethyl acetate (9:1 v/v).1HNMR (300 MHz, CDCl3): δ = 2.16(s, 3H), 2.36 (s, 3H), 7.17-7.28 (m, 3H), 7.80 (dd, J = 6 Hz, J = 3 Hz, 2H), 7.97 (dd, J = 3 Hz, J = 3 Hz, 2H). 13C-NMR (75 MHz, CDCl3): δ = 17.56, 20.83, 123.74, 129.15, 130.29, 130.37, 130.95, 132.05, 133.27, 134.29, 136.72, 167.47. LRMS EI (m/z): [M+] calc'd for C16H13NO2 251.09, observed 251.10 m/z.
2-(perfluorophenyl)isoindoline-1,3-dione 7. Yield=7 0.04 g (20%). Rf= 0.18, hexane/ethyl acetate (9:1 v/v). 1H-NMR (300 MHz, CDCl3): δ = 7.87 (dd, J = 3 Hz, J = 3 Hz, 2H), 8.01 (dd, J = 3 Hz, J = 3 Hz, 2H). 13C-NMR (75 MHz, CDCl3): δ = 123.89, 123.91, 124.35, 124.5, 125.82, 128.13, 131.69, 134.62, 134.88, 134.95, 135.05, 138.27. LRMS EI (m/z): [M+] calc'd for C14H4 F5NO2 313.02, observed 313.0 m/z
2-(2, 5-difluorophenyl) isoindoline-1, 3-dione 8. Yield = 0.0917g (53 %). Rf = 0.19, hexane/ethyl acetate (9:1 v/v). 1HNMR (300 MHz, CDCl3): δ = 7.12-7.26 (m, 3H), 7.82 (dd, J = 3 Hz, J = 3 Hz, 2H), 7.98 (dd, J = 3 Hz, J = 3 Hz, 2H). 13C NMR (75 MHz, CDCl3): δ = 116.00, 116.30, 116.64, 124.10, 128.44, 131.77, 134.67, 166.06, 167.22. LRMS EI (m/z): [M+] calc'd for C14H7F2NO2 259.04, observed 259.1 m/z.
2-(2, 5-bis (trifluoromethyl) phenyl) isoindoline-1, 3-dione 9. Yield= 0.2441 g (24%). Rf = 0.2187, hexane/ethyl acetate (9:1 v/v) . 1H-NMR (300 MHz, CDCl3): δ = 7.22-7.26 (m, 1H), 7.67 (s, 1H), 7.82-8.02 (m, 5H). 13C-NMR (75 MHz, CDCl3): δ= 123.89, 124.25, 127.18, 128.13, 129.00, 130.48, 131.72, 134.63, 134.82, 138.27, 166.66. LRMS EI (m/z): [M+] calc'd for C16H7 F6NO2 359.0, observed 359.0 m/z.
2-(3, 4-dichlorophenyl) isoindoline-1,3-dione 10. Yield = 0.1118 g (56 %). Rf = 0.19, hexane/ethyl acetate (9:1 v/v). 1HNMR (300 MHz, CDCl3): δ = 7.37 (dd, J = 8.6, 2.4 Hz, 1H), 7.58 (d, J = 8.6 Hz, 1H), 7.64 (d, J = 2.4 Hz, 1H), 7.86 – 7.79 (m, 2H), 7.98 – 7.94 (m, 2H). 13C-NMR (75 MHz, CDCl3): δ = 124.01, 125.53, 128.16, 130.70, 131.05, 131.43, 132.12, 133.02, 134.77, 166.63. LRMS EI (m/z): [M+] calc'd for C14H7F2NO2 291.0, observed 291.0 m/z.
2-(2,3-dichlorophenyl)isoindoline-1,3-dione 11. Yield = 0.034 g (17 %). Rf = 0.09, hexane/ethyl acetate (9:1 v/v). 1HNMR (300 MHz, CDCl3): δ = 7.27-7.4 (m, 2H), 7.59-7.62 (m, 1H), 7.83 (dd, J = 3, 3 Hz, 2H), 7.99 (dd, J = 3 Hz, 3 Hz, 2H). 13C-NMR (75 MHz, CDCl3): δ = 124.11, 127.71, 128.97, 131.32, 131.49, 131.75, 132.32, 134.35, 134.66, 166.37. LRMS EI (m/z): [M+] calc'd for C14H7F2NO2 291.0, observed 291.0 m/z.
14 and 15 Yield = 0.1287 g (70 %, 14:15 = 2:3). Rf = 0.1714, hexane/ethyl acetate (9:1 v/v) . 1H-NMR (300 MHz, CDCl3): δ = 2.13 (s, 2H), 2.34 (s, 3H), 3.78 (dd, J = 9.6, 2.1 Hz, 6H), 6.75 (d, J = 2.5 Hz, 1H), 6.95 (d, J = 8.4 Hz, 2H), 7.07 (s, 1H), 7.20 – 7.30 (m, 3H), 7.76-7.82 (m, 4H), 7.91 – 7.99 (m, 4H). 13C-NMR (75 MHz, CDCl3): δ = 17.15, 20.40, 55.45, 55.91, 112.02, 113.85, 115.65, 119.75, 123.66, 123.80, 128.29, 130.38, 131.16, 131.70, 131.96, 132.24, 134.10, 134.36, 153.22, 158.27, 167.30, 167.53. LRMS EI (m/z): [M+] calc'd for C14H9NO2 267.09, observed 267.10 m/z.
12 and 13. Yield = 0.1333 g (80 %, 12:13 = 3:4). Rf = 0.22 , hexane/ethyl acetate (9:1 v/v) . 1H-NMR (300 MHz, CDCl3): δ = 2.08 (s, 3H), 2.31 (s, 5H), 2.35 (s, 3H), 7.23 – 7.28 (m, 7H), 7.76 – 7.81 (m, 4H), 7.93-7.98 (m, 4H). 13C-NMR (75 MHz, CDCl3): δ = 14.67, 19.55, 19.91, 20.45, 123.67, 123.77, 124.16, 126.28, 127.76, 129.12, 130.29, 130.48, 131.00, 131.88, 132.06, 134.29, 135.13, 137.07, 137.66, 138.43, 167.57. LRMS EI (m/z): [M+] calc'd for C14H9NO2 251.09, observed 251.10 m/z.
17, 18 and 19 Yield = 0.128 g (75 %, 17:18:19 = 4:3:4). Rf = 0.1666, hexane/ethyl acetate (9:1 v/v). 1H-NMR (300 MHz, CDCl3): δ = 2.17 (s, 9H), 2.38 (s, 7H), 7.32 – 6.99 (m, 11H), 7.76 – 7.84 (m, 6H), 7.92 – 8.00 (m, 6H). 13C-NMR (75 MHz, CDCl3): δ = 17.94, 18.09, 21.21, 21.31, 123.73, 123.79, 124.52, 127.65, 127.85, 128.45, 128.49, 129.48, 130.16, 131.91, 132.07, 134.26, 134.33, 136.16, 136.86, 138.93, 139.50, 167.55. LRMS EI (m/z): [M+] calc'd for C14H9NO2 251.09, observed 251.10 m/z.
1-phenylpyrrolidine-2,5-dione 20. Yield = 0.0982 g (83 %). The NMR spectra matched with that of previously published.17 Rf = 0.44 hexane/ethyl acetate (1:1 v/v) . 1H-NMR (400 MHz, CDCl3): δ = 2.9(s, 4H), 7.28 (d, J = 7.2 Hz, 2H),7.39-7.4 (m,1H), 7.49-7.5(m,2H). 13C-NMR (100 MHz, CDCl3): δ = 28.4, 126.4, 128.6, 129.2, 131.8, 176.2. LRMS EI (m/z): [M+] calc'd for C10H9 NO2 175.06, observed 175.10 m/z.
5-nitro-2-phenylisoindoline-1, 3-dione 22. Yield= 0.0557 g (40 %). Rf = 0.33, hexane/ethyl acetate (8:2 v/v). 1H-NMR (300 MHz, CDCl3): δ = 7.26-7.55 (m, 5H), 8.17 (d, J = 9Hz, 1H), 8.68 (d, J = 6Hz, 1H), 8.78 (s, 1H). 13C-NMR (75 MHz, CDCl3): δ= 119.21, 125.04, 126.36, 128.75, 129.35, 129.62, 133.13, 151.82, 164.93. LRMS EI (m/z): [M+] calc'd for C14H8 N2O4 268.05, observed 268.0 m/z.
N-phenyl-1, 8-naphthalimide 23. Yield = 0.0356 g (27 %). Rf = 0.34, hexane/ethyl acetate (7:3 v/v). 1H-NMR (300 MHz, CDCl3): δ = 7.32-7.59 (m, 5H), 7.78-7.83(m, 2H), 8.28 (d, J = 9 Hz, 2H), 8.66 (d, J = 6 Hz, 2H). 13C-NMR (75 MHz, CDCl3): δ = 122.81, 127.05, 128.61, 128.73, 129.42, 131.64, 134.30, 135.41, 164.39. LRMS EI (m/z): [M+] calc'd for C18H11 NO2, 273.08, observed 273.10 m/z.
N-phenyl-1, 1-Dioxo-1, 2-benzothiazol-3-one 24. Yield = 0.037 g (26 %). Rf = 0.26, hexane/ethyl acetate (7:3 v/v) 1HNMR (300 MHz, CDCl3): δ = 7.55 (s, 5H), 7.87-8.01(m, 3H), 8.15 (d, J = 9 Hz, 1H). 13C-NMR (75 MHz, CDCl3): δ = 120.75, 121.25, 125.65, 127.16, 128.69, 128.75, 129.94, 130.13, 134.49, 135.12, 137.56, 158.40. LRMS EI (m/z): [M+] calc'd for C13H9 NO3S, 259.03, observed 259.0 m/z.
Representative Procedure with Stoichiometric Arene
A magnetically stirred solution of phthalimide (0.68 mmol), iodobenzene diacetate (1.7 mmol), and the simple arene substrate (2.0 mmol) in 4 mL of acetonitrile was microwave heated at 145 °C for 3h. The excess solvent from the mixture was removed at reduced pressure by rotary evaporation, and the crude product was purified by column chromatography.
2-(2, 3, 4, 5, 6-pentamethylphenyl) isoindoline-1, 3-dione 6 Yield = 0.085 g (43%). Rf = 0.23, hexane/ethyl acetate (9:1 v/v). 1H-NMR (300 MHz, CDCl3): δ = 2.06-2.26 (m, 15H), 7.79 (dd, J = 3 Hz, 3 Hz, 2H), 7.97 (dd, J = 3 Hz, 3 Hz, 2H). 13C-NMR (75 MHz, CDCl3): δ = 15.51, 16.81, 17.04, 123.76, 126.99, 131.77, 131.97, 133.57, 134.26, 136.9, 167.81. LRMS EI (m/z): [M+] calculated for C19H19NO2 293.14, observed 293.15 m/z.
2-(5-tert-butyl-2-methoxyphenyl) isoindoline-1,3-dione 16. Yield = 0.117 g (56%), Rf = 0.11, hexane/ethyl acetate (9:1 v/v) 1H-NMR (300 MHz, CDCl3): δ = 1.29-1.32 (m, 9H), 3.77 (s, 3H), 6.98 (d, J = 9 Hz, 1H), 7.25 (m, 1H) 7.44 (dd, J = 3 Hz, 3 Hz, 1 H) 7.77 (dd, J = 3 Hz, 3 Hz, 2H), 7.94 (dd, J = 3 Hz, 3 Hz, 2H). 13C-NMR (75 MHz, CDCl3): δ = 31.42, 34.17, 55.87, 111.64, 119.51, 123.61, 126.98, 132.29, 134.06, 143.74, 153.05, 167.55. LRMS EI (m/z): [M+] calc'd for C19H19NO3 309.14, observed 309.12 m/z
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the National Science Foundation (CAREER 0847222), and the National Institutes of Health (NIGMS, 1R15GM097708-01).
Footnotes
Supporting Information. Complete experimental details, compound characterization, analysis of product purity and both 1H and 13C-NMR data for novel compounds. This material is available free of charge via the Internet at http://pubs.acs.org
REFERENCES
- 1.For recent reviews describing oxidative C-C formation, see: Cho SH, Kim JY, Kwak J, Chang S. Chem. Soc. Rev. 2011;40:5068–5083. doi: 10.1039/c1cs15082k.Yeung CS, Dong VM. Chem. Rev. 2011;111:1215. doi: 10.1021/cr100280d.McGlacken GP, Bateman LM. Chem. Soc. Rev. 2009;38:2447. doi: 10.1039/b805701j.
- 2.For recent reviews describing C-N formation via nitreneoid intermediates, see: Davies HML, Manning JR. Nature. 2008;451:417. doi: 10.1038/nature06485.Müller P, Fruit C. Chem. Rev. 2003;103:2905. doi: 10.1021/cr020043t.
- 3.For a partial review, see: ref. 1a, pp. 12-16.
- 4.For recent examples of Cu and Pd-catalyzed aminations, see: Tsang WCP, Zheng N, Buchwald SL. J. Am. Chem. Soc. 2005;127:14560. doi: 10.1021/ja055353i.Thu H-Y, Yu W-Y, Che CM. J. Am. Chem. Soc. 2006;128:9048. doi: 10.1021/ja062856v.Tsang WCP, Munday RH, Brasche G, Zheng N, Buchwald SL. J. Org. Chem. 2008;73:7603. doi: 10.1021/jo801273q.Li B, Tian S, Fang Z, Shi Z. Angew. Chem. Int. Ed. 2008;47:1115. doi: 10.1002/anie.200704092.Jordan-Hore JA, Johansson CCC, Beck EM, Gaunt MJ. J. Am. Chem. Soc. 2008;130:16184. doi: 10.1021/ja806543s.Miura T, Murakami M. Chem. Lett. 2009;38:328.John A, Nicholas KM. J. Org. Chem. 2011;76:4158. doi: 10.1021/jo200409h.Xiao B, Gong T-J, Xu J, Liu Z-J, Liu L. J. Am. Chem. Soc. 2011;133:1466. doi: 10.1021/ja108450m.Sun K, Li Y, Xiong T, Zhang J, Zhan Q. J. Am. Chem. Soc. 2011;133:1694. doi: 10.1021/ja1101695.Yoo EJ, Ma S, Mei T-S, Chan KSL, Yu J-Q. J. Am. Chem. Soc. 2011;133:7652. doi: 10.1021/ja202563w.
- 5.For a recent examples of a metal-free amination, see: Cho SH, Yoon J, Chang S. J. Am. Chem. Soc. 2011;133:5696. doi: 10.1021/ja111652v.Lamani M, Prabhu KR. Iodine-Catalyzed Amination of Benzoxazoles: A Metal-Free Route to 2-Aminobenzoxazoles under Mild Conditions. J. Org. Chem. 2011 doi: 10.1021/jo201402a. [Online early access]. DOI: 10.1021/jo201402a. PublishedOnline: Aug 25, 2011.
- 6.a Monguchi D, Fujiwara T, Furukawa H, Mori A. Org. Lett. 2009;11:1607. doi: 10.1021/ol900298e. [DOI] [PubMed] [Google Scholar]; b Wang Q, Schreiber SL. Org. Lett. 2009;11:5178. doi: 10.1021/ol902079g. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cho SH, Kim JY, Lee SY, Chang S. Angew. Chem., Int. Ed. 2009;48:9127. doi: 10.1002/anie.200903957. [DOI] [PubMed] [Google Scholar]; d Kim JY, Cho SH, Joseph J, Chang S. Angew. Chem., Int. Ed. 2010;49:9899. doi: 10.1002/anie.201005922. [DOI] [PubMed] [Google Scholar]; e Wang J, Hou J-T, Wen J, Zhang J, Yu X-Q. Chem. Commun. 2011;47:3652. doi: 10.1039/c0cc05811d. [DOI] [PubMed] [Google Scholar]
- 7.While preparing this manuscript, two communications were published that describe metal-free intermolecular C-H amination: Antonchick AP, Samanta R, Kulikov K, Lategahn J. Angew. Chem. Int. Ed. 2011;50:8605. doi: 10.1002/anie.201102984.Kim HJ, Kim J, Cho SH, Chang S. J. Am. Chem. Soc. 2011;133:16382. doi: 10.1021/ja207296y.
- 8.For examples, see: Stuart DR, Fagnou K. Science. 2007;316:1172. doi: 10.1126/science.1141956.Dwight TA, Rue NR, Charyk D, Josselyn R, DeBoef B. Org. Lett. 2007;9:3137. doi: 10.1021/ol071308z.Yeung CS, Zhao X, Borduas N, Dong VM. Chem. Sci. 2010;1:331.Potavathri S, Pereira KC, Gorelsky SI, Pike A, LeBris AP, DeBoef B. J. Am. Chem. Soc. 2010;132:14676. doi: 10.1021/ja107159b.
- 9.a Campeau L, Parisien M, Jean A, Fagnou K. J. Am. Chem. Soc. 2006;128:581. doi: 10.1021/ja055819x. [DOI] [PubMed] [Google Scholar]; b Pascual S, de Mendoza P, Echavarren AM. Org. Biomol. Chem. 2007;5:2727. doi: 10.1039/b707940k. [DOI] [PubMed] [Google Scholar]
- 10.Malamidou-Xenikaki E, Spyroudis S, Tsanakopoulou M, Hadjipavlou-Litina D. J. Org. Chem. 2009;74:7315. doi: 10.1021/jo9013063. [DOI] [PubMed] [Google Scholar]
- 11.a Du Y, Liu R, Linn G, Zhao K. Org. Lett. 2006;8:5919. doi: 10.1021/ol062288o. [DOI] [PubMed] [Google Scholar]; b Yu W, Du Y, Zhao K. Org. Lett. 2009;11:2417. doi: 10.1021/ol900576a. [DOI] [PubMed] [Google Scholar]
- 12.Kita Y, Tohma H, Hatanaka K, Takada T, Fujita S, Mitoh S, Sakurai H, Oka SJ. Am. Chem. Soc. 1994;116:3684. [Google Scholar]
- 13.Kita Y, Takada T, Tohma H. Pure Appl. Chem. 1996;68:627. [Google Scholar]
- 14.Capitosti SM, Hansen TP, Brown ML. Bioorg. Med. Chem. 2004;12:327. doi: 10.1016/j.bmc.2003.11.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.