

Abstract
In allopatric populations, geographical separation simultaneously isolates the entire genome, allowing genetic divergence to accumulate virtually anywhere in the genome. In sympatric populations, however, the strong divergent selection required to overcome migration produces a genetic mosaic of divergent and non-divergent genomic regions. In some recent genome scans, each divergent genomic region has been interpreted as an independent incidence of migration/selection balance, such that the reduction of gene exchange is restricted to a few kilobases around each divergently selected gene. I propose an alternative mechanism, ‘divergence hitchhiking’ (DH), in which divergent selection can reduce gene exchange for several megabases around a gene under strong divergent selection. Not all genes/markers within a DH region are divergently selected, yet the entire region is protected to some degree from gene exchange, permitting genetic divergence from mechanisms other than divergent selection to accumulate secondarily. After contrasting DH and multilocus migration/selection balance (MM/SB), I outline a model in which genomic isolation at a given genomic location is jointly determined by DH and genome-wide effects of the progressive reduction in realized migration, then illustrate DH using data from several pairs of incipient species in the wild.
Keywords: speciation genetics, adaptation, genome scan, sympatric speciation, genomic divergence, insects
1. Introduction
In geographically separated (allopatric) populations, a physical barrier to gene flow simultaneously blocks gene flow across the entire genome. This makes speciation straightforward because genetic divergence by any mechanism can readily accumulate. In contrast, speciation-with-gene-flow is controversial [1–3] because population genetic models suggest that recombination nearly always foils the joint evolution of genes affecting adaptive divergence and assortative mating (e.g. [4], review in [3], pp. 131–137).
Even among skeptics, however, there is general consensus that certain conditions can minimize the disruptive effects of between-population recombination. For example, either mate choice that is correlated with a trait under divergent selection or habitat choice in taxa that mate locally can limit the opportunity for between-population recombination [1,2,5,6]. Speciation in sympatry is also facilitated when divergently selected traits involve relatively few genes of large effect, because this focuses the effects of selection and reduces the number of sites subject to recombination [6,7]. Even so, between-population recombination is still considered a major impediment to speciation-with-gene-flow. As a consequence, tight linkage (see glossary) or a structural reduction in recombination such as a chromosomal inversion are usually thought to be required for coordinated evolution at key genes during speciation-with-gene-flow [8–11].
This paper concerns a mechanism called ‘divergence hitchhiking’ (DH) in which gene exchange between diverging populations is reduced over genomic regions of several megabases as a collateral effect of strong divergent selection on genes involved in local adaptation [12]. Without requiring any structural reduction in recombination, DH produces blocks of genomic isolation (see glossary) in the very genomic regions most involved in local adaptation, facilitating the divergence of other loosely linked genes and reducing the destructive effects of between-population recombination.
In DH regions, genomic isolation and genomic divergence (see glossary) become uncoupled, much as they are during allopatric speciation. Although gene exchange between populations is partially suppressed across an entire DH region, only genes or markers that are tightly linked to a direct target of divergent selection, or that experience lineage sorting during the population split will initially be divergent. As time passes, however, secondary divergence (see glossary) can accumulate in DH regions by independent selective sweeps or genetic drift at genes unaffected by divergent selection, and this process may even be accelerated by the reduced effective population size (Ne) in DH regions owing to divergent selection [13].
2. Multilocus migration/selection balance and divergence hitchhiking are alternative visions of genomic divergence during speciation-with-gene-flow
Genetic divergence in the face of gene flow is governed by the balance between the homogenizing effects of migration and the divergent effects of selection. Recent discussions of divergence-with-gene-flow [9,14–16] appear to consider migration/selection balance as the primary mechanism of genomic divergence. In addition, Lewontin & Krakauer's [17] suggestion that high FST can be used to identify divergently selected genes has motivated a spate of genome scans in which it is widely assumed that each high-FST marker is either a direct target of divergent selection or is very closely linked to one (figure 1a and [16,18–21]). Because genomic isolation in migration/selection balance is restricted to a few kilobases around the site of divergent selection, gene exchange is implicitly assumed to continue between divergently selected regions (outliers).
Figure 1.
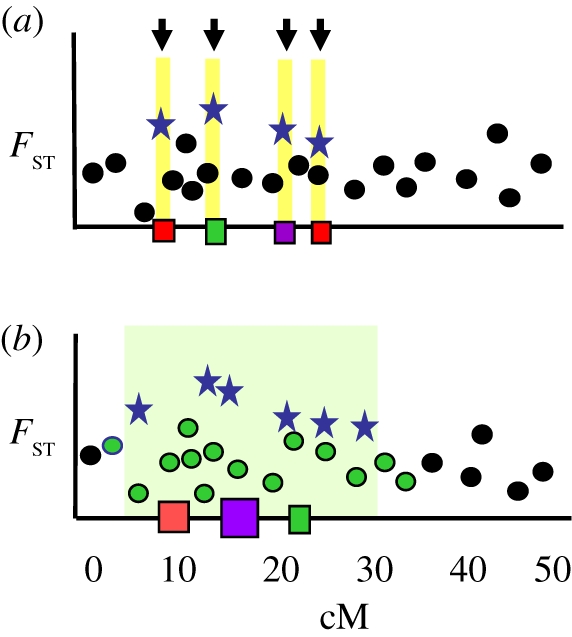
Alternate interpretations of outliers. (a) In the conventional view, each outlier marks an individual candidate gene under migration/selection balance (arrowed yellow bars). Between candidate gene regions, low FST markers are assumed to experience gene exchange. (b) A region of divergence hitchhiking (DH; green shading) around a cluster of divergently selected QTL is recognized by the associated cluster of FST outliers. Low FST markers within DH regions are assumed to be protected from gene exchange. Red, green and purple squares, QTLs for different traits; blue star, class1; green circle, class2; black circle, class 3.
In a genome scan, samples from divergent populations are genotyped at many loci, then FST outliers are identified [22–25] and used to identify candidate genes (figure 1a; [19,20]). Despite widespread enthusiasm for this approach, it has some pitfalls. First, on average in a genome scan, about 8 per cent of tested markers are FST outliers [14,26]. Thus, if each outlier is an independent target of selection, a very large number of loci must be affected by strong enough divergent selection to block gene flow. Secondly, various demographic events and aspects of population structure can elevate FST and increase the frequency of false positives [24,25,27,28]. This makes it vital to couple genome scans with other methods [24], such as identifying outliers at which allelic differences correlate with environmental variables [29,30], combining genome scans with ecological genetic manipulations [31], or choosing markers in genes with functional effects that are relevant to the particular situation [30–32]. Performing a genome scan with markers chosen from a quantitative trait locus (QTL) map (see glossary) reveals FST outliers linked to bona fide candidate genes by their proximity to QTLs that affect phenotypic traits involved in population divergence [12,26,28,33–35]. Indeed, QTL maps provide a much-needed connection between population genomic analyses and adaptive phenotypic evolution in the wild.
3. In divergence hitchhiking, a block of genomic isolation forms around one or a few genes under strong divergent selection
When populations diverge during local adaptation, random mating is disrupted and the opportunity for between-population recombination is reduced. Divergent selection against the ‘wrong’ locally adapted alleles in migrants then spills over onto neighbouring genes, because the lack of free between-population recombination keeps neutral loci from separating from the one with the disadvantageous allele. In relatively small populations without much migration [15,27], strong divergent selection can limit introgression and realized gene exchange for several centimorgans (cM) around a divergently selected gene (figure 2, modified from [27]). In such regions, loosely linked QTLs (see glossary) under weaker divergent selection may then receive enough protection from gene exchange to diverge. Because each additional QTL increases the extent of genomic isolation, this can launch a cascade of divergence at QTLs within a genomic region of 10 Mb or more (figure 2; [36]; see §8d). The reduction of between-population recombination in a DH region protects sets of locally adapted alleles from disruption without requiring tight linkage, a chromosomal inversion or location near a centromere.
Figure 2.

Relationship between the maximum FST that can be maintained by DH at equilibrium and the map distance from a selected gene, for two intensities of divergent selection (modified from [27], for a population with Ne = 1000 and m = 0.001). Hypothetical FST estimates for different markers in a genome scan are overlaid; open circles are FST outliers, filled circles are non-outliers. Although the curves in this figure are often thought to illustrate the predicted FST values under DH, many markers will have lower FST than predicted if the sampled populations are not at equilibrium. This is illustrated here by the mixture of FST outliers and non-outliers near the selected gene. If the selected gene is not at the end of the chromosome, the curves and estimated FST values would be symmetrical around it. Black line, strong local selection; grey line, moderate local selection; dotted line, no local selection.
The recruitment of multiple QTLs into a DH region may explain a baffling observation: QTLs that affect phenotypic differences between domesticated varieties and their wild ancestors [37–40], ecotypes [33,41–43] or locally adapted species [44] are often clustered into large blocks within the genome. In domesticated chickens, genetic correlations among key traits are thought to be caused by a core of pleiotropic or tightly linked loci surrounded by variable numbers of loosely linked QTLs over 20–30 cM or more [38]. Wright et al. [38] call these QTL blocks ‘domestication clusters’. Multilocus migration/selection balance (MM/SB) cannot explain this clustering without invoking some structural reduction in recombination. In contrast, clusters of QTLs associated with phenotypic divergence are a hallmark of DH, and they are expected under the intense divergent selection associated with domestication.
The reduction of between-population recombination around a gene under divergent selection determines the maximum FST that can be maintained by DH at equilibrium (figure 2, modified from [27]). As the distance between a neutral locus and a selected gene increases, the genomic isolation declines and less divergence (i.e. a smaller FST) can be maintained (although some FST values could exceed this maximum due to selection or stochastic effects). The establishment of a DH region does not automatically cause all genes/markers to diverge to the levels shown in figure 2—the partial genomic isolation in a DH region simply maintains divergence that occurs through standard evolutionary mechanisms. In other words, DH creates genomic isolation, but it does not directly produce genomic divergence.
Blurring this distinction has led some investigators to interpret the curves in figure 2 as the predicted FST values under DH around a divergently selected gene. However, most of the recently diverged taxa studied with genome scans will not be at equilibrium, so many sites within a cluster of outliers will not yet have reached the maximum level of divergence that can be maintained by DH. For this reason, we expect genome scans in most wild populations to reveal clusters of outliers or divergent regions around divergently selected genes/QTLs, and we also expect these clusters to contain non-divergent regions. Under divergent selection, the evolutionary unit becomes a cluster of genes/QTLs that diverge coordinately, and not an individual divergent region.
The name ‘divergence hitchhiking’ is meant to distinguish this process from traditional within-population hitchhiking [45], not to suggest that large blocks of continuous genetic divergence between incipient species will be formed. Traditional hitchhiking is a within-population process in which neutral sites tightly linked to a selected gene are swept with it to high frequency. By contrast, DH is a strictly between-population process in which genes over a much larger genomic region (of the order of megabases rather than kilobases) are protected from between-population recombination. This allows them to diverge coordinately as they ‘hitchhike’ with one another without disruption during the evolution of local adaptation.
Although Charlesworth et al. [27] considered the strength of selection required to produce DH to be extreme by population genetic standards (i.e. s = 0.1–0.5), this is not unreasonable for sympatric populations under divergent resource-based selection. For example, when wild-collected pea aphids collected from alfalfa and red clover were experimentally ‘migrated’ to the alternate host plant, their mean fecundity was only 17 per cent of what they would have enjoyed on their usual host, making phenotypic selection on habitat choice approximately sphenotype = 0.83 [46]. Dividing this phenotypic selection among 4.5 QTLs (the average number affecting each trait in the second generation QTL map; [47]), the selection per QTL is strong enough to initiate a DH region (sQTL ∼ 0.18) if Ne is relatively small and there is not much migration [15,27].
In both DH and MM/SB, genes/markers that are tightly linked to the target of selection are likely to diverge with it, so that individual divergent regions in both mechanisms should be about the same size (a few kilobases). However, under MM/SB, divergent regions are independent, and non-divergent genes in between are thought to remain subject to gene exchange. In contrast, a DH region is expected to include multiple divergent regions interspersed with non-outliers, which are interpreted as ancestral polymorphisms.
4. Genomic isolation increases genome-wide as realized migration declines, augmenting effects of divergence hitchhiking
As allelic changes throughout the genome increase phenotypic divergence between sympatric populations, the rate of ‘realized’ migration declines, reducing the opportunity for between-population recombination equally across the genome. Realized migration is calculated as the nominal rate of migration discounted by various sequentially acting components of reproductive isolation. For example, a successful migrant must move to the other population, survive, mate and produce F1 progeny that also survive to mate and reproduce. The resulting genome-wide reduction in gene exchange (which Feder et al. [48] call genomic hitchhiking (GH)) then interacts with the localized genomic isolation from DH, as described in §5.
5. Outlining a model for divergence hitchhiking and the spread of genomic isolation during speciation-with-gene-flow
The evolution of genomic isolation during speciation-with-gene-flow involves a progressive and predictable reduction of between-race recombination, the primary cause of gene exchange between divergent populations. Here, I outline how DH and the reduction of realized migration may interact to reduce between-population recombination across the genome. This is far from a full analysis because my goal is primarily to stimulate thought on how the spread of genomic isolation might be quantified.
Extending the model of Kobayashi & Telschow ([49], eqns 17 and 31), the effective between-population recombination rate at a given genomic location and time, re(g,t), is obtained by discounting the within-population recombination rate by the product of localized effects from DH (left-hand term) and the genome-wide effect of declines in realized migration (GH, right-hand bracket):
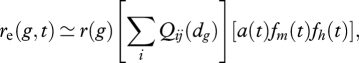 |
5.1 |
where r(g) is the nominal within-population recombination rate (as estimated from a linkage map), and Qij is the reduction in between-race recombination at site g from the ith QTL under divergent selection, as a function dg of its map distance to g. The effects of QTL in the same DH region are considered here to be additive, although there may be circumstances in which they have multiplicative effects.
The reduction of successful migration (GH, right-hand bracket) occurs through the cumulative effects of allelic change at all QTLs that affect ecologically based reproductive isolation, such that a(t) = the frequency of movement to the alternate habitat at time t due to effects of all QTLs at time t; fm(t), fh(t) = the mean fitness of migrants or F1 hybrids (respectively) relative to residents in the alternate habitat owing to allelic effects at all QTLs at time t.
I suggest that DH and GH have multiplicative effects on the realized rate of between-population recombination because they act sequentially (i.e. there has to be successful migration for recombination to occur in an F1). This would cause genomic isolation in DH regions to increase quickly relative to isolation in regions affected only by GH. The extent of genomic isolation in DH and GH regions will be most different early in the process of divergence, when there may be a few DH regions around strongly selected genes but not much overall reduction in successful migration. This is a crucial time for speciation-with-gene-flow, and it is then that DH plays its major role by facilitating the retention of genetically correlated sets of alleles at QTLs within DH regions. It is also the stage of speciation-with-gene-flow at which Ne is likely to be at a minimum. This early protection of sets of locally adapted alleles from the destructive effects of between-population recombination may be what allows speciation-with-gene-flow to continue until successful migration dwindles and the entire genome becomes isolated.
This model suggests that genomic isolation will spread much more quickly across the genome during speciation-with-gene-flow than will genomic divergence. Early in the process, genomic divergence becomes concentrated in DH regions because they are the focus of divergent selection. This selection also decreases Ne in DH regions, accelerating secondary divergence through drift or lineage sorting. Not only does genomic divergence outside DH regions begin later because of ongoing gene flow, it accumulates more slowly than divergence in DH regions because it results primarily from within-population selective sweeps and genetic drift rather than divergent selection. Thus, the clustered pattern of FST outliers and divergent genomic regions that is the signature of DH is likely to remain visible for quite a long time after gene exchange has ceased.
6. Are conditions during early speciation-with-gene-flow likely to produce divergence hitchhiking?
Feder & Nosil [15] suggest that DH may have limited applicability during speciation-with-gene-flow because the reduction of between-population recombination appears to be most extensive when Ne is moderate (approx. 1000) and migration is relatively low (m = 0.001) [15,27]. Their view [15] appears to be that speciation in sympatry is most likely to occur in large populations (i.e. Ne ∼ 10 000–100 000) with high rates of migration (m ∼ 0.01–0.1). This raises some important ecological and genetic issues: what are the demographic conditions that typify diverging populations at early stages of speciation? Is the extent of DH determined by the Ne for neutral loci or by the reduced Ne caused by selection in DH regions? What is the appropriate measure of effective population size for a local population responding to divergent selection? It seems possible that during the early stages of speciation in sympatry, the neighbourhood size (the effective size of the population that actually interacts) could affect the extent of DH more than Ne for the entire species, since individuals in different locations may be completely unaffected by divergent selection. Finally, if two incipient species have unequal population sizes (which seems very likely when a new habitat is first colonized), Ne can be dramatically reduced, as when the sex ratio is unequal. For example, in a population of Ne = 10 000 in which 1000 individuals split off into a new habitat, Ne = 4N1N2/(N1 + N2) = 3600. If only 100/10 000 initially occupy the new habitat, Ne = 396. Clearly, we need to learn more about the typical demographic situation early in speciation-with-gene-flow, because the small mathematical parameter space in which DH is effective [15,48] may actually describe the very conditions under which DH is most likely to occur.
7. Conflicts between multilocus migration/selection balance and divergence hitchhiking as explanations for patterns of genomic divergence during speciation
(a). Is there gene flow at non-divergent markers between FST outliers?
In MM/SB, gene exchange is blocked only at the target of selection and a few linked neighbours, leaving non-divergent genes/markers between outliers subject to ongoing gene flow. Divergent regions under migration/selection balance are expected to be small, and their boundaries are often identified on a sequence or map as the first non-outlier marker on either side of a candidate gene or FST outlier (figure 1a; [19,21,50]).
In DH, however, genomic isolation extends over regions that are much larger (megabases of sequence) than those expected under MM/SB (measured in kilobases). Within DH regions, low FST values of markers between divergent regions are attributed to ancestral polymorphism rather than to gene flow, and even genes/markers that are unaffected by divergent selection are partially protected from gene exchange.
(b). What curbs the effects of recombination in multilocus migration/selection balance?
If each ‘genomic island of divergence’ represents an independent incidence of divergent selection [51], the classic problem of recombination remains. With ongoing gene flow between ‘islands’, what prevents between-population recombination from shuffling locally adapted alleles and preventing divergence? In MM/SB, the usual explanation is that multilocus divergence is concentrated in genomic regions of low recombination [8,10,11,52]. In contrast, DH regions can reduce gene exchange at any genomic location harbouring a gene or QTL under strong divergent selection. Borrowing the terminology of Michel et al. [51], one could consider DH regions as ‘continents of genomic isolation’ within which ‘islands of genomic divergence’ can be maintained (see §8d and figure 3).
Figure 3.

Probable DH regions in threespine stickleback. (a) Genome scan of divergence on LGIV between freshwater and oceanic threespine stickleback (reproduced from figs. 7 & 8 in [53], except for the addition of the QTLs and proposed DH regions). The candidate gene regions are shown as yellow bars; Class1 SNPs appear as blue dots. The purple square marks the location of the gene Eda. Green and red squares mark QTL affecting body shape [54]. Regions of DH (green shading) are arbitrarily extended to the edge of each cluster of SNP outliers. (b) LGVIII from the same study [53]. Blue square denotes a candidate gene at marker stn90; Class1 SNPs appear as blue dots; from [55].
(c). Why are divergent quantitative trait loci and FST outliers often clustered within the genome?
Recent genome scans in wild populations have revealed clusters of outliers/candidate genes that are not associated with either a centromere or a known chromosomal rearrangement [51,53,56], with no discussion of how they might be maintained. Since DH can cause clusters of divergence without any structural reduction of recombination, these clusters may be DH regions. Once a DH region is established, the recruitment of additional QTLs [36] and secondary divergence from other causes can form a cluster of divergence, which is maintained over the long term.
(d). Does the frequency of FST outliers estimate the number of genes under divergent selection?
This idea is a logical extension of MM/SB, in which every outlier is assumed to represent an independent target of divergent selection [20,51,57,58]. However, for this to be true, hundreds or thousands of genes must experience strong enough divergent selection to block migration and become significant outliers. Before evaluating this assertion, I review how phenotypic selection is thought to act during speciation in sympatric populations.
Speciation-with-gene-flow is considered to be most likely when relatively few genes are under divergent selection [6,7] and when there are multiple forms of ecologically based reproductive isolation involving a variety of different phenotypic traits (multifarious selection, see glossary; sensu Rice & Hostert [1]). Genomic regions (QTLs) containing one or more genes affecting these traits can be identified using QTL mapping [59]. Because the strength of phenotypic selection on a given trait is spread among the QTLs that affect it, divergent selection on an individual QTL is always weaker than phenotypic selection on the trait. In this setting, could divergent selection block gene flow at enough genes to account for the number of outliers in a typical genome scan (on average, about 8 per cent of tested markers [14,26])?
Butlin [60] estimated that there may be over 1000 outliers (and the same number of divergently selected genes) between ecotypes of the intertidal snail Littorina. With the generous assumption that 10 traits (i.e. habitat choice or shell shape) affect ecologically based reproductive isolation between the ecotypes, then phenotypic selection on each trait would be divided among 100 genes. Even if divergent phenotypic selection on each trait is intense, say sphenotype = 0.9 (as it is on habitat choice in the pea aphid host races [34]), selection on each gene would be only (sgene ∼ 0.009), which is too weak to block gene flow and produce an FST outlier under migration/selection balance.
In contrast, strong divergent selection on a handful of genes/QTLs can produce a DH region. Although genes/markers within the region that diverge secondarily under the protection of genomic isolation may become outliers, they will not necessarily tag a divergently selected gene.
8. Identifying and testing divergence hitchhiking in wild populations
DH produces genomic isolation across tens of megabases or centimorgans, but because genomic isolation is not directly visible on a genome scan, the existence of DH is controversial [15,26,61,62]. The identification of DH regions is indirect, based on the fact that divergent genes/markers can only accumulate in regions where gene exchange is reduced. Because clusters of FST outliers cannot be maintained without some genomic isolation, they provide a useful signature of DH. However, we have little information about outlier clustering in wild populations, because recent genome scans have focused instead on the size of divergent regions and their locations relative to candidate genes or regions of reduced recombination. Recent genome scans with tests for the existence of DH regions have yielded the following conclusions.
(a). Regions of genomic divergence are too small to be due to divergence hitchhiking
Most investigators appear to expect that under DH, FST values for every gene/marker around a selected gene or QTL will be elevated according to the pattern in figure 2. Thus, the small observed size of individual regions of divergence has been used as evidence against DH [26,61,62]. However, the smooth profile of declining FST values suggested by the Charlesworth model [27] is only expected at equilibrium, which is unlikely to have been attained in recently diverged wild populations. The expected signal of DH is therefore not a large genomic region of continuous divergence, but a cluster of individually small divergent regions. Within these clusters, low FST markers are expected to be interspersed among outliers (figures 1b and 2).
(b). FST is not spatially autocorrelated in genomic regions with divergent markers
This is an inappropriate test for the same reason as above. In addition, FST estimates at individual markers are highly variable, so that it is better to make inferences based on outlier status than on estimated FST values. Finally, depending on the scale, tests for spatial autocorrelation in divergence [61,62] may not be significant if many non-divergent markers are found within a cluster of outliers.
(c). No FST clusters are found
There are several reasons for the apparent absence of outlier clusters: (i) DH is never expected between allopatric populations [26,34], (ii) if too small a sample from the divergent populations is genotyped, errors in FST values may mask the true pattern of divergence, (iii) genome scans with less than several hundred markers are unlikely to resolve enough outliers to recognize a cluster [61,62], (iv) an outlier cluster cannot be recognized if the genomic region analysed is smaller than the probable size of the cluster [55,63], and (v) outlier analyses are best performed on recently diverged taxa [23,24]. In genome scans of taxa that diverged more than 500 000 years ago [61,62], high levels of divergence may make the clusters of FST outliers that tag DH regions indistinct, and the few FST outliers that are detected may reflect recent selective sweeps instead of the initial patterns of genomic divergence. In such taxa, outliers are unlikely to cluster around QTLs for traits that were important in the original divergence [61,62].
(d). Example 1
To illustrate how clusters of FST outliers can be used to identify probable regions of DH, I re-interpreted the study of Hohenlohe et al. [53] in which FST between freshwater and oceanic threespine sticklebacks was estimated for approximately 45 000 markers localized on the genome sequence. The authors identified narrow regions of significant divergence on seven of the 21 linkage groups, and suggested that these harbour candidate genes (yellow bars in original figs. 7 & 8 [53], reproduced here in figure 3). Although the authors did not interpret the genomic pattern of outlier single nucleotide polymorphisms (SNPs; blue dots on the scans in figure 3), divergent SNPs are clearly clustered around the proposed candidate gene regions (and are surprisingly absent in other locations). This clustered pattern strongly suggests regions of DH around the candidate genes (indicated by green shading, figure 3). To estimate the size of these proposed DH regions, I used recombination rates from Hohenlohe et al. [64]. On LGIV, the proposed DH region (approx. 23.8 cM) contains Eda, a gene under divergent selection due to its effects on the size of the lateral plates [65]. The other proposed DH region on LGIV (approx. 40.5 cM) contains QTLs affecting divergently selected shape differences between the forms [54]. On LGVIII, a single DH region (7.3 cM) is proposed around a candidate gene that also affects the lateral plates [55].
These proposed regions of DH in sticklebacks illustrate how clusters of outliers tend to occur around known candidate genes, while being virtually absent elsewhere. This example also shows that non-outliers are interspersed in these clusters. Finally, these DH regions are comparable in size to estimates made for other systems [12]: approximately 20 cM for pea aphids, and approximately 32 cM for lake whitefish.
9. Distinguishing divergence hitchhiking from multilocus migration/selection balance using analyses of markers in different genomic locations
Under DH, the evolutionary histories of neutral markers depend upon their genomic location relative to DH regions. By comparing population genetic analyses of mapped markers from different locations, we can test predictions that distinguish DH from MM/SB. The three marker classes are (see also figure 1):
— Class1. Markers that are significantly divergent between populations, i.e. FST outliers.
— Class2. Non-divergent markers with low FST interspersed among outliers in a DH region.
— Class3. Non-divergent markers with low FST located at least 5 cM from the nearest DH region.
(a). Example 2
Because selection accelerates the progress to monophylly [66], outliers are likely to provide a clearer picture of phylogenetic or population structure after adaptive divergence than non-outlier markers (examples in [12,18,50]). This allows us to test the prediction that low FST markers within DH regions (Class2) are protected from gene flow. With reduced gene exchange, Class2 markers can begin to become concordant with the genealogical pattern defined by the outliers [34], yielding a picture of population structure that is more similar to that from the outliers than to results from the Class3 markers. In contrast, under MM/SB, Class2 and Class3 markers should reveal the same population structure because they are assumed to be equally subject to ongoing gene flow.
This test was applied to the pea aphid host races by genotyping 100 individuals of each race from our NY study site for mapped microsatellite markers, and estimating FST outliers with FDIST2 [22]. We chose five unlinked markers of each class, because STRUCTURE can produce spurious results with linked markers [67]. Results of separate STRUCTURE analyses on each marker class (figure 4) show that despite the low mean FST of both Class2 and Class3 markers, the Class2 markers reveal the population structure of the divergently selected host races almost as clearly as the outliers (Class1). By contrast, the Class3 markers are unable to resolve the adaptive divergence between the host races, as predicted for markers outside DH regions that are likely to experience ongoing gene flow. These results strongly contradict the prediction from MM/SB that all low FST markers are equally exposed to gene exchange.
Figure 4.

Comparative analysis of population structure in the pea aphid host races on alfalfa and clover (STRUCTURE, K = 2). The proportional representation of the two clusters in each of 100 pea aphid genotypes from alfalfa is shown on the left side of each panel; results for aphids collected from red clover are on the right. Marker classes are defined in the text.
10. Conclusions
DH is a simple genetic mechanism with great explanatory power. It reduces the extent to which recombination foils the process of speciation-with-gene-flow, explains why so many FST outliers are typically found in genome scans and provides a mechanism by which divergent selection produces genomic clusters of QTLs and outliers. Just as reproductive isolation is the core of speciation at the population level, the origin and spread of genomic isolation is the key to the genomic mechanisms of speciation. By using mapped markers in genome scans of wild populations and interpreting patterns of genomic divergence as well as the size of individual divergent regions, the genetic mechanisms of speciation-with-gene-flow will be clarified.
Acknowledgements
I am grateful to Paul Hohenlohe and Bill Cresko for allowing me to use their genome scans, and to Gina Conte, Jeff Feder, Tom Kocher (and laboratory), Loren Rieseberg, Sebastien Renaut and Dolph Schluter for astute comments and criticisms. Support was provided by NSF DEB0528288 and USDA-NIFA Competitive Grant 2006-35607-16707.
Glossary
- Between-population recombination
recombination between chromosomes from divergent populations during meiosis in an F1 or backcross, by which locally adapted alleles are transferred into the alternate genetic background.
- DH
the coordinated evolution of multiple genes in a genomic region extending over several megabases due to partial protection from between-population recombination and gene exchange; caused by spill-over effects of strong divergent selection in small subdivided populations.
- FST outlier
marker or gene at which FST (the standardized variance of allele frequency between populations) is significantly larger or smaller than expected under neutrality [22]. Here, we consider markers with large FST values; evidence of exposure to divergent selection [17].
- Genomic isolation
reproductive isolation at the genome level; protection from between-population recombination and gene exchange. Evolves either one locus at a time (under migration/selection balance), or in blocks (through DH). Not equivalent to genomic divergence, but a pre-requisite for it.
- Genomic divergence
significant difference in allele frequency between taxa. In sympatric populations, requires genomic isolation from either migration/selection balance or DH.
- Loosely linked
genes that are a few hundred kilobases to several megabases apart; can diverge coordinately within DH regions.
- Multifarious selection
phenotypic selection on multiple components of reproductive isolation that block gene flow in different ways [1].
- Multilocus mutation/selection balance
genetic divergence at multiple loci (and tightly linked markers) caused by independent incidences of divergent selection that is strong enough to balance the effects of gene flow.
- Secondary divergence
genetic divergence within a partially genomically isolated DH region by mechanisms other than divergent selection. May begin as soon as a region of DH is established around a gene/QTL under strong divergent selection.
- Tightly linked
genes that are so close together on a chromosome that they are very unlikely to be separated by recombination; of the order of a few kilobases apart.
- QTL
Quantitative Trait Locus; a genomic region (not a gene) that affects a quantitative (continuously varying) trait. We focus on QTLs for traits under divergent selection.
- QTL map
a linkage map on which QTLs are localized by statistically associating phenotypic differences among individuals from a mapping population with differences in their allele frequencies at markers localized on a linkage map [57].
References
- 1.Rice W. R., Hostert E. E. 1993. Laboratory experiments on speciation: what have we learned in 40 years? Evolution 47, 1637–1653 10.2307/2410209 (doi:10.2307/2410209) [DOI] [PubMed] [Google Scholar]
- 2.Via S. 2001. Sympatric speciation in animals: the ugly duckling grows up. Trends Ecol. Evol. 16, 381–390 10.1016/S0169-5347(01)02188-7 (doi:10.1016/S0169-5347(01)02188-7) [DOI] [PubMed] [Google Scholar]
- 3.Coyne J. A., Orr H. A. 2004. Speciation. Sunderland, MA: Sinauer Associates [Google Scholar]
- 4.Felsenstein J. 1981. Skepticism toward Santa Rosalia, or why are there so few kinds of animals? Evolution 35, 124–138 10.2307/2407946 (doi:10.2307/2407946) [DOI] [PubMed] [Google Scholar]
- 5.Schluter D. 2001. Ecology and the origin of species. Trends Ecol. Evol. 16, 372–380 10.1016/S0169-5347(01)02198-X (doi:10.1016/S0169-5347(01)02198-X) [DOI] [PubMed] [Google Scholar]
- 6.Gavrilets S. 2004. Fitness landscapes and the origin of species. Princeton, NJ: Princeton University Press [Google Scholar]
- 7.Gavrilets S., Vose A. 2007. Case studies and mathematical models of ecological speciation. II. Palms on an oceanic island. Mol. Ecol. 16, 2910–2921 10.1111/j.1365-294X.2007.03304.x (doi:10.1111/j.1365-294X.2007.03304.x) [DOI] [PubMed] [Google Scholar]
- 8.Noor M. A. F., Bennett S. M. 2009. Islands of speciation or mirages in the desert? Examining the role of restricted recombination in maintaining species. Heredity 103, 439–444 10.1038/hdy.2009.151 (doi:10.1038/hdy.2009.151) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pinho C., Hey J. 2010. Divergence with gene flow: models and data. Annu. Rev. Ecol. Evol. Syst. 41, 215–230 10.1146/annurev-ecolsys-102209-144644 (doi:10.1146/annurev-ecolsys-102209-144644) [DOI] [Google Scholar]
- 10.Hoffman A. A., Rieseberg L. H. 2008. Revisiting the impact of inversions in evolution: from population genetic markers to drivers of adaptive shifts and speciation? Annu. Rev. Ecol. Evol. Syst. 39, 21–42 10.1146/annurev.ecolsys.39.110707.173532 (doi:10.1146/annurev.ecolsys.39.110707.173532) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feder J. L., Roethele J. B., Filchak K. 2003. Evidence for inversion polymorphism related to sympatric host race formation in the apple maggot fly, Rhagoletis pomonella. Genetics 163, 939–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Via S., West J. 2008. The genetic mosaic suggests a new role for hitchhiking in ecological speciation. Mol. Ecol. 17, 4334–4345 10.1111/j.1365-294X.2008.03921.x (doi:10.1111/j.1365-294X.2008.03921.x) [DOI] [PubMed] [Google Scholar]
- 13.Charlesworth B. 2009. Effective population size and patterns of molecular evolution and variation. Nat. Rev. Genet. 10, 195–205 10.1038/nrg2526 (doi:10.1038/nrg2526) [DOI] [PubMed] [Google Scholar]
- 14.Stinchcombe J. R., Hoekstra H. E. 2008. Combining population genomics and quantitative genetics: finding the genes underlying ecologically important traits. Heredity 100, 158–170 10.1038/sj.hdy.6800937 (doi:10.1038/sj.hdy.6800937) [DOI] [PubMed] [Google Scholar]
- 15.Feder J. L., Nosil P. 2010. The efficacy of divergence hitchhiking in generating genomic islands during ecological speciation. Evolution 64, 1729–1747 10.1111/j.1558-5646.2009.00943.x (doi:10.1111/j.1558-5646.2009.00943.x) [DOI] [PubMed] [Google Scholar]
- 16.Storz J. 2005. Using genome scans of DNA polymorphism to infer adaptive population divergence. Mol. Ecol. 14, 671–688 10.1111/j.1365-294X.2005.02437.x (doi:10.1111/j.1365-294X.2005.02437.x) [DOI] [PubMed] [Google Scholar]
- 17.Lewontin R. C., Krakauer J. 1973. Distribution of gene frequency as a test of the theory of selective neutrality of polymorphisms. Genetics 74, 175–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilding C. S., Butlin R. K., Grahame J. 2001. Differential gene exchange between parapatric morphs of Littorina saxatillis detected using AFLP markers. J. Evol. Biol. 14, 611–619 10.1046/j.1420-9101.2001.00304.x (doi:10.1046/j.1420-9101.2001.00304.x) [DOI] [Google Scholar]
- 19.Galindo J., Grahame J. W., Butlin R. K. 2010. An EST-based genome scan using 454 sequencing in the marine snail Littorina saxatilis. J. Evol. Biol. 23, 2004–2016 10.1111/j.1420-9101.2010.02071.x (doi:10.1111/j.1420-9101.2010.02071.x) [DOI] [PubMed] [Google Scholar]
- 20.Stapley J. A., et al. 2010. Adaptation genomics: the next generation. Trends Ecol. Evol. 25, 705–772 10.1016/j.tree.2010.09.002 (doi:10.1016/j.tree.2010.09.002) [DOI] [PubMed] [Google Scholar]
- 21.Wood H. M., Grahame J. W., Humphray S., Rogers J., Butlin R. K. 2008. Sequence differentiation in regions identified by a genome scan for local adaptation. Mol. Ecol. 17, 3123–3135 10.1111/j.1365-294X.2008.03755.x (doi:10.1111/j.1365-294X.2008.03755.x) [DOI] [PubMed] [Google Scholar]
- 22.Beaumont M. A., Balding D. J. 2004. Identifying adaptive genetic divergence among populations from genome scans. Mol. Ecol. 13, 969–980 10.1111/j.1365-294X.2004.02125.x (doi:10.1111/j.1365-294X.2004.02125.x) [DOI] [PubMed] [Google Scholar]
- 23.Vitalis R., Dawson K., Boursot P., Belkhir K. 2003. DetSel 1.0: a computer program to detect markers responding to selection. J. Hered. 94, 429–431 10.1093/jhered/esg083 (doi:10.1093/jhered/esg083) [DOI] [PubMed] [Google Scholar]
- 24.Vasemagi A., Primmer C. R. 2005. Challenges for indentifying functionally important genetic variation: the promise of combining complementary research strategies. Mol. Ecol. 14, 3623–3642 10.1111/j.1365-294X.2005.02690.x (doi:10.1111/j.1365-294X.2005.02690.x) [DOI] [PubMed] [Google Scholar]
- 25.Excoffier L., Hofer T., Foll M. 2009. Detecting loci under selection in a hierarchically structured population. Heredity 103, 285–298 10.1038/hdy.2009.74 (doi:10.1038/hdy.2009.74) [DOI] [PubMed] [Google Scholar]
- 26.Strasburg J. L., Sherman N. A., Wright K. M., Moyle L. C., Willis J. H., Rieseberg L. H. 2012. What can patterns of differentiation across plant genomes tell us about adaptation and speciation? Phil. Trans. R. Soc. B 367, 364–373 10.1098/rstb.2011.0199 (doi:10.1098/rstb.2011.0199) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Charlesworth B., Nordborg M., Charlesworth D. 1997. The effects of local selection, balanced polymorphism and background selection on equilibrium patterns of genetic diversity in subdivided populations. Genet. Res. 70, 155–174 10.1017/S0016672397002954 (doi:10.1017/S0016672397002954) [DOI] [PubMed] [Google Scholar]
- 28.Bierne N., Welch J., Loire E., Bonhomme F., David P. 2011. The coupling hypothesis: why genome scans may fail to map local adaptation genes. Mol. Ecol. 20, 2044–2072 10.1111/j.1365-294X.2011.05080.x (doi:10.1111/j.1365-294X.2011.05080.x) [DOI] [PubMed] [Google Scholar]
- 29.Manel S., Conord C., Despres L. 2009. Genome scan to assess the respective role of host-plant and environmental constraints on the adaptation of a widespread insect. BMC Evol. Biol. 9, 288. 10.1186/1471-2148-9-288 (doi:10.1186/1471-2148-9-288) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mariac C., et al. 2011. Genetic basis of pearl millet adaptation along an environmental gradient investigated by a combination of genome scan and association mapping. Mol. Ecol. 20, 80–91 10.1111/j.1365-294X.2010.04893.x (doi:10.1111/j.1365-294X.2010.04893.x) [DOI] [PubMed] [Google Scholar]
- 31.Turner T. L., von Wettberg E. J., Nuzhdin S. V. 2008. Genomic analysis of differentiation between soil types reveals candidate genes for local adaptation in Arabidopsis lyrata. PLoS ONE 3, e3183. 10.1371/journal.pone.0003183 (doi:10.1371/journal.pone.0003183) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeFaveri J., Shikano T., Shimada Y., Goto A., Merila J. 2011. Global analysis of genes involved in freshwater adaptation in threespine sticklebacks (Gasterosteus aculeatus). Evolution 65, 1800–1807 10.1111/j.1558-5646.2011.01247.x (doi:10.1111/j.1558-5646.2011.01247.x) [DOI] [PubMed] [Google Scholar]
- 33.Rogers S. M., Bernatchez L. 2007. The genetic architecture of ecological speciation and the association with signatures of selection in natural lake whitefish (Coregonus sp. Salmonidae) species pairs. Mol. Biol. Evol. 24, 1423–1438 10.1093/molbev/msm066 (doi:10.1093/molbev/msm066) [DOI] [PubMed] [Google Scholar]
- 34.Via S. 2009. Natural selection in action during speciation. Proc. Natl Acad. Sci. USA 106, 9339–9946 10.1073/pnas.0901397106 (doi:10.1073/pnas.0901397106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Renaut S., Maillet N., Normandeau E., Sauvage C., Derome N., Rogers S. M., Bernatchez L. 2012. Genome-wide patterns of divergence during speciation: the lake whitefish case study. Phil. Trans. R. Soc. B 367, 354–363 10.1098/rstb.2011.0197 (doi:10.1098/rstb.2011.0197) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mather K. 1983. Response to selection. In The genetics and biology of Drosophila, vol. 3c (eds Ashburner M., Carson H. L., Thompson J. N.), pp. 155–221 London, UK: Academic Press [Google Scholar]
- 37.Burke J. M., Tang S., Knapp S. J., Rieseberg L. H. 2002. Genetic analysis of sunflower domestication. Genetics 161, 1257–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wright D., Rubin C.-J., Martinez Barrio A., Schütz K., Kerje S., Brändström H., Kindmark A., Jensen P., Andersson L. 2010. The genetic architecture of domestication in the chicken: effects of pleiotropy and linkage. Mol. Ecol. 19, 5140–5156 10.1111/j.1365-294X.2010.04882.x (doi:10.1111/j.1365-294X.2010.04882.x) [DOI] [PubMed] [Google Scholar]
- 39.Albert F. W., et al. 2009. Genetic architecture of tameness in a rat model of animal domestication. Genetics 182, 541–554 10.1534/genetics.109.102186 (doi:10.1534/genetics.109.102186) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baack E. J., Sapir Y., Chapman M. A., Burke J. H., Rieseberg L. H. 2007. Selection on domestication traits and quantitative trait loci in crop-wild sunflower hybrids. Mol. Ecol. 17, 666–677 10.1111/j.1365-294X.2007.03596.x (doi:10.1111/j.1365-294X.2007.03596.x) [DOI] [PubMed] [Google Scholar]
- 41.Hawthorne D. J., Via S. 2001. Genetic linkage facilitates ecological specialization and reproductive isolation in pea aphids. Nature 412, 904–907 10.1038/35091062 (doi:10.1038/35091062) [DOI] [PubMed] [Google Scholar]
- 42.Turner T. L., Levine M. I., Eckert M. L., Begun D. 2008. Genomic analysis of adaptive differentiation in Drosophila melanogaster. Genetics 179, 455–473 10.1534/genetics.107.083659 (doi:10.1534/genetics.107.083659) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Merrill R. M., VanSchooten B., Scott J. A., Jiggins C. D. 2011. Pervasive genetic associations between traits causing reproductive isolation in Heliconius butterflies. Proc. R. Soc. B. 278, 511–518 10.1098/rspb.2010.1493 (doi:10.1098/rspb.2010.1493) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim S. C., Rieseberg L. H. 1999. Genetic architecture of species differences in annual sunflowers: implications for adaptive trait introgression. Genetics 153, 965–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Begun D. J., Aquadro C. F. 1992. Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature 356, 519–520 10.1038/356519a0 (doi:10.1038/356519a0) [DOI] [PubMed] [Google Scholar]
- 46.Via S., Bouck A. C., Skillman S. 2000. Reproductive isolation between sympatric races of pea aphids. II. Selection against migrants and hybrids in the parental environment. Evolution 54, 1626–1637 [DOI] [PubMed] [Google Scholar]
- 47.Via S., Conte G. L., Mason-Foley C., Mills K., Lew J. Submitted Coupling a QTL map with a genome scan reveals evidence for divergence hitchhiking during incipient speciation. [Google Scholar]
- 48.Feder J. L., Gejji R., Yeaman S., Nosil P. 2012. Establishment of new mutations under divergence and genomic hitchhiking. Phil. Trans. R. Soc. B 367, 461–474 10.1098/rstb.2011.0256 (doi:10.1098/rstb.2011.0256) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kobayashi Y., Telschow A. 2010. The concept of effective recombination rate and its application in speciation theory. Evolution 65, 617–628 10.1111/j.1558-5646.2010.01156.x (doi:10.1111/j.1558-5646.2010.01156.x) [DOI] [PubMed] [Google Scholar]
- 50.Ting C.-T., Tsaur S.-C., Wu C.-I. 2000. The phylogeny of closely related species as revealed by the genealogy of a speciation gene, Odysseus. Proc. Natl Acad. Sci. USA 97, 5313–5316 10.1073/pnas.090541597 (doi:10.1073/pnas.090541597) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Michel A. P., Sim S., Powell T. H. Q., Taylor M. S., Nosil P., Feder J. L. 2010. Widespread genomic divergence during sympatric speciation. Proc. Natl Acad. Sci. USA 107, 9724–9729 10.1073/pnas.1000939107 (doi:10.1073/pnas.1000939107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Turner T. L., Hahn M. W., Nuzhdin S. V. 2005. Genomic islands of speciation in Anopheles gambiae. PLoS Biol. 3, e285. 10.1371/journal.pbio.0030285 (doi:10.1371/journal.pbio.0030285) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hohenlohe P. A., Bassham S., Etter P. D., Stiffler N., Johnson E. A., Cresko W. A. 2010. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet. 6, e1000862. 10.1371/journal.pgen.1000862 (doi:10.1371/journal.pgen.1000862) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Albert S. Y. K., Sawaya S., Vines T. H., et al. 2007. The genetics of adaptive shape shift in stickleback: pleiotropy and effect size. Evolution 62, 76–85 [DOI] [PubMed] [Google Scholar]
- 55.Makinen H. S., Shikano T., Manuel Cano J., Merila J. 2008. Hitchhiking mapping reveals a candidate genomic region for natural selection in three-spined stickleback chromosome VIII. Genetics 178, 453–465 10.1534/genetics.107.078782 (doi:10.1534/genetics.107.078782) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lawniczak M. K. N., et al. 2010. Widespread divergence between incipient Anopheles gambiae species revealed by whole genome sequences. Science 330, 512–514 10.1126/science.1195755 (doi:10.1126/science.1195755) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Egan S. P., Nosil P., Funk D. J. 2008. Selection and genomic differentiation during ecological speciation: isolating the contributions of host association via a comparative genome scan of Neochlamisus bebbianae leaf beetles. Evolution 62, 1162–1181 10.1111/j.1558-5646.2008.00352.x (doi:10.1111/j.1558-5646.2008.00352.x) [DOI] [PubMed] [Google Scholar]
- 58.Willig E.-M., Bentzen P., van Oosterhout C., Hoffmann M., Cable C., Breden F., Weigel D., Dreyer C. 2010. Genome-wide single nucleotide polymorphism reveals population history and adaptive divergence in wild guppies. Mol. Ecol. 19, 968–984 10.1111/j.1365-294X.2010.04528.x (doi:10.1111/j.1365-294X.2010.04528.x) [DOI] [PubMed] [Google Scholar]
- 59.MacKay T. F. C., Stone E. A., Ayroles J. 2009. The genetics of quantitative traits: challenges and prospects. Nat. Rev. Genet. 10, 565–577 10.1038/nrg2612 (doi:10.1038/nrg2612) [DOI] [PubMed] [Google Scholar]
- 60.Butlin R. 2008. Population genomics and speciation. Genetica 138, 409–418 10.1007/s10709-008-9321-3 (doi:10.1007/s10709-008-9321-3) [DOI] [PubMed] [Google Scholar]
- 61.Strasburg J. L., Scotti-Saintagne C., Scotti I., Lai Z., Rieseberg L. H. 2009. Genomic patterns of adaptive divergence between chromosomally differentiated sunflower species. Mol. Biol. Evol. 26, 1341–1355 10.1093/molbev/msp043 (doi:10.1093/molbev/msp043) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scascitelli M., Whitney K. D., Randell R. A., King M., Buerkle C. A., Rieseberg L. H. 2010. Genome scan of hybridizing sunflowers from Texas (Helianthus annuus and H. debilis) reveals asymmetric patterns of introgression and small islands of genomic differentiation. Mol. Ecol. 19, 521–541 10.1111/j.1365-294X.2009.04504.x (doi:10.1111/j.1365-294X.2009.04504.x) [DOI] [PubMed] [Google Scholar]
- 63.Carneiro M., Blanco-Aguiar J. A., Villafuerte R., Ferrand N., Nachman M. W. 2010. Speciation in the european rabbit (Oryctolagus cuniculus): islands of differentiation on the X chromosome and autosomes. Evolution 64, 3443–3460 10.1111/j.1558-5646.2010.01092.x (doi:10.1111/j.1558-5646.2010.01092.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hohenlohe P. A., Bassham S., Currey M., Cresko W. A. 2012. Extensive linkage disequilibrium and parallel adaptive divergence across threespine stickleback genomes. Phil. Trans. R. Soc. B 367, 395–408 10.1098/rstb.2011.0245 (doi:10.1098/rstb.2011.0245) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Le Rouzic A., Østbye K., Klepaker T. O., Hansen T. M., Bernatchez L., Schluter D., Vollestad A. 2011. Strong and consistent natural selection associated with armour reduction in sticklebacks. Mol. Ecol. 20, 2483–2493 10.1111/j.1365-294X.2011.05071.x (doi:10.1111/j.1365-294X.2011.05071.x) [DOI] [PubMed] [Google Scholar]
- 66.Avise J. C. 2000. Phylogeography: the history and formation of species. Cambridge, MA: Harvard University Press [Google Scholar]
- 67.Pritchard J. K., Stephens M., Donnelly P. 2000. Inference of population structure using multilocus genotype data. Genetics 155, 945–949 [DOI] [PMC free article] [PubMed] [Google Scholar]