

Abstract
Introduction
Nondystrophic myotonia (NDM) is caused by mutations in muscle chloride and sodium channels. Currently there is no standardized instrument for documenting symptom frequency and severity in NDM.
Methods
Subjects used an automated interactive telephone-based voice response diary (IVR) to record frequency and severity of stiffness, weakness, pain, and tiredness once a week for 8 weeks following their baseline visits.
Results
Here we describe the IVR and report data on 76 subjects for a total of 385 person-weeks. Overall there were 5.1 calls per subject. Forty-eight subjects called in 5 or more times, and 14 called in 8 times. Stiffness was both the most frequent and severe symptom. Warm-up and handgrip myotonia were associated with higher severity scores for stiffness.
Discussion
IVR is a convenient technology to allow patient reporting of repeated and real-time symptom frequency and severity, and is being used in a trial of mexiletine in NDM.
Search terms: myotonia, nondystrophic myotonia, patient reported outcome measures, ion channel gene defects, muscle disease
Introduction
Non-dystrophic myotonia (NDM) is caused by mutations in the skeletal muscle sodium and chloride channels [1-7], and includes disorders such as myotonia congenita, paramyotonia congenita, potassium aggravated myotonia, and hyperkalemic periodic paralysis. The hallmark of NDM is myotonia, impaired muscle relaxation following voluntary or evoked contraction. Myotonia can be associated with stiffness, weakness, and pain. Although there are some classic phenomenon associated with NDM—reduction of symptoms with repetition (warm-up) in myotonia congenita, transient paresis in autosomal recessive myotonia congenita, and worsening of symptoms with repetition (paramyotonia) in paramyotonia congenita—in practice there is considerable clinical overlap between different genetic subtypes[5, 8, 9]. Diagnosis is based on clinical examination, electrophysiological studies, and, ultimately, genetic testing. Myotonic dystrophy type 2 (DM2) is caused by an unstable expansion of a CCTG tetraplet and can have a similar clinical presentation to NDM. Unlike myotonic dystrophy type 1 which presents with significant weakness and muscle atrophy, DM2 can present primarily with myotonia[10].
Although many promising drugs have been evaluated for the treatment of myotonia, there are no FDA approved treatments for NDM[11]. In the Cochrane review looking at treatment in myotonic disorders, one of the major problems identified with studies to date has been lack of clearly defined outcome measures[12]. Although there have been a number of genotype phenotype studies, there is no gold standard for either quantitative or qualitative assessment of myotonia. Most studies that include patient-reported symptoms simply state how many subjects reported a given symptom during a particular study visit or whether a given symptom got worse, remained the same, or improved[13, 14]. In fact, it is unclear how quantitative measures of myotonia relate to patient perception of improving or worsening symptoms.
Increasingly, patient-reported outcome measures (PROs) are being used to help characterize patients’ experience of their disorder directly [15-18]. PROs have the advantage of recording the patient experience as it occurs, without the bias of interpretation by an interviewer. These measures can be collected in many formats, from pen and paper questionnaires, to interactive call-in responses, and they can be used to establish outcomes as diverse as quality of life and symptom severity. The U.S. Food and Drug Administration has increasingly been encouraging industry to adopt PROs in public forums, and it has published guidelines for the use of PRO measures by industry[19]. This report describes our initial use of an automated interactive voice response diary (IVR) of real time and repeated patient-reported symptom frequency and severity in a natural history study, which includes how the system works, as well as initial results of symptom frequency and severity for NDM. This IVR system is currently being used as the primary endpoint measure in an ongoing phase II trial of mexiletine for NDM.
Methods
The NDM subjects enrolled in this study (n=76) were all part of the Consortium for Clinical Investigation of Neurological Channelopathies (CINCH) Group’s Non-dystrophic Myotonia: Genotype-Phenotype Correlation and Longitudinal Study, sponsored by the NIH Office of Rare Diseases Research. Subjects were recruited from 6 academic centers across the United States, Canada, and United Kingdom between 2006 to 2009. All sites received approval by their IRB for investigations involving human subjects, and informed consent was obtained from all study participants. For subjects less than 18 years of age patient assent and parental consent were obtained. Inclusion criteria were: age greater than 6 years; clinical symptoms or signs suggestive of myotonic disorders; presence of myotonic discharges on electromyography; and persistence of symptoms and signs after discontinuation of medications that produce myotonia (fibrate acid derivatives, hydroxymethylglutaryl CoA reductase inhibitors, chloroquine, and colchicine). In addition subjects had to have the absence of features suggestive of myotonic dystrophy (ptosis, temporal wasting, mandibular weakness, and cataracts occurring before the age 50, evidence of multisystem involvement). Exclusion criteria were: inability or unwillingness to provide informed consent, or other neurologic conditions that might affect the assessment of the study measurements.
All subjects underwent a baseline visit which included a symptom questionnaire, physical examination, and electrophysiological studies. Genetic testing was performed on all participants. For this study, subjects were grouped into chloride channel mutations, sodium channel mutations, DM2, and no mutation detected. Genetic testing was based on previously published mutations (OMIM # 255700, 160800, 118425, 168300, 170500, 608390, 602668), and subjects with no mutations detected may turn out to have novel mutations.
Interactive Voice Response system (see Figure 1)
Figure 1.
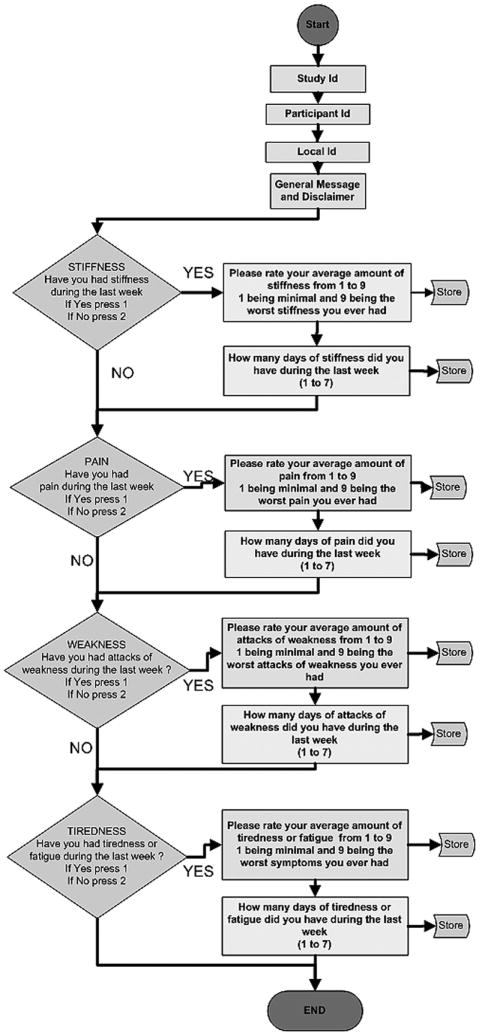
Flow diagram illustrating the subject call-in process for the IVR. All subjects are identified by entering a participant ID. Then they are asked to enter 1 for yes or 2 for no whether they had a given symptom (stiffness, pain, weakness, tiredness) in the preceding week. Second tier questions ask them to estimate the number of days in a given week the symptom is experienced and then to estimate the symptom severity on a 1-9 ranked scale.
At the baseline visit, participants were given a brief orientation to an automated interactive voice response (IVR) system, and were expected to call in once weekly for 8 consecutive weeks. As part of the IVR system orientation, subjects were given the toll-free phone number, a unique identification number, and an instruction sheet for use of the telephone-based IVR data collection system. When subjects first use the system by dialing the toll-free phone number, they hear a brief welcome message, a reminder that their participation is voluntary, and are instructed to press specified keys on the telephone number pad to begin reporting their symptoms. This IVR system assessed symptom severity and frequency in four categories: muscle stiffness, weakness, pain, and tiredness. When subjects called in they were asked to answer four core questions: whether or not they experienced stiffness, weakness, pain, or tiredness (yes/no) in the prior week. If they gave a positive response to any of the core questions, second tier questions asked them to rate the frequency they had each symptom (days/week) and to rank the severity of each symptom on a scale of 1-9, 1 being minimal and 9 being the worst ever experienced. Before moving on to each element of the IVR the automated system asks participants if the information they have entered is correct. As the interest here was in creating a real-time voice response diary of symptom frequency and severity, once entered there was no mechanism to edit an IVR entry. This approach was taken to reduce “editing” of entries after the fact, introducing “back-filling” bias seen in traditional paper diaries [16, 17]. The subject response data were immediately stored on an Oracle database. After the baseline visit a reminder was sent in the mail to the participants after 1 week. A phone call reminder to complete the IVR was placed after the 4th week.
Statistical Considerations
If there was more than one call into the system in a given week and the adjacent week had no calls, the average severity score and number of days was extrapolated into the two week period. In cases where the number of days reported exceeded 7 days, it was reduced to 7 days. Compliance was considered 100% if a subject called in at least once each week for the first 8 weeks.
Severity data fit a normal distribution and standard deviation both within subject and between subjects was estimated. Median data is presented with quartile data in parenthesis (1st quartile = Q1 = 25% of data, 3rd quartile = Q3 = 75% of data). When comparing genetic subtypes, a mixed model was used (Laird and Ware) when fitting the severity scores and regressing on various covariates using the maximum likelihood method. The subject characteristics were entered as fixed effects, while each subject was considered to have a random intercept effect. The Wald test was used to compute significance level for the fixed effect covariates.
Results
76 subjects (42 male, and 34 female) had 8 week follow-up data available at the time of this report. The mean age was 45.7 years, the median 46 years, and the range 12-79 years. The mean age of stiffness symptom onset was 13.7 years. Twenty-five subjects (32.9%) had sodium channel mutations, 27 (35.5%) chloride channel mutations, 5 (6.6%) DM2 mutations, and 19 (25.0%) had no mutations detected (Table 1).
Table 1.
Demographics. 76 subjects, 385 person-weeks recorded.
| Demographics | |
|---|---|
| Factor | No. (%) |
|
| |
| Subjects | 76 (100.0) |
|
| |
| Male | 42 (55.3) |
| Female | 34 (44.7) |
|
| |
| Mean Age (yrs.) | 45.7 |
| Median Age | 46 |
| Range | 12-79 |
|
| |
| Mean Age at Diagnosis (yrs.) | 31.2* |
|
| |
| Sodium Channel Mutations | 25 (32.9%) |
| Chloride Channel Mutations | 27 (35.5%) |
| DM2 | 5 (6.6%) |
| No Mutation Detected | 19 (25.0) |
Based on 23 subjects reporting
The 76 participants accounted for 385 person-weeks reported during the first 8 weeks after the baseline visit. Overall there were 5.07 calls per participant (63.4%). Forty-eight (63.2%) subjects called in 5 or more times, and 14 (18.4%) called in all 8 weeks (Table 2). Comparing compliance for weeks 5-8 (month 2) to compliance for weeks 1-4 (month 1) there was a median of 1 fewer calls per participant (Q1, Q3: 0, 2).
Table 2.
Number of weeks a call was recorded by the IVR system. Total number reporting 76. Forty-eight subjects called in 5 or more times (63.2%), and 14 called in 8 times (18.4% of the total population).
| Number of Weeks a Call was recorded by IVR System | ||
|---|---|---|
| No. of Weeks | Frequency | Percent |
| 0 | 2 | 2.6 |
| 1 | 6 | 7.9 |
| 2 | 4 | 5.3 |
| 3 | 7 | 9.2 |
| 4 | 9 | 11.8 |
| 5 | 11 | 14.5 |
| 6 | 14 | 18.4 |
| 7 | 9 | 11.8 |
| 8 | 14 | 18.4 |
| Total | 76 | 100.0 |
The weekly frequency of symptoms was: stiffness 344 (89.4%), weakness 243 (63.1%), tiredness 267 (69.4%), and pain 242(62.9%) (Figure 2A). Weekly reporting included the number of days for which each subject experienced a given symptom. The median numbers of days per week reported for each symptom were: stiffness 5 (Q1, Q3: 2, 7), weakness 2 (Q1, Q3: 0, 5), tiredness 3 (Q1, Q3: 0, 6), and pain 2 (Q1, Q3: 0, 7).
Figure 2.
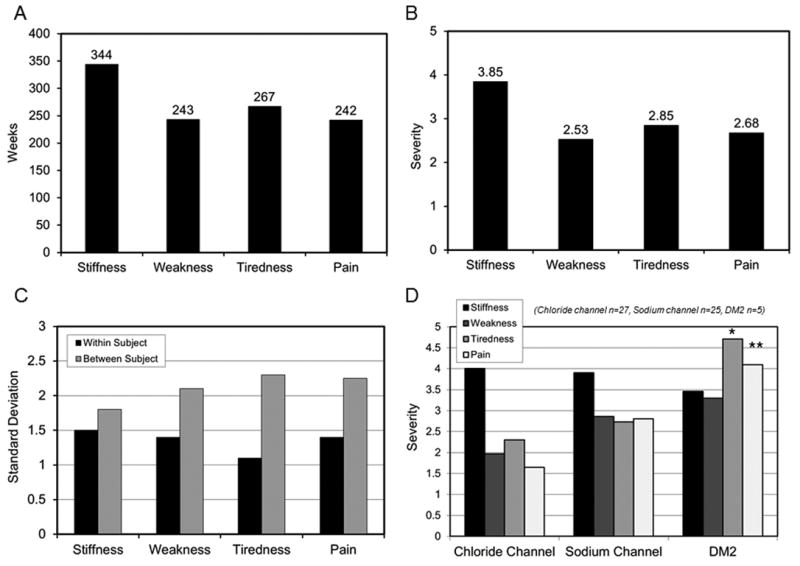
IVR—frequency and severity of reported symptoms. A. Weekly frequency of reported symptoms (n=76, total of 385 person-weeks recorded). Subjects were considered to have the given symptom if they answered yes to the first tier question whether they experienced a given symptom in the prior week. Stiffness was the most frequently reported symptom at 344 weeks (89.4% of total person-weeks reported). B. Mean symptom severity (n=76, total of 385 person-weeks recorded). If subjects experienced a given symptom in the prior week they were asked to rate severity on a 1-9 ranked scale, 1 being minimal and 9 being the worst ever experienced. If a symptom was not experienced, severity was assumed to be 0. Stiffness was the most severe symptom recorded with a mean of 3.85. C. Standard deviation for symptom reported (n=76, total 385 person-weeks recorded). The within subject standard deviation was less than between subject standard deviation for all symptoms reported. The standard deviation was relatively high compared to the mean for each symptom category and may reflect natural variation in week-to-week symptomology in subjects with NDM. D. Mean severity score by genetic mutation. There were significantly higher severity scores for tiredness (p=0.04) and pain (p=0.007) for DM2 compared to chloride channel mutations. * = p< 0.05, ** p< 0.01, *** = p< 0.001.
For each week a given symptom was reported, the patient was also asked to rate the severity of the symptom on a 1-9 scale. The median symptom severity for each symptom was: stiffness 4 (Q1, Q3: 2, 6), weakness 2 (Q1, Q3: 0, 5), tiredness 3 (Q1, Q3: 0, 5), and pain 3 (Q1, Q3: 0, 5). The mean symptom severity was stiffness 3.85, weakness 2.53, tiredness 2.85 and pain 2.68 (Figure 2B). Figure 2C shows the within-subject and between-subject standard deviation (SD) for symptom severity. The within-subject SDs were 1.50 for stiffness, 1.41 for weakness, 1.13 for tiredness, and 1.38 for pain. The between subject SDs were 1.82 for stiffness, 2.08 for weakness, 2.35 for tiredness, and 2.25 for pain. The within subject SD was less than the between subject SD for all symptoms reported.
Stratifying the study population by genetic mutation, there were significantly higher severity scores for tiredness and pain for DM2 compared to chloride channel mutations (Figure 2D). When tiredness severity scale was regressed on the genetic mutation the mean estimates were 4.71 for DM2 and 2.30 for chloride channel mutation (p = 0.04). When the dependent variable was pain, the mean estimates were 4.09 for DM2, and 1.71 for chloride channel mutation (p = 0.007). Of note, stiffness did not differ significantly in either severity or frequency by genetic subtype.
For subjects who had handgrip myotonia on clinical examination there was an average increase in the severity score for stiffness of 1.12 (p=0.02). For subjects who had warm-up on clinical examination there was an average increase in severity score for stiffness of 0.99 (p=0.03). There was no association between any symptom category on the IVR and eye closure myotonia, clinical measures of paramyotonia or electromyography myotonia grade for any muscle tested.
Discussion
We report the use of an automated telephone call-in diary of symptom severity and frequency in subjects with NDM. Traditionally, subject compliance for paper-based diaries has varied widely. One study noted a compliance rate of 90%, but actual on-time compliance as low as 11%[18], and other studies have reported compliance between 11% and 20%[16]. In this study, we report a mean 5.07 out of 8 possible calls (63.4%) per participant for the IVR.
The closest gold-standard for patient reported outcomes is the paper diary. Designing a study that would compare the IVR system with a paper diary is in our opinion not feasible, because it would require micro-managing the subject on a daily basis. Any validity study would require recording the outcome for a specific time period in both systems, while at the same time recording their outcomes independently of each other. We anticipate that patients would use the diary as a script to make their IVR calls. Once this occurs the disconcordance would simply represent phone keying errors and would not reflect any error on the part of the diary.
In addition, we do not consider the paper diary a gold-standard in the sense that it is more accurate than most methods. To the contrary, it has been documented to be flawed in many ways. It is well documented that patients who do not have access to their diary for an extended period have tried to complete the diary by recall (back-filling). In addition patients can fill in multiple forms prior to the time they are due (forward-filling)[17, 18]. Furthermore, most diaries allow the patient to see what they have recorded previously and may cause a subliminal tendency to maintain consistency.
The IVR system represents a unique medium for patients to document their outcomes. The natural benefit has been that a phone (land-line or cell) would be available to patients more often than a paper diary. The IVR call is date-stamped and would prevent forward- or back-filling of forms. In addition this automated approach reduces the time burden and potential for mistakes seen in transcription of traditional paper forms. Based on the experience in this natural history trial, when we designed the ongoing randomized control trial of mexiletine in NDM we made adjustments to the protocol so that patients call in daily rather than weekly. We designed an automated reminder system to patients for their daily calls, and if they did not call on a particular day we contacted them again directly.
No previous studies have detailed the frequency and severity of symptoms related to myotonia on a weekly basis over a two month period. A recent paper described 62 patients with NDM from the Netherlands and suggested that subjects with chloride channel mutations reported weakness significantly more than subjects with sodium channel mutations, and subjects with sodium channel mutations reported “painful myotonia” more than chloride channel mutations[14]. Interestingly, in our analysis of the IVR there was no significant difference in patient-reported weakness between genetic subtypes. Pain and tiredness appear to correlate with mutations in DM2. Overall, we found stiffness was the most frequent and severe symptom reported, and it did not differ significantly between genetic subtypes.
NDM is classically considered a chronic condition, although day-to-day and even hour-to-hour variations in the degree of myotonia are often reported. A study that examined a quantitative measure of handgrip myotonia in myotonic dystrophy reported daily test-retest variability of 22.6% and same day intertrial coefficient of variation of 33.2%[20]. Our study supports this notion. We found variability in weekly symptom severity, regardless of symptom category. However, subjects report stiffness 89.4% of the time, with a median daily frequency of 5 days per week. And higher severity scores for stiffness on the IVR, in particular, appear to be associated with warm-up and handgrip myotonia seen on clinical examination.
Based on the severity score for stiffness and standard deviations reported here, a two-period cross-over study design would provide 87% to 93% statistical power to detect a change in the severity scale of 2/3 of a standard deviation with complete outcome data from 60 participants. Some degree of over-sampling would be required to assure that 60 subjects provided complete outcome data. The power estimate is based on Monte Carlo simulations using a mixed linear model rounding the random variable to the nearest integer from 1 to 9[21].
Currently, stiffness on the IVR is being used as a primary outcome measure in an FDA Orphan Drug grant supported phase II placebo-controlled cross-over study examining the effects of mexiletine in NDM.
Acknowledgments
The authors wish to acknowledge Jeff Krischer and Joseph Gomes (USF) for their vision and development of the IVR system.
Disclosures The NDM Genotype-Phenotype Correlation and Longitudinal Study and the IVR evaluation project described was funded by the Consortium of Clinical Investigation on Neurologic Channelopathies grant number U54RR019482 from the NIH and Office of Rare Diseases. The views expressed in written materials or publications do not necessarily reflect the official policies of the Department of Health and Human Services; nor does mention by trade names, commercial practices, or organizations imply endorsement by the U.S. Government.
Additional funding was provided in part by the University of Kansas Medical Center GCRC grant M01 RR 023940 NCRR/NIH, and the University of Rochester CTSA grant UL1 RR 024160 NCRR/NIH. The initial development of the IVR system was supported by Department of Defense award #DAMD 17-01-2-0056.
Rachel Richesson and Brian Bundy’s contributions to this work, and the development of the system were supported by Grant Number RR019259 from the NCRR, an NIH component, and the Office of Rare Diseases. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NCRR or ORD or NIH.
Abbreviations
- NDM
Nondystrophic Myotonia
- IVR
Interactive Voice Response Diary
- DM2
Myotonic Dystrophy Type 2
- FDA
Food and Drug Association
- PRO
Patient-Reported Outcomes
- CINCH
Consortium of Clinical Investigation on Neurologic Channelopathies
Additional Authors Appendix
The Consortium of Clinical Investigation on Neurologic Channelopathies (CINCH)
Steering Committee:Principal Investigator: Richard J Barohn, MD (Kansas City, KS); Site Principal Investigators: Anthony Amato, MD (Boston, MA), Robert C Griggs, MD (Rochester, NY), Stephen C Cannon, MD, PhD (Dallas, TX), Michael Hanna, MD (London, UK), Shannon Venance, MD PhD (London, ON); Chief Statistician: Brian N Bundy, PhD (Tampa, FL); Study Coordinator at Lead Site: Laura Herbelin, BS (Kansas City, KS); CINCH Lead Research Coordinator: Kimberly Hart (Rochester, NY); and CINCH Program Manager: Barbara Herr, MS (Rochester, NY). Investigators/CINCH Trainees: Yunxia Wang, MD (Kansas City, KS), Jeffrey Statland, MD (Rochester, NY), Doreen Fiahlo, MD (London, UK), Emma Matthews, MD (London, UK), Mohammad Salajegheh, MD (Co-Principal investigator, Boston, MA), Ronan Walsh, MD (Boston, MA), Jaya Trivedi, MD (Co-Principal Investigator, Dallas, TX), Angelika Hahn, MD (Co-Principal Investigator, London, ON), James Cleland, MD (Rochester, NY). Data and Technology Coordinating Center Principal Investigator: Jeffrey Krischer, PhD (Tampa, FL). Clinical Evaluators: Laura Herbelin, BS (Kansas City, KS), Merideth Donlan PT, (Boston, MA), Rhonda McLin, PTA(Dallas, TX), Shree Pandya, PT, MS, Katy Eichinger, PT, DPT, NCS (Rochester NY), Karen Findlater, PT (London, ON) Liz Dewar PT (London UK). Clinical Coordinators: Laura Herbelin, BS (Kansas City, KS), Kristen Whiteside (Boston, MA), Nina Gorham (Dallas, TX), Kori Ladonna (London, ON), Jennifer Verheyden (London, ON), Kimberly Hart (Rochester, NY). ParticipatingMembers of the Data and Technology Coordinating Center: Joseph Gomes (Tampa, FL), Bonnie Patterson, BA CCRP (Tampa, FL), David Cuthbertson, MS (Tampa, FL), Rachel Richesson, PhD, MPH (Tampa, FL), and Jennifer Lloyd (Tampa, FL).
References
- 1.Cannon SC. Spectrum of sodium channel disturbances in the nondystrophic myotonias and periodic paralyses. Kidney Int. 2000;57(3):772–9. doi: 10.1046/j.1523-1755.2000.00914.x. [DOI] [PubMed] [Google Scholar]
- 2.Colding-Jorgensen E. Phenotypic variability in myotonia congenita. Muscle Nerve. 2005;32(1):19–34. doi: 10.1002/mus.20295. [DOI] [PubMed] [Google Scholar]
- 3.Ptacek L. The familial periodic paralyses and nondystrophic myotonias. Am J Med. 1998;105(1):58–70. doi: 10.1016/s0002-9343(98)00123-5. [DOI] [PubMed] [Google Scholar]
- 4.Ptacek LJ, Johnson KJ, Griggs RC. Genetics and physiology of the myotonic muscle disorders. N Engl J Med. 1993;328(7):482–9. doi: 10.1056/NEJM199302183280707. [DOI] [PubMed] [Google Scholar]
- 5.Fialho D, et al. Chloride channel myotonia: exon 8 hot-spot for dominant-negative interactions. Brain. 2007;130(Pt 12):3265–74. doi: 10.1093/brain/awm248. [DOI] [PubMed] [Google Scholar]
- 6.Hoffman EP, Wang J. Duchenne-Becker muscular dystrophy and the nondystrophic myotonias. Paradigms for loss of function and change of function of gene products. Arch Neurol. 1993;50(11):1227–37. doi: 10.1001/archneur.1993.00540110101010. [DOI] [PubMed] [Google Scholar]
- 7.Vicart S, et al. Human skeletal muscle sodium channelopathies. Neurol Sci. 2005;26(4):194–202. doi: 10.1007/s10072-005-0461-x. [DOI] [PubMed] [Google Scholar]
- 8.Matthews E, et al. What causes paramyotonia in the United Kingdom? Common and new SCN4A mutations revealed. Neurology. 2008;70(1):50–3. doi: 10.1212/01.wnl.0000287069.21162.94. [DOI] [PubMed] [Google Scholar]
- 9.Rudel R, Ricker K, Lehmann-Horn F. Genotype-phenotype correlations in human skeletal muscle sodium channel diseases. Arch Neurol. 1993;50(11):1241–8. doi: 10.1001/archneur.1993.00540110113011. [DOI] [PubMed] [Google Scholar]
- 10.Meola G, Moxley RT., 3rd Myotonic dystrophy type 2 and related myotonic disorders. J Neurol. 2004;251(10):1173–82. doi: 10.1007/s00415-004-0590-1. [DOI] [PubMed] [Google Scholar]
- 11.Cleland JC, Griggs RC. Treatment of neuromuscular channelopathies: current concepts and future prospects. Neurotherapeutics. 2008;5(4):607–12. doi: 10.1016/j.nurt.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trip J, et al. Drug treatment for myotonia. Cochrane Database Syst Rev. 2006;(1):CD004762. doi: 10.1002/14651858.CD004762.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kwiecinski H, Ryniewicz B, Ostrzycki A. Treatment of myotonia with antiarrhythmic drugs. Acta Neurol Scand. 1992;86(4):371–5. doi: 10.1111/j.1600-0404.1992.tb05103.x. [DOI] [PubMed] [Google Scholar]
- 14.Trip J, et al. Redefining the clinical phenotypes of non-dystrophic myotonic syndromes. J Neurol Neurosurg Psychiatry. 2009 doi: 10.1136/jnnp.2008.162396. [DOI] [PubMed] [Google Scholar]
- 15.Chang CH. Patient-reported outcomes measurement and management with innovative methodologies and technologies. Qual Life Res. 2007;16(Suppl 1):157–66. doi: 10.1007/s11136-007-9196-2. [DOI] [PubMed] [Google Scholar]
- 16.Gwaltney CJ, Shields AL, Shiffman S. Equivalence of electronic and paper-and-pencil administration of patient-reported outcome measures: a meta-analytic review. Value Health. 2008;11(2):322–33. doi: 10.1111/j.1524-4733.2007.00231.x. [DOI] [PubMed] [Google Scholar]
- 17.Palmblad M, Tiplady B. Electronic diaries and questionnaires: designing user interfaces that are easy for all patients to use. Qual Life Res. 2004;13(7):1199–207. doi: 10.1023/B:QURE.0000037501.92374.e1. [DOI] [PubMed] [Google Scholar]
- 18.Stone AA, et al. Patient non-compliance with paper diaries. BMJ. 2002;324(7347):1193–4. doi: 10.1136/bmj.324.7347.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guidance for industry: patient-reported outcome measures: use in medical product development to support labeling claims: draft guidance. Health Qual Life Outcomes. 2006;4:79. doi: 10.1186/1477-7525-4-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moxley RT, 3rd, et al. Computerized hand grip myometry reliably measures myotonia and muscle strength in myotonic dystrophy (DM1) Muscle Nerve. 2007;36(3):320–8. doi: 10.1002/mus.20822. [DOI] [PubMed] [Google Scholar]
- 21.Laird NM, Ware JH. Random-effects models for longitudinal data. Biometrics. 1982;38(4):963–74. [PubMed] [Google Scholar]