

Abstract
Erythrocytes, via release of ATP in areas of low oxygen (O2) tension, are components of a regulatory system for the distribution of perfusion in skeletal muscle ensuring optimal O2 delivery to meet tissue needs. In type 2 diabetes (DM2), there are defects in O2 supply to muscle as well as a failure of erythrocytes to release ATP. The goal of this study was to ascertain if a phosphodiesterase 3 (PDE3) inhibitor, cilostazol, would rescue low O2-induced ATP release from DM2 erythrocytes and, thereby, enable these cells to dilate isolated erythrocyte-perfused skeletal muscle arterioles exposed to decreased extraluminal O2. Erythrocytes were obtained from healthy humans (HH; n = 12) and humans with DM2 (n = 17). We determined that 1) PDE3B is similarly expressed in both groups, 2) mastoparan 7 (Gi activation) stimulates increases in cAMP in HH but not in DM2 erythrocytes, and 3) pretreatment of DM2 erythrocytes with cilostazol resulted in mastoparan 7-induced increases in cAMP not different from those in HH cells. Most importantly, cilostazol restored the ability of DM2 erythrocytes to release ATP in response to low O2. In contrast with perfusion with HH erythrocytes, isolated hamster retractor muscle arterioles perfused with DM2 erythrocytes constricted in response to low extraluminal PO2. However, in the presence of cilostazol (100 μM), DM2 erythrocytes induced vessel dilation not different from that seen with HH erythrocytes. Thus rescue of low O2-induced ATP release from DM2 erythrocytes by cilostazol restored the ability of erythrocytes to participate in the regulation of perfusion distribution in skeletal muscle.
Keywords: microcirculation, red blood cell, oxygen transport
blood flow within the peripheral microvasculature must be actively regulated to provide optimal delivery of O2 and nutrients. Although there is significant evidence implicating a defect in both macrovascular and microvascular control in individuals with type 2 diabetes, a full understanding of the mechanisms that contribute to this defect remains elusive. It was reported that, in humans with type 2 diabetes, both endothelium-dependent and endothelium-independent vasodilatation is impaired (2, 15, 19, 37, 38). It has also been suggested that there is reduced nitric oxide (NO) synthesis (19), increased NO degradation (20, 37), and/or abnormalities in the vascular smooth muscle (38) in these individuals. Although these factors may contribute, none of these mechanisms is sufficient to explain the critical failure to match O2 delivery with metabolic need in skeletal muscle in humans with type 2 diabetes (10, 20, 29).
In skeletal muscle, entry of erythrocytes into a region of increased tissue oxygen need (exposure to low O2) results in release of both O2 and ATP (3, 12, 17). As a result, this mobile cell is able to alter the distribution of perfusion to adequately supply tissue O2 requirements (5, 7–9,, 11, 18, 21, 22, 30). Using a computational model, we predicted that a defect in this mechanism for regulated ATP release from erythrocytes could contribute to impaired O2 delivery in skeletal muscle (10, 29). In humans with type 2 diabetes, defects in perfusion contribute to peripheral vascular dysfunction (19, 37, 38). Erythrocytes from humans with type 2 diabetes release significantly less ATP in response to exposure to low PO2 (31, 32, 36), and these erythrocytes do not stimulate dilation of isolated skeletal muscle arterioles exposed to reduced extraluminal O2, a model of increased O2 demand (29).
Several components of a signaling pathway for low O2-induced ATP release that requires increases in cAMP have been defined (1, 13, 14, 23, 26–28). The cAMP generated in this pathway is hydrolyzed by phosphodiesterase 3 (PDE3) (1, 13, 14) making inhibition of the activity of this PDE a potential target for the restoration of ATP release from erythrocytes of humans with type 2 diabetes. Here we investigated the hypothesis that the PDE3 inhibitor cilostazol will rescue low O2-induced ATP release from erythrocytes of humans with type 2 diabetes, enabling these cells to stimulate dilation of isolated skeletal muscle arterioles. First, we measured PDE3B expression in erythrocytes of both healthy humans and humans with type 2 diabetes. We then determined whether pretreatment with cilostazol alters cAMP levels and/or ATP release from type 2 diabetes erythrocytes. Finally, we examined the ability of cilostazol to restore the ability of erythrocytes of humans with type 2 diabetes to stimulate dilation of isolated skeletal muscle arterioles when these vessels are exposed to reduced extraluminal O2.
MATERIALS AND METHODS
Isolation of erythrocytes.
Blood was obtained from healthy volunteers (n = 12) and patients with type 2 diabetes (n = 17) by venipuncture using a syringe containing heparin (500 units). Immediately after collection of blood, erythrocytes were isolated by centrifugation at 500 g at 4°C for 10 min with the supernatant and buffy coat removed by aspiration. Packed erythrocytes were resuspended and washed three times in a physiological salt solution containing (in mM) 4.7 KCl, 2.0 CaCl2, 1.2 MgSO4, 140.5 NaCl, 21.0 Tris-base, and 5.5 dextrose with 0.5% BSA, pH adjusted to 7.4. Erythrocytes isolated in this fashion contain less than 1 leukocyte per 50 high-power fields (8–10 leukocytes/mm3) and are devoid of platelets. Erythrocytes were prepared on the day of use.
Preparation of erythrocyte membranes.
Washed erythrocytes were diluted 1:100 with ice-cold hypotonic buffer containing 5 mM Tris·HCl and 2 mM EDTA (pH 7.4) and stirred vigorously at 4°C for 20 min. The lysate was centrifuged at 23,000 g for 15 min at 4°C. The supernatant was removed and discarded. The pellet containing the erythrocyte membranes was washed two times with ice-cold buffer and centrifuged. The membranes were resuspended in ice cold buffer and frozen at −80°C. Membrane protein concentrations were determined using BCA protein assay (Pierce).
Western analysis.
Membranes were solubilized in SDS buffer containing 8% SDS, 60% glycerol, 0.25 M Tris·HCl (pH 6.8), 0.004% bromophenol blue, and 400 mM dithiothreitol, boiled, and loaded onto a precast 5% gel (Bio-Rad) for PDE3B or a precast 4–20% gel (Bio-Rad) for β-actin and subjected to electrophoresis. Proteins were transferred to a polyvinylidene difluoride (PVDF) membrane in buffer containing 25 mM Tris-base, 192 mM glycine, and 10% methanol. Membranes were blocked overnight with 5% nonfat dry milk in PBS containing 0.1% Tween-20 at pH 7.4. The PVDF membranes were then immunoblotted with a primary polyclonal antibody directed against the first 300 amino acid residues of human PDE3B (H-300; Santa Cruz Biotechnology) or β-actin (Sigma) in PBS containing 1% nonfat dry milk and 0.1% Tween 20 at pH 7.4. The PVDF membranes were incubated with an appropriate secondary antibody conjugated to horseradish peroxidase. Labeled proteins were visualized using enhanced chemiluminescence. Equal amounts of membrane proteins were loaded onto gels. Amounts of PDE3B protein present in individual membrane preparations were calculated as the ratio of the density of that protein to that of β-actin, as determined by densitometry.
Measurement of cAMP.
Washed erythrocytes were diluted to a 50% hematocrit in a buffer that contained (in mM) 21.0 Tris-base, 4.7 KCl, 2.0 CaCl2, 140.5 NaCl, 1.2 MgSO4, 5.5 dextrose, and 0.5% bovine albumin fraction V, final pH 7.4. The erythrocytes were incubated with mastoparan 7 (MAS7; BioMol) with and without cilostazol (Sigma), and the reaction was halted by the addition of 4 ml of ice cold ethanol containing 1 mM HCl. The mixture was centrifuged at 14,000 g for 10 min at 4°C. The supernatant was removed and stored overnight at −20°C to precipitate the remaining proteins and then centrifuged at 3,700 g for 10 min at 4°C. The final supernatant was removed and dried under vacuum centrifugation. The dried sample was reconstituted in assay buffer (GE Healthcare). The concentration of cAMP was determined using an enzyme immunoassay. The amount of cAMP was normalized to that present in 1010 erythrocytes.
Measurement of ATP.
ATP was measured using the luciferin-luciferase technique (30). A 200 μl sample of a 20% erythrocyte suspension was injected into a cuvette containing 100 μl of 10 mg/ml crude firefly tail extract (Sigma) and 100 μl of 0.5 mg/ml D-Luciferin (Research Products International). The light emitted from the reaction was detected using a luminometer (Turner Designs, TD 20/20). A standard curve was obtained for each experiment. Cell counts were obtained from the suspension of erythrocytes, and amounts of ATP measured were normalized to ATP released from 4 × 108 erythrocytes. The peak ATP value was compared with an ATP standard curve generated on the day of each study.
Measurement of total intracellular ATP.
A 50 μl sample of erythrocytes (20% hematocrit) was lysed in distilled water at room temperature. ATP in the lysate, diluted 8,000-fold, was measured using the ATP assay. The values were normalized to the ATP concentration per erythrocyte as determined by direct cell counting.
Measurement of hemoglobin.
Erythrocyte suspensions used to measure ATP were centrifuged at 500 g for 10 min at 4°C. The amount of hemoglobin present in the supernatant was determined by measurement of absorbance at 405 nm. If any increase in free hemoglobin was detected, the data were excluded from analysis.
Determination of ATP release from erythrocytes in response to exposure to reduced O2 tension.
Washed erythrocytes were diluted to a 20% hematocrit in a Ringers buffer containing bicarbonate containing (in mM) 4.7 KCl, 2.0 CaCl2, 140.5 NaCl, 1.2 MgSO4, 5.5 glucose, 21.4 NaHCO3, and 0.5% BSA, pH 7.4 at 37°C. Erythrocytes were equilibrated for 30 min with a gas mixture containing 15% O2, 6% CO2, and balance N2 (pH = 7.41 ± 0.03, Pco2 = 36 ± 2 mmHg and Po2 = 107 ± 5 mmHg; normoxia) in a thin-film tonometer (model 237; Instrumentation Laboratory) (4) in the absence and presence of 100 μM cilostazol (Sigma) or its vehicle, dimethyl formamide. The gas mixture was then changed to one containing 0% O2, 6% CO2, and balance N2 (pH = 7.42 ± 0.02, Pco2 = 38 ± 2 mmHg and Po2 = 10 ± 1 mmHg; low O2 tension). The concentration of ATP released from erythrocytes was determined during normoxia and following a 10-min exposure of erythrocytes to low Po2. The pH, Po2, and Pco2 were determined during exposure to each gas mixture using a blood gas analyzer (model pHOx; Nova Biomedical).
Isolation, cannulation, and perfusion of arterioles from hamster skeletal muscle.
Male golden hamsters (94 ± 4 g; n = 21) were anesthetized with pentobarbital sodium (6.5 mg/100 g ip). The trachea was cannulated to ensure a patent airway for spontaneous breathing. The right-cheek pouch retractor muscle was surgically exteriorized, as described by Sullivan and Pittman (35). Unbranched segments of first- and second order arterioles, ∼1,000 μm in length, were surgically removed from the ventral side of the muscle. The vessel was trimmed and cleared of connective tissue while immersed in cold (4°C), modified Ringers buffer containing (in mM) 144.0 NaCl, 3.0 KCl, 2.5 CaCl2, 1.5 MgSO4, 5.0 glucose, 2.0 pyruvate, 0.02 EDTA, 2.0 MOPS, 1.21 NaH2PO4, and 1% BSA (dialyzed for 48 h against distilled water and 48 h against MOPS-Ringers) with the pH adjusted to 7.4. The vessel was then transferred to an organ bath (2.5 ml) mounted on a microscope stage containing the Ringers buffer described above but without albumin.
Isolated arterioles were cannulated using concentric glass pipettes mounted on micromanipulators attached to the microscope base. Each end of the vessel was, in turn, aspirated into the holding pipette using a controlled vacuum and cannulated with the perfusion pipette filled with the albumin containing Ringers buffer, as described above. The vessel (initial diameter: 34 ± 3 μm) was held in place suspended between the two pipettes. Following stabilization, intraluminal pressure was increased to 60 mmHg resulting in a maximal diameter of 76 ± 3 μm. Bath temperature was then increased to 37°C, and the vessel was allowed to develop spontaneous myogenic tone (34 ± 2%). Viability was determined by the demonstration of constriction in response to alkaline pH (7.59) of 15 ± 2% and dilation to an acidic pH (6.80) of 30 ± 3%. The vessel was viewed using a Zeiss Axiovert 100 inverted microscope with long working-distance objectives (10× and 20×). The microscope image was recorded using a high-resolution, closed-circuit video system consisting of a charge-coupled device video camera (model 72; Dage-MTI), video monitor (PVM-137; Sony), DV HDD DVD Recorder (SR-DVM600; JVC), and a time-date generator (WJ-810; Panasonic). Vessel diameter was determined offline using both an automated system (Diamtrax, version 3.5) and direct measurement, using a video caliper (model 308; Colorado Video). The Po2 in the microscope chamber was measured using an oxygen microelectrode (MI 730; Microelectrodes) polarized to −0.7 V and a chemical microsensor (Diamond Electro-Tech).
Vessels were perfused with albumin containing Ringers buffer or buffer containing fully oxygenated washed human (healthy human or type 2 diabetes) erythrocytes (hematocrit 17.5%) without and with cilostazol (10 or 100 μM) at 3 μl/min, using a three syringe microinjection pump (model CMA/100; CMA/Microdialysis). The pump was configured so that the perfusate could be instantly switched by means of a microswitch. Initially, the buffer surrounding the vessel was equilibrated with room air (Po2 = 147 ± 2 mmHg). After stability was achieved and vessel diameter recorded, the buffer in the vessel chamber was replaced with buffer equilibrated with 100% nitrogen (Po2 = 17 ± 2 mmHg). Vessel diameter was again recorded. The chamber buffer was then returned to one equilibrated with room air, and the vessel was allowed to stabilize. The perfusate was switched, and the sequence of normal and low Po2 exposures were repeated. Care was taken to ensure that the erythrocytes were well distributed within the syringe. The integrity of the vessel at the end of the experiment was confirmed by a second pH test.
Data analysis.
Statistical significance was determined using an ANOVA. In the event that the F ratio indicated a change had occurred, a Fisher's least significant difference test was performed to identify individual differences between groups. In some cases, a paired t-test was used as appropriate. Results are reported as means ± SE.
Institutional approvals.
The protocol for blood collection was approved by the Institutional Review Board of Saint Louis University. All record keeping was in strict compliance with Health Insurance Portability and Accountability Act (HIPAA) regulations. The protocol for the use of hamsters was approved by the Institutional Animal Care and Use Committee of Saint Louis University.
RESULTS
Characteristics of subjects.
Patients with type 2 diabetes were identified by physicians in the Endocrinology Clinic of Saint Louis University. Healthy volunteers were faculty and students at Saint Louis University School of Medicine. A history form that included a detailed listing of all medical conditions, medications, and age was completed for each subject. In the case of humans with type 2 diabetes, the degree of glycemic control was determined by measurement of hemoglobin A1C (HbA1c; Bayer) at the time of blood removal. The mean age for the healthy humans (n = 12, 5 males, 7 females) and humans with type 2 diabetes (n = 17, 8 males, 9 females) was 36 ± 6 years (range 22 to 61) and 57 ± 4 years (range 20 to 77), respectively. The average HbA1c of humans with type 2 diabetes was 8.5 ± 0.5% (range 4.7 to 11.1%). Erythrocyte ATP content in healthy humans and humans with type 2 diabetes did not differ and was 4.1 ± 0.3 and 3.4 ± 0.6 mM/cell, respectively. Patients with type 2 diabetes were treated with insulin (8), oral hypoglycemic agents (11), lipid-lowering agents (6), β-receptor antagonists (2), angiotensin converting enzyme inhibitors (7) or angiotensin receptor antagonists (1), calcium channel blockers (5), diuretics (4), and low dose aspirin (6). It is not possible to withdraw such medications from humans with type 2 diabetes for the purpose of this study. However, there are no published reports that, after washing of erythrocytes, any of the medications listed above alter ATP release from these cells.
Identification of PDE3B as a component of erythrocyte membranes.
Our laboratory has reported previously that PDE3B is a component of the membranes of healthy human and rabbit erythrocytes (13). Here we sought to determine whether PDE3B is also expressed in membranes of erythrocytes of humans with type 2 diabetes. Using Western analysis and an antibody previously shown to detect human PDE3B (13), we determined that PDE3B is expressed in type 2 diabetes erythrocyte membranes (Fig. 1A). Importantly, amounts of PDE3 present in healthy human erythrocyte membranes (n = 7) were not different from amounts in type 2 diabetes erythrocytes (n = 8; Fig. 1B).
Fig. 1.
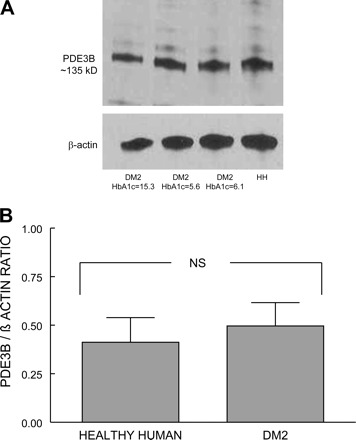
A: identification of phosphodiesterase (PDE)3B in human erythrocyte membranes. Solubilized membranes of healthy human (HH) and type 2 diabetes (DM2) erythrocytes were probed with an antibody generated against the first 300 amino acid residues of human PDE3B or β-actin (representative of 7 and 8 individual membrane preparations from healthy human and type 2 diabetes erythrocytes, respectively). B: determination of PDE3B content in erythrocyte membranes. Quantification by densitometric scanning corrected for the amount of β-actin in each sample. Values are means ± SE. NS, no significant difference.
Effect of the direct activator of Gi, mastoparan 7 (MAS 7), on cAMP levels in erythrocytes in that absence and presence of the PDE3 inhibitor, cilostazol.
Low O2-induced ATP release from erythrocytes occurs via a signaling pathway that involves activation of the heterotrimeric G protein Gi (23, 26) and increases in intracellular cAMP levels (28). As noted above, PDE3 activity was reported to hydrolyze cAMP in this pathway (1, 14). The Gi activator, MAS 7, was shown to stimulate increases in both cAMP and ATP release from healthy human erythrocytes, but not from type 2 diabetes erythrocytes (31, 32). Here we report that MAS 7 (10 μM) increases cAMP in healthy human erythrocytes in the absence and presence of cilostazol (n = 5; Fig. 2A). In contrast, in type 2 diabetes erythrocytes, cAMP increased significantly only in the presence of cilostazol (n = 5; Fig. 2B). The concentration of cilostazol used (100 μM) had no effect on baseline cAMP levels in either group. These results support the hypothesis that PDE3 activity is present in type 2 diabetes erythrocytes and demonstrate that inhibition of this PDE results in increases in cAMP associated with activation of Gi, a component of the signaling pathway for low O2-induced ATP release.
Fig. 2.
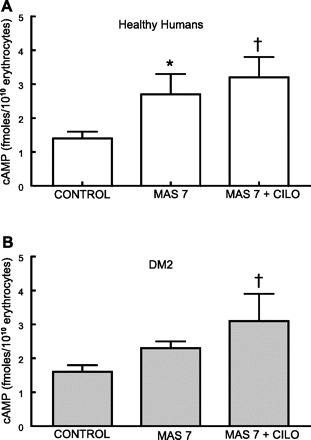
Effect of mastoparan 7 (MAS 7; 10 μM) on cAMP levels in healthy human (A) and DM2 erythrocytes (B) in the absence and presence of cilostazol (Cilo; 100 μM). Erythrocytes were incubated with Cilo for 30 min before addition of MAS 7, and the reaction was stopped after an additional 15 min. Values are means ± SE. *Different from respective control (P < 0.05); †different from respective control (P < 0.01).
Effect of cilostazol on low O2-induced ATP release from type 2 diabetes erythrocytes.
We next determined whether cilostazol can, in addition to increasing cAMP levels, rescue the ability of type 2 diabetes erythrocytes to release ATP in response to exposure to low O2. In contrast with erythrocytes of healthy humans (n = 7), erythrocytes of humans with type 2 diabetes (n = 7; HbA1c = 7.0 ± 0.5) failed to release ATP when exposed to reduced O2 (Fig. 3). The failure of type 2 diabetes erythrocytes to release ATP could not be attributed to decreased ATP content of the cells. Total ATP values were 4.1 ± 0.3 and 3.4 ± 0.6 mM in healthy human and type 2 diabetes erythrocytes, respectively, and were not significantly different. Importantly, the ability to release ATP in response to low O2 was rescued by pretreatment of the same type 2 diabetes erythrocytes with cilostazol (100 μM).
Fig. 3.
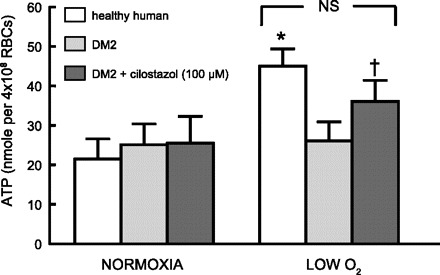
Effect of exposure to reduced O2 tension on ATP release from erythrocytes of healthy humans (n = 7) and humans with DM2 (n = 10). In a tonometer, isolated erythrocytes (hematocrit 20%) were exposed to gas mixtures containing either 15% O2, 6% CO2, and balance nitrogen (Po2 = 107 ± 5 mmHg) or 0% O2, 6% CO2, balance nitrogen (Po2 = 10 ± 1 mmHg). ATP release was determined after a 30-min equilibration with 15% O2 and 10 min after exposure to 0% O2. In separate studies, type 2 diabetes erythrocytes were exposed to identical gas compositions in the absence and presence of the PDE3 inhibitor cilostazol (100 μM). Values are means ± SE. *Greater than respective normoxia (15% O2) value (P < 0.05); †different from respective normoxia value and from DM2 erythrocytes in the absence of cilostazol. RBCs, red blood cells.
Effect of cilostazol on response of isolated, arterioles perfused with type 2 diabetes erythrocytes to reduced extraluminal O2 tension.
To investigate the effect of impaired low O2-induced ATP release from type 2 diabetes erythrocytes on the ability of vessels to dilate in response to reduced extraluminal O2, hamster skeletal muscle arterioles were isolated and perfused with 1) buffer alone (n = 6), 2) buffer containing healthy human erythrocytes (n = 5), or 3) buffer containing erythrocytes of humans with type 2 diabetes (n = 10; HbA1C = 9.2 ± 0.7). When perfused with buffer, decreases in extraluminal O2 did not stimulate dilation of perfused vessels (Table 1). However, when perfused with buffer containing fully oxygenated healthy human erythrocytes, the vessels dilated in response to the same stimulus (Table 1, Fig. 4). In contrast, arterioles perfused with buffer containing similarly well-oxygenated type 2 diabetes erythrocytes constricted upon exposure to reduced extraluminal O2 (Table 1, Fig. 4). Importantly, the ability of type 2 diabetes erythrocytes to dilate arterioles in response to this physiological stimulus was rescued by pretreatment with the PDE3 inhibitor cilostazol in a concentration-dependent manner. In three vessels, pretreatment of type 2 diabetes erythrocytes with 10 μM cilostazol reversed the constriction to low O2, but a significant dilation was not seen (Fig. 4). However, in the presence of 100 μM cilostazol, low Po2-induced dilation was not different from that observed when similar vessels were perfused with buffer containing healthy human erythrocytes (Fig. 4). Neither cilostazol nor its vehicle (dimethyl formamide; data not shown) had any effect on the response of arterioles perfused with buffer alone to reduced extraluminal O2 (Table 1).
Table 1.
Diameters of isolated perfused skeletal muscle arterioles during exposure to extraluminal normoxia and low Po2
| Arteriolar Diameter, μm |
|||
|---|---|---|---|
| Perfusate | n | Normoxia | Low Po2 |
| Buffer | 6 | 59.7 ± 7 | 59.3 ± 7 |
| Buffer +100 μM cilostazol | 6 | 62.2 ± 9 | 62.6 ± 9 |
| Healthy human erythrocytes | 5 | 47.9 ± 7 | 52.1 ± 6† |
| Type 2 diabetes erythrocytes | 7 | 45.1 ± 4 | 37.2 ± 4† |
| Type 2 diabetes erythrocytes +100 μM cilostazol | 7 | 56.1 ± 8 | 61.2 ± 7*† |
Values are means ± SE.
Different from diameter in absence of cilostazol;
different from normoxia.
Fig. 4.
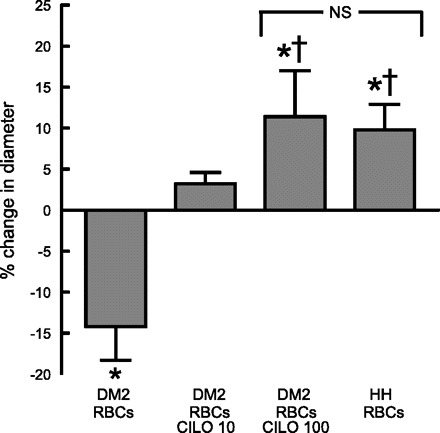
Effect of reduced extraluminal O2 tension on dilation of isolated skeletal muscle arterioles perfused with erythrocytes (RBCs). Isolated arterioles were exposed to either extraluminal normoxia (room air, Po2 = 147 ± 2 mmHg) or reduced O2 tension (Po2 = 17 ± 2 mmHg) and perfused with buffer containing well-oxygenated erythrocytes from either healthy humans (n = 5) or humans with DM2 (n = 10, HbA1c = 8.3 ± 0.8). In some studies type 2 diabetes erythrocytes were pretreated with cilostazol (Cilo) at a concentration of either 10 μM (n = 3) or 100 μM (n = 7). Values are means ± SE. *Different from value during extraluminal normoxia (P < 0.05); †different from vessels perfused with type 2 diabetes erythrocytes in the absence of Cilo (P < 0.05).
DISCUSSION
Normal skeletal muscle function requires that perfusion be appropriately distributed within the tissue to provide optimal levels of both O2 and nutrients (11, 12, 22, 25). This highly regulated process requires both a sensor of tissue O2 need and an affector of alterations in perfusion to meet that specific need. Several studies have suggested that healthy human erythrocytes can serve both roles through hemoglobin saturation-dependent release of the vasodilator ATP (3, 12, 17). Should this mechanism be impaired, normal muscle physiology could be adversely affected. Here we have confirmed the previous finding that erythrocytes of humans with type 2 diabetes neither release ATP nor stimulate dilation of isolated skeletal muscle arterioles exposed to low extraluminal O2 (25, 29–31).
A signaling pathway for reduced O2-induced ATP release from erythrocytes has been proposed (Fig. 5) (13, 23, 26–28). Under this construct, exposure of erythrocytes to reduced O2 results in hemoglobin desaturation and an associated conformational change in the hemoglobin molecule (17, 34). Although the mechanism is not fully understood, this change in hemoglobin saturation/conformation results in activation of the heterotrimeric G protein Gi and increases in cAMP (34). The PDE that hydrolyzes cAMP in this pathway is PDE3. Although PDE3B has been identified as a component of the membranes of healthy human erythrocytes (14), its presence in erythrocytes of humans with type 2 diabetes had not been determined. Here we report that PDE3B is present in membranes of erythrocytes of humans with type 2 diabetes (Fig. 1A) with the amount present not different from that found in erythrocytes of healthy humans (Fig. 1B).
Fig. 5.
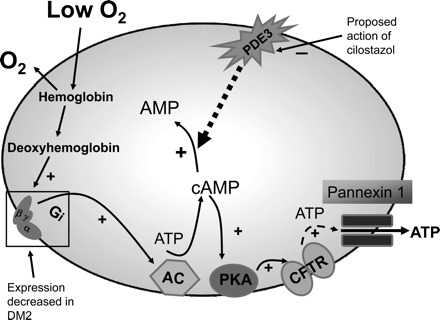
Proposed model of the mechanism by which cilostazol increases cAMP accumulation and ATP release from type 2 diabetes erythrocytes. Exposure of healthy human erythrocytes to reduced extracellular oxygen (O2) tension results in release of O2 from hemoglobin (desaturation) and activation of a signaling pathway for ATP release. This pathway requires an increase in cAMP that is regulated by PDE3 activity. The conduit for ATP release is pannexin 1 (33). In erythrocytes of humans with type 2 diabetes, expression of Gi and ATP release in response to exposure to reduced O2 are decreased. Cilostazol, by inhibiting the degradation of cAMP in this pathway, enhances low O2-induced ATP release. AC, adenylyl cyclase; CFTR, cystic fibrosis transmembrane conductance regulator; (+)activation; (−)inhibition.
Erythrocytes of humans with type 2 diabetes have reduced expression of Gαi2, one of the isoforms of the heterotrimetic G protein activated by the desaturation of hemoglobin (31, 32). Because increases in cAMP are required for erythrocyte ATP release, decreased cAMP synthesis and/or increased activity of the PDE responsible for hydrolysis cAMP in this pathway would result in decreased ATP release. When incubated with the direct activator of Gi, mastoparan 7 (MAS7), erythrocytes of humans with type 2 diabetes display impaired cAMP generation and ATP release compared with cells of healthy humans (31, 32). The PDE3 inhibitor cilostazol restored the ability of erythrocytes of humans with type 2 diabetes to release ATP in response to MAS7 (25). The latter finding suggested that cilostazol increased ATP release via the inhibition of cAMP degradation. Here we demonstrate that MAS7 does increase cAMP levels in healthy human erythrocytes and that cilostazol potentiates that response (Fig. 2A). Of greater importance, in erythrocytes of humans with type 2 diabetes, cilostazol restored MAS7-induced increases in cAMP (Fig. 2B) to levels not different from those seen in healthy humans.
Although definition of the effect of cilostazol on pharmacologically induced ATP release is useful for the understanding of the mechanism of action of the PDE3 inhibitor, of greater importance is the effect of cilostazol on ATP release in response to exposure of erythrocytes to a physiological stimulus such as reduced O2 tension. We confirmed our previous finding that, in contrast with cells of healthy humans, erythrocytes of humans with type 2 diabetes do not release ATP when exposed to reduced O2 (Fig. 3). However, here we demonstrated that pretreatment of these type 2 diabetes erythrocytes with cilostazol rescues the ability of these cells to release ATP in response to this stimulus (Fig. 3).
To determine the physiological importance of this effect of cilostazol, we determined whether restoring the ability of type 2 diabetes erythrocytes to release ATP enables these cells to stimulate dilation of perfused arterioles exposed to reduced extraluminal O2 tension; that is, can cilostazol restore the ability of type 2 diabetes erythrocytes to act as regulators of perfusion distribution? When isolated skeletal muscle arterioles were perfused with buffer containing erythrocytes of humans with type 2 diabetes, the vessels constricted in response of reduced oxygen tension (Fig. 4). This is in marked contrast with the significant dilation induced by this stimulus when vessels were perfused with buffer containing healthy human erythrocytes (Table 1, Fig. 4). However, when these vessels were perfused with buffer containing type 2 diabetes erythrocytes that were pretreated with 100 μM cilostazol, the dilation in response of reduced extraluminal O2 was not different from that seen in vessels perfused with healthy human erythrocytes in the absence of cilostazol (Fig. 4). Cilostazol did not alter the response to vessels perfused with buffer in the absence of erythrocytes (Table 1). One interpretation of these findings is that restoration of the capacity of type 2 diabetes erythrocytes to release ATP upon exposure to low oxygen tension by cilostazol reestablished the ability of these cells to stimulate low extraluminal O2-induced dilation of arterioles. This action of cilostazol would enable type 2 diabetes erythrocytes to affect appropriate changes in perfusion of skeletal muscle to match O2 delivery with need.
Peripheral vascular disease is a major contributor to the significant morbidity and mortality associated with type 2 diabetes. Cilostazol is used clinically to reduce the symptoms of intermittent claudication in individuals with peripheral vascular disease (12a, 16, 24). Although its role as a PDE inhibitor is well accepted, the mechanism by which it attenuates claudication is not well understood. Here we propose a mechanism by which cilostazol, by restoring low PO2-induced ATP release from type 2 diabetes erythrocytes, could aid in ameliorating the consequences of the maldistribution of perfusion in skeletal muscle. Although further research is required to establish the clinical usefulness of this approach, our findings suggest that inhibition of PDE3 can restore the ability of erythrocytes to serve as sensors and affectors of alterations in perfusion to meet tissue need and that such drugs could have significant therapeutic value in treating and/or preventing peripheral vascular disease, a significant complication of type 2 diabetes.
GRANTS
This work was supported by grants from the American Diabetes Association (RA-133 and BS-150) and the National Heart, Lung, and Blood Institute (HL-089094 and HL-089125) and the President's Research Fund of Saint Louis University. A portion of this work has been published previously as an abstract (FASEB J, March 17, 2011 25:1023.14).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: R.S.S., A.H.S., C.G.E., and M.L.E. conception and design of research; R.S.S., E.A.B., D.A., and M.L.E. performed experiments; R.S.S., E.A.B., D.A., and M.L.E. analyzed data; R.S.S., A.H.S., C.G.E., and M.L.E. interpreted results of experiments; R.S.S. and M.L.E. prepared figures; R.S.S., E.A.B., D.A., A.H.S., C.G.E., and M.L.E. drafted manuscript; R.S.S., A.H.S., C.G.E., and M.L.E. edited and revised manuscript; R.S.S., E.A.B., D.A., A.H.S., C.G.E., and M.L.E. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank J. L. Sprague for inspiration.
REFERENCES
- 1. Adderley SP, Sprague RS, Stephenson AH, Hanson MS. Regulation of cAMP by phosphodiesterases in erythrocytes. Pharmacol Rep 62: 475–482, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bagi Z, Koller A, Kaley G. Superoxide-NO interaction decreases flow- and agonist-induced dilations of coronary arterioles in type 2 diabetes mellitus. Am J Physiol Heart Circ Physiol 285: H1404–H1410, 2003 [DOI] [PubMed] [Google Scholar]
- 3. Bergfeld GR, Forrester T. Release of ATP from human erythrocytes in response to a brief period of hypoxia and hypercapnea. Cardiovasc Res 26: 40–47, 1992 [DOI] [PubMed] [Google Scholar]
- 4. Chalmers C, Bird BD, Whitwam JG. Evaluation of a new thin film tonometer. Br J Anaesth 46: 253–259, 1974 [DOI] [PubMed] [Google Scholar]
- 5. Collins DM, McCullough WT, Ellsworth ML. Conducted vascular responses: communication across the capillary bed. Microvas Res 56: 43–53, 1998 [DOI] [PubMed] [Google Scholar]
- 7. Dietrich HH, Ellsworth ML, Sprague RS, Dacey RG., Jr Red blood cell regulation of microvascular tone through adenosine triphosphate. Am J Physiol Heart Circ Physiol 278: H1294–H1298, 2000 [DOI] [PubMed] [Google Scholar]
- 8. Dietrich HH, Horiuchi T, Xiang C, Hongo K, Falck JR, Dacey RG., Jr Mechanism of ATP-induced local and conducted vasomotor responses in isolated rat cerebral penetrating arterioles. J Vasc Res 46: 253–264, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dufour SP, Patel RP, Brandon A, Teng X, Pearson J, Barker H, Ali L, Yuen AH, Smolenski RT, González-Alonso J. Erythrocyte-dependent regulation of human skeletal muscle blood flow: role of varied oxyhemoglobin and exercise on nitrite, S-nitrosohemoglobin, and ATP. Am J Physiol Heart Circ Physiol 299: H1936–H1946, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ellis CG, Goldman D, Hanson MS, Stephenson AH, Milkovich S, Benlamri A, Ellsworth ML, Sprague RS. Defects in oxygen supply to skeletal muscle of prediabetic ZDF rats. Am J Physiol Heart Circ Physiol 298: H1661–H1670, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ellsworth ML. The red blood cell as an oxygen sensor: what is the evidence. Acta Physiol Scand 168: 551–559, 2000 [DOI] [PubMed] [Google Scholar]
- 12. Ellsworth ML, Forrester T, Ellis CG, Dietrich HH. The erythrocyte as a regulator of vascular tone. Am J Physiol Heart Circ Physiol 269: H2155–H2161, 1995 [DOI] [PubMed] [Google Scholar]
- 12a. Grouse JR, 3rd, Allen MC, Elam MB. Clinical manifestations of atherosclerotic peripheral vascular disease and the role of cilostazol in treatment of intermittent claudication. J Clin Pharmacol 42: 1291–1298, 2002 [DOI] [PubMed] [Google Scholar]
- 13. Hanson MS, Stephenson AH, Bowles EA, Sridharan M, Adderley S, Sprague RS. Phosphodiesterase 3 is present in rabbit and human erythrocytes and its inhibition potentiates iloprost-induced increases in cAMP. Am J Physiol Heart Circ Physiol 295: H786–H793, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hanson MS, Stephenson AH, Bowles EA, Sprague RS. Insulin inhibits human erythrocyte cAMP accumulation and ATP release: role of phosphodiesterase 3 and phosphoinositide 3-kinase. Exp Biol Med (Maywood) 235: 256–262, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hsueh WA, Quinones MJ. Role of endothelial dysfunction in insulin resistance. Am J Cardiol 92: 10j–17j, 2003 [DOI] [PubMed] [Google Scholar]
- 16. Jaff MR. Pharmacotherapy for peripheral arterial disease: emerging therapeutic options. Angiology 53: 627–633, 2002 [DOI] [PubMed] [Google Scholar]
- 17. Jagger JE, Bateman RM, Ellsworth ML, Ellis CG. Role of erythrocyte in regulating local O2 delivery mediated by hemoglobin oxygenation. Am J Physiol Heart Circ Physiol 280: H2833–H2839, 2001 [DOI] [PubMed] [Google Scholar]
- 18. McCullough WT, Collins DM, Ellsworth ML. Arteriolar responses to extracellular ATP in striated muscle. Am J Physiol Heart Circ Physiol 272: H1886–H1891, 1997 [DOI] [PubMed] [Google Scholar]
- 19. McVeigh GE, Brennan GM, Johnston BJ, McGrath LT, Henry WR, Andrews JW, Hayes JR. Impaired endothelium-dependent and independent vasodilation in patients with type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia 35: 771–776, 1992 [DOI] [PubMed] [Google Scholar]
- 20. Meyer JA, Froelich JM, Reid GE, Karunarathne WKA, Spence DA. Metal-activated C-peptide facilitates glucose clearance and release of a nitric oxide stimulus via the GLUT1 transporter. Diabetologia 52: 175–182, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mortensen SP, González-Alonso J, Nielsen JJ, Saltin B, Hellsten Y. Muscle interstitial ATP and norepinephrine concentrations in the human leg during exercise and ATP infusion. J Appl Physiol 107: 1757–1762, 2009 [DOI] [PubMed] [Google Scholar]
- 22. Mortensen SP, Thaning P, Nyberg M, Saltin B, Hellsten Y. Local release of ATP into the arterial inflow and venous drainage of human skeletal muscle: insight from ATP determination with the intravascular microdialysis technique. J Physiol 589: 1847–1857, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Olearczyk JJ, Stephenson AH, Lonigro AJ, Sprague RS. Heterotrimeric G protein Gi is involved in a signal transduction pathway for ATP release from erythrocytes. Am J Physiol Heart Circ Physiol 286: H940–H945, 2004 [DOI] [PubMed] [Google Scholar]
- 24. Regensteiner JG, Hiatt WR. Current medical therapies for patients with peripheral arterial disease: a critical review. Am J Med 57: 49–57, 2002 [DOI] [PubMed] [Google Scholar]
- 25. Sprague RS, Bowles EA, Achilleus D, Ellsworth ML. Erythrocytes as controllers of perfusion distribution in the microvasculature of skeletal muscle. Acta Physiol (Oxf) 202: 285–292, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sprague RS, Bowles EA, Olearczyk JJ, Stephenson AH, Lonigro AJ. The role of G protein β-subunits in the release of ATP from human erythrocytes. J Physiol Pharmacol 53: 667–674, 2002 [PubMed] [Google Scholar]
- 27. Sprague RS, Ellsworth ML, Stephenson AH, Kleinhenz ME, Lonigro AJ. Deformation-induced ATP release from red blood cells requires cystic fibrosis transmembrane conductance regulator activity. Am J Physiol Heart Circ Physiol 275: H1726–H1732, 1998 [DOI] [PubMed] [Google Scholar]
- 28. Sprague RS, Ellsworth ML, Stephenson AH, Lonigro AJ. Participation of cAMP in a signal-transduction pathway relating erythrocyte deformation to ATP Release. Am J Physiol Cell Physiol 281: C1158–C1164, 2001 [DOI] [PubMed] [Google Scholar]
- 29. Sprague RS, Goldman D, Bowles EA, Achilleus D, Stephenson AH, Ellis CG, Ellsworth ML. Divergent effects of low O2 tension and iloprost on ATP release from erythrocytes of humans with type 2 diabetes: implications for O2 supply to skeletal muscle. Am J Physiol Heart Circ Physiol 299: H566–H573, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sprague RS, Hanson MS, Achilleus D, Bowles EA, Stephenson AH, Sridharan M, Adderley S, Ellsworth ML. Rabbit erythrocytes release ATP and dilate skeletal muscle arterioles in the presence of reduced oxygen tension. Pharm Res 295: H786–H793, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sprague RS, Stephenson AH, Bowles EA, Stumpf MS, Lonigro AJ. Reduced expression of Gi in erythrocytes of humans with diabetes type 2 is associated with impairment of both cAMP generation and ATP release. Diabetes 55: 3588–3593, 2006 [DOI] [PubMed] [Google Scholar]
- 32. Sprague RS, Stephenson AH, Bowles EA, Stumpf MS, Ricketts G, Lonigro AJ. Expression of the heterotrimeric G protein Gi and ATP release are impaired in erythrocytes of humans with diabetes mellitus. Adv Exp Med Biol 588: 207–216, 2006 [DOI] [PubMed] [Google Scholar]
- 33. Sridharan M, Adderley SP, Bowles EA, Egan TM, Stephenson AH, Ellsworth ML, Sprague RS. Pannexin 1 is the conduit for low oxygen tension-induced ATP release from human erythrocytes. Am J Physiol Heart Circ Physiol 299: H1146–H1152, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sridharan M, Sprague RS, Adderley SP, Bowles EA, Ellsworth ML, Stephenson AH. Diamide decreases deformability of rabbit erythrocytes and attenuates low oxygen tension-induced ATP release. Exp Biol Med (Maywood) 235: 1142–1148, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sullivan SM, Pittman RN. Hamster retractor muscle: a new preparation for intravital microscopy. Microvasc Res 23: 329–335, 1982 [DOI] [PubMed] [Google Scholar]
- 36. Wasanthi S, Spence DM. Simultaneous determination of cell aging and ATP release from erythrocytes and its implications in type 2 diabetes. Analytica Chimica Acta 618: 227–233, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Williams SB, Cusco JA, Roddy MA, Johnstone MT, Creager MA. Impaired nitric oxide-mediated vasodilation in patients with non-insulin-dependent diabetes mellitus. J Am Coll Cardiol 27: 567–574, 1996 [DOI] [PubMed] [Google Scholar]
- 38. Yugar-Toledo JC, Tanus-Santos JE, Sabha M, Sousa MG, Cittadino M, Tácito LH, Moreno H., Jr Uncontrolled hypertension, uncompensated type II diabetes, and smoking have different patterns of vascular dysfunction. Chest 125: 823–830, 2004 [DOI] [PubMed] [Google Scholar]