

Abstract
During B lymphopoiesis, Igk recombination requires pre-B cell receptor (pre-BCR) expression and escape from interleukin 7 receptor (IL-7R) signaling. By activating the transcription factor STAT5, IL-7R signaling maintains proliferation and represses Igk germline transcription by unknown mechanisms. We demonstrate that STAT5 tetramer bound the Igk intronic enhancer (Eκi), leading to recruitment of the histone methyltransferase Ezh2. Ezh2 marked H3K27me3 throughout Jκ to Cκ. In the absence of Ezh2, IL-7 failed to repress Igk germline transcription. H3K27me3 modifications were lost after termination of IL-7R–STAT5 signaling and E2A bound Eκi, resulting in acquisition of H3K4me1 and H4Ac. Genome-wide analyses revealed a STAT5 tetrameric binding motif associated with transcriptional repression. These data demonstrate how IL-7R signaling represses Igk germline transcription and provide a general model for STAT5-mediated epigenetic transcriptional repression.
INTRODUCTION
B lymphopoiesis is driven by the sequential rearrangement and expression of immunoglobulin heavy (Igμ) and then light (Igκ followed by Igλ) chains. Each recombination constitutes a discrete transition in which rearrangements capable of supporting expression of a surface receptor are selected for further development1. Igμ assembles with surrogate light chain and Igα-Igβ to form a pre-B cell receptor (pre-BCR) that first expands pre-B cells bearing a single in-frame heavy chain rearrangement and then initiates Igk recombination2-4. Initial clonal proliferation occurs in the context of bone marrow-derived signals such as interleukin 7 (IL-7)5,6. However, this cooperation is transient and B cell progenitors must exit cell cycle before initiating Igk recombination7.
Igk recombination requires that the locus be accessible to the recombinase activation gene proteins (Rags)8,9 and Igk germline transcription correlates with accessibility and precedes recombination10,11. Deletion of either the intronic Igk enhancer (Eκi) within the Jκ-Cκ intron or, to a lesser degree, the 3′ Cκ enhancer (3′Eκ) diminishes Igk germline transcription and recombination12-14, while deletion of both enhancers completely blocks Igk rearrangement13. In vivo experiments have demonstrated that binding of the transcription factor E2A to two sites within Eκi (E-boxes κE1 and κE2) are required for Igk transcription and rearrangement15-18. In contrast, the interferon regulator factors (IRFs) 4 and 8 bind the 3′Eκi and are necessary for Igk recombination in vivo and for progression beyond the large pre-B cell stage19-21.
Igk germline transcription, and the initiation of Igk recombination, is also associated with the acquisition of activating histone post-translational modifications (PTMs)19,22,23. E2A binding correlates with acquiring the activating marks histone 3 acetylation (H3Ac) and H3 lysine 4 tri-methylation (H3K4me3) at the Jλ segments24, and genome-wide, E2A binding at enhancers is associated with increased H3K4me1 (ref. 25). Furthermore, the E-boxes contained within Eκi are necessary for Jκ to acquire open chromatin marks in pre-B cells23. Histone PTMs are particularly important for Ig gene recombination as Rag2 is recruited to and activated by H3K4me3 (refs. 26,27) providing a direct link between PTMs and recombination.
We have recently demonstrated that pre-BCR mediated Erk activation increases the level of nuclear E2A available for binding Eκi16. Pre-BCR signaling is also associated with the Igk locus acquiring the epigenetic marks of open chromatin23. These data are consistent with observations that expression of the pre-BCR directs both cell cycle exit and the induction of Igk recombination16,28-30 at the pre-B cell stage.
The pre-BCR-mediated differentiation program is antagonized by IL-7R signaling, which promotes proliferation and represses Igk recombination. Downstream of the IL-7R, activated STAT5 enhances transcription of the cell cycle effector cyclin D3 while repressing Igk germline transcription16,31. Pre-B cells must escape the effects of IL-7R signaling to effectively initiate Igk recombination. Escape can be regulated through intrinsic mechanisms32 and through extrinsic mechanisms including movement of pre-B cells along chemokine gradients into IL-7 deficient niches in the bone marrow33.
An important facet of this interplay between the IL-7R and the pre-BCR is STAT5-mediated repression of Igk recombination16,31. STAT5 binds directly to Eκi and can prevent E2A recruitment16,19. However, it is not clear if this apparent competition is sufficient to explain STAT5-mediated repression. Herein, we demonstrated that STAT5 binds as a tetramer to Eκi and enabled recruitment of the histone methyltransferase (HMT) Ezh2 (enhancer of zeste homolog 2) that decorated the Eκi, the Jκ cluster and Cκ with the repressive mark H3K27me3 (ref. 22). Genome-wide analyses indicated that the Igk locus provides an example of a general mechanism by which STAT proteins can directly repress locus accessibility and transcription.
RESULTS
STAT5 binding at κS2 is functionally important
IL-7R-mediated STAT5 activation represses Igk transcription in pro-B cells by binding directly to the Eκi, and this is associated with inhibition of E2A recruitment16,31. Examination of Eκi (MGI:1354193) using the TFSEARCH program revealed two potential STAT binding sites (κS1 and κS2) that closely match the STAT consensus binding sequence (5′-TTCNNNGAA-3′), referred to as Interferon-gamma activated sequence, GAS)(Fig. 1a). κS1 (5′-TTCTTGGTA-3′) was identified 60 bp upstream of the NF-κB binding site17,34. The κS2 site (5′-TTCCGAGAG-3′) overlapped with the NF-κB binding site and was only 2 bp upstream of the core κE1 site (Fig. 1a)31.
Figure 1.
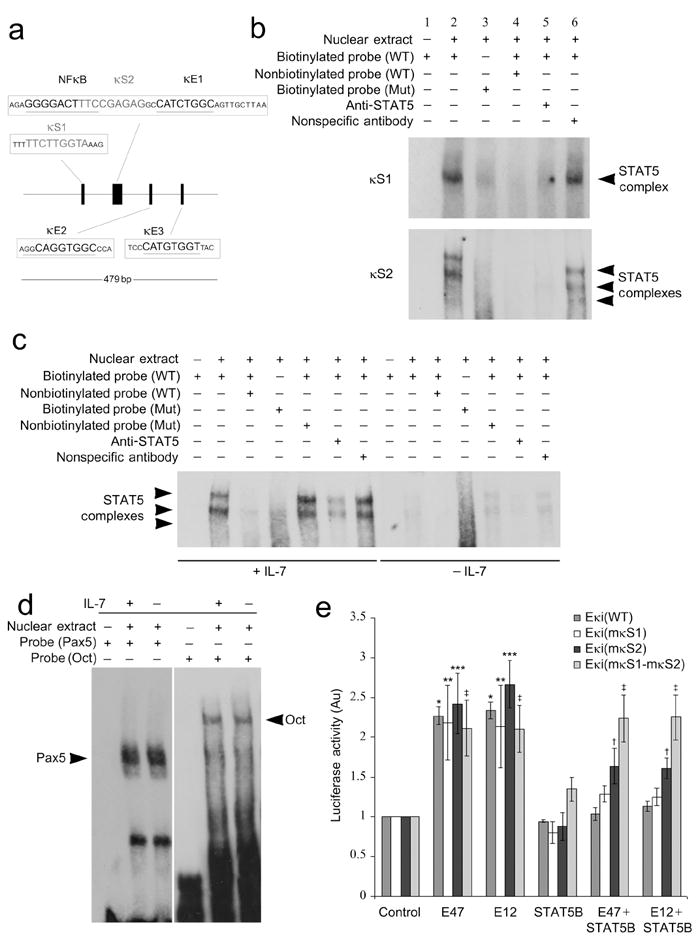
STAT5 binding at κS2 in Eκi is functionally important. (a) Diagram of the Igk intronic enhancer (Eκi) and its functional motifs. The NF-κB site and the three E-boxes (κE1-3) are shown along with the two putative STAT5 binding sites, κS1 and κS2. (b) EMSAs using nuclear extracts from Irf4-/-Irf8-/- pre-B cells cultured in IL-7 (10 ng/ml) for 24 h, assayed with probes corresponding to wild-type (WT) and mutated κS1 and κS2 binding sites. (c) EMSAs with nuclear extracts from Irf4-/-Irf8-/- pre-B cells cultured in high (10 ng/ml) or low (0.1 ng/ml) IL-7, assayed with probes corresponding to wild-type and mutated κS2 binding sites. (d) EMSAs with nuclear extracts from Irf4-/-Irf8-/- pre-B cells cultured in high (10 ng/ml, +) or low (0.1 ng/ml, −) IL-7, assayed with probes corresponding to Oct or Pax5 binding sites. (e) Cos7 cells were transfected with a pGL3-promoter luciferase reporter plasmid containing wild-type Eκi, mutant κS1, mutant κS2, or κS1-κS2 mutant enhancers. Plasmids encoding pRL-TK (Renilla luciferase) and indicated molecules or appropriate control plasmids were transfected as indicated. After 48 h, dual luciferase assays were performed on cell lysates (average ± s.d, n=3). *P<0.001, versus Eκi(WT) control; ** P<0.01, versus Eκi(mκS1) control; *** P<0.001, versus Eκi(mκS2) control; †P<0.01, versus Eκi(mκS2) control; ‡P<0.001, versus Eκi(mκS1-mκS2) control. All data representative of three independent experiments.
To determine if either of these κS sites could bind STAT5, we performed electrophoretic mobility-shift assays (EMSAs) with nuclear extracts from Irf4-/-Irf8-/- pre-B cells cultured with IL-7 and biotinylated oligonucleotide probes corresponding to κS1 and κS2 (Fig. 1b). We detected one complex bound to the wild-type (WT) κS1 probe while two major low electrophoretic mobility complexes and one minor high electrophoretic mobility complex bound the WT κS2 probe. These complexes were attenuated by addition of STAT5-specific antibody, excess unlabeled WT oligonucleotides, or by mutation of the corresponding putative STAT5 binding site. These data indicate that one STAT5 containing complex detectably binds κS1 while three distinct STAT5 containing complexes bind κS2.
To examine the role of IL-7R signaling on STAT5 binding to Eκi we performed EMSAs with nuclear extracts from Irf4-/-Irf8-/- pre-B cells cultured in high (+IL-7, 10 ng/ml) or low (-IL-7, 0.1 ng/ml) IL-7 and biotinylated oligonucleotide probes corresponding to the κS2 site (Fig. 1c) or probes corresponding to binding sites for the octamer binding protein (Oct) or Pax 5 (Fig. 1d). As expected, in the presence of IL-7 STAT5-containing complexes bound the WT but not the mutant κS2 probe. These complexes were attenuated by addition of STAT5-specific antibody or excess unlabeled WT oligonucleotides. In contrast, the complex was not diminished by the addition of unlabeled oligonucleotide containing mutations in the κS2 binding motif or by addition of isotype-matched control antibodies (Fig. 1c). Similar results were obtained when a probe corresponding to the κS1 was used (data not shown). IL-7 did not affect the binding of complexes to either the Oct or Pax5 probes (Fig. 1d).
We next examined the capacity of STAT5 to repress Eκi enhancer activity. We cloned Eκi into a pGL3 vector containing a SV40 promoter-luciferase gene cassette. This Eκi(WT) reporter or mutants in which either κS1 (Eκi(mκS1)), κS2 (Eκi(mκS2)) or both κS1 and κS2 (Eκi(mκS1- mκS2)) were mutated were co-transfected into Cos7 cells with pRL-TK (Renilla luciferase, internal control) and combinations of plasmids encoding E47, E12, constitutively active (CA)-STAT5B or a control plasmid16. Forty-eight hours after transfection, luciferase activity was measured by a dual-luciferase reporter assay. Expression of E47 or E12 enhanced Eκi activity while co-expression of CA-STAT5B repressed E2A-mediated Eκi induction (Fig. 1e). Mutation of κS1 alone did not inhibit the ability of STAT5 to repress Eκi; however, mutating κS2 or both κS1 and κS2 progressively inhibited the ability of STAT5 to repress E2A-mediated Eκi activation (Fig. 1e). These data indicate that κS2 is more important than κS1 for STAT5-mediated repression of Eκi.
Competition between STAT5 and E2A
We speculated that STAT5 might repress Eκi by competing with E2A for binding. If so, increasing the distance between the κS2 and κE1 binding sites should mitigate STAT5-mediated Eκi repression. Therefore, we created mutants of Eκi(WT) in which spacers of three, six or nine adenines (As) were introduced between the κS2 and κE1 sites (Eκi(κS2-3A-κE1), Eκi(κS2-6A-κE1), and Eκi(κS2-9A-κE1), respectively) (Fig. 2a). WT or mutant reporters were then co-transfected into Cos7 cells with plasmids encoding pRL-TK, E47, CA-STAT5B, or a control plasmid singly or in combination. We found that increasingly longer adenine spacers progressively attenuated STAT5-mediated repression such that with the 9A spacer no significant repression was observed (Fig. 2b). Similar results were observed when a plasmid encoding E12 was used (Supplementary Fig. 1a). Overall, these data are consistent with physical competition between STAT5 and E2A.
Figure 2.
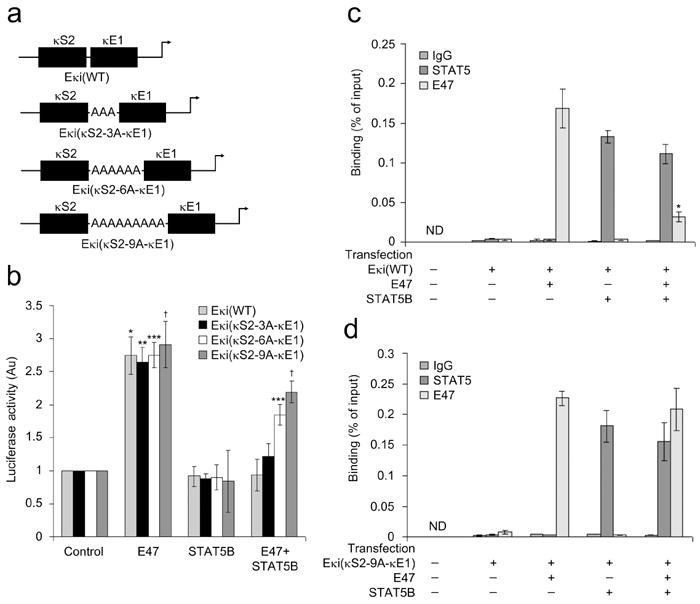
STAT5 and E2A compete for binding at their respective Eκi sites. (a) Schematic representation of the 3, 6 and 9 adenine oligonucleotide spacers (between κS2 and κE1) expressed in pGL3-promoter luciferase plasmids. (b) Cos7 cells were transfected with the pGL3-promoter luciferase reporter plasmids described in (a) along with plasmids encoding pRL-TK (Renilla luciferase) and indicated molecules or appropriate control plasmids. After 48 h, dual luciferase assays were performed on cell lysates (average ± s.d, n=3). *P<0.001, versus Eκi(WT) control; ** P<0.001, versus Eκi(κS2-3A-κE1) control; *** P<0.001, versus Eκi(κS2-6A-κE1) control; †P<0.001, versus Eκi(κS2-9A-κE1) control. (c,d) ChIP assay of nuclear preparations from Cos7 cells transfected as in (b) with (c) Eκi(WT) or (d) Eκi(κS2-9A-κE1)(average ± s.d, n=3). ChIPs were performed with antibodies specific for STAT5, E47 or control IgG1 and assayed with PCR primers specific for the Eκi fragment in the reporter plasmids. *P<0.001, versus E47 binding in only E47 expressed cells.
We next directly examined if STAT5 competed with E2A at Eκi. DNA from Cos7 cells transfected as above and expressing either the Eκi(WT) or Eκi(κS2-9A-κE1) reporters was isolated. Samples were immunoprecipitated with either antibodies specific for STAT5 or E47, and immunoprecipitations were subjected to quantitative PCR with primers specific for the Eκi reporter. In cells expressing Eκi(WT), both E47 and CA-STAT5B bound the enhancer when expressed individually (Fig. 2c). However, when both E47 and CA-STAT5B were co-expressed, only CA-STAT5B binding was preserved. Consistent with direct competition at WT Eκi, both CA-STAT5B and E47 bound to Eκi(κS2-9A-κE1) when co-expressed (Fig. 2d).
E2A activates Igk transcription through both the κE1 and κE2 sites17. Therefore, to determine if STAT5 repressed E2A-mediated Eκi activation through either κE1 or κE2, Cos7 cells were transiently transfected with Eκi(WT), Eκi(mκE1), or Eκi(mκE2) in the presence of ectopically expressed E47, CA-STAT5B, or both E47 and CA-STAT5B (Supplementary Fig. 1b). Interestingly, STAT5 repressed both κE1 and κE2-directed E2A-mediated activation. STAT5 is unlikely to repress κE2-mediated activation by direct competition, as κE2 is not in proximity to either κS1 or κS2. Therefore, we next examined if STAT5 could epigenetically regulate Eκi.
Epigenetic repression of Eκi by STAT5
Initiation of Igk transcription in vivo is associated with activating histone marks at Jκ genes23. However, epigenetic regulation of Eκi has not been fully characterized19. Therefore, we first examined the epigenetic regulation of Eκi during B lymphopoiesis. Pro-B (B220+CD43hiIgM-Lin-), large pre-B (B220+CD43lo/-IgM-Lin-FSChi), small pre-B (B220+CD43lo/-IgM-Lin-FSClo), and immature B (B220+CD43-IgM+IgD-Lin-) cells were isolated by flow cytometry from WT bone marrow, and quantitative chromatin immunoprecipitations (ChIPs) were performed16 with antibodies specific for H4Ac, the enhancer specific mark H3K4me1 (ref. 35), and H3K27me3, and PCR primers specific for Eκi. As expected, in pro- and large pre-B cells in which Igk transcription is quiescent, repressive H3K27me3 marks predominated (Fig. 3a). Upon transition to the small pre-B cell stage and the initiation of Igk transcription36, marks of active enhancers (H4Ac and H3K4me1) predominated.
Figure 3.
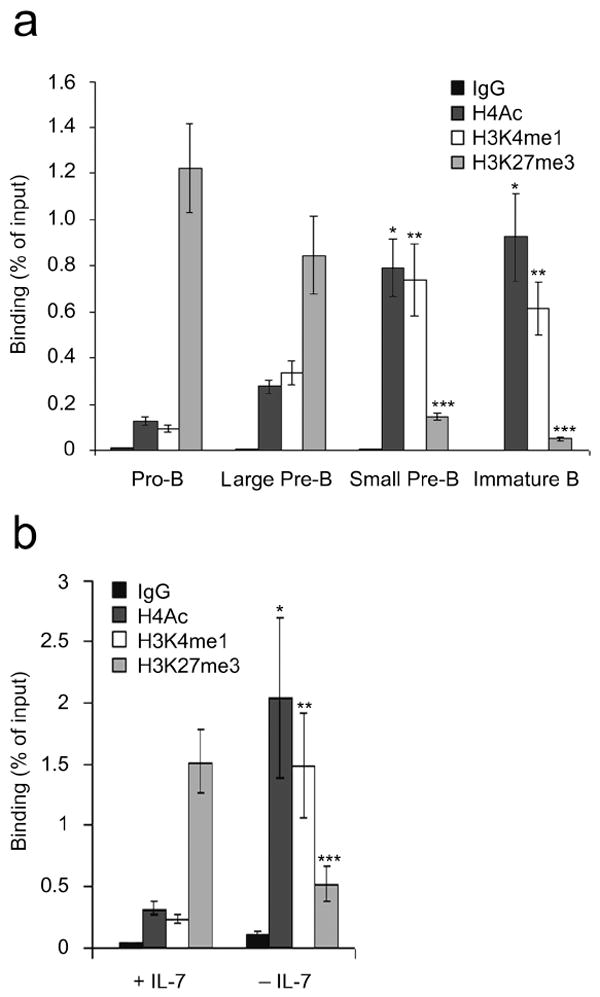
Epigenetic regulation of Eκi during B lymphopoiesis. (a) Wild-type pro-B, large pre-B, small pre-B and immature B cells were isolated by flow cytometry and assayed by ChIP using antibodies specific for H4Ac, H3K4me1, H3K27me3 or control IgG1. qPCR was performed with primers specific for Eκi (average ± s.d, n=3). *P<0.001, versus pro-B H4Ac; **P<0.001, versus pro-B H3K4me1; ***P<0.001, versus pro-B H3K27me3. (b) Irf4-/-Irf8-/- pre-B cells cultured for 48 h in high (+) or low (-) IL-7 were analyzed as in (a) (average ± s.d, n=3). *P<0.001, versus +IL-7 H4Ac; **P<0.001, versus +IL-7 H3K4me1; ***P<0.001, versus +IL-7 H3K27me3.
We next asked whether IL-7R signaling could regulate Eκi accessibility. In cultures with IL-7, Irf4-/-Irf8-/- B cell progenitors resemble proliferating pre-B cells. However, after withdrawal of IL-7 they exit the cell cycle and initiate Igk recombination19. Therefore, Irf4-/-Irf8-/- pre-B cells were cultured in high IL-7 (+IL-7, 10 ng/ml) or low IL-7 (-IL-7, 0.1 ng/ml) and ChIPs were performed as above. As was observed in primary pro- and large pre-B cells, H3K27me3 predominated at Eκi in Irf4-/-Irf8-/- pre-B cells cultured in high IL-7 (Fig. 3b). Upon IL-7 withdrawal, H3K27me3 was attenuated and Eκi associated histones became decorated with H4Ac and H3K4me1. These data suggest that the IL-7R epigenetically regulates Eκi.
Since IL-7 modulates the epigenetic histone marks associated with Eκi, we next examined if this function of IL-7 could be supplanted by activated STAT5. Cultured Irf4-/-Irf8-/- pre-B cells were infected with control retrovirus (MIGR1-IRES-GFP+) or retrovirus encoding CA-STAT5B, and GFP+ cells were isolated by flow cytometry and re-cultured in low IL-7. Cell aliquots were then analyzed in ChIP assays as described above. As expected, in mock-infected cells Eκi was marked with H4Ac and HeK4me1 (Fig. 4a). However, in STAT5B expressing cells Eκi was decorated predominantly with H3K27me3. These results indicate that downstream of IL-7R signaling, STAT5 maintains H3K27me3 at Eκi and prevents the acquisition of markers of open chromatin associated with entry into the small pre-B cell pool.
Figure 4.
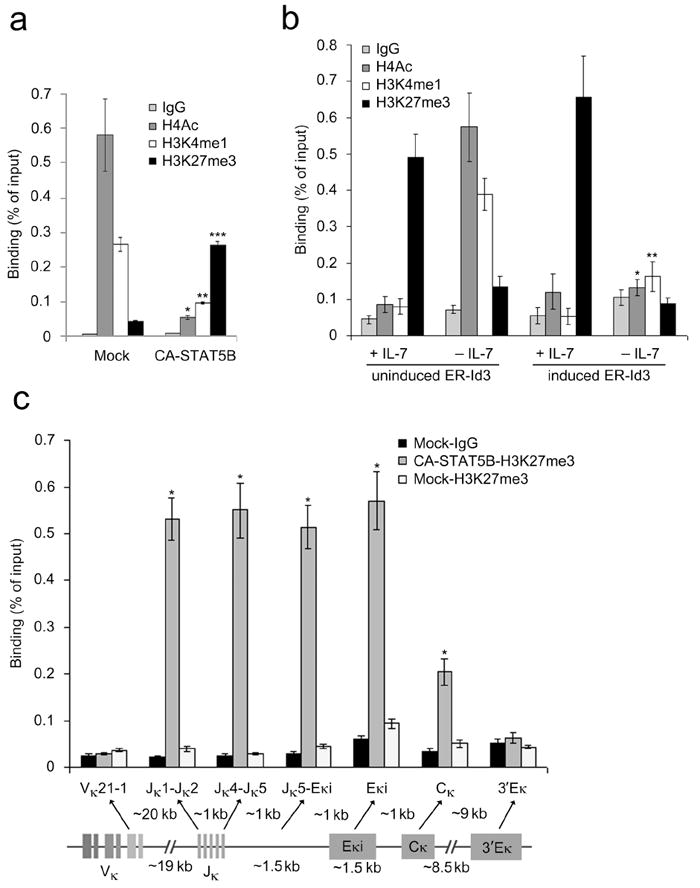
STAT5 and E2A mediate repressive and activation marks respectively from Jκ through Cκ. (a) ChIP assays of Irf4-/-Irf8-/- pre-B cells expressing CA-STAT5B or vector alone and cultured for 48 h in low IL-7. ChIP using antibodies specific for H4Ac, H3K4me1, H3K27me3 or control IgG1 followed by qPCR for Eκi (average ± s.d, n=3). *P<0.001, versus mock H4Ac; **P<0.001, versus mock H3K4me1; ***P<0.001, versus mock H3K27me3. (b) ChIP analysis of Irf4-/-Irf8-/- pre-B cells expressing inducible Id3 (ER-Id3) cultured for 48 h in high (+) or low (-) IL-7 and mock treated or induced for 48 h with 4-OH-tamoxifen (1 μM). Assayed as in (a)(average ± s.d, n=3). *P<0.001, versus uninduced -IL-7 H4Ac; **P<0.001, versus uninduced −IL-7 H3K4me1. (c) ChIP analysis of Irf4-/-Irf8-/- pre-B cells expressing CA-STAT5B or vector alone, cultured for 48 h in low IL-7 and assayed as in (a)(average ± s.d, n=3). Different regions of Igk locus in the H3K27me3 ChIP fractions were determined by qPCR using non overlapping primer sets designed to detect indicated segments. *P<0.001, versus mock-H3K27me3 in respective regions.
Epigenetic activation of Eκi by E2A
Downstream of the pre-BCR, Erk activation coordinately induces E2A and represses the E2A inhibitor Id3 (ref. 16). The resulting increase in free nuclear E2A can then bind Igk enhancers and activate Igk transcription15,17,37,38. Therefore, we next examined whether the acquisition of active histone modifications at Eκi following IL-7 withdrawal was dependent upon E2A. Irf4-/-Irf8-/- pre-B cells expressing a retroviral encoded fusion protein of the estrogen receptor and Id3 (ER-Id3) were cultured in high (+IL-7, 10 ng/ml) or low (-IL-7, 0.1 ng/ml) IL-7 in the presence or absence of 4-OH-tamoxifen (Tx). ChIP assays were then performed with antibodies specific for H4Ac, H3K4me1, and H3K27me3, and PCR primers specific for Eκi. In the presence of IL-7, in both Tx (induced) or vehicle only (uninduced) treated Irf4-/-Irf8-/- pre-B cells, the histones in the Eκi region were primarily marked with H3K27me3 (Fig. 4b). However, upon IL-7 withdrawal in Tx treated cells, Eκi lacked both activating and repressive histone modifications. These data indicate that the balance between STAT5 and E2A determines the epigenetic status of Eκi.
STAT5 binding confers H3K27me3 repressive marks
The Eκi is positioned approximately 1.5 kb downstream of the 3′ Jκ gene cluster and 1kb upstream of Cκ (Fig. 4c). This close positioning raised the possibility that STAT5 recruitment to Eκi could regulate the accessibility of these flanking regions. To test this, Irf4-/-Irf8-/- pre-B cells expressing control or CA-STAT5B retrovirus were cultured in low IL-7, and the Jκ, Cκ, and Eκi regions were interrogated for H3K27me3 using ChIPs. In CA-STAT5B expressing cells, we observed significant levels of H3K27 trimethylation throughout the Jκ and Cκ regions. In contrast, expression of CA-STAT5B did not enhance H3K27me3 in the Vκ segments or the 3′ Eκ. These data suggested that direct binding of STAT5 to Eκi epigenetically represses Igk transcription.
Tetrameric STAT5 binding to Eκi in κS2 recruits Ezh2
Our results demonstrated that, although STAT5 has no intrinsic HMT activity, STAT5 targets H3K27me3 to the Igk locus. In lymphocytes, two polycomb group (pcG) family members, Ezh 1 and 2, are known to transmethylate H3K27me2 to generate H3K27me3 (ref. 39). Of these, only Ezh2 is abundantly expressed during B lymphopoiesis where it plays an obligatory role40. To assess the ability of STAT5 to recruit Ezh2, Irf4-/-Irf8-/- pre-B cells were cultured in high or low IL-7. Nuclear extracts were then subjected to ChIP with STAT5-specific antibody. Immunoprecipitations were resolved by SDS-PAGE, transferred to membranes, and probed with STAT5 or Ezh2-specific antibodies. In high IL-7, but not low IL-7, both STAT5 and Ezh2 were readily detected in STAT5 ChIPs (Fig. 5a) even though both molecules were readily detected in the low IL-7 samples. Similar results were obtained when cultured Rag-2-/- pro-B cells were used (data not shown). In contrast, if STAT5 was immunoprecipitated from nuclear lysates, in the absence of chromatin crosslinking, there was not a detectable association with Ezh2 (Fig. 5b). These data suggest that, when bound to chromatin DNA, STAT5 can recruit Ezh2.
Figure 5.
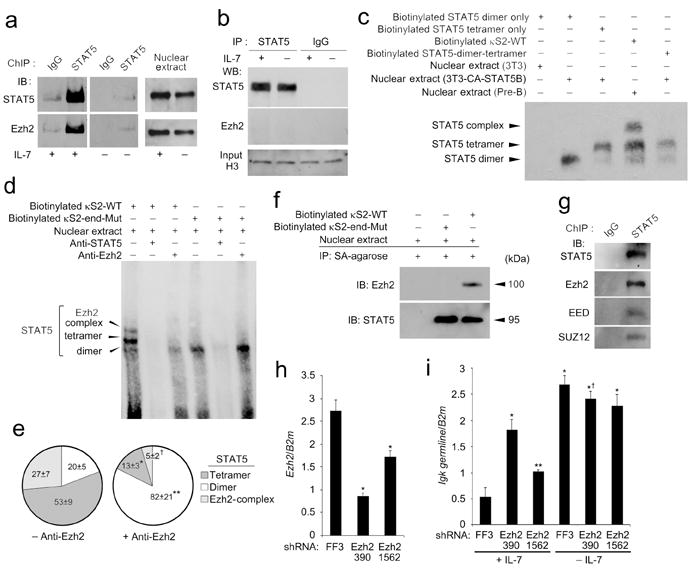
Tetrameric STAT5 binding to κS2 recruits Ezh2 and repress Igk germline transcription. (a) ChIP was performed from Irf4-/-Irf8-/- pre-B cells cultured in high IL-7 (+) or low (-) IL-7 using anti-STAT5 or control antibodies, resolved by SDS-PAGE and membranes probed with anti-STAT5 or anti-Ezh2. Immunoblotting nuclear extract aliquots with anti-STAT5 or anti-Ezh2 demonstrated similar availability of both molecules from cells cultured in high or low IL-7. (b) Immunoprecipitations from Irf4-/-Irf8-/- pre-B cell nuclear preparations were performed as in (a) but without DNA crossing (no formaldehyde). Supernatants from nuclear lysate immunoprecipitations were immunoblotted with anti-histone H3 antibody as a loading control. (n = 2) (c) EMSAs of nuclear extracts from Irf4-/-Irf8-/- pre-B cells cultured in high IL-7 for 24 h or 3T3 cells transduced with CA-STAT5. Extracts were probed with biotinylated κS2(WT) or oligonucleotides known to bind STAT5 dimers, tetramers or dimers and tetramers. (d) EMSA of nuclear extracts from Irf4-/-Irf8-/- pre-B cells cultured in high IL-7 assayed with biotinylated κS2-WT and κS2-end-Mut probes. (e) Quantitative analysis of experiments represented in (d) (n = 3) obtained using κS2-WT probe. Relative average density (±s.d.) of indicated electrophoretic bands compared to total densities for all three bands (displayed as a percentage) in either the presence or absence of anti-Ezh. *, **, and † all P<0.001 comparing indicated relative band density with or without anti-Ezh2. (f) Biotinylated κS2-WT and κS2-end-Mut probes were precipitated from nuclear extracts from Irf4-/-Irf8-/- pre-B cells cultured in high IL-7 with strepavidin Agarose. Samples were resolved by SDS-PAGE and membranes probed with anti-STAT5 and anti-Ezh2. (g) ChIP was performed from Irf4-/-Irf8-/- pre-B cells cultured in high IL-7 using anti-STAT5 or control antibodies, resolved by SDS-PAGE and membranes probed with antibodies specific for STAT5, Ezh2, anti-EED, or SUZ12. (h) qPCR of Ezh2 mRNA expression in Irf4-/-Irf8-/- pre-B cells expressing shRNA for firefly luciferase (sh-FF3; control) and two shRNAs targeting Ezh2 (sh-Ezh2-390 and sh-Ezh2-1562)(average ± s.d, n=3). *P<0.001, versus sh-FF3. (i) qPCR of Igk germline transcription in Irf4-/-Irf8-/- pre-B cells expressing indicated shRNAs in high or low IL-7 for 48 h (average ± s.d, n=3). *P<0.001, versus sh-FF3 +IL-7; **P<0.05 versus sh-FF3 +IL-7; †P<0.01, versus sh-Ezh2-390 +IL-7. Except were indicated, all experiments are representative of three independent experiments.
These observations raised a central question: how could STAT5 recruit Ezh2 to Eκi, and act as a repressor, when it usually functions as a potent transcriptional activator? Conventionally, STAT proteins are recruited as dimers to TTCN3GAA-containing GAS motifs41. However, STAT5 proteins can also bind as a dimer of dimers (tetramer) formed through homotypic N-terminal domain interactions42. In vivo, STAT5 tetramers have been associated with aberrant transcriptional patterns and leukemogenesis43. In vitro, STAT5 tetramers are recruited to DNA sequences containing tandem GAS motifs in which partial GAS elements were separated by 4-19 nucleotides44. Examination of the κS2 site revealed 3′ sequences containing partial GAS motifs (TCT, TTG, and TAA) 4-12 nucleotides downstream (Fig. 1a). Therefore, we determined if STAT5 bound κS2 as a tetrameric complex.
In EMSAs using pre-B cell nuclear preparations, three STAT5 containing complexes with different electrophoretic mobilities bound κS2 (Fig. 1b,c). To compare these to complexes formed by STAT5 dimers or tetramers, nuclear preparations from CA-STAT5 over-expressing fibroblasts (3T3) were incubated with nucleotide probes known to bind only STAT5 dimers, STAT5 tetramers, or known to bind both dimers and tetramers44 (Supplementary Table 1). The results showed that the fastest migrating κS2-bound complex obtained from Irf4-/-Irf8-/- nuclear preparations corresponded to the STAT5 dimer while the intermediate complex corresponded to the STAT5 tetramer (Fig. 5c). κS2 also formed a third distinct slowly migrating complex.
We next examined the relationships between the different κS2 binding complexes and Ezh2 recruitment. Pre-incubation with STAT5-specific antibody abolished all three κS2 binding complexes while anti-Ezh2 diminished only the two more slowing migrating complexes (Figs. 5d,e). Tetrameric, but not dimeric, STAT5 binding is predicted to be dependent upon the 3’ partial GAS motifs. Indeed, mutation of these motifs (κS2-end-Mut)(Supplementary Table 1) disrupted the two electrophoretically slower complexes but not the putative dimer complex. Finally, we directly examined the ability of κS2(WT) and κS2-end-Mut probes to precipitate STAT5 and Ezh2 from Irf4-/-Irf8-/- nuclear preparations. Although both sequences bound STAT5, only the WT κS2 site also detectably precipitated Ezh2 (Fig. 5f). These data suggest that STAT5 tetramers, bound to the tetrameric motif, recruit Ezh2.
Ezh2 is usually recruited to DNA in a multimeric complex referred to as the polycomb repressive complex 2 (PRC2) which contains, among other components, EED and SUZ12 (ref. 39). Therefore, STAT5 ChIPs from Irf4-/-Irf8-/- pre-B cells cultured in IL-7 were probed in immunoblots with EED or SUZ12-specific antibodies. We found that both components, along with STAT5 and Ezh2, were readily detected in immunoblots of the STAT5 ChIPs (Fig. 5g). These data suggest that Ezh2 is recruited to chromatin-bound STAT5 as part of the PRC2 complex.
We next examined the role of Ezh2 in IL-7 mediated repression of Igk germline transcription. Small hairpin RNAs (shRNA) predicted to target Ezh2 were cloned into the miR30-based retroviral vector, expressed in Irf4-/-Irf8-/- cells, and GFP+ cells were isolated by flow cytometry. Two shRNAs, 390 and 1562, inhibited Ezh2 expression by 73% and 37% respectively (Fig. 5h). Expression of either shRNA inhibited IL-7-mediated Igk germline transcription repression while cells expressing 390 demonstrated an almost complete loss of repression (Fig. 5i). In conjunction with the findings described above, these data indicated that STAT5-mediated recruitment of Ezh2 to Eκi is the primary mechanism by which the IL-7R represses Igk germline transcription.
Tetrameric STAT5-mediated H3K27me3 is not limited to Igk
Our data revealed that tetrameric STAT5 binding to Eκi recruits Ezh2, which marks Igk with H3K27me3 and represses germline transcription. To determine if this is a unique feature of Igk transcriptional regulation, or is a more broadly applicable feature of STAT5 biology, we performed ChIPs with STAT5 or H3K27me3-specific antibodies using nuclear preparations from Rag2-/- pro-B cells cultured in IL-7. Immunoprecipitations were then subjected to deep sequencing (ChIP-Seq). We obtained 554 peaks for STAT5 and 34,778 peaks for H3K27me3. Examination of the Igk locus revealed a strong single STAT5 peak at Eκi (Fig. 6a). There was a corresponding H3K27me3 peak with additional flanking peaks covering the Jκ and Cκ regions. However, there were no other significant STAT5 or H3K27me3 peaks throughout the Igk locus including 5′ to the Vκ cluster or at the 3′Eκ (Fig. 6a and data not shown). These findings, which corroborate results obtained using CA-STAT5 transfected Irf4-/-Irf8-/- pre-B cells (Fig. 4c), indicate that the Igk locus is only marked with H3K27me3 at sites near where STAT5 recruits Ezh2. Consistent with previous results45, the Igh locus had STAT5 binding sites in the VH cluster but no significant H3K27me3 marks (Fig. 6b). De novo prediction of motifs within the STAT5 peaks revealed a strong association with canonical STAT5 binding sites (P = 5.6 × 10-32) (Fig. 6c). Comparison of the distribution of H3K27me3 and STAT5 peaks identified 53 peaks that overlapped by two or more base pairs (Fig. 6d). Of these, 47 were within 25 kb of the known transcriptional start sites of 44 genes (Supplementary Table 2). Analysis of these STAT5 binding sites demonstrated that all were predicted to have tetrameric STAT5 binding sites (Supplementary Table 3). Alignment of each motif (Supplementary Table 4) revealed an average STAT5 tetrameric motif containing two consensus STAT5 motifs (5′-TTCN3GAA-3′) usually separated by 3-8 nucleotides (Fig. 6e). Interestingly, all sequences had an incomplete 3′ GAS motif even though some 5′ sequences were canonical GAS motifs (Supplementary Table 3). These results indicate that STAT5 recruitment to tetrameric binding sites is associated with H3K27 trimethylation in vivo.
Figure 6.
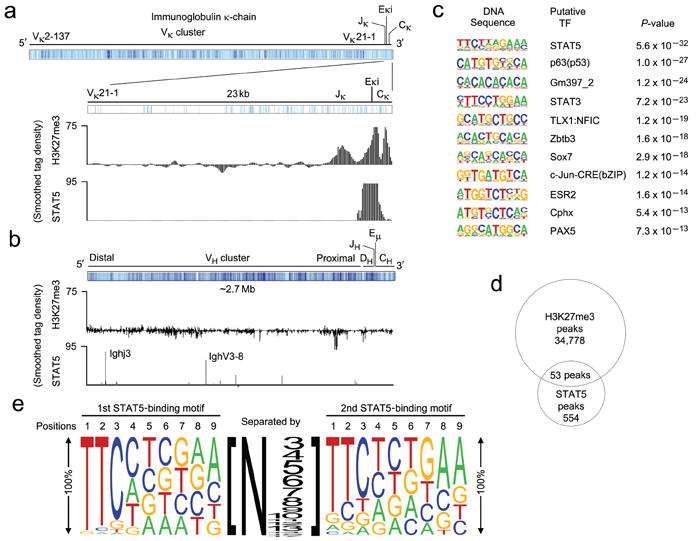
Tetrameric STAT5 binding mediates H3K27 trimethylation in vivo. (a-b) ChIP-seq analysis of Rag2-/- pro-B cell populations expanded in vitro for 2 days in the presence of IL-7 (10 ng/ml). ChIPs were performed using anti-STAT5 and anti-H3K27me3. (a) Schematic of the Igk locus noting the location of Vκ, Jκ, Eκi and Cκ gene segments (mm9 chromosome 6: 70,653,572-70,676,748). The regions of detected H3K27me3 and STAT5 binding are provided. (b) Schematic of the Igh locus noting the location of VH, DH, JH, Eμ, and CH gene segments (mm9 chromosome 12: 114,496,979-117,248,165). The regions of detected H3K27me3 and STAT5 binding are provided. (a,b) Results are presented as a smoothed tag density where tag = sequence read. (c) Conserved de novo DNA sequence motifs identified among STAT5-bound regions. (d) Venn diagram showing the common peaks obtained from STAT5 and H3K27me3 ChIP-seq analysis. (e) Predicted tetrameric STAT5 binding motif determined from the gene regulatory regions demonstrating STAT5 and H3K27me3 common peaks (Supplementary Tables 3,4). The size of the letter at each position represents the percentage of presence of that nucleotide in that position. The size of the number represents the number of the nucleotides present between two STAT5 binding motifs.
Concurrent STAT5 and H3K27me3 peaks predict repression
We next analyzed the relationship between the co-occurrence of STAT5 and H3K27me3 peaks and transcriptional regulation using data available through the ImmGen Consortium (www.ImmGen.org), which contains expression data on 40 of the 44 genes identified above. Expression was normalized to that observed in pro-B cells, and then fluctuations, as a function of development, were assessed. Most genes co-targeted by STAT5 and H3K27me3 remained unchanged throughout later stages of development (Fig. 7a). In contrast, five genes were significantly upregulated after the large pre-B cell stage (Igk, Brwd1, Adar, Wnt16 and Dmrt2) with Igk and Brwd1 being strongly induced.
Figure 7.
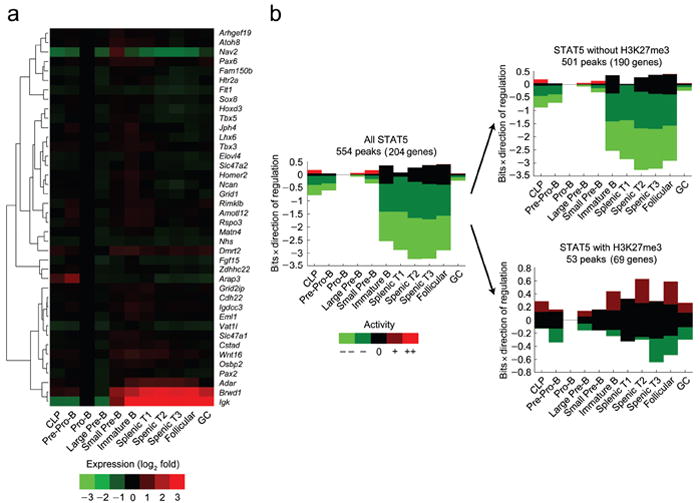
H3K27 trimethylation correlates with STAT5 target genes that are repressed throughout B cell development. (a) Heat map of genes targeted by both STAT5 and H3K27me3 indentified in Fig. 6 representing fold changes in expression (log2) as a function of B cell development and maturation relative to the pro-B cell stage (ImmGen Consortium). (b) EMBER analysis combining STAT5 and H3K27me3 ChIP-seq results with total mouse genome expression microarrays from the ImmGen Consortium. Predominant expression patterns of genes within 100 kb of STAT5 binding with or without coincidental H3K27me3 marks were assessed. Coincidence required a minimum of 2 bp overlap. In some instances more than one gene is within 100 kb of a peak. For example, 69 genes are within 100 kb of the 53 STAT5 and H3K27me3 coincident peaks. All indicated B cell developmental stages are compared to expression at pro-B cell stage. Change in mean expression behavior is categorized as: ≤−3s.d. (--); between −1 and −3s.d. (-); between −1 and 1s.d. (0); between 1 and 3s.d. (+); greater than 3s.d., with s.d. defined as the sum of the two standard deviations calculated for the experimental replicates at the two conditions.
The above statistics were limited to genes that were physically close to the overlapping STAT5 and H3K27me3 peaks. Because regulatory interactions can be long-ranged, we also used Expectation Maximization of Binding and Expression Profiles (EMBER), a method that integrates ChIP-seq and gene expression data to infer gene targets at distances up to 100 kb from transcription binding sites using an unsupervised machine learning algorithm. Using EMBER, we integrated our STAT5 and H3K27me3 ChIP-seq data with B cell developmental stage expression data (ImmGen Consortium). Data were normalized to expression detected at the pro-B cell stage. These analyses demonstrated that STAT5 binding at the pro-B cell state, irrespective of H3K27me3 marks, was associated with subsequent gene downregulation (Fig. 7b, first panel and Supplementary Fig. 2). This finding is consistent with an overall activating role for STAT5. For genes associated with STAT5 binding but not H3K27me3 marks (Fig. 7b, top panel and Supplementary Fig. 3), the pattern of expression was similar to that associated with STAT5 binding irrespective of H3K27me3. In contrast, STAT5 binding sites co-occurring with H3K27me3 marks (Fig. 7b bottom panel and Supplementary Fig. 4) were associated with stable patterns of expression. These data suggest that the co-occurrence of STAT5 binding and H3K27me3 is usually associated with a stable or repressed gene expression profile.
DISCUSSION
Herein, we provide a bi-molecular model in which the relative binding of two transcription factors, STAT5 and E2A, to Eκi control the initiation of Igk germline transcription in pre-B cells. Central to this model is our demonstration that STAT5 binds as a tetramer to Eκi and recruits the PRC2 which contains Ezh2. Ezh2 then marks Eκi, as well as the adjacent Jκ cluster and Cκ, with H3K27me3. While other areas of the Igk locus are not marked with H3K27me3, loss of Ezh2 is sufficient for escape from IL-7-mediated Igk repression. This STAT5-mediated repression mechanism ensures that Igk germline transcription is silenced as long as B cell progenitors are productively receiving proliferative signals through the IL-7R. Upon escape from IL-7R signaling, and the loss of activated STAT5, H3K27me3 is lost. The transition to open chromatin is regulated by pre-BCR-mediated induction of free nuclear E2A, which binds Eκi and promotes acquisition of H3K4me1 and H4Ac. In the absence of both STAT5 and E2A binding, the Eκi was devoid of the assayed histone modifications. These data suggest that STAT5 and E2A binding, and the complexes they recruit, are sufficient to account for transition of the Eκi region from a silenced to an active state46. This conclusion is consistent with previous studies demonstrating promiscuous Igk germline transcription in Stat5-/- early B cell progenitors31 and observations that both E2A24 and the Eκi23 are required for opening chromatin at Igk.
The epigenetic status of the Eκi region was controlled by two transcription factors, providing an explanation for the tight correlation between Igk germline transcription and accessibility to recombination11. In our genome-wide studies, the H3K27me3 mark conferred by STAT5 and Ezh2 binding correlates with transcriptional repression. It is interesting that STAT5 binding was associated with discrete H3K27me3 peaks over Eκi as well as Jκ and Cκ. This suggests that the chromatin in this region is not linear and that the Jκ and Cκ regions might be positioned over Eκi.
In bulk populations of B cell progenitors, the transition between a repressed and active Igk locus appeared complete with little evidence of substantial heterogeneity. These data are consistent with recent observations that both Igk alleles are transcriptionally active before recombination10. Presumably, the mechanisms described here, which control Igk transcription and accessibility, do not restrict recombination to one allele (allelic exclusion)36. Furthermore, the strong co-segregation of the H3K4me1 and H4Ac marks suggest that there is not an intermediate “poised” state for Eκi in the context of Igk recombination in pre-B cells22,46.
Assessment of the relationships between STAT5 and H3K27me3 across the genome of proliferating pro-B cells revealed that STAT5 likely functions as a repressor of other genes. Analysis of H3K27me3 and STAT5 co-incident peaks within putative regulatory regions of genes revealed that all had tandem GAS motifs, and in all, the 3′ GAS was a partial motif. However, overall the most common nucleotides at each position constituted a canonical GAS motif. These data provide in vivo evidence that STAT5 recruitment to tetrameric binding sites in multiple genes is associated with epigenetic repression. Furthermore, the high correlation between STAT5 binding to tetrameric sites and H3K27me3 marks suggests that tetrameric binding to specific DNA sequences may be necessary for epigenetic repression.
The consequences of STAT5 mediated repression may vary depending upon the context in which STAT5 binding occurs. While Igk transcription is induced upon entry into the pre-B cell pool, many genes binding both STAT5 and H3K27me3 in pro-B and large pre-B cells remained relatively repressed throughout development and peripheral maturation. This may reflect the fact that, in many cases, H3K27me3 is a stable mark as compared to other histone PTMs47 or that STAT5-mediated silencing co-occurs at some sites with other more stable mechanisms of gene repression.
Our data suggest that tetrameric STAT5 recruits Ezh2 which represses a significant subset of genes regulated by STAT5 during B lymphopoiesis. Furthermore, we predict that this mechanism is relevant to T lymphopoiesis as the TCRα enhancer contains potential STAT5 tetrameric sites abutting several conserved DNA binding motifs48. However, it is unlikely that, in general, the presence of STAT5 tetramers, or of tetrameric binding sites, is sufficient to predict gene repression. This is suggested by observations that STAT5 tetramers can be associated with gene activation and multilineage leukemias43. Furthermore, recent evidence indicates that STAT5 can also repress transcription by recruiting a histone deacetylase and by competing for binding with STAT349. These examples indicate that understanding how and when STAT5 represses transcription will require a detailed understanding of how STAT5 recruits PRC2 and other repressive complexes.
METHODS
Mice
Wild-type (WT), Irf4-/-Irf8-/- (C57BL/6) and Rag2-/- (BALB/c) mice were housed in the animal facilities of the University of Chicago. Mice were used at 6-10 weeks of age and experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of the University of Chicago.
Isolation, culture and flow cytometry of bone marrow B cell progenitors
Rag2-/- pro-B and Irf4-/-Irf8-/- pre-B cells were isolated from bone marrow using a magnetic-activated cell sorter separation column (MACS; Miltenyi Biotec) to isolate CD19+ cells. Rag2-/- pro-B cells were cultured in complete Opti-MEM containing 7.5% (vol/vol) FBS and IL-7 (10 ng/ml)50. Irf4-/-Irf8-/- pre-B cells (>99% pure) were overlaid on OP9 stromal cells in complete medium with 10 ng/ml (high) or 0.1 ng/ml (low) IL-7 (ref. 19).
Bone marrow was collected from WT mice and cells were resuspended in staining buffer (3% (vol/vol) FBS in PBS). Erythrocytes were lysed and cells stained50 with antibodies against CD11c (HL3), NK1.1 (PK136), TCRβ (H57-597), Ter119 (TER-119), Mac-1 (M1/70), Gr-1 (RB6-8C5), CD43 (S7), IgM (R6-60.2), IgD (11-36), pre-BCR (SL156), CD19 (1D3), and B220 (RA3-6B2) (all from BD Pharmingen). Antibodies were directly coupled to fluorescein isothiocyanate, phycoerythrin, phycoerythrin-indotricarbocyanine, allophycocyanin, efluor 450, or biotin. Pro-B (B220+CD43hiIgM-Lin-), large pre-B (B220+CD43lo/-IgM-Lin-FSChi), small pre-B (B220+CD43lo/-IgM-Lin-FSClo), and immature B (B220+CD43-IgM+IgD-Lin-) cells were isolated by cell sorting (MoFlo or FACS Aria).
Short hairpin RNA
shRNA oligonucleotides (97 mer) for Ezh2 were designed using http://katahdin.cshl.org. These shRNA oligonucleotides were cloned into a miR30-based retroviral vector expressing GFP. Targeting sequences are listed in Supplementary Table 1.
Retroviral transduction
The cDNA encoding mouse CA-STAT5B or human ER-Id3 were sub-cloned into MIGR1 (ref. 16). Retroviruses, including the shEzh2 constructs, were produced by transient transfection in PLAT-E packaging cell lines. Infection of B cell progenitors or 3T3 cells was performed as described16. After 48 h, GFP+ cells were isolated by cell sorting, and B cell progenitors were cultured in complete medium with high IL-7 (10 ng/ml) or low IL-7 (0.1 ng/ml). Id3 was induced by treating cultures with 4-OH-tamoxifen (1 μM) 48 h before assay.
EMSA
Nuclear extracts were prepared as described19. Prior to the addition of biotin-labeled double-stranded DNA probe, 5 μg nuclear extract was incubated for 20 min on ice in 20 μl reaction buffer containing 1X binding buffer, 1 μg double-stranded poly(dI-dC), 2.5% (vol/vol) glycerol, 0.05% (vol/vol) Nonidet-P40, and 1 μg BSA. Samples were incubated for 20 min at 22°C with biotin-labeled probes (20 fmol/each). For competition experiments, nuclear extracts were pre-incubated for 20 min on ice with 100-fold molar excess unlabeled, double-stranded oligonucleotides. In super-shift experiments, the extracts were pre-incubated for 60 min on ice with anti-STAT5 (sc-835; Santa Cruz Biotechnology) or anti-Ezh2 (4905; Cell Signaling). Protein-DNA complexes were separated by electrophoresis on 6% nondenaturing TBE gels (Invitrogen) and visualized with a LightShift Chemiluminescent EMSA kit according to the manufacturer’s procedure (Pierce). Oligonucleotide probes used are provided in Supplementary Table 1.
ChIP, quantitative PCR analysis and nuclear immunoprecipitations
A ChIP assay kit was used according to the manufacturer’s instructions (Upstate Biotechnology). Immunoprecipitations were performed with anti-STAT5 (sc-835x), anti-E47 (sc763x; both from Santa Cruz Biotechnology), Rabbit IgG (011-000-003; Jackson Immunoresearch Labs Inc), anti-acetyl-Histone H4 (06-866), anti-monomethyl-Histone H3-lys4 (07-436), and anti-trimethyl-Histone H3-Lys27 (07-449; all three from Millipore). Purified DNA was then analyzed by quantitative real-time PCR (primers, Supplementary Table 1). A total volume of 25 μl containing 1 μl DNA template, 0.5 μM each primer, and SYBR Green PCRMaster Mix (Applied Biosystems) was analyzed in quadruplicate. Gene expression was analyzed with an ABI PRISM 7300 Sequence Detector and ABI Prism Sequence Detection Software version 1.9.1 (Applied Biosystems). To assess association of proteins with nuclear STAT5 not bound to chromatin, ChIPs were performed as above but in the absence of the DNA cross-linker formaldehyde. Precipitations were then immunblotted with STAT5 or Ezh2-specific antibodies as indicated. Supernatants were immunoblotted with histone H3-specific antibody (9715; Cell Signaling).
ChIP-Sequencing
Rag2-/- pro-B cells were cultured for 48 h in 10 ng/ml IL-7. Chromatin from 4 × 107 cells was used for each ChIP experiment. Antibodies to STAT5 (sc-835x; Santa Cruz Biotechnology) and H3K27me3 (07-449; Millipore) were used, which yielded approximately 140 ng and 320 ng DNA, respectively. DNA libraries were prepared from the sheared chromatin (200-600bp) and were sequenced using an Illumina Genome Analyzer II. Raw sequence data were produced using the Illumina CASAVA1.7 analysis pipeline, using the “eland-extended” option. Fastq-formatted, single-end, 36 bp reads were aligned to the mm9 reference genome (National Center for Biotechnology Information build mm9_NCBI_build_37.1) using Bowtiev0.12.7 alignment software (http://bowtie-bio.sourceforge.net/index.shtml), and only reads with unique matches were retained (Supplementary Table 5). MACSv1.3.7 (http://liulab.dfci.harvard.edu/MACS/) was used to identify peaks (tag size = 36, band width = 100, model fold = 2, P-value cutoff = 1 × 10-5), and HOMER software (http://biowhat.ucsd.edu/homer/chipseq/peakMotifs.html) was used for de novo prediction of motifs within the peaks. The STAT5 and H3K27me3 peaks were presented as “smoothed tag density”, where tag=sequence read, which were calculated by a program (http://compbio.med.harvard.edu/Supplements/ChIP-seq/). To identify the overlapping peaks of STAT5 and H3K27me3, sequence reads were aligned with the UCSC genome browser (http://genome.ucsc.edu).
Comparison of ChIP-Seq and mRNA expression
To determine which genes were targeted by STAT5 binding and how those genes behaved over the course of B cell development, we employed Expectation Maximization of Binding and Expression Profiles (EMBER). EMBER integrates transcription factor binding data with DNA microarray gene expression data and uses an unsupervised learning algorithm to infer the genes targeted by the transcription factor. This is done by defining a set of pair-wise comparisons (here, between developmental stages), discretizing the changes in expression as in the caption to Fig. 7, and searching for over-represented patterns among these data for the genes within 100 kb of the transcription factor binding sites. Only genes that match an expression pattern were selected; therefore, not all transcription factor-binding sites were assigned to a target gene. Utilizing EMBER, we compared our STAT5 and H3K27me3 ChIP-Seq from Rag2-/- pro-B cells with total genome microarray expression data of all B cell developmental stages obtained from the ImmGen consortium (http://www.ImmGen.org). We applied EMBER to all STAT5 binding sites, as well as STAT5 binding sites with or without coincident binding of H3K27me3 (coincidence defined by two peaks with a minimal overlap of 2 bp).
Immunoblotting
ChIP or streptavidin(SA)-Agarose (20347; Thermo Scientific) immunoprecipitated samples were resolved by 4-20% SDS-PAGE and then transferred to Immuno-Blot PVDF membranes (BioRad)50. Blots were probed with antibody specific for STAT5 (9363S; Cell Signaling or sc-835; Santa Cruz Biotechnology), Ezh2 (4905, Cell Signaling), SUZ12 (17-661; Millipore) or EED (17-10034; Millipore).
Luciferase assays
Cos7 (kidney fibroblast) cells were transduced with a pGL3-promoter-luciferase construct containing Eκi(WT), Eκi(mκS1), Eκi(mκS2), or Eκi(mκS1-mκS2) enhancer elements and pRL-TK (Renilla luciferase, internal control) along with mammalian expression vector constructs for E47 (pHbAP-E47), E12 (pHbAP-E12), or CA-STAT5B (pcDNA3.1-CA-STAT5B) singly or in combination (using pHbAPneo and pcDNA3.1 as controls). Luciferase activity was determined using a Dual-Luciferase Reporter Assay System (Promega) and a 2020n luminometer (Turner Biosystems) configured for dual assays.
Statistical analysis
Data were analyzed with the unpaired t-test and analysis of variance, followed by the test of least-significant difference for comparisons within and between groups. All categories in each analyzed experimental panel were compared; significant P values (<0.05) are provided. All P values <0.001 were rounded to facilitate comparisons between results.
Supplementary Material
Supplementary Figure 1. STAT5 and E12 compete for binding at respective sites of Eκi. (a) Cos7 cells were transfected with the pGL3-promoter luciferase reporter plasmids described in the figure along with plasmids encoding pRL-TK (Renilla luciferase) and indicated molecules or appropriate control plasmids. After 48 hours, dual luciferase assays were performed on cell lysates (average ± s.d, n=3). *P<0.001 versus Eκi(WT) control; ** P<0.001 versus Eκi(κS2-3A-κE1) control; *** P<0.001 versus Eκi(κS2-6A-κE1) control; †P<0.001 versus Eκi(κS2-9A-κE1) control. (b) Cos7 cells were transfected with the pGL3-promoter luciferase reporter plasmid containing Eκi(WT) or mutants as indicated along with plasmids encoding pRL-TK (Renilla luciferase) and indicated molecules or appropriate control plasmids. After 48 hours, dual luciferase assays were performed on cell lysates (average ± s.d, n=3). *P<0.001 versus Eκi(WT) control; ** P<0.001 versus Eκi(κS2-9A-κE1) control; *** P<0.01 versus Eκi(mκE1) control; †P<0.001 versus Eκi(mκE2) control.
Supplementary Figure 2. STAT5 binding, irrespective of H3K27me3, is transcriptionally activating during the pro-B stage. Heat map of behavioral activity of genes identified by EMBER analysis as being within 100 kb of total STAT5 binding sites and fitting a dominant expression pattern. All indicated B cell developmental stages are compared to expression levels at pro-B cell stage. Fold change in expression behavior is categorized as: --, decreased expression by ≥3; -, decreased expression by >1 but <3; 0, no change; +, increased expression by >1 but <3; ++, increased expression by ≥3. Results are shown as mean±s.d.
Supplementary Figure 3. STAT5 binding without H3K27me3 overlap resembles total STAT5 analysis and is transcriptionally activating during the pro-B stage. Behavioral heat map of genes selected by EMBER analysis as being within 100 kb of STAT5 binding sites without coincidental H3K27me3 marks and fitting a dominant expression pattern. Coincidence required a minimum of 2 bp overlap. All indicated B cell developmental stages are compared to expression levels at pro-B cell stage. Fold change in expression behavior was categorized as: --, decreased expression by ≥3; -, decreased expression by >1 but <3; 0, no change; +, increased expression by >1 but <3; ++, increased expression by ≥3. Results are shown as mean±s.d.
Supplementary Figure 4. STAT5 binding with co-occurrence of H3K27me3 marks stably repressed genes throughout development. Heat map of behavioral activity of EMBER selected genes fitting an over-represented expression pattern. Assessed genes were within 100 kb of STAT5 binding sites with coincidental H3K27me3 marks. Coincidence required a minimum of 2 bp overlap. All indicated B cell developmental stages are compared to expression levels at pro-B cell stage. Fold change in expression behavior is categorized as: --, decreased expression by ≥3; -, decreased expression by >1 but <3; 0, no change; +, increased expression by >1 but <3; ++, increased expression by ≥3. Results are shown as mean±s.d.
Acknowledgments
We thank H. Singh (Genentech) and U. Storb (University of Chicago) for helpful discussions. We also thank R. Duggan and D. Leclerc for cell-sorting services. This work benefited from data assembled by the ImmGen consortium. This work was supported by National Institute of Health grant GM088847 (to M.R.C.), a Department of Energy Computational Science Graduate Fellowship to M.M-C. and the Chicago NIH Systems Biology Center (A.R.D., P50 GM081892). B.L.K. was supported by the Leukemia and Lymphoma Society and the NIH (CA099978).
Footnotes
AUTHOR CONTRIBUTIONS: M.M. designed, carried out, and analyzed most of the experiments performed. He also prepared the first draft of the paper; S.E.P. assisted in the design and analysis of many experiments; M.M-C performed the comparison of mRNA expression and ChIP-seq data assisted by K.M.H.; E.T.B. assisted in the Chip-seq analysis. B.L.K. provided E2A-specific reagents and contributed to the design of some experiments; A.R.D oversaw the analysis comparing microarray and ChIP-seq data; M.R.C. oversaw the entire project and prepared the final manuscript.
References
- 1.Clark MR, Cooper AB, Wang L, Aifantis I. The pre-B cell receptor in B cell development: recent advances, persistent questions and conserved mechanisms. Curr Top Microbiol Immunol. 2005;290:87–104. doi: 10.1007/3-540-26363-2_5. [DOI] [PubMed] [Google Scholar]
- 2.Pelanda R, Braun U, Hobeika E, Nussenzweig MC, Reth M. B cell progenitors are arrested in maturation but have intact VDJ recombination in the absence of Ig-alpha and Ig-beta. J immunol. 2002;169:865–72. doi: 10.4049/jimmunol.169.2.865. [DOI] [PubMed] [Google Scholar]
- 3.Shimizu T, Mundt C, Licence S, Melchers F, Martensson I. VpreB1/VpreB2/lambda 5 triple-deficient mice show impaired B cell development but functional allelic exclusion of the IgH locus. J Immunol. 2002;168:6286–6293. doi: 10.4049/jimmunol.168.12.6286. [DOI] [PubMed] [Google Scholar]
- 4.Herzog S, Reth M, Jumaa H. Regulation of B-cell proliferation and differentiation by pre-B-cell receptor signalling. Nat Rev Immunol. 2009;9:195–205. doi: 10.1038/nri2491. [DOI] [PubMed] [Google Scholar]
- 5.Erlandsson L, et al. Both the pre-BCR and the IL-7Ralpha are essential for expansion at the pre-BII cell stage in vivo. Eur J Immunol. 2005;35:1969–76. doi: 10.1002/eji.200425821. [DOI] [PubMed] [Google Scholar]
- 6.Fleming HE, Paige CJ. Pre-B cell receptor signaling mediates selective response to IL-7 at the pro-B to pre-B cell transition via an ERK/MAP kinase-dependent pathway. Immunity. 2001;15:521–531. doi: 10.1016/s1074-7613(01)00216-3. [DOI] [PubMed] [Google Scholar]
- 7.Zhang L, Reynolds TL, Shan S, Desiderio S. Coupling of V(D)J recombination to cell cycle suppresses genomic instability and lymphoid tumorigenesis. Immunity. 2011;34:163–174. doi: 10.1016/j.immuni.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alt FW, Blackwell TK, Yancopoulos GD. Development of the primary antibody repertoire. Science. 1987;238:1079–87. doi: 10.1126/science.3317825. [DOI] [PubMed] [Google Scholar]
- 9.Schlissel MS, Stanhope-Baker P. Accessibility and the developmental regulation of V(D)J recombination. Semin Immunol. 1997;9:161–70. doi: 10.1006/smim.1997.0066. [DOI] [PubMed] [Google Scholar]
- 10.Amin RH, et al. Biallelic, ubiquitous transcription from the distal germline Ig-kappa locus promoter during B cell development. Proc Nat Acad Sci, USA. 2009;106:522–527. doi: 10.1073/pnas.0808895106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schlissel MS. Regulation of activation and recombination of the murine Ig-kappa locus. Immunol Rev. 2004;200:215–223. doi: 10.1111/j.0105-2896.2004.00157.x. [DOI] [PubMed] [Google Scholar]
- 12.Gorman JR, et al. The Ig(kappa) 3’ enhancer influences the ratio of Ig(kappa) versus Ig(lambda) B lymphocytes. Immunity. 1996;5:241–52. doi: 10.1016/s1074-7613(00)80319-2. [DOI] [PubMed] [Google Scholar]
- 13.Inlay M, Alt FW, Baltimore D, Xu Y. Essential roles of the kappa light chain intronic enhancer and 3’ enhancer in kappa rearrangement and demethylation. Nat Immunol. 2002;3:463–8. doi: 10.1038/ni790. [DOI] [PubMed] [Google Scholar]
- 14.Xu Y, Davidson L, Alt FW, Baltimore D. Deletion of the Ig-kappa light chain intronic enhancer/matrix attachment region impairs but does not abolish V kappa J kappa rearrangement. Immunity. 1996;4:377–85. doi: 10.1016/s1074-7613(00)80251-4. [DOI] [PubMed] [Google Scholar]
- 15.Lazorchak AS, Schlissel MS, Zhuang Y. E2A and IRF-4/Pip promote chromatin modification and transcription of the immunoglobulin kappa locus in pre-B cells. Mol Cell Biol. 2006;26:810–821. doi: 10.1128/MCB.26.3.810-821.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mandal M, et al. Ras orchestrates cell cycle exit and light chain recombination during early B cell development. Nat Immunol. 2009;10:1110–1117. doi: 10.1038/ni.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inlay MA, Tian H, Lin T, Xu Y. Important roles for E protein binding sites within the immunoglobulin k chain intronic enhance in avtivating V-kappaJ-kappa rearrangement. J Exp Med. 2004;200:1205–1211. doi: 10.1084/jem.20041135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bain G, et al. E2A proteins are required for proper B cell development and initiation of immunoglobulin gene rearrangements. Cell. 1994;79:885–892. doi: 10.1016/0092-8674(94)90077-9. [DOI] [PubMed] [Google Scholar]
- 19.Johnson K, et al. Regulation of immunoglobulin light-chain recombination by the transcription factor IRF-4 and the attenuation of interleukin-7 signaling. Immunity. 2008;28:335–45. doi: 10.1016/j.immuni.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 20.Lu R, Kay L, Lancki DW, Singh H. IRF-4,8 orchestrate the pre-B to B transition in lymphocyte development. Genes & Dev. 2003;17:1703–1708. doi: 10.1101/gad.1104803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma S, Turetsky A, Trinh L, Lu R. IFN regulatory factor 4 and 8 promote Ig light chain kappa locus activation in pre-B cell development. J Immunol. 2006;177:7898–904. doi: 10.4049/jimmunol.177.11.7898. [DOI] [PubMed] [Google Scholar]
- 22.Cedar H, Bergman Y. Epigenetics of haematopoietic cell development. Nat Rev Immunol. 2011;11:478–487. doi: 10.1038/nri2991. [DOI] [PubMed] [Google Scholar]
- 23.Xu C-R, Feeney AJ. The epigenetic profile of Ig genes is dynamically regulated during B cell differentiation and is modulated by pre-B cell receptor signaling. J Immunol. 2009;182:1362–1369. doi: 10.4049/jimmunol.182.3.1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beck K, Peak MM, Ota T, Nemazee D, Murre C. Distinct roles for E12 and E47 in B cell specification and the sequential rearrangement of immunoglobulin light chain loci. J Exp Med. 2009;206:2271–2284. doi: 10.1084/jem.20090756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin YC, et al. A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat Immunol. 2010;11:635–643. doi: 10.1038/ni.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y, Subrahmanyam R, Chakroborty T, Sen R, Desiderio S. A plant homeodomain in RAG-2 that binds Hypermethylated lysine 4 of histone H3 is necessary for efficient antigen-receptor-gene rearrangement. Immunity. 2007;27:561–571. doi: 10.1016/j.immuni.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ji Y, et al. The in vivo pattern of binding of RAG1 and RAG2 to antigen receptor loci. Cell. 2010;141:419–431. doi: 10.1016/j.cell.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flemming A, Brummer T, Reth M, Jumaa H. The adaptor protein SLP-65 acts as a tumor suppressor that limits pre-B cell expansion. Nat Immunol. 2003;4:38–43. doi: 10.1038/ni862. [DOI] [PubMed] [Google Scholar]
- 29.Xu S, Lee KG, Huo J, Kurosaki T, Lam KP. Combined deficiencies in Bruton tyrosine kinase and phospholipase Cgamma2 arrest B-cell development at a pre-BCR+ stage. Blood. 2007;109:3377–84. doi: 10.1182/blood-2006-07-036418. [DOI] [PubMed] [Google Scholar]
- 30.van Loo PF, Dingjan GM, Maas A, Hendriks RW. Surrogate-light-chain silencing is not critical for the limitation of pre-B cell expansion but is for the termination of constitutive signaling. Immunity. 2007;27:1–13. doi: 10.1016/j.immuni.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 31.Malin S, et al. Role of STAT5 in controlling cell survival and immunoglobulin gene recombination during pro-B cell development. Nat Immunol. 2010;11:171–179. doi: 10.1038/ni.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herzog S, et al. SLP-65 regulates immunoglobulin light chain gene recombination through the PI(3)K-PKB-Foxo pathway. Nat Immunol. 2008;9:623–31. doi: 10.1038/ni.1616. [DOI] [PubMed] [Google Scholar]
- 33.Tokoyoda K, Egawa T, Sugiyama T, Choi BI, Nagasawa T. Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity. 2004;20:335–344. doi: 10.1016/j.immuni.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 34.Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46:705–16. doi: 10.1016/0092-8674(86)90346-6. [DOI] [PubMed] [Google Scholar]
- 35.Heintzman ND, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;46:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vettermann C, Schlissel MS. Allelic exclusion of immunoglobulin genes: models and mechanisms. Immunol Rev. 2010;237:22–42. doi: 10.1111/j.1600-065X.2010.00935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quong MW, et al. Receptor editing and marginal zone B cell development are regulated by the helix-loop-helix protein, E2A. J Exp Med. 2004;199:1101–12. doi: 10.1084/jem.20031180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Romanow WJ, et al. E2A and EBF act in synergy with the V(D)J recombinase to generate a diverse immunoglobulin repertoire in nonlymphoid cells. Mol Cell. 2000;5:343–53. doi: 10.1016/s1097-2765(00)80429-3. [DOI] [PubMed] [Google Scholar]
- 39.Margueron R, Reinberg D. The polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Su IH, et al. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol. 2003;4:124–31. doi: 10.1038/ni876. [DOI] [PubMed] [Google Scholar]
- 41.Stocklin E, Wissler M, Gouilleux F, Groner B. Functional interactions between Stat5 and the glucocorticoid receptor. Nature. 1996;383:726–728. doi: 10.1038/383726a0. [DOI] [PubMed] [Google Scholar]
- 42.Kornfeld J-W, et al. The different functions of Stat5 and chromatin alteration through Stat5 proteins. Front Biosci. 2008;13:6237–6254. doi: 10.2741/3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moriggl R, et al. Stat5 tetramer formation is associated with leukemogenesis. Cancer Cell. 2005;7:87–99. doi: 10.1016/j.ccr.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 44.Soldaini E, et al. DNA binding site selection of dimeric and tetrameric Stat5 proteins reveals a large repertoire of divergent tetrameric Stat5a binding sites. Mol Cell Biol. 2000;20:389–401. doi: 10.1128/mcb.20.1.389-401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bertolino E, et al. Regulation of interleukin 7-dependent immunoglobulin heavy-chain variable gene rearrangements by transcription factor STAT5. Nat Immunol. 2005;6:836–843. doi: 10.1038/ni1226. [DOI] [PubMed] [Google Scholar]
- 46.Northrup DL, Zhao K. Application of ChIP-seq and related techniques to the study of immune function. Immunity. 2011;34:830–842. doi: 10.1016/j.immuni.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zee BM, et al. In vivo residue-specific histone mehtylation dynamics. J Biol Chem. 2010;285:3341–3350. doi: 10.1074/jbc.M109.063784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spicuglia S, et al. TCRalpha enhancer activation occurs via a conformational change of a pre-assembled nucleoprotein complex. EMBO J. 2000;19:2034–2045. doi: 10.1093/emboj/19.9.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang X-P, et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. 2011;12:247–254. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cooper AB, et al. A unique function for cyclin D3 in early B cell development. Nat Immunol. 2006;7:489–497. doi: 10.1038/ni1324. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. STAT5 and E12 compete for binding at respective sites of Eκi. (a) Cos7 cells were transfected with the pGL3-promoter luciferase reporter plasmids described in the figure along with plasmids encoding pRL-TK (Renilla luciferase) and indicated molecules or appropriate control plasmids. After 48 hours, dual luciferase assays were performed on cell lysates (average ± s.d, n=3). *P<0.001 versus Eκi(WT) control; ** P<0.001 versus Eκi(κS2-3A-κE1) control; *** P<0.001 versus Eκi(κS2-6A-κE1) control; †P<0.001 versus Eκi(κS2-9A-κE1) control. (b) Cos7 cells were transfected with the pGL3-promoter luciferase reporter plasmid containing Eκi(WT) or mutants as indicated along with plasmids encoding pRL-TK (Renilla luciferase) and indicated molecules or appropriate control plasmids. After 48 hours, dual luciferase assays were performed on cell lysates (average ± s.d, n=3). *P<0.001 versus Eκi(WT) control; ** P<0.001 versus Eκi(κS2-9A-κE1) control; *** P<0.01 versus Eκi(mκE1) control; †P<0.001 versus Eκi(mκE2) control.
Supplementary Figure 2. STAT5 binding, irrespective of H3K27me3, is transcriptionally activating during the pro-B stage. Heat map of behavioral activity of genes identified by EMBER analysis as being within 100 kb of total STAT5 binding sites and fitting a dominant expression pattern. All indicated B cell developmental stages are compared to expression levels at pro-B cell stage. Fold change in expression behavior is categorized as: --, decreased expression by ≥3; -, decreased expression by >1 but <3; 0, no change; +, increased expression by >1 but <3; ++, increased expression by ≥3. Results are shown as mean±s.d.
Supplementary Figure 3. STAT5 binding without H3K27me3 overlap resembles total STAT5 analysis and is transcriptionally activating during the pro-B stage. Behavioral heat map of genes selected by EMBER analysis as being within 100 kb of STAT5 binding sites without coincidental H3K27me3 marks and fitting a dominant expression pattern. Coincidence required a minimum of 2 bp overlap. All indicated B cell developmental stages are compared to expression levels at pro-B cell stage. Fold change in expression behavior was categorized as: --, decreased expression by ≥3; -, decreased expression by >1 but <3; 0, no change; +, increased expression by >1 but <3; ++, increased expression by ≥3. Results are shown as mean±s.d.
Supplementary Figure 4. STAT5 binding with co-occurrence of H3K27me3 marks stably repressed genes throughout development. Heat map of behavioral activity of EMBER selected genes fitting an over-represented expression pattern. Assessed genes were within 100 kb of STAT5 binding sites with coincidental H3K27me3 marks. Coincidence required a minimum of 2 bp overlap. All indicated B cell developmental stages are compared to expression levels at pro-B cell stage. Fold change in expression behavior is categorized as: --, decreased expression by ≥3; -, decreased expression by >1 but <3; 0, no change; +, increased expression by >1 but <3; ++, increased expression by ≥3. Results are shown as mean±s.d.