

Abstract
The folliculin gene (FLCN), also known as BHD, is the only known susceptibility gene for Birt-Hogg-Dubé syndrome. BHDS is the autosomal dominant predisposition to the development of follicular hamartomas, lung cysts, spontaneous pneumothorax, and/or kidney neoplasms. To date, 53 unique germline mutations have been reported. FLCN mutation detection rate is 88%. FLCN encodes a predicted 579-amino acid protein, designated folliculin that is highly conserved between humans and homologs in mice, Drosophila, and C. elegans. We developed the first online database detailing all FLCN variants identified in our laboratory and reported in the literature. The FLCN database applies, and assists researchers in applying HGVS nomenclature guidelines. To date, the FCLN database includes 84 variants: 53 unique germline mutations and 31 SNPs. The majority of FLCN germline mutations are predicted to produce a truncated folliculin, resulting in loss of function. The FLCN mutations consist of: 45% (24/53) deletions, 32% (17/53) substitutions (10 putative-splice site, 5 nonsense, and 2 missense), 15% (8/53) duplications, 6% (3/53) insertion/deletions and 2% (1/53) insertions. The database strives to systematically unify current knowledge of FLCN variants and will be useful to geneticists and genetic counselors while also providing a rapid and systematic resource for investigators.
Keywords: folliculin, FLCN, Birt-Hogg-Dube syndrome, Fibrofolliculoma, mutation database
Introduction
Birt–Hogg–Dubé syndrome (BHDS; MIM# 135150) is an autosomal dominantly inherited genodermatosis that predisposes to the development of cutaneous hamartomas (fibrofolliculomas and trichodiscomas/angiofibromas), kidney neoplasms, lung cysts, and spontaneous pneumothorax [Birt, et al., 1977; Toro et al., 1999; Toro et al. 2008]. The BHDS locus was mapped to the short arm of chromosome 17 (17p11.2) [Schmidt, et al., 2001]. In 2002, germline mutations in the folliculin gene (approved gene symbol FLCN; MIM# 607273; GenBank accession number AF517523; also known as BHD) were identified as responsible for BHDS susceptibility [Nickerson et al., 2002]. FLCN is composed of 14 exons. Researchers at the National Cancer Institute previously reported 36 unique FLCN germline mutations in 102 BHDS families [Nickerson et al., 2002; Schmidt et al., 2005; Toro et al., 2008], and 17 additional mutations have been reported by other investigators (Figure 1) [Khoo et al., 2002; van Steensel et al., 2007; Leter et al., 2008; Gunji et al., 2007; Graham et al., 2005; Painter et al., 2005; Kawasaki et al., 2005; Lamberti et al., 2005; Bessis et al., 2006; Murakami et al., 2007; da Silva et al., 2003; Toro et al., 2008; Woodward et al., 2008; Kim eh et al., 2008; Fuentes et al.,2009; Palmirotta et al. 2008. Germline insertion or deletion of a cytosine in the hypermutable polycytosine (C8) tract in exon 11 of the gene has been detected in 53% of BHDS families and is suggested as a mutation “hot spot” [Schmidt et al., 2005; Toro et al., 2008]. The majority of FLCN germline mutations are predicted to truncate the FLCN protein, folliculin [Nickerson et al., 2002; Schmidt et al., 2005; Toro et al. 2008]. Germline FLCN mutations have been identified in the majority of patients with multiple skin fibrofolliculomas, and the relatives of probands have often been subsequently diagnosed with skin fibrofolliculomas, suggesting that many more cases are currently unrecognized. Recognition of BHDS is especially pertinent because patients with BHDS have a 50-fold and 7-fold increased risk for the development of spontaneous pneumothorax and renal tumors, respectively, compared with family members unaffected with BHDS [Zbar et al., 2002].
Figure 1.
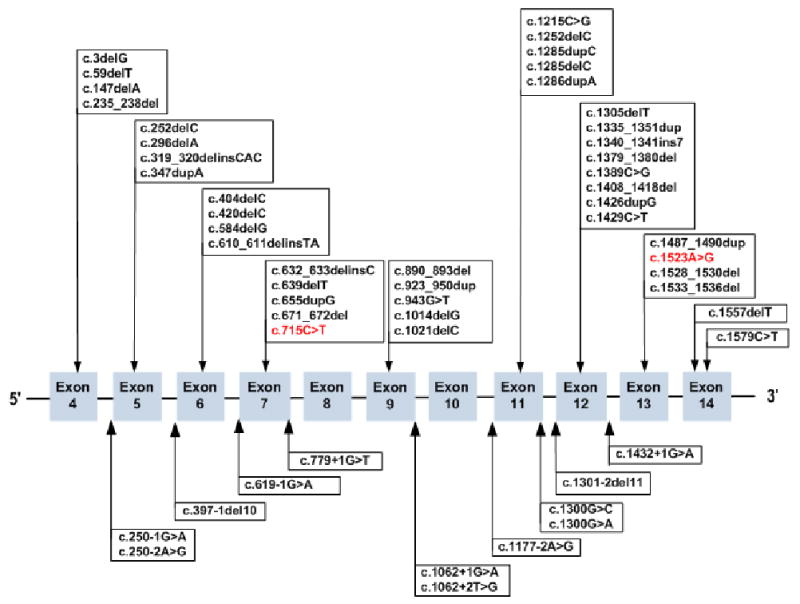
FLCN mutations in the database. Exon distribution of FLCN mutations associated with Birt-Hogg-Dubé Syndrome. Missense mutations are shown in red. FLCN frameshift, missense, and nonsense mutations are illustrated in the top panel and splice site mutations are shown in the lower panel. The nucleotide numbering is based on the published GenBank sequence NM_144997.5 and nucleotide +1 is the adenine of the ATG initiation codon.
BHDS is associated with a histological spectrum of renal tumors ranging from hybrid oncocytic (67%) to chromophobic (23%) to oncocytic (3%) renal cell carcinoma (RCC) [Pavlovich et al. 2005]. Clear cell RCC (3%) has also been reported in a few BHDS cases. Somatic FLCN mutations or loss of heterozygosity (LOH) at the FLCN locus has been detected in at least one of the RCCs in all BHDS patients tested and 70% of RCCs either had a FLCN somatic mutation (53%) or LOH (17%) in the second FLCN copy [Vocke et al., 2005]. Interestingly, 75% of second hit somatic mutations were frameshifts, while no missense mutations were identified [Vocke et al., 2005]. Inactivation of both copies of FLCN occurred in several histologic subtypes of RCC, suggesting that inactivation of FLCN occurs at an early stage of renal tumorigenesis. Renal tumors in Nihon rats and German Shepherd dogs with renal cystadenocarcinoma and nodular dermatofibrosis (RCND) show biallelic inactivation in Flcn by LOH or point mutation [Okimoto et al., 2004; Lingaas et al., 2008].
The high frequency of second hits in FLCN and its orthologs in tumors supports that FLCN is a tumor-suppressor gene.
Other pathology associated with BHDS includes: oral papules [Nadershahi et al., 1997; Toro et al., 1999], parotid oncocytoma [Schmidt et al., 2005; Toro et al., 2008]; lipoma, angiolipoma, and collagenoma [Toro et al., 1999]; cutaneous neurothekeoma and meningioma [Vincent et al., 2003], multinodular goiter [Drummond et al., 2002; Welsch et al., 2005], and chorioretinal lesions [Walter et al., 1997; Godbolt et al., 2003]. Whether these clinical phenotypes are part of the l spectrum of BHDS needs to be investigated in further studies.
Folliculin is predicted to encode a 579 amino acid protein that is highly conserved among species. FLCN mRNA is expressed in a variety of tissues including stromal cells, the distal nephron of the kidney, type I pneumocytes of the lung, and the skin and its appendages [Warren et al., 2004]. Recent studies have provided some insight into the functions of folliculin in the mTOR pathway. A novel folliculin-interacting protein, FNIP1, binds to 5′-AMP activated protein kinase, a negative regulator of mTOR. FLCN phosphorylation is facilitated by FNIP1 and is reduced by inhibitors of mTOR and AMPK activity, implicating FLCN/FNIP1 in the AMPK and mTOR signalling pathways [Baba et al., 2006]. Hartman and co-workers showed that downregulation of FLCN in mammalian cells reduces the phosphorylation of ribosomal protein S6, an indicator of TORC1 activity [Hartman et al., 2009].
van Slegtenhorst and co-workers deleted the FLCN also known as BHD) homolog in Schizosaccharomyces pombe and using expression profiling showed that six permease and transporter genes, known to be down-regulated in Δtsc1 and Δtsc2, were up-regulated in Δbhd [van Slegtenhorst et al., 2007]. Loss of Flcn sensitized yeast to rapamycin-induced increases in permease expression levels and rapamycin-induced lethality in Δflcn yeast expressing the hypomorphic Rhb1 allele [van Slegtenhorst et al., 2007]. These results suggest that Flcn activates Tor2. Hartman et al. also generated a knockout mouse with targeted inactivation of the Flcn gene that developed spontaneous oncocytic renal cysts and tumors that resemble the RCCs in BHDS patients. The renal tumors exhibit low levels of phosphorylation of ribosomal protein S6. Altogether these studies showed that folliculin and TSC1/TSC2 share a common pathway but opposite roles in TORC1 [Hartman et al., 2009].
Hereditary multifocal RCNDin German shepherd dogs [Lium et al., 1985; Jonasdottir et al., 2000] and hereditary renal carcinoma in the Nihon rat [Hino et al., 2001; Okimoto et al., 2000] are naturally occurring animal models for BHDS. Germline mutations in the FLCN orthologous genes have been identified in affected animals with these renal cancer syndromes. A missense mutation in exon 7 in the canine Flcn [Lingaas et al., 2003], and an insertion of a cytosine in a C5 tract in the Nihon rat Flcn [Okimoto et al., 2004] have been reported.
Since before 2008 there was no sequence variation database for FLCN and there are many inconsistencies in the reporting of FLCN mutations, we constructed a FLCN online database in August 2008 for The Inaugural Birt-Hogg- Dubé Symposium in Denmark and recently updated it (March, 30, 2009). The database characterizes all reported FLCN sequence variants reported to date. The purpose of the database is to systematically unify all current genetic knowledge of FLCN variants.
Construction of Database
The FLCN mutation database (http://www.skingenedatabase.com) is based on the recently described Leiden Open Variation Database (LOVD) system and it was created using version 2 of the LOVD software [Fokkema et al., 2005]. LOVD software allows the easy creation and maintenance of a fully web-based gene sequence variation database [Bayley et al., 2008]. LOVD is platform-independent and uses PHP and MySQL open source software. The basic gene-centered and modular design of the database follows the recommendations of the Human Genome Variation Society (HGVS) and focuses on the collection of DNA sequence variations. The LOVD platform can include clinical data, and database contents may be downloaded and imported into a spreadsheet for further analysis [Bayley et al., 2008].
Nomenclature
We encourage researchers to submit newly identified variants to the database. Currently all database content is derived from published, peer-reviewed literatures. Unfortunately, the mutation nomenclature currently used for FLCN in publications is inconsistent and uses a variety of annotations rarely in agreement with HGVS guidelines. One of the aims of the HGVS and the FLCN database is to establish a consistent nomenclature. We recommend that DNA variants submitted to the FLCN database be described in accordance with the most up-to-date nomenclature guidelines. Recommendations of the HGVS can be found online at HGVS.org. The Mutalyzer program (http://www.LOVD.nl/mutalyzer/) using name generator or batch checker was used to check sequence variant descriptions [Wildeman et al., 2008].
The HGVS-recommended cDNA numbering starts from the first ATG of the full coding sequence. Protein reference sequences should represent the primary translation product. Currently many variants are present in the literature under several titles, and the correct identification of intronic variants is especially problematic. The FLCN mutation database will accept all variants and will assist in assigning the correct nomenclature. Due to inconsistency of the FLCN mutation nomenclature, an extra column including the originally reported variant description(s) at the cDNA level is included for those variants that have been reassigned.
Mutations
The term ‘mutation’ is reserved for those variants in which there is a reasonable suspicion that they are deleterious. All variants that disrupt the reading frame, affect highly conserved residues (conserved by sequence alignments), or disrupt the consensus donor or acceptor splice sites (GT-AG), and are not found in healthy controls, should be suspected as potentially pathogenic. Thus, current knowledge suggests that most missense changes will be pathogenic, but this is not always the case. Inclusion of sequence variants in the FLCN mutation database does not imply that there is convincing evidence for pathogenicity. Ideally, several lines of evidence should support publications which present mutations as “pathogenic”, including screening DNA from a panel of >100 healthy individuals, describing the nature of the amino acid substitution (conservative/non-conservative) and the significance of the position in the protein (evolutionary conservation or occurrence in a known functional domain). It is important to note whether the mutation has previously been reported, if it has been found in several families, and if it segregates with disease within the family. However, none of these factors is definitive and each variant must be considered on its merits. Unfortunately, most mutations are currently reported without this accompanying analysis, and many have been identified in a single case or family. Thus, caution should be exercised when attempting to derive clinically relevant information from the database.
SNP
DNA variation in populations is generally benign and as such should be termed a single nucleotide polymorphism (SNP). Non-conserved missense variants and potential splice site mutations that do not disrupt the consensus splice sites are considered to be SNPs or rare variants of uncertain significance. Polymorphisms, including intronic variants, synonymous (silent) variants, nonsynonymous missense variants found in a healthy control panel are included as SNPs in the database unless accompanied by clear evidence of pathogenic potential. Further information can be included under “remarks and references”, such as the country of origin of the patient or study, the number of healthy controls tested for the variant, and any other supporting evidence.
How to submit a variant and use the database
The FLCN mutation database allows researchers to view variant information and deposit sequence variants. The database describes mutations exon by exon, summarizing submitted mutations in a single table. The researcher will first see the general information of the summary page about the gene, the database, and links to other sites related to the variant (Figure 2).
Figure 2.
The FLCN mutation database home page.
Researchers can conduct different types of searches or import data into a spreadsheet for further analysis. The sequence variants are ordered by position relative to the cDNA reference sequence (Figure 3). Tables can be selected for viewing both specific variants and all reported variants. Variants can be re-ordered by database ID (DB-ID), exon, previous description, mutation type, and the authors who reported the variant.
Figure 3.
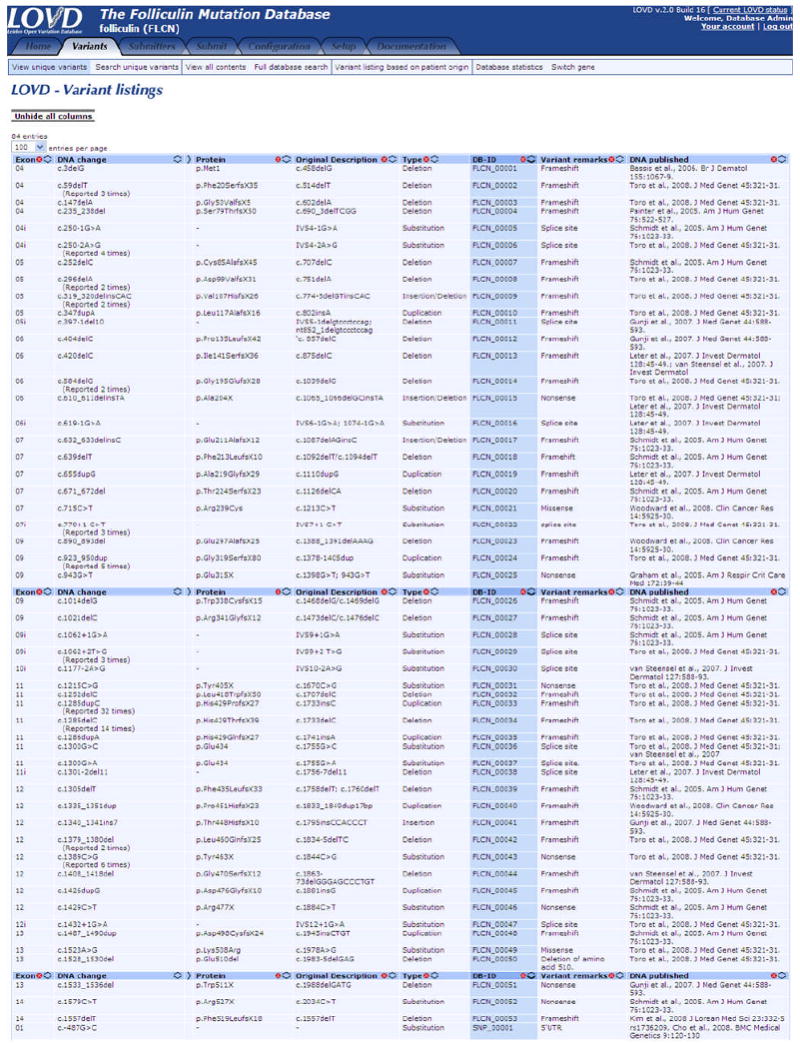
Overview of the FLCN unique variant page
We have created a map that contains the amino acids sequence encoded by the nucleotide sequence (NM_144997.5) of folliculin and indicates the intron-exon boundaries (Figure 4). The map is a useful tool for submitters in determining the location of their variant and in using the proper HGVS nomenclature. In addition, the FLCN map (Figure 4) indicates the position of the 53 unique FLCN mutations in the online database.
Figure 4.

Map of FLCN amino acid and nucleotide sequences (NM_144997.5) including the intron-exon boundaries (shown in blue). The position of the 53 unique FLCN mutations is shown in red. The number in red corresponds to the variant ID (FLCN_00001 to FLCN_00053) in the FLCN mutation database. Splice site mutations are marked by green boxes and the affected splice site is marked by a heavy red bar. The FLCN coding sequence and its deduced amino acid sequence are obtained using NCBI ORF Finder (Open Reading Frame Finder).
To submit the variant online, a new submitter will first be asked to register so that contact information is available for reference purposes and clarification of submitted details. After registering, he or she can submit the variant online. Then, researchers need to provide essential necessary information for mutation databases as described by the Human Genome Organization Mutation Database Initiative including an exact molecular description of the variant (DNA-level), and details about the source of the material and detection method used [Claustres et al., 2002; Bayley et al., 2008]. New submitted variants are given a unique identifier. After the curator's approval, the new variant is added to the database and all connected web pages are instantly updated.
Discussion
To date, the FCLN database includes 84 variants: 53 unique germline mutations and 31 SNPs. Of 53 unique germline mutations, 36 were reported by NCI groups, and 17 were reported by other investigators worldwide (Figure 1). The frequency of FLCN mutations types consists of 45% (24/53) deletion, 32% (17/53) substitutions, 15% (8/53) duplications, 6% (3/53) insertion/deletions, and 2% (1/53) insertions. One of the 3 insertion/deletions causes a nonsense change. Among 17 mutations that were substitutions, 10 affect putative splice sites, 5 cause nonsense changes, and 2 were missense mutations. Although uncommon, germline FLCN missense mutations may occur in BHDS. The missense mutation (Lys508Arg) was confirmed by two independent laboratories and it was not present in DNA from 160 unrelated control individuals [Toro et al., 2008]. ClustalW multiple protein sequence alignments showed that amino acid Lys 508 of folliculin is highly conserved in chimpanzee, horse, mouse, rat, dog, cow, chicken, zebrafish and sea urchin. In addition, a block of continuous high conservation was shown between amino acids Leu507 to Val515. The conservation across species suggests an important biological role for Lys508 amino acid in folliculin. The second FLCN missense mutation (c.715C>T; p.Arg239Cys) was reported in a woman with BHDS but no family history of RCC (Woodward et al., 2008). The Arg239 residue is also conserved in FLCN homologs. The substitution c.715C>T in exon 7 of FLCN was not observed in 280 control chromosomes and mutation prediction programs indicated a strong likelihood that it represents a mutation (Woodward et al., 2008). The presence of missense mutation in exon 7 of FLCN is not surprising since a missense mutation in exon 7 (His255Arg) in the canine FLCN ortholog is the disease-associated germline mutation in hereditary multifocal RCND in German shepherd dogs [Lingaas et al., 2003]. The His255Arg mutation confers an amino acid change in a highly conserved region of canine folliculin.
No mutation has yet been identified in exons 8 and 10, and no large deletions have been reported but this probably reflects limited effort in this direction to date. Exon 12 was the most frequent site of mutations including 3 deletions, 2 nonsense, 2 duplications and 1 insertion. The hot spot mutation (c.1285dupC/1285delC) was the most common FLCN mutation reported to date [Schmidt et al., 2005; Toro et al., 2008]. The most 5′ FLCN mutation reported occurred at nucleotide 3 (c. 3delG) in exon 4 affecting the initiator codon of FLCN [Bessis et al., 2006]. The most 3′ mutation reported occurred at nucleotide 1579 (c.1579 C>T) in exon 14 and was predicted to produce a premature termination codon at position 527 [Schmidt et al., 2005]. There are previous reports of families with FLCN mutations and a prominent and almost exclusive history of spontaneous pneumothorax inherited in an autosomal dominant fashion [Painter et al., 2005; Graham et al., 2005].
The database contains 30 FLCN SNPs that were recently reported [Cho et al., 2008]. Among these 30 SNPs, three were coding variants (c.1269 G>A; p.His423His), (c.1278 G>A; p.IIe426IIe), (c.1333 C>T; p.Ala445Thr) and two of these three were synonymous (p.His423His and p.IIe426IIe). The non-synonymous variant (p.Ala445Thr) was not predicted to have significant functional consequences based on PolyPhen analysis [Sunyaev et al., 2001] and it was located in a region that is poorly conserved. Of the remaining SNPs, 4 were in the 5′ untranslated region (UTR) and 23 were in the intronic region. Except for the two synonymous variants, all of the reported FLCN SNPs reported by Cho and co-workers, including those in the 5′UTR, mapped to regions of low conservation [Cho et al., 2008].
Conclusion
We developed a simple online database of FLCN gene variants. It provides a complete and updated summary of all reported FLCN mutations and SNPs. Since the FLCN gene was recently identified, we presume that only a fraction of all FLCN mutations associated with BHDS have been identified. To date, there is no evidence of genotype-phenotype correlation associated with specific mutation, type of mutation, or location in the gene [Toro et al. 2008]. This database will assist with increasing knowledge of FLCN mutations and with developing insight into phenotypes and FLCN protein function. Importantly, every FLCN mutation found in a patient increases confidence in the pathogenic role of that variant, improving the accuracy of genetic testing. We hope that the FLCN mutation database, which strives to systematically unify all current genetic knowledge of FLCN variants, will increase the confidence of clinical geneticists and physicians, will provide a convenient resource for research scientists, and may eventually assist in gaining novel insights into FLCN and BHDS.
We encourage researches to submit newly identified variants to the database. Currently all database content is derived from published, peer-reviewed literature. We anticipate that in the future, a large number of variants will be submitted directly to the database. The FLCN mutation database (http://www.skingenedatabase.com) is freely accessible and all researchers may submit new sequence variants online.
Acknowledgments
We would like to thank the Myrovlytis Trust, the BHDS patients, their families, and physicians and members of the American Academy of Dermatology. The content of this article was presented at at the Inagural Birt-Hogg-Dube Symposium, Roskilde, Denmark, September 3, 2008 supported by a travel grant from the Myrovlytis Trust. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government. This research was supported in part by the Intramural Research Program of the NCI and private donation of Dr. Jorge Toro. P.B. is an HHMI-NIH Research Scholar.
Contract grant sponsor: This article is a US Government work, and, as such, is in the public domain in the United States of America.
Footnotes
Communicated by Garry R. Cutting
References
- Baba M, Hong SB, Sharma N, Warren MB, Nickerson ML, Iwamatsu A, Esposito D, Gillette WK, Hopkins RF, 3rd, Hartley JL, Furihata M, Oishi S, Zhen W, Burke TR, Jr, Linehan WM, Schmidt LS, Zbar B. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci USA. 2006;103:15552–15557. doi: 10.1073/pnas.0603781103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayley JP, Launonen V, Tomlinson IPM. The FH mutation database: an online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med Genet. 2008;9:1–9. doi: 10.1186/1471-2350-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessis D, Giraud S, Richard S. A novel familial germline mutation in the initiator codon of the BHD gene in a patient with Birt-Hogg-Dubé syndrome. Br J Dermatol. 2006;155:1067–1069. doi: 10.1111/j.1365-2133.2006.07449.x. [DOI] [PubMed] [Google Scholar]
- Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113:1674–1677. [PubMed] [Google Scholar]
- Bønsdorff TB, Jansen JH, Lingaas F. Second hits in the FLCN gene in a hereditary renal cancer syndrome in dogs. Mamm Genome. 2008;19:121–6. doi: 10.1007/s00335-007-9088-3. [DOI] [PubMed] [Google Scholar]
- Claustres M, Horaitis O, Vanevski M, Cotton RGH. Time for a unified system of mutation description and reporting: a review of locus-specific mutation databases. Genome Research. 2002;12:680–688. doi: 10.1101/gr.217702. [DOI] [PubMed] [Google Scholar]
- Cho MH, Klanderman BJ, Litonjua AA, Sparrow D, Silverman EK, Raby BA. Folliculin mutations are not associated with severe COPD. BMC Med Gene. 2008;9:120. doi: 10.1186/1471-2350-9-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva NF, Gentle D, Hesson LB, Morton DG, Latif F, Maher ER. Analysis of the Birt-Hogg-Dubé (BHD) tumour suppressor gene in sporadic renal cell carcinoma and colorectal cancer. J Med Genet. 2003;40:820–4. doi: 10.1136/jmg.40.11.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond C, Grigoris I, Dutta B. Birt-Hogg-Dubé syndrome and multinodular goitre. Australas J Dermatol. 2002;43:301–304. doi: 10.1046/j.1440-0960.2002.00618.x. [DOI] [PubMed] [Google Scholar]
- Fuertes I, Mascaró-Galy JM, Ferrando J. t-Hogg-Dubé Syndrome in a Patient with Cutaneous Symptoms and a c.1429 C > T;p.R477X Mutation in Exon 12 of the Folliculin Gene. ActasDermosifiliogr. 2009;100:227–30. [PubMed] [Google Scholar]
- Fokkema IFAC, den Dunnen JT, Taschner PEM. LOVD: Easy creation of a locus-specific sequence variation database using an “LSDB-in-a-Box” approach. Human Mutation. 2005;26:63–68. doi: 10.1002/humu.20201. [DOI] [PubMed] [Google Scholar]
- Godbolt AM, Robertson IM, Weedon D. Birt-Hogg-Dubé syndrome. Australas J Dermatol. 2003;44:52–56. doi: 10.1046/j.1440-0960.2003.00638.x. [DOI] [PubMed] [Google Scholar]
- Graham RB, Nolasco M, Peterlin B, Garcia CK. Nonsense mutations in folliculin presenting as isolated familial spontaneous pneumothorax in adults. Am J Respir Crit Care Med. 2005;172:39–44. doi: 10.1164/rccm.200501-143OC. [DOI] [PubMed] [Google Scholar]
- Gunji Y, Akiyoshi T, Sato T, Kurihara M, Tominaga S, Takahashi K, Seyama K. Mutations of the Birt Hogg Dube gene in patients with multiple lung cysts and recurrent pneumothorax. J Med Genet. 2007;44:588–93. doi: 10.1136/jmg.2007.049874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman TR, Nicolas E, Klein-Szanto A, Al-Saleem T, Cash TP, Simon MC, Henske EP. The role of the Birt-Hogg-Dubé protein in mTOR activation and renal tumorigenesis. Oncogene. 2009 doi: 10.1038/onc.2009.14. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hino O, Okimoto K, Kouchi M, Sakurai J. A novel renal carcinoma predisposing gene of the Nihon rat maps on chromosome 10. Jpn J Cancer Res. 2001;92:1147–1149. doi: 10.1111/j.1349-7006.2001.tb02133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonasdottir TJ, Mellersh CS, Moe L, Heggebo R, Gamlem H, Ostrander EA, Lingaas F. Genetic mapping of a naturally occurring hereditary renal cancer syndrome in dogs. Proc Natl Acad Sci USA. 2000;97:4132–4137. doi: 10.1073/pnas.070053397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H, Sawamura D, Nakazawa H, Hattori N, Goto M, Sato-Matsumura KC, Akiyama M, Shimizu H. Detection of 1733insC mutations in an Asian family with Birt-Hogg-Dubé syndrome. Br J Dermatol. 2005;152:142–145. doi: 10.1111/j.1365-2133.2004.06283.x. [DOI] [PubMed] [Google Scholar]
- Khoo SK, Giraud S, Kahnoski K, Chen J, Motorna O, Nickolov R, Binet O, Lambert D, Friedel J, Levy R, Ferlicot S, Wolkenstein P, Hammel P, Bergerheim U, Hedblad MA, Bradley M, Teh BT, Nordenskjold M, Richard S. Clinical and genetic studies of Birt-Hogg-Dubé syndrome. J Med Genet. 2002;39:906–912. doi: 10.1136/jmg.39.12.906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EH, Jeong SY, Kim HJ, Kim YC. A case of Birt-Hogg-Dubé syndrome. J Korean Med Sci. 2008;23:332–5. doi: 10.3346/jkms.2008.23.2.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberti C, Schweiger N, Hartschuh W, Schulz T, Becker-Wegerich P, Küster W, Rütten A, Sauerbruch T, Ruzicka T, Kruse R. Birt-Hogg-Dubé syndrome: germline mutation in the (C)8 mononucleotide tract of the BHD gene in a German patient. Acta Derm Venereol. 2005;85:172–173. [PubMed] [Google Scholar]
- Leter EM, Koopmans AK, Gille JJ, van Os TA, Vittoz GG, David EF, Jaspars EH, Postmus PE, van Moorselaar RJ, Craanen ME, Starink TM, Menko FH. Birt-Hogg-Dubé syndrome: clinical and genetic studies of 20 families. J Invest Dermatol. 2008;128:45–49. doi: 10.1038/sj.jid.5700959. [DOI] [PubMed] [Google Scholar]
- Lingaas F, Comstock KE, Kirkness EF, Sorensen A, Aarskaug T, Hitte C, Nickerson ML, Moe L, Schmidt LS, Thomas R, Breen M, Galibert F, Zbar B, Ostrander EA. A mutation in the canine BHD gene is associated with hereditary multifocal renal cystadenocarcinoma and nodular dermatofibrosis in the German shepherd dog. Hum Mol Genet. 2003;12:3043–3053. doi: 10.1093/hmg/ddg336. [DOI] [PubMed] [Google Scholar]
- Lium B, Moe L. Hereditary multifocal renal cystadenocarcinomas and nodular dermatofibrosis in the German shepherd dog: macroscopic and histopathologic changes. Vet Pathol. 1985;22:447–455. doi: 10.1177/030098588502200503. [DOI] [PubMed] [Google Scholar]
- Murakami T, Sano F, Huang Y, Komiya A, Baba M, Osada Y, Nagashima Y, Kondo K, Nakaigawa N, Miura T, Kubota Y, Yao M, Kishida T. Identification and characterization of Birt-Hogg-Dubé associated renal carcinoma. J Pathol. 2007;211:524–531. doi: 10.1002/path.2139. [DOI] [PubMed] [Google Scholar]
- Nadershahi NA, Wescott WB, Egbert B. Birt-Hogg-Dubé syndrome: a review and presentation of the first case with oral lesions. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;83:496–500. doi: 10.1016/s1079-2104(97)90152-9. [DOI] [PubMed] [Google Scholar]
- Nickerson ML, Warren MB, Toro JR, Matrosova V, Glenn G, Turner ML, Duray P, Merino M, Choyke P, Pavlovich CP, Sharma N, Walther M, Munroe D, Hill R, Maher E, Greenberg C, Lerman MI, Linehan WM, Zbar B, Schmidt LS. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2002;2:157–164. doi: 10.1016/s1535-6108(02)00104-6. [DOI] [PubMed] [Google Scholar]
- Okimoto K, Kouchi M, Kikawa E, Toyosawa K, Koujitani T, Tanaka K, Matsuoka N, Sakurai J, Hino O. A novel “Nihon” rat model of a Mendelian dominantly inherited renal cell carcinoma. Jpn J Cancer Res. 2000;91:1096–1099. doi: 10.1111/j.1349-7006.2000.tb00890.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okimoto K, Sakurai J, Kobayashi T, Mitani H, Hirayama Y, Nickerson ML, Warren MB, Zbar B, Schmidt LS, Hino O. A germ-line insertion in the Birt-Hogg-Dubé (BHD) gene gives rise to the Nihon rat model of inherited renal cancer. Proc Natl Acad Sci USA. 2004;101:2023–2027. doi: 10.1073/pnas.0308071100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmirotta R, Donati P, Savonarola A, Cota C, Ferroni P, Guadagni F. Birt-Hogg-Dubé (BHD) syndrome: report of two novel germline mutations in the folliculin (FLCN) gene. Eur J Dermatol. 2008;18:382–6. doi: 10.1684/ejd.2008.0431. [DOI] [PubMed] [Google Scholar]
- Painter JN, Tapanainen H, Somer M, Tukiainen P, Aittomaki K. A 4-bp deletion in the Birt-Hogg-Dube gene (FLCN) causes dominantly inherited spontaneous pneumothorax. Am J Hum Genet. 2005;76:522–527. doi: 10.1086/428455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlovich CP, Grubb RL, 3rd, Hurley K, Glenn GM, Toro J, Schmidt LS, Torres-Cabala C, Merino MJ, Zbar B, Choyke P, Walther MM, Linehan WM. Evaluation and management of renal tumors in the Birt-Hogg-Dubé syndrome. J Urol. 2005;173:1482–1486. doi: 10.1097/01.ju.0000154629.45832.30. [DOI] [PubMed] [Google Scholar]
- Schmidt LS, Nickerson ML, Warren MB, Glenn GM, Toro JR, Merino MJ, Turner ML, Choyke PL, Sharma N, Peterson J, Morrison P, Maher ER, Walther MM, Zbar B, Linehan WM. Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dube syndrome. Am J Hum Genet. 2005;76:1023–1033. doi: 10.1086/430842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt LS, Warren MB, Nickerson ML, Weirich G, Matrosova V, Toro JR, Turner ML, Duray P, Merino M, Hewitt S, Pavlovich CP, Glenn G, Greenberg CR, Linehan WM, Zbar B. Birt-Hogg-Dubé syndrome, a genodermatosis associated with spontaneous pneumothorax and kidney neoplasia, maps to chromosome 17p11.2. Am J Hum Genet. 2001;69:876–882. doi: 10.1086/323744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SR, Zhen W, Zheng Z, Wang H, Oh SW, Liu W, Zbar B, Schmidt LS, Hou SX. The Drosophila homolog of the human tumor suppressor gene BHD interacts with the JAK-STAT and Dpp signaling pathways in regulating male germline stem cell maintenance. Oncogene. 2006;25:5933–5941. doi: 10.1038/sj.onc.1209593. [DOI] [PubMed] [Google Scholar]
- Sunyaev S, Ramensky V, Koch I, Lathe W, 3rd, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Hum Mol Genet. 2001;10:591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- Toro JR, Glenn G, Duray P, Darling T, Weirich G, Zbar B, Linehan WM, Turner ML. Birt-Hogg-Dubé syndrome: a novel marker of kidney neoplasia. Arch Dermatol. 1999;135:1195–1202. doi: 10.1001/archderm.135.10.1195. [DOI] [PubMed] [Google Scholar]
- Toro JR, Wei MH, Glenn GM, Weinreich M, Toure O, Vocke C, Turner M, Choyke P, Merino MJ, Pinto PA, Steinberg SM, Schmidt LS, Linehan WM. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet. 2008;45:321–331. doi: 10.1136/jmg.2007.054304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Slegtenhorst M, Khabibullin D, Hartman TR, Nicolas E, Kruger WD, Henske EP. The Birt-Hogg-Dube and tuberous sclerosis complex homologs have opposing roles in amino acid homeostasis in Schizosaccharomyces pombe. J Biol Chem. 2007;282:24583–24590. doi: 10.1074/jbc.M700857200. [DOI] [PubMed] [Google Scholar]
- van Steensel MA, Verstraeten VL, Frank J, Kelleners-Smeets NW, Poblete-Gutiérrez P, Marcus-Soekarman D, Bladergroen RS, Steijlen PM, van Geel M. Novel mutations in the BHD gene and absence of loss of heterozygosity in fibrofolliculomas of Birt-Hogg-Dubé patients. J Invest Dermatol. 2007;127:588–593. doi: 10.1038/sj.jid.5700592. [DOI] [PubMed] [Google Scholar]
- Vincent A, Farley M, Chan E, James WD. Birt-Hogg-Dubé syndrome: two patients with neural tissue tumors. J Am Acad Dermatol. 2003;49:717–719. doi: 10.1067/s0190-9622(03)01583-4. [DOI] [PubMed] [Google Scholar]
- Vocke C, Yang Y, Pavlovich CP, Schmidt LS, Nickerson ML, Torres-Cabala CA, Merino MJ, Walther MM, Zbar B, Linehan WM. High frequency of somatic frameshift BHD gene mutations in Birt-Hogg-Dubé-associated renal tumors. J Natl Cancer Inst. 2005;97:931–935. doi: 10.1093/jnci/dji154. [DOI] [PubMed] [Google Scholar]
- Walter P, Kirchhof B, Korge B, Heimann K. Flecked chorioretinopathy associated with Birt-Hogg-Dubé syndrome. Graefes Arch Clin Exp Ophthalmol. 1997;235:359–361. doi: 10.1007/BF00937284. [DOI] [PubMed] [Google Scholar]
- Warren MB, Torres-Cabala CA, Turner ML, Merino MJ, Matrosova VY, Nickerson ML, Ma W, Linehan WM, Zbar B, Schmidt LS. Expression of Birt-Hogg-Dubé mRNA in normal and neoplastic human tissues. Mod Pathol. 2004;17:998–1011. doi: 10.1038/modpathol.3800152. [DOI] [PubMed] [Google Scholar]
- Welsch MJ, Krunic A, Medenica MM. Birt-Hogg-Dubé Syndrome. Int J Dermatol. 2005;44:668–673. doi: 10.1111/j.1365-4632.2004.02095.x. [DOI] [PubMed] [Google Scholar]
- Wildeman M, van Ophuizen E, den Dunnen JT, Taschner PE. Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum Mutat. 2008;29:6–13. doi: 10.1002/humu.20654. [DOI] [PubMed] [Google Scholar]
- Woodward ER, Ricketts C, Killick P, Gad S, Morris MR, Kavalier F, Hodgson SV, Giraud S, Bressac-de Paillerets B, Chapman C, Escudier B, Latif F, Richard S, Maher ER. Familial non-VHL clear cell (conventional) renal cell carcinoma: clinical features, segregation analysis, and mutation analysis of FLCN. Clin Cancer Res. 2008;14:5925–5930. doi: 10.1158/1078-0432.CCR-08-0608. [DOI] [PubMed] [Google Scholar]
- Zbar B, Alvord WG, Glenn G, Turner M, Pavlovich CP, Schmidt L, Walther M, Choyke P, Weirich G, Hewitt SM, Duray P, Gabril F, Greenberg C, Merino MJ, Toro J, Linehan WM. Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dubé syndrome. Cancer Epidemiol Biomarkers Prev. 2002;11:393–400. [PubMed] [Google Scholar]