

Abstract
The multifunctional nucleolar proteins, nucleophosmin (NPM) and the tumor suppressor ARF, have been assigned numerous roles in diverse cellular processes impacting cellular proliferation, tumorigenesis and apoptosis. In addition, both proteins have been linked to the oncogenic function of c-Myc, a transcription factor that drives the majority of human cancers. Both proteins are induced by oncogenic c-Myc, but have opposing outcomes. Whereas loss of ARF accelerates c-Mycinduced tumorigenesis, NPM overexpression enhances c-Myc transformation. Accordingly, ARF expression is lost in many tumors, while NPM expression is elevated. Previously, we demonstrated that ARF interacts directly with c-Myc, leading to inhibition of its transforming activity while enhancing its apoptotic activity, independently of p53. We have recently shown that NPM also binds directly to c-Myc, but with opposite effects compared to ARF. NPM dramatically enhances the oncogenic activity of c-Myc, independently of ARF and p53. In tumor cells, the ARF-p53 pathway is often inactivated while NPM is elevated. However, when NPM and ARF are both expressed with oncogenic c-Myc the outcome of the interactions becomes more complex, since NPM and ARF also interact directly and NPM controls ARF localization. In this report we demonstrate that in the presence of ARF, NPM overexpression dramatically inhibits c-Myc-induced p53-independent apoptosis, while enhancing proliferation and transformation. We find that NPM sequesters ARF in nucleoli, blocking the relocalization of ARF to the nucleoplasm caused by activation of c-Myc. Therefore, the fate of a cell to undergo apoptosis or become transformed is dependent on this complex interacting network of oncogenic and tumor suppressor proteins.
Key words: c-Myc, NPM/B23, ARF, cell proliferation, cell transformation, apoptosis, cancer
The transcription factor c-Myc has an essential role in the control of cellular proliferation, transformation and apoptosis and the disruption of this regulation has been shown to lead to tumorigenesis. Deregulated c-Myc expression has been demonstrated in many types of human cancer.1 The ability of c-Myc to drive proliferation and transformation is thought to be mediated by numerous target genes involved in a multitude of cellular processes, including cell cycle regulation and metabolism. The most well characterized target genes are regulated by c-Myc through a canonical E box sequence (CACGTG), but c-Myc is also known to induce and repress many genes by several less characterized noncanonical mechanisms.1 In addition to its well-known function as a crucial regulator of cellular proliferation in normal and neoplastic cells, c-Myc can also either induce or sensitize cells to apoptosis upon removal of growth factors. Following growth factor withdrawal, c-Myc expression is downregulated and normal cells exit the cell cycle, whereas enforced c-Myc expression under these conditions induces apoptosis.2 c-Myc-triggered apoptosis provides an intrinsic failsafe program to limit unchecked cell cycle progression under inappropriate conditions. A major mechanism mediating this response is the induction of the tumor suppressor ARF by oncogenic signals, resulting in apoptosis via the ARF-Mdm2-p53 axis that controls the levels of p53 protein. Deregulated c-Myc leads to increased ARF expression, which binds to Mdm2 and inhibits its activity. Mdm2, a p53 E3 ubiquitin ligase, degrades p53 protein and thus ARF induction results in increased stability of p53, leading to apoptosis.3 Consequently, tumors from cells overexpressing c-Myc often have mutations that disable the ARF-Mdm2-p53 axis.2
c-Myc-induced apoptosis can also be p53-independent, determined by the cell type and apoptotic stimuli. We found that c-Myc can induce apoptosis independently of p53 in mouse embryo fibroblasts (MEFs) with high ARF, but cannot do so efficiently in cells lacking ARF.4 Upon c-Myc activation, endogenous ARF relocalizes to the nucleoplasm, directly binds to c-Myc, and inhibits the canonical transcriptional activity of c-Myc.4,5 In addition, endogenous or exogenous ARF inhibits c-Myc-induced hyperproliferation and transformation, while enhancing c-Myc-induced apoptosis.4 We have also shown that ARF is recruited with c-Myc to several canonical target gene promoters.4 In addition, ARF does not appear to affect repression of c-Myc target genes,4 but may actually enhance a noncanonical transcriptional activity of c-Myc mediating p53-independent apoptosis. In contrast, another study found c-Myc localized in nucleoli with ARF in transient assays and thus hypothesized that ARF inactivates c-Myc by sequestering it to nucleoli.6 We have also shown that high transient ARF over-expression can lead to c-Myc nucleolar localization, depending on the ratio of exogenous ARF to c-Myc proteins.5 However, we have not observed this phenomenon in cells with high endogenous ARF (p53−/− MEFs) or in cells stably expressing exogenous ARF. Therefore, our model is that c-Myc activation causes a relocalization of nucleolar ARF to the nucleoplasm where ARF controls the transcriptional activity and target gene expression of c-Myc.
Our findings suggest that ARF is sequestered in nucleoli until oncogenic activation. This would explain how p53−/− MEFs with high levels of nucleolar ARF proliferate at a similar rate compared to p53−/−/ARF−/− double knockout (DKO) MEFs, but when c-Myc is activated proliferation is inhibited and apoptosis is induced. These results are relevant to an ongoing controversy concerning the function of nucleolar ARF. The “active nucleolar ARF” hypothesis contends that high nucleolar ARF is functional in growth inhibition and is supported by data showing that nucleolar ARF inhibits proliferation of p53−/−/ARF−/−/Mdm2−/− triple knockout (TKO) MEFs,7 inhibits rRNA processing,8 and inhibits nucleophosmin,9,10 Mdm2,11,12 and E2F.13 The opposing “active nucleoplasmic ARF” hypothesis is supported by our data showing ARF relocalization to the nucleoplasm to inhibit c-Myc,4 other data showing that nucleoplasmic ARF inhibits growth and stabilizes p53 without relocalization of Mdm2 to nucleoli,14–16 and that ARF is relocalized to the nucleoplasm upon UV radiation/DNA damage-driven apoptosis mediated by interaction with c-Jun and controlled by JNK phosphorylation of c-Jun.17,18 As suggested by our results, one explanation for these opposing hypotheses is that high transiently expressed exogenous ARF can cause a relocalization of its binding partners, such as c-Myc, to nucleoli, perhaps causing deleterious effects on cell growth that are not normally observed with high endogenous ARF in stable cell lines. Alternatively, ARF may play an active role in both the nucleoli and nucleoplasm under specific circumstances, such as senescence when endogenous ARF is dramatically elevated, but the evidence suggests that cells can sustain high levels of endogenous ARF until oncogene activation or DNA damage causes relocalization to the nucleoplasm, resulting in apoptosis or growth inhibition.
Nucleophosmin (NPM) is another major nucleolar protein that has roles in proliferation, tumorigenesis and apoptosis. Unlike ARF, NPM, also called B23, NO38 or numatrin, is abundant and ubiquitous. It was originally found to be involved in ribosomal biogenesis, but has roles in pre-ribosomal RNA processing, ribosome assembly and transport, DNA duplication, nucleolus-nucleoplasm-cytoplasm protein trafficking as a molecular chaperone, transcriptional regulation and centrosome duplication.19 NPM has a clear role in proliferation and has been described as a proto-oncogene. NPM levels are tightly regulated during proliferation and differentiation. Increased levels of NPM are found in highly proliferating cells compared to normal resting cells. NPM is frequently deregulated and overexpressed in several different types of cancer and it has been proposed as a marker for gastric, colon, ovarian and prostate carcinomas. In fact, in some cancers, the levels of NPM expression correlate with the stage of tumor progression. NPM is also involved in chromosomal translocations in hematological tumors, where it forms chimeric proteins with different partners such as ALK (anaplastic lymphoma kinase), RARα (retinoic acid receptor-α) and MLF1 (myelodysplasia/myeloid leukemia factor 1).19
The main biological effects of NPM overexpression or deregulation are increased cellular proliferation and cell growth, and the inhibition of differentiation and apoptosis.19 Overexpressed NPM has been shown to enhance myc/ras cotransformation in MEFs, while inhibiting oncogene-induced apoptosis and senescence.20 The mechanism that mediates the role of NPM in proliferation and tumorigenesis is unclear. Some have proposed that the enhancement of ribosomal biogenesis by NPM or its interaction with DNA polymerase-α mediates the increased proliferation and transformation.19 Recently, we have demonstrated that endogenous and exogenous NPM directly interact with c-Myc and regulate c-Myc target gene expression at the promoter. NPM expression was found to be necessary for efficient induction of canonical c-Myc target genes, while NPM overexpression enhanced the induction of c-Myc target genes.21 We also showed that overexpressed NPM dramatically stimulates c-Myc-induced hyperproliferation and transformation in p53−/−/ARF−/− DKO MEFs, while endogenous NPM is essential for these oncogenic activities.21 Although NPM is mostly nucleolar, we did observe colocalization of NPM and c-Myc in the nucleoplasm of a subset of cells,21 suggesting that the mechanisms controlling NPM shuttling are critical for c-Myc function. Interestingly, the dramatic effects of NPM on c-Myc were not due to a significant increase in total levels of NPM. This suggests that the deregulation of NPM, due to enforced expression, causes disruption of the temporal regulation of nucleoplasmic NPM, allowing a sustained interaction with c-Myc.
Considering that both ARF and p53 have been shown to bind to NPM,19 we performed our experiments described above in MEFs lacking endogenous ARF and p53 to eliminate the possibility that the observed biological effects were due to NPM interactions with p53 or ARF instead of with c-Myc alone.21 In addition, these conditions would reflect the environment in many tumors that have increased c-Myc and NPM with inactivation of the ARF-Mdm2-p53 axis. Now, to examine the effects of NPM on c-Myc-induced apoptosis, we used p53−/− MEFs. These cells have a high level of endogenous ARF, which allows efficient c-Myc-induced p53-independent apoptosis.4 We compared MEFs expressing the chimeric c-MycER protein with and without overexpressed NPM. Expression of the c-MycER and NPM proteins was confirmed by immunoblot analyses (data not shown). As previously observed,4 activation of c-MycER in media containing low serum induced apoptosis in the p53−/− MEFs (Fig. 1A). Apoptosis was confirmed by the appearance of activated caspase-3 (data not shown). In contrast, the induction of apoptosis by c-MycER in the p53−/− MEFs was dramatically inhibited by overexpression of NPM (Fig. 1A). Since NPM controls ARF nucleolar localization, we examined the effects of NPM overexpression on ARF localization upon c-MycER activation. Figure 1B shows that c-MycER activation induced the translocalization of ARF to the nucleoplasm, as previously observed.5 In contrast, when NPM is overexpressed, ARF remains in the nucleoli upon c-MycER activation (Fig. 1C). This outcome is similar to our previous observation that c-Myc fails to induce apoptosis efficiently in cells lacking ARF and p53,4 supporting the idea that ARF is necessary for efficient p53-independent c-Myc-induced apoptosis in fibroblasts. Taken together, these results suggest that NPM is a negative regulator of c-Myc-induced apoptosis in fibroblasts, which is also in agreement with several studies showing that NPM inhibits apoptosis induced by a number of factors, including c-Myc, hypoxia and UV irradiation.19
Figure 1.
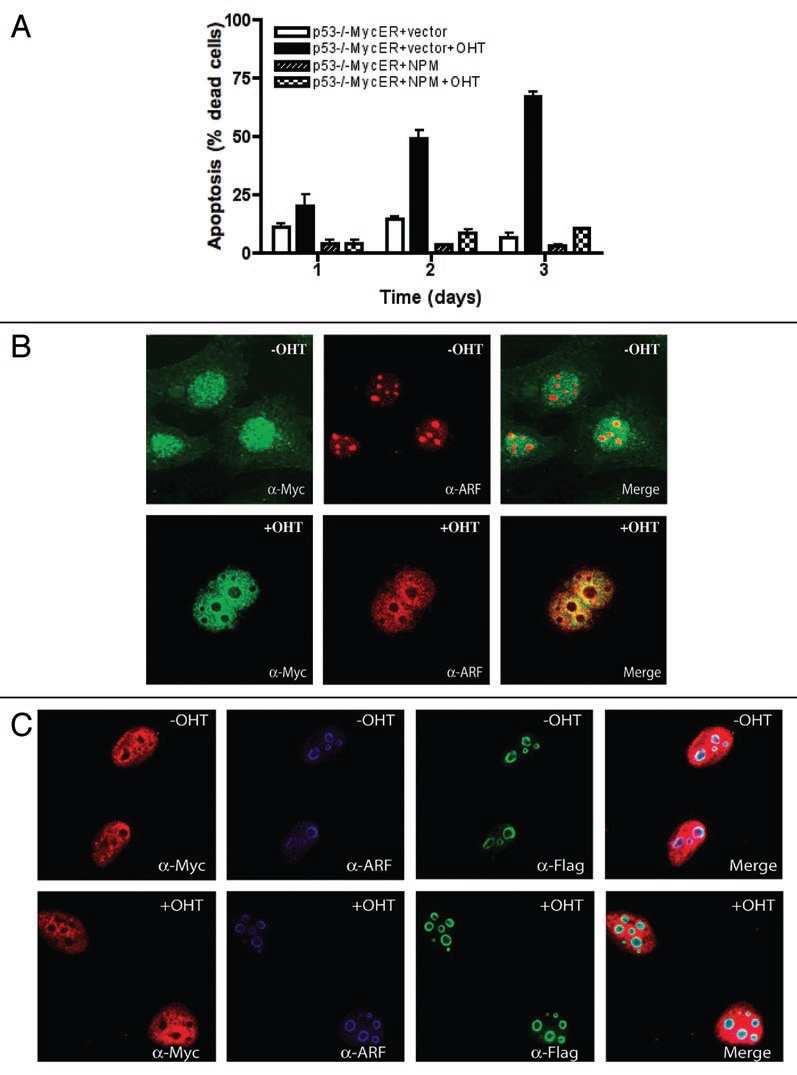
NPM inhibits p53-independent c-Myc-induced apoptosis. (A) p53−/− MycER MEFs with or without exogenous NPM and with or without c-MycER activation (+/− OHT) were assayed for apoptotic cell death as described previously.4 (B) Activated c-Myc induces the translocation of ARF to the nucleoplasm. p53−/− MycER MEFs with or without activation of c-MycER (+/− OHT) immunostained for c-Myc and ARF. (C) NPM blocks c-Myc translocation of ARF. p53−/− MycER MEFs expressing exogenous NPM with or without activation of c-MycER (+/− OHT) immunostained for c-Myc, ARF and NPM-Flag.
Since the overexpression of NPM inhibited the ability of c-Myc to induce apoptosis in p53−/− MEFs in low serum (Fig. 1A) and enhanced the ability of c-Myc to induce hyperproliferation in DKO MEFs,21 we next examined the effects of NPM on c-Myc-induced hyperproliferation in p53−/− MEFs that have high levels of ARF. As shown in Figure 2A, there was no hyperproliferation when c-Myc was activated, even in media with high serum, suggesting inhibition of c-Myc proliferative activity by ARF as previously observed.4 In contrast, overexpression of NPM caused a substantial enhancement of c-Myc-induced hyperproliferation. This may be explained by the observation that NPM sequesters ARF in the nucleoli and blocks ARF's regulatory function (Fig. 1C). The enhancement of c-Myc-induced hyperproliferation by NPM was not as dramatic as observed in DKO MEFs lacking ARF.21 We next examined whether NPM also enhances anchorage-independent proliferation of p53−/− MEFs. Figure 2B shows that overexpression of NPM enhances c-Myc-induced anchorage-independent growth in p53−/− MEFs by approximately two fold, albeit less efficiently than in DKO MEFs.21 Since NPM sequesters ARF in nucleoli, the muted enhancement of c-Myc proliferative activity by NPM may be due to less nucleoplasmic NPM available to interact with c-Myc. Taken together, our results suggest that overexpression or deregulation of NPM, which is often observed in tumors, can counter the anti-proliferative, pro-apoptotic effects of ARF on c-Myc activity.
Figure 2.

NPM stimulates c-Myc oncogenic activity in the presence of ARF. (A) NPM stimulates c-Myc-induced hyperproliferation that is suppressed by ARF. Proliferation assay of p53−/− MycER MEFs with or without NPM-Flag and with or without c-MycER activation (+/− OHT) was performed as described previously.21 (B) NPM stimulation of c-Myc-induced anchorage-independent growth. p53−/− MycER MEFs with or without exogenous NPM and with or without c-MycER activation (+/− OHT) were plated in soft agar and analyzed for colony growth after 4 weeks.
Since NPM controls ARF localization, ARF is likely held in a complex with NPM in the nucleolus of cells without overexpressed NPM or c-Myc. It is unknown how activation of c-Myc causes the relocalization of ARF to the nucleoplasm. It seems unlikely that the nucleoplasmic c-Myc could directly compete for ARF binding with the abundant, nucleolar NPM, but it may compete with a specific pool of NPM or act when NPM levels are suppressed. Alternatively, c-Myc activation may indirectly cause an alteration of NPM, perhaps a post-translational modification that “releases” ARF from NPM. ARF itself has been proposed to stimulate sumoylation of NPM that is critical for NPM localization and the interaction of NPM with Rb.22 In addition, ARF-stimulated sumoylation protects NPM from apoptotic caspase-3-mediated degradation.22 Alternatively, ARF has been suggested to have a negative effect on NPM, by stimulating NPM proteasomal degradation by 3–4 fold9 and impeding NPM shuttling and sequestering NPM in the nucleolus.23 Although ARF is more stable in the nucleolus, most likely due to the absence of proteasomes in nucleoli,24 our data would suggest that NPM sequesters ARF in the nucleolus, but that it primarily regulates c-Myc function in the nucloplasm. Considering that NPM is an abundant protein and only a minor fraction of NPM binds to ARF, while a significant proportion of ARF binds to NPM, it is unclear how ARF can dominate the function of NPM, unless there is a transiently large amount of ARF when the levels of NPM are low.
The majority of studies suggest a role for NPM in proliferation and inhibition of apoptosis. Proliferation is severely impaired and apoptosis is induced in NPM-deficient cells,25 and downregulation of NPM mRNA delays cell cycle progression and entry of cells into mitosis.26 In contrast to our findings that c-Myc-induced hyper-proliferation and transformation are impaired in both NPM−/−/p53−/− cells and in cells where NPM expression has been knocked down by siRNA,21 Colombo et al.27 showed that c-Myc-induced transformation was actually enhanced in NPM−/−/p53−/− cells. The different outcomes induced by c-Myc are not easily resolved, but a major difference between the two studies is that Colombo et al. used a constituitive c-Myc expression vector while we used the inducible c-MycER. Recently, Bonetti et al.28 showed that loss of NPM or expression of a NPM cytoplasmic mutant associated with acute myelogenous leukemia (AML) results in c-Myc protein stabilization through direct effects on the E3 ubiqutin ligase SCF (Fbw7γ) supporting the hypothesis that loss of NPM enhanced c-Myc-induced transformation. However, in contrast, we did not observe any significant differences in the proteolysis of c-Myc protein in the absence of NPM or when NPM was overexpressed.21 One possible explanation for enhanced proliferative activity of the cytoplasmic NPM mutants is that they cause a delocalization of ARF to the cytoplasm.29 This would result in a sequestered ARF, resulting in enhanced c-Myc transformation. This again suggests that NPM inhibits ARF function.
In apparent contrast with our results is the finding that NPM+/− mice show accelerated Myc-induced lymphomagenesis.25 However, loss of NPM does cause genomic instability and aneuploidy is observed in NPM+/− mice, leading to increased susceptibility to c-Myc-induced lymphomagenesis.25 Thus, haploinsufficiency may disrupt the function of NPM in maintaining proper centrosome numbers and genetic stability, but still is abundant enough to interact with c-Myc. In support of this idea, the stability of ARF is unaffected in NPM+/− MEFs as compared to NPM−/− MEFs, suggesting that the levels of NPM in NPM+/− MEFs are sufficient to interact with ARF.19
c-Myc has the ability to induce hyperproliferation/transformation; while at the same time it can also induce apoptosis through both p53-dependent and p53-independent pathways. At first glance, this appears to be a contradiction, but oncogene activation would be expected to induce a cellular response to eliminate these potentially harmful cells. Such a tumor surveillence mechanism is manifested by the elevation of ARF expression. As illustrated in Figure 3, oncogenic c-Myc induces the expression of ARF, which binds and inhibits Mdm2, the negative regulator of p53, leading to the activation of p53 and cell cycle arrest or apoptosis. In addition, ARF acts independently of p53 via a negative feedback mechanism in which ARF binds to c-Myc directly to inhibit c-Myc-induced hyperproliferation and transformation and mediates c-Myc-induced apoptosis through an unknown non-canonical mechanism. Oncogenic c-Myc also induces NPM expression, which then binds to c-Myc and stimulates the activity of c-Myc through a positive feedback mechanism. In addition, overexpressed or deregulated NPM sequesters ARF in nucleoli, blocking the ability of ARF to interact with c-Myc and other ARF binding factors, such as Mdm2. Therefore, overexpressed NPM controls c-Myc function by at least two distinct mechanisms-enhancing c-Myc's oncogenic activity by binding directly as a cofactor and sequestering an inhibitor of c-Myc's oncogenic activity. In normal cells with p53, the interactions and outcomes become more complex and unresolved, since NPM has shown to have both positive and negative effects on p53.19 In tumors that have lost p53, but still retain an active ARF, overexpression of NPM, frequently observed in cancer, would represent another oncogenic mechanism to counter the tumor suppressive activity of ARF. Major unanswered questions remain about the molecular mechanisms that control the interactions of these proteins, such as post-translational modifications and localization, the specific c-Myc molecular changes caused by ARF or NPM binding, and the effects of p53 on c-Myc function, with and without ARF and NPM.
Figure 3.

Schematic illustration of the interaction network between c-Myc, NPM, p53 and ARF. Dashed lines represent effects on expression levels and solid lines represent direct protein interactions.
Acknowledgements
Work described in this review was funded through grants RO1 CA109586 and CA125760 from NCI to S.R.H. and P50 CA095103 from NCI. We also thank David Boone and Dr. Christine Eischen for critical review of the manuscript.
Abbreviations
- NPM
nucleophosmin
- ARF
alternative reading frame
- DKO
double knockout
- TKO
triple knockout
- MEF
mouse embryo fibroblasts
- OHT
hydroxytamoxifen
- ER
estrogen receptor
- Mdm2
mouse double minute 2
- YY1
yin yang 1 transcription factor
- IRF-1
interferon regulatory factor 1
- NFκB
nuclear factor of kappa light polypeptide gene enhancer in B-cells
- Gadd45
growth arrest and DNA-damage-inducible protein
- Rb
retinoblastoma
- ALK
anaplastic lymphoma kinase
- RARα
retinoic acid receptor-α
- MLF1
myelodysplasia/myeloid leukemia factor 1
- JNK
c-Jun N-terminal kinase
Footnotes
Previously published online as a Cell Cycle E-publication: http://www.landesbioscience.com/journals/cc/article/9418
References
- 1.Oster SK, Ho CS, Soucie EL, Penn LZ. The myc oncogene: Marvelously complex. Adv Cancer Res. 2002;84:81–154. doi: 10.1016/s0065-230x(02)84004-0. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman B, Liebermann DA. Apoptotic signaling by c-Myc. Oncogene. 2008;27:6462–6472. doi: 10.1038/onc.2008.312. [DOI] [PubMed] [Google Scholar]
- 3.Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, et al. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998;12:2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qi Y, Gregory MA, Li Z, Brousal JP, West K, Hann SR. p19(ARF) directly and differentially controls the functions of c-Myc independently of p53. Nature. 2004;431:712–717. doi: 10.1038/nature02958. [DOI] [PubMed] [Google Scholar]
- 5.Gregory MA, Qi Y, Hann SR. The ARF tumor suppressor: keeping Myc on a leash. Cell Cycle. 2005;4:249–252. [PubMed] [Google Scholar]
- 6.Datta A, Nag A, Pan W, Hay N, Gartel AL, Colamonici O, et al. Myc-ARF (alternate reading frame) interaction inhibits the functions of Myc. J Biol Chem. 2004;279:36698–36707. doi: 10.1074/jbc.M312305200. [DOI] [PubMed] [Google Scholar]
- 7.Weber JD, Jeffers JR, Rehg JE, Randle DH, Lozano G, Roussel MF, et al. p53-independent functions of the p19(ARF) tumor suppressor. Genes Dev. 2000;14:2358–2365. doi: 10.1101/gad.827300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sugimoto M, Kuo ML, Roussel MF, Sherr CJ. Nucleolar Arf tumor suppressor inhibits ribosomal RNA processing. Mol Cell. 2003;11:415–424. doi: 10.1016/s1097-2765(03)00057-1. [DOI] [PubMed] [Google Scholar]
- 9.Itahana K, Bhat KP, Jin A, Itahana Y, Hawke D, Kobayashi R, et al. Tumor suppressor ARF degrades B23, a nucleolar protein involved in ribosome biogenesis and cell proliferation. Mol Cell. 2003;12:1151–1164. doi: 10.1016/s1097-2765(03)00431-3. [DOI] [PubMed] [Google Scholar]
- 10.Bertwistle D, Sugimoto M, Sherr CJ. Physical and functional interactions of the Arf tumor suppressor protein with nucleophosmin/B23. Mol Cell Biol. 2004;24:985–996. doi: 10.1128/MCB.24.3.985-996.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tao W, Levine AJ. p19(ARF) stabilizes p53 by blocking nucleo-cytoplasmic shuttling of Mdm2. Proc Natl Acad Sci USA. 1999;96:6937–6941. doi: 10.1073/pnas.96.12.6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol. 1999;1:20–26. doi: 10.1038/8991. [DOI] [PubMed] [Google Scholar]
- 13.Martelli F, Hamilton T, Silver DP, Sharpless NE, Bardeesy N, Rokas M, et al. p19ARF targets certain E2F species for degradation. Proc Natl Acad Sci USA. 2001;98:4455–4460. doi: 10.1073/pnas.081061398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Llanos S, Clark PA, Rowe J, Peters G. Stabilization of p53 by p14ARF without relocation of MDM2 to the nucleolus. Nat Cell Biol. 2001;3:445–452. doi: 10.1038/35074506. [DOI] [PubMed] [Google Scholar]
- 15.Lin AW, Lowe SW. Oncogenic ras activates the ARF-p53 pathway to suppress epithelial cell transformation. Proc Natl Acad Sci USA. 2001;98:5025–5030. doi: 10.1073/pnas.091100298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korgaonkar C, Zhao L, Modestou M, Quelle DE. ARF function does not require p53 stabilization or Mdm2 relocalization. Mol Cell Biol. 2002;22:196–206. doi: 10.1128/MCB.22.1.196-206.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee C, Smith BA, Bandyopadhyay K, Gjerset RA. DNA damage disrupts the p14ARF-B23(nucleophosmin) interaction and triggers a transient subnuclear redistribution of p14ARF. Cancer Res. 2005;65:9834–9842. doi: 10.1158/0008-5472.CAN-05-1759. [DOI] [PubMed] [Google Scholar]
- 18.Yogev O, Saadon K, Anzi S, Inoue K, Shaulian E. DNA damage-dependent translocation of B23 and p19 ARF is regulated by the Jun N-terminal kinase pathway. Cancer Res. 2008;68:1398–1406. doi: 10.1158/0008-5472.CAN-07-2865. [DOI] [PubMed] [Google Scholar]
- 19.Grisendi S, Mecucci C, Falini B, Pandolfi PP. Nucleophosmin and cancer. Nat Rev Cancer. 2006;6:493–505. doi: 10.1038/nrc1885. [DOI] [PubMed] [Google Scholar]
- 20.Li J, Sejas DP, Burma S, Chen DJ, Pang Q. Nucleophosmin suppresses oncogene-induced apoptosis and senescence and enhances oncogenic cooperation in cells with genomic instability. Carcinogenesis. 2007;28:1163–1170. doi: 10.1093/carcin/bgm025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Z, Boone D, Hann SR. Nucleophosmin interacts directly with c-Myc and controls c-Myc-induced hyperproliferation and transformation. Proc Natl Acad Sci USA. 2008;105:18794–18799. doi: 10.1073/pnas.0806879105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu H, Tan BC, Tseng KH, Chuang CP, Yeh CW, Chen KD, et al. Nucleophosmin acts as a novel AP2alpha-binding transcriptional corepressor during cell differentiation. EMBO Rep. 2007;8:394–400. doi: 10.1038/sj.embor.7400909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brady SN, Yu Y, Maggi LB, Jr, Weber JD. ARF impedes NPM/B23 shuttling in an Mdm2-sensitive tumor suppressor pathway. Mol Cell Biol. 2004;24:9327–9338. doi: 10.1128/MCB.24.21.9327-9338.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lam YW, Lamond AI, Mann M, Andersen JS. Analysis of nucleolar protein dynamics reveals the nuclear degradation of ribosomal proteins. Curr Biol. 2007;17:749–760. doi: 10.1016/j.cub.2007.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grisendi S, Bernardi R, Rossi M, Cheng K, Khandker L, Manova K, et al. Role of nucleophosmin in embryonic development and tumorigenesis. Nature. 2005;437:147–153. doi: 10.1038/nature03915. [DOI] [PubMed] [Google Scholar]
- 26.Jiang PS, Yung BY. Downregulation of nucleophosmin/B23 mRNA delays the entry of cells into mitosis. Biochem Biophys Res Commun. 1999;257:865–870. doi: 10.1006/bbrc.1999.0551. [DOI] [PubMed] [Google Scholar]
- 27.Colombo E, Marine JC, Danovi D, Falini B, Pelicci PG. Nucleophosmin regulates the stability and transcriptional activity of p53. Nat Cell Biol. 2002;4:529–533. doi: 10.1038/ncb814. [DOI] [PubMed] [Google Scholar]
- 28.Bonetti P, Davoli T, Sironi C, Amati B, Pelicci PG, Colombo E. Nucleophosmin and its AML-associated mutant regulate c-Myc turnover through Fbw7gamma. J Cell Biol. 2008;182:19–26. doi: 10.1083/jcb.200711040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254–266. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]