

Abstract
Spondyloepimetaphyseal dysplasia with joint laxity (SEMDJL), leptodactylic (lepto-SEMDJL) or Hall type, is an autosomal-dominant skeletal dysplasia manifesting with short stature, joint laxity with dislocation(s), limb malalignment, and spinal deformity. Its causative gene mutation has not yet been discovered. We captured and sequenced the exomes of eight affected individuals in six unrelated kindreds (three individuals in a family and five simplex individuals). Five novel sequence variants in KIF22, which encodes a member of the kinesin-like protein family, were identified in seven individuals. Sanger sequencing of KIF22 confirmed that c.443C>T (p.Pro148Ser) cosegregated with the phenotype in the affected individuals in the family; c.442C>T (p.Pro148Leu) or c.446G>A (p.Arg149Gln) was present in four of five simplex individuals, but was absent in unaffected individuals in their family and 505 normal cohorts. KIF22 mRNA was detected in human bone, cartilage, joint capsule, ligament, skin, and primary cultured chondrocytes. In silico analysis of KIF22 protein structure indicates that Pro148 and Arg149 are important in maintaining hydrogen bonds in the ATP binding and motor domains of KIF22. We conclude that these mutations in KIF22 cause lepto-SEMDJL.
Main Text
Spondyloepimetaphyseal dysplasia with joint laxity (SEMDJL), leptodactylic (lepto-SEMDJL) or Hall type (MIM 603546), is a rare but distinct entity of the spondyloepimetaphyseal dysplasias group. Hall et al.1 first described this disease entity in 1998, differentiating it from spondylar and nasal alterations with striated metaphyses (SPONASTRIME) dysplasia (MIM 271510) and SEMDJL, Beighton type (MIM 271640). Since then, several series of affected individuals have been reported.2, 3, 4, 5, 6, 7, 8 Subsequently, the nosology was confirmed as lepto-SEMDJL or SEMDJL Hall type.9, 10 It is considered an autosomal-dominant or possibly X-linked dominant trait on the basis of studies of three families in which it was inherited either from mother to son and/or daughter6, 7 or from father to daughter.3
Lepto-SEMDJL is characterized by short stature, distinctive midface retrusion, progressive knee malalignment (genu valgum and/or genu varum), generalized ligamentous laxity, and mild spine deformity, but intellectual development is not impaired.1, 5, 6, 7 Radiographic characteristics include significantly retarded epiphyseal ossification that evolves into epiphyseal dysplasia and precocious osteoarthritis, metaphyseal irregularities and vertical striations, constricted femur neck, slender metacarpals/metatarsals, and mild thoracolumbar kyphosis or scoliosis with normal or mild platyspondyly5, 7(Figure 1).
Figure 1.
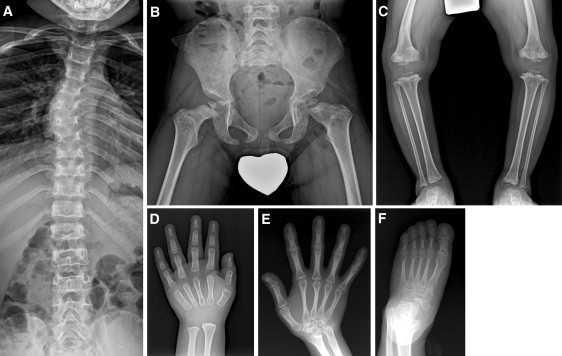
Radiographic Characteristics of Lepto-SEMDJL
(A–D) Spine, pelvis, knee, and hand of individual S-1 at age 4 show epimetaphyseal dysplasias with constricted femur neck, metaphyseal vertical streaks at the knee and ankle, genu varum deformity, and mild platyspondyly with thoracic scoliosis. Nonossified epiphyseal ossifications at the phalanges and carpal bones suggest remarkably retarded bone age.
(E and F) Hand and foot of individual F-1 (II-5 in Figure 2) at age of 45 show persistently slender metacarpals and metatarsals. Carpal and tarsal bones are small and dysplastic.
This study was approved by the institutional review boards of the participating institutions, and informed consent was obtained from all subjects. We performed genomic analysis on three affected individuals and an unaffected father from one family (Figure 2) and on five simplex individuals with lepto-SEMDJL to identify the disease-causing mutations. All subjects were of Korean origin and clinical and radiographic findings of five individuals were reported previously.7 An additional three individuals were included in this study and clinical features of the eight affected individuals are summarized in Table 1. They all fulfilled the diagnostic criteria for lepto-SEMDJL on the basis of skeletal survey.
Figure 2.
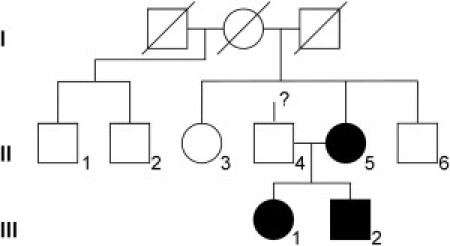
Pedigree of the Familial Case
Symbols in black indicate affected individuals.
Table 1.
Clinical Summary of the Patients
|
Familial Cases |
Simplex Cases |
|||||||
|---|---|---|---|---|---|---|---|---|
|
F-1 (II-5a) |
F-2 (III-1a) |
F-3(III-2a) |
S-1 |
S-2 |
S-3 |
S-4 |
S-5 |
|
| Gender | F | F | M | M | M | F | F | F |
| Height | ||||||||
| Age of examination (in years) | 45 | 11 | 8 | 4 | 13 | 12 | 4 | 13 |
| Z value | −9.2 | −4.9 | −4.4 | −3.0 | −1.7 | −3.2 | −4.2 | −5.3 |
| Birth weight (kg) | (low) | 2.0 | 2.75 | 2.91 | 2.8 | 3.3 | 2.9 | 3.3 |
| Clinical Findings | ||||||||
| Midface retrusion | obvious | obvious | obvious | obvious | mild | mild | obvious | obvious |
| Radial head (sub)luxation | yes | yes | yes | yes | nob | noc | yes | yes |
| Knee | genu valgum, bilateral | genu varum, bilateral | genu valgum, bilateral | genu varum, bilateral | genu valgum with patellar dislocation, bilateral | genu valgum, right > left | windswept deformity | genu varum, mild |
| Hip | dislocation, right | none | none | none | none | none | subluxation, right | none |
| Spine deformity | scoliosis | none | none | kyphoscoliosis, mild | none | scoliosis, mild | kyphosis, mild | mild kyphosis, resolved |
| Laryngotra-cheomalacia | yes | yes | yes | yes | denied | denied | yes | denied |
| Other | strabismus | epilepsyd | ||||||
| Individual numbering in Kim et al.7 | 2 | 3 | 1 | new | new | 4 | new | 6 |
In Figure 2.
Cubitus varus with elbow flexion contracture.
Mild flattening of the capitellum only.
Benign Rolandic epilepsy, controlled by medication.
We first screened by using a high-resolution microarray (SurePrint G3 Human CGH Microarray Kit, 1X1M, Agilent Technologies, Santa Clara, CA), which did not reveal any pathogenic copy-number variations at 2.1 kb overall median probe spacing in all affected individuals. We then performed whole-exome sequencing of eight affected individuals and the unaffected father of the familial case. The exome was captured with the Agilent SureSelect Human All Exon Kit covering 38 Mb (Agilent, Santa Clara, CA) and sequenced with the Illumina Genome Analyzer II platform (Illumina, San Diego, CA). We obtained a mean coverage depth of the exome ranging from 39.5× to 60.8×, which is of sufficient depth to interrogate the exons for mutations (Table S1, available online).
Alignment of the affected individual sequences was performed with Burrows Wheeler Aligner11 and Samtools12. Regions near short indels were realigned with IndelRealigner function in Genome Analysis Toolkit13 with default parameters. Single nucleotide variants (SNVs) and indels affecting coding sequence or splicing sites were identified with Unified Genotype and annotated by Genomic Annotator.14 All genomic changes were filtered against of dbSNP (build 132), 1000 Genomes Project (February 28, 2011, releases for SNVs, February 16, 2011, release for indels), and 18 genomes from healthy ethnic Korean individuals.15
A total of 5.1 Gb sequence mapped uniquely per individual as paired-end 76 bp reads to the human reference genome. More than 97% of the targeted bases were covered to pass our thresholds for variant calling at this depth of coverage (Table S1). In the familial case, 97 novel candidate variants in 86 genes were inherited from the mother to the two affected offspring (Figure 2 and Table S1). Next, we sought to discover any gene with recurrent mutations between the family and the five simplex individuals. Surprisingly, KIF22 (KINESIN family member 22 [MIM 603213]) was found to harbor sequence variants in seven of eight affected individuals, including three known SNPs (rs67578835, rs235648, and rs2450399) and five novel variants (Figure 3 and Table 2). All 14 exons and their flanking intron sequences of KIF22 were amplified for Sanger sequencing from genomic DNA of the eight affected individuals, nine unaffected family members, and 505 healthy Korean individuals (Table S2 for primers sequence). Of five novel sequence variants, c.695G>A (p.Arg232Gln) in individual S-3 and c.1677+124A>T in individual S-4 were considered as polymorphisms because they were inherited from unaffected mothers (Table 2), and the latter was also found at a low frequency in the normal Korean cohort database (Table S3).
Figure 3.
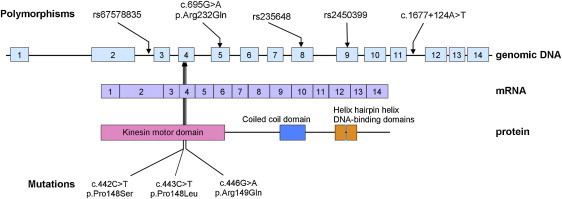
KIF22 Mutations and Polymorphisms Identified in Individuals with Lepto-SEMDJL
Three pathogenic variants of KIF22, p.Pro148Ser, p.Pro148Leu, and p.Arg149Gln, are located at the exon 4, substituting Pro148 or Arg149.
Table 2.
Summary of Sequence Variations in KIF22 in Lepto-SEMDJL Patients
| Individual |
Familial Cases |
Simplex Cases |
||||||
|---|---|---|---|---|---|---|---|---|
| F-1 (II-5a) | F-2 (III-1a) | F-3 (III-2a) | S-1 | S-2 | S-3 | S-4 | S-5 | |
| KIF22 Sequence Variants | ||||||||
| Mutation | c.442C>T (p.Pro148Ser) | c.442C>T (p.Pro148Ser) | c.442C>T (p.Pro148Ser) | c.446G>A (p.Arg149Gln) | none (none) | c.446G>A (p.Arg149Gln) | c.443C>T (p.Pro148Leu) | c.443C>T (p.Pro148Leu) |
| Polymorphism | rs235648 rs2450399 | rs235648 rs2450399 | rs235648 rs2450399 | rs235648 rs2450399 rs67578835 | rs235648 rs2450399 | c.695G>A (p.Arg232Gln) rs235648 rs2450399 | rs235648 rs2450399 c.1677+124>T | rs235648 rs2450399 |
| Parental Genotypes | ||||||||
| Mother | n.a. | individual F-1 | individual F-1 | rs235648 rs2450399 rs67578835 | rs235648 rs2450399 rs67578835 | rs235648 rs2450399 c.695G>A (p.Arg232Gln) | rs235648 rs2450399 c.1677+124>T | rs235648 rs2450399 |
| Father | n.a. | rs235648 rs2450399 | rs235648 rs2450399 | rs235648 rs2450399 | n.a.b | n.a.b | rs235648 rs2450399 | rs235648 rs2450399 |
In Figure 2.
n.a. is used as an abbreviation for not available.
One of the three novel heterozygous sequence variations at residues Pro148 or Arg149 was found in the three affected individuals of the familial case and four of five simplex individuals and not in 1,010 control chromosomes from normal cohorts. The sequence variation, c.442C>T (p.Pro148Ser) cosegregated with the phenotype in the familial case. The sequence variations c.446G>A (p.Arg149Gln) in individual S-1 and c.443C>T (p.Pro148Leu) in individuals S-4 and S-5 were results of de novo mutation (Table 2). We therefore conclude that these sequence variants in KIF22 are the strongest candidate mutations for autosomal-dominant lepto-SEMDJL (Table S3).
The mouse Kif22 is expressed in various tissues including thymus, spleen, testis, and fetal liver16. Because lepto-SEMDJL involves primarily the connective tissues, we examined the expression of Kif22/KIF22 in various tissues, including bone and cartilage, of mouse and human by RT-PCR with gene-specific primers (Table S2). In C57BL mice, Kif22 mRNA was expressed in bone, cartilage, liver, ovary, small intestine, and spleen (Figure S1A). KIF22 mRNA was detected in bone, cartilage, joint capsule, ligament, skin, and primary cultured chondrocytes harvested from human donors (Figure S1B).
Kinesin binds to ATP and hydrolyzes it to provide energy for molecular motors, and many regions in kinesin are involved in ATP binding.17 Although a crystal structure of KIF22 (Protein Data Bank number 3BFN) is available, there are many missing structural parts, including one α-helix (from sequence number 231 to 241, NRTVGATRLNQ) that is known to be critical to ATP binding.17 An additional reference structure, (3KIN, a homodimer structure for kinesin motor and neck domain with bound ADP) was selected to predict the structures of missing α-helix part in KIF22 with MODELER18 (Figure S2A). The amino acid substitutions were applied to the modeled structure and the side chains of the mutated residues were optimized with FoldX,19 and the structural changes due to the mutations were investigated by the molecular dynamics (MD) simulations with GROMACS.20 Structural analysis and visualization were performed with PyMOL.21
A helix-loop-helix structure adjacent to the ATP binding site spans from129 to 156 amino acid residues containing Pro148 and Arg149 (Figure S2B). The loop structure adjacent to the ATP binding site is stabilized by hydrogen bonds among its residues, for example, Leu138 with Gly145, Pro144 with Ser140, and Gln143 and Glu142 with Ser140 as shown in Figure S1B. Loop residues also interact with the adjacent α-helix and other residues near the loop region; for example, Gln143 with His135 and Gly145 with Arg149.
In the MD simulation on the p.Pro148Leu mutation, we found two novel hydrogen bonds, which were absent in wild-type (Figures 4A and 4B). The hydrogen bond between NH of Arg149 and C = O of Gly145 is the expected regular hydrogen bond between ith and (i+4)th residues in the helix structure. The other hydrogen bond was formed between NH of Leu148 and C = O of Leu138. These two new hydrogen bonds slightly pull the loop structure away from ATP binding pocket, damaging ATP binding and probably resulting in the loss of motor function. We also obtained identical results for p.Pro148Ser.
Figure 4.
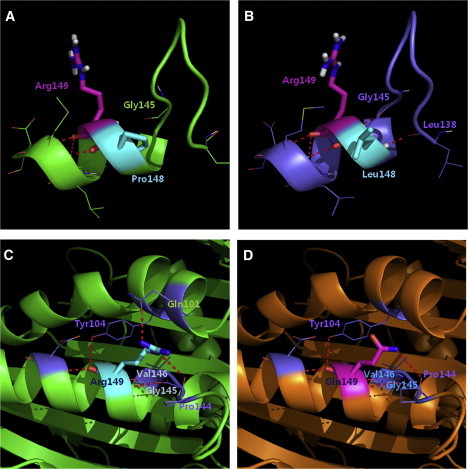
3D Structure of KIF22 and the Effect of Mutations
(A) The wild-type structure was obtained by running MD simulation.
(B) The mutant structure with Pro148Leu mutation from MD simulation revealed two new hydrogen bonds; the hydrogen bond between NH of Arg148 and C = O of Gly145, and the hydrogen bond between NH of Leu148 and C = O of Leu138.
(C) Arg149 forms a hydrogen bond with Gln101 and Tyr104 residues in an α-helix located near ATP binding site and also with loop residue Pro144.
(D) The p.Arg149Gln mutation would disrupt the hydrogen bonds with the α-helix of Gln101 and Tyr104. The red dotted line symbolizes a hydrogen bond.
With regard to the effect of the mutation, p.Arg149Gln, Arg149 forms a hydrogen bond with Gln101 and Tyr104 residues in an α-helix located near ATP binding site and also with the loop residue Pro144 (Figure 4C). The p.Arg149Gln mutation would disrupt the hydrogen bonds with the α-helix of Gln101 and Tyr104 (Figure 4D). Analogous to mutations in Pro148, the connection between two α-helices would be loosened to move away from the ATP binding pocket.
An α-helix spanning from 231 to 241 contains a sequence variation, p.Arg232Gln of individual S-3, which was considered as a polymorphism. Arg232 forms a hydrogen bond with residue Ser243 in an α-helical structure that is part of the ATP binding pocket (Figure S2C). Although the mutation p.Arg232Gln breaks a hydrogen bond between Arg149 and Ser242, it creates a new hydrogen bond between Gln232 and Arg242, maintaining proper interaction with the α-helix (Figure S2D), and hence does not seem to change the KIF22 structure significantly.
Although the role of KIF22 in humans is not thoroughly investigated, animal models with mutations in KIF22 provide valuable information about the function of this gene. Xkid, a Xenopus ortholog of human KIF22, was degraded in anaphase by ubiquitin-mediated proteolysis, and degradation was necessary to support the chromosome movement at anaphase.22, 23 Loss of Xkid function in the stage of anaphase chromosome compaction resulted in the formation of multinucleated oocytes and embryonic death.23 NOD, a Drosophila ortholog of KIF22, is required for the proper segregation of chromosomes. The role of KIF22 in the development of the skeletal system has not been elucidated yet. However, the formation of primary cilia, another key role for kinesin family proteins, could be crucial. Many members of the kinesin family control the formation of primary cilia, which deliver the signals from extracellular cues for development and tissue homeostasis. Kif3a,24 Kif7,25 and Kif2426 support the formation of primary cilia in keratinocytes, retinal cells, and chondrocytes, respectively. Kif24, a kinesin-13 subfamily member, plays a role in regulating centriolar length and ciliogenesis. Loss of Kif24 from cycling cells resulted in aberrant cilia assembly but did not promote growth of abnormally long centrioles.27 Mutations in KIF7 were reported in human Joubert syndrome, and knockdown of KIF7(MIM 611254) caused problems in cilia formation, centrosomal duplication, and golgi network25. Knockout of Kif3a in the mouse resulted in depletion of cilia and postnatal dwarfism because of premature loss of the growth plate, probably a result of reduced proliferation and accelerated hypertrophic differentiation of chondrocytes.28 Kif3a regulates important developmental processes such as skeletogenesis28 and skin differentiation26 through the formation of primary cilium. Their deficiency causes abnormal topography of hedgehog signaling, growth plate dysfunction, and nonphysiologic responses and processes in perichondrial tissues, including ectopic cartilage formation and excessive intramembranous ossification.28 It is likely that the specific mutations of KIF22 identified in individuals with lepto-SEMDJL could interfere with hedgehog signaling as in Kif3a knock-out mice resulting in osteochondrodysplasia phenotype.
We did not detect any mutation of KIF22 in individual S-2 by either exome or Sanger sequencing, and no notable genomic imbalance greater than 2.1 kb was detected by aCGH analysis, either (data not shown). This individual showed a relatively mild clinical phenotype, including moderate short stature (z = −1.7 at age 13 years) and equivocal midface retrusion, suggesting the possibility of a mutation in another gene related to KIF22.
Given the mutations in KIF22 identified in this cohort, the KIF22 expression pattern in connective tissues, and the predicted protein structural changes caused by these mutations, we conclude that the specific mutations of KIF22 affecting amino acid 148 or 149 are pathogenic for lepto-SEMDJL. The mechanism by which these mutations lead to disease is likely related to motor dysfunction of KIF22, but additional studies will be needed to elucidate the KIF22 function in the skeletal system and to understand the molecular pathogenesis of lepto-SEMDJL associated with KIF22 mutation.
Acknowledgments
The authors deeply appreciate Andrea Superti-Furga and Sheila Unger for reviewing the affected individuals, Murim Choi and Charles Lee for reading the manuscript, and Soong Deok Lee for helping DNA preparation. This study was supported by a grant from the Korea Healthcare Technology Research and Development Project, Ministry for Health, Welfare and Family Affairs, Republic of Korea (No A080588).
Published online: December 8, 2011
Footnotes
Supplemental Data include two figures and three tables and can be found with this article online at http://www.cell.com/AJHG/.
Contributor Information
Woong-Yang Park, Email: wypark@snu.ac.kr.
Tae-Joon Cho, Email: tjcho@snu.ac.kr.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project (February 28, 2011 releases for SNPs, February 16, 2011 release for indels), http://www.1000genome.org
Consensus Coding Sequence Region (CCDS) database, http://www.ncbi.nlm.nih.gov/projects/CCDS/
GROMACS, http://www.gromacs.org
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Pymol, http://www.pymol.org
UCSC database, version hg19 (NCBI Build 37, 1 Feb 2009), http://genome.ucsc.edu/
Accession Numbers
Two novel polymorphisms detected in this study were registered to dbSNP as ss469414041 (c.695G>A, p.Arg232Gln) and ss469414042 (c.1677+124A > T). The three sequence variations considered pathogenic were registered to dbSNP, as ss475871913 (c.443C>T, p.Pro148Ser), ss475871912 (c.442C>T, p.Pro148Leu), and ss475871914 (c.446G>A, p.Arg149Gln).
Supplemental Data
References
- 1.Hall C.M., Elçioglu N.H., Shaw D.G. A distinct form of spondyloepimetaphyseal dysplasia with multiple dislocations. J. Med. Genet. 1998;35:566–572. doi: 10.1136/jmg.35.7.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rossi M., De Brasi D., Hall C.M., Battagliese A., Melis D., Sebastio G., Andria G. A new familial case of spondylo-epi-metaphyseal dysplasia with multiple dislocations Hall type (leptodactylic form) Clin. Dysmorphol. 2005;14:13–18. [PubMed] [Google Scholar]
- 3.Hall C.M., Elcioglu N.H., MacDermot K.D., Offiah A.C., Winter R.M. Spondyloepimetaphyseal dysplasia with multiple dislocations (Hall type): Three further cases and evidence of autosomal dominant inheritance. J. Med. Genet. 2002;39:666–670. doi: 10.1136/jmg.39.9.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mégarbané A., Ghanem I., Le Merrer M. Spondyloepimetaphyseal dysplasia with multiple dislocations, leptodactylic type: Report of a new patient and review of the literature. Am. J. Med. Genet. A. 2003;122A:252–256. doi: 10.1002/ajmg.a.20262. [DOI] [PubMed] [Google Scholar]
- 5.Nishimura G., Honma T., Shiihara T., Manabe N., Nakajima E., Adachi M., Mikawa M., Fukushima Y., Ikegawa S. Spondyloepimetaphyseal dysplasia with joint laxity leptodactylic form: Clinical course and phenotypic variations in four patients. Am. J. Med. Genet. A. 2003;117A:147–153. doi: 10.1002/ajmg.a.10927. [DOI] [PubMed] [Google Scholar]
- 6.Park S.M., Hall C.M., Gray R., Firth H.V. Persistent upper airway obstruction is a diagnostic feature of spondyloepimetaphyseal dysplasia with multiple dislocations (Hall type) with further evidence for dominant inheritance. Am. J. Med. Genet. A. 2007;143A:2024–2028. doi: 10.1002/ajmg.a.31857. [DOI] [PubMed] [Google Scholar]
- 7.Kim O.H., Cho T.J., Song H.R., Chung C.Y., Miyagawa S., Nishimura G., Superti-Furga A., Unger S. A distinct form of spondyloepimetaphyseal dysplasia with joint laxity (SEMDJL)-leptodactylic type: Radiological characteristics in seven new patients. Skeletal Radiol. 2009;38:803–811. doi: 10.1007/s00256-009-0671-4. [DOI] [PubMed] [Google Scholar]
- 8.Holder-Espinasse M., Fayoux P., Morillon S., Fourier C., Dieux-Coeslier A., Manouvrier-Hanu S., Le Merrer M., Hall C.M. Spondyloepimetaphyseal dysplasia (Hall type) with laryngeal stenosis: A new diagnostic feature? Clin. Dysmorphol. 2004;13:133–135. doi: 10.1097/01.mcd.0000130236.02981.c5. [DOI] [PubMed] [Google Scholar]
- 9.Superti-Furga A., Unger S. Nosology and classification of genetic skeletal disorders: 2006 revision. Am. J. Med. Genet. A. 2007;143:1–18. doi: 10.1002/ajmg.a.31483. [DOI] [PubMed] [Google Scholar]
- 10.Warman M.L., Cormier-Daire V., Hall C., Krakow D., Lachman R., LeMerrer M., Mortier G., Mundlos S., Nishimura G., Rimoin D.L., et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am. J. Med. Genet. A. 2011;155A:943–968. doi: 10.1002/ajmg.a.33909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zanders S., Ma X., Roychoudhury A., Hernandez R.D., Demogines A., Barker B., Gu Z., Bustamante C.D., Alani E. Detection of heterozygous mutations in the genome of mismatch repair defective diploid yeast using a Bayesian approach. Genetics. 2010;186:493–503. doi: 10.1534/genetics.110.120105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ju Y.S., Kim J.I., Kim S., Hong D., Park H., Shin J.Y., Lee S., Lee W.C., Kim S., Yu S.B., et al. Extensive genomic and transcriptional diversity identified through massively parallel DNA and RNA sequencing of eighteen Korean individuals. Nat. Genet. 2011;43:745–752. doi: 10.1038/ng.872. [DOI] [PubMed] [Google Scholar]
- 16.Tokai N., Fujimoto-Nishiyama A., Toyoshima Y., Yonemura S., Tsukita S., Inoue J., Yamamota T. Kid, a novel kinesin-like DNA binding protein, is localized to chromosomes and the mitotic spindle. EMBO J. 1996;15:457–467. [PMC free article] [PubMed] [Google Scholar]
- 17.Kozielski F., Sack S., Marx A., Thormählen M., Schönbrunn E., Biou V., Thompson A., Mandelkow E.M., Mandelkow E. The crystal structure of dimeric kinesin and implications for microtubule-dependent motility. Cell. 1997;91:985–994. doi: 10.1016/s0092-8674(00)80489-4. [DOI] [PubMed] [Google Scholar]
- 18.Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 19.Schymkowitz J., Borg J., Stricher F., Nys R., Rousseau F., Serrano L. The FoldX web server: An online force field. Nucleic Acids Res. 2005;33(Web Server issue):W382–W388. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A.E., Berendsen H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 21.The PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC.
- 22.Funabiki H., Murray A.W. The Xenopus chromokinesin Xkid is essential for metaphase chromosome alignment and must be degraded to allow anaphase chromosome movement. Cell. 2000;102:411–424. doi: 10.1016/s0092-8674(00)00047-7. [DOI] [PubMed] [Google Scholar]
- 23.Ohsugi M., Adachi K., Horai R., Kakuta S., Sudo K., Kotaki H., Tokai-Nishizumi N., Sagara H., Iwakura Y., Yamamoto T. Kid-mediated chromosome compaction ensures proper nuclear envelope formation. Cell. 2008;132:771–782. doi: 10.1016/j.cell.2008.01.029. [DOI] [PubMed] [Google Scholar]
- 24.Verhey K.J., Kaul N., Soppina V. Kinesin assembly and movement in cells. Annu Rev Biophys. 2011;40:267–288. doi: 10.1146/annurev-biophys-042910-155310. [DOI] [PubMed] [Google Scholar]
- 25.Dafinger C., Liebau M.C., Elsayed S.M., Hellenbroich Y., Boltshauser E., Korenke G.C., Fabretti F., Janecke A.R., Ebermann I., Nürnberg G., et al. Mutations in KIF7 link Joubert syndrome with Sonic Hedgehog signaling and microtubule dynamics. J. Clin. Invest. 2011;121:2662–2667. doi: 10.1172/JCI43639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ezratty E.J., Stokes N., Chai S., Shah A.S., Williams S.E., Fuchs E. A role for the primary cilium in Notch signaling and epidermal differentiation during skin development. Cell. 2011;145:1129–1141. doi: 10.1016/j.cell.2011.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kobayashi T., Tsang W.Y., Li J., Lane W., Dynlacht B.D. Centriolar kinesin Kif24 interacts with CP110 to remodel microtubules and regulate ciliogenesis. Cell. 2011;145:914–925. doi: 10.1016/j.cell.2011.04.028. [DOI] [PubMed] [Google Scholar]
- 28.Koyama E., Young B., Nagayama M., Shibukawa Y., Enomoto-Iwamoto M., Iwamoto M., Maeda Y., Lanske B., Song B., Serra R., Pacifici M. Conditional Kif3a ablation causes abnormal hedgehog signaling topography, growth plate dysfunction, and excessive bone and cartilage formation during mouse skeletogenesis. Development. 2007;134:2159–2169. doi: 10.1242/dev.001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.