

Abstract
Spondyloepimetaphyseal dysplasia with joint laxity, leptodactylic type (lepto-SEMDJL, aka SEMDJL, Hall type), is an autosomal dominant skeletal disorder that, in spite of being relatively common among skeletal dysplasias, has eluded molecular elucidation so far. We used whole-exome sequencing of five unrelated individuals with lepto-SEMDJL to identify mutations in KIF22 as the cause of this skeletal condition. Missense mutations affecting one of two adjacent amino acids in the motor domain of KIF22 were present in 20 familial cases from eight families and in 12 other sporadic cases. The skeletal and connective tissue phenotype produced by these specific mutations point to functions of KIF22 beyond those previously ascribed functions involving chromosome segregation. Although we have found Kif22 to be strongly upregulated at the growth plate, the precise pathogenetic mechanisms remain to be elucidated.
Main Text
Heritable disorders of skeletal growth and development have revealed a surprising variety of underlying molecular mechanisms, bringing this clinical and diagnostically difficult field to the front of molecular genetics research.1, 2 Genes responsible for these disorders might code for extracellular structural proteins, enzymes responsible for the synthesis or degradation of matrix components, hormones and signal transmission factors, nuclear transcription factors, intracellular cytoskeletal proteins, structural proteins of the endoplasmic reticulum, noncoding RNAs, and most recently, genes involved in ciliary assembly and transport. Here we report that mutations in KIF22 (aka KID [MIM 603213]), which encodes a monomeric kinesin,3, 4, 5 are the cause of spondyloepimetaphyseal dysplasia with joint laxity, leptodactylic type (lepto-SEMDJL; aka SEMD, Hall type [MIM 603546]). This implicates this class of molecules in the pathogenesis of human skeletal dysplasias and suggests a hitherto unknown role for KIF22 in skeletal growth and homeostasis.
Lepto-SEMDJL is characterized by a flat face, perinatal onset of short stature with shortening of both the trunk and the limbs, generalized joint laxity with multiple dislocations, and progressive scoliosis and limb deformity.6 The radiographic pattern is that of a spondyloepimetaphyseal dysplasia with moderately flattened vertebral bodies, striated metaphyses, and small and fragmented epiphyses with delayed maturation. The most distinctive features for differential diagnosis are the slender metacarpals and phalanges (“leptodactyly,” meaning slender fingers) and the progressive degeneration of carpal bones; however, the latter two features are evident only in older children and young adults. The soft consistency of cartilage in the airways leads to laryngotracheomalacia with proneness to respiratory obstruction and inspiratory stridor in infancy and childhood.7, 8, 9 Although the majority of cases have been sporadic in their families, dominant inheritance has been documented.8, 10, 11, 12 The condition is likely to be both under- and misdiagnosed because the specific radiographic findings appear only in late childhood.
The pathogenesis of lepto-SEMDJL has remained obscure. Disturbed formation of the extracellular matrix was suggested by the observation of highly abnormal collagen fibers in a tendon biopsy of an affected individual (Figure 1). This, and some phenotypic overlap with two other conditions characterized by generalized bone dysplasia and joint laxity, namely spondyloepiphyseal dysplasia congenita (a dominant collagen 2 disorder [MIM 183900]) and pseudoachondroplasia (a dominant disorder associated with mutations in cartilage oligomeric matrix protein [MIM 177170]) had led to the investigation of these genes in a few cases, with negative results.
Figure 1.
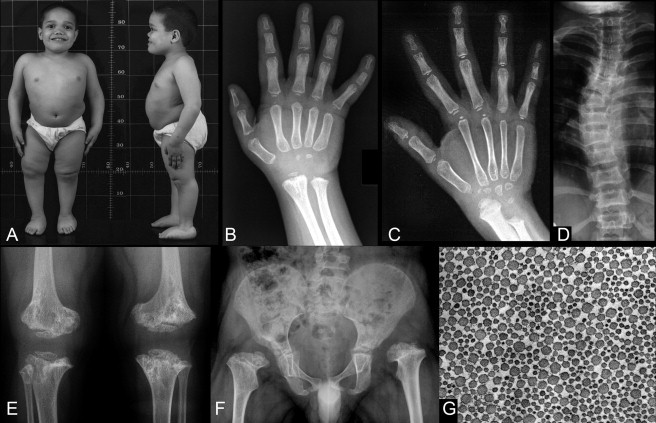
Morphologic Features of Lepto-SEMDJL
(A, B, D, E and F) are all from subject 2 (family 1) at age 7.
(A) In this boy, stature is markedly below the normal range, with short-trunk type disproportion. There is frontal bossing with flattening of the face and a sunken nasal bridge. There is left hip subluxation (F), leg length difference, and right genu varum (A). Joint laxity is indicated by the scoliosis and the flat feet.
(B) The hand radiographs of this boy; there is a very marked delay in the maturation of all epiphyseal centers and of the carpal bones, as well as metaphyseal irregularities at the distal radius and ulna.
(C) The hand X-ray of an unrelated boy, age 10. Also in this individual, there is a marked delay in all secondary ossification centers and there is shortening of the distal ulna. The proximal phalanges and the metacarpals are slender; this feature, leptodactyly, that becomes apparent only over time, is characteristic for this bone dysplasia.
(D) The moderate platyspondyly and the scoliosis (ligamentous laxity).
(E) The marked dysplasia of the metaphyses at the knee (distal femur, proximal tibia) and at the same time the small and dysplastic epiphyses.
(F) A similar pattern at the proximal femurs with shortening of the femoral necks and the presence of epiphyses that are barely visible and markedly small for age. The acetabula are not well developed; they are less well developed on the left than on the right; the left hip is subluxated because of the acetabular dysplasia and associated ligamentous laxity.
(G) An electron microscopy image of a tendon biopsy section (subject 6 in Table 1) at right angle to the collagen fiber (magnification, approximately 5000×). The diameter of the fibers shows a significant variability.
We studied a cohort of 32 affected individuals with lepto-SEMDJL from 20 families of different ethnic origins (Table 1). The study was approved by the cantonal ethic committee of Lausanne, Switzerland. In 20 individuals from eight families, the condition was inherited in an autosomal dominant manner, whereas there was no family history of disease in 12 individuals. Clinical and radiographic features common to all affected individuals are summarized in Figure 1. A significant proportion of cases presented laryngotracheomalacia. We performed whole-exome sequencing by using DNA from five unrelated lepto-SEMDJL individuals (subjects 2, 6, 7, 30, and 32 in Table 1) and 14 unrelated controls to identify gene variants that were present in affected individuals and absent in controls. Exome capture utilized the SureSelectXT Human All Exon 50Mb kit (Agilent Technologies) following the manufacturer's protocol, except that we used adapters with 3 bp barcodes13 to allow multiplexing of samples during capture. Captures were performed in seven independent reactions with two to four samples per reaction with 300–500 ng DNA per sample and then combined into one molarity-balanced library on which we performed 75 bp paired-end sequencing in seven lanes of one Illumina HiSeq2000 flow cell. Sequence reads were debarcoded with Novobarcode (Novocraft Technologies), aligned to the reference genome (hg19) with BWA,14 and culled of PCR duplicate reads with SAMtools.15 Variant bases were called with SAMtools/BCFtools, and annotated with ANNOVAR,16 filtered by presence in dbSNP132, 1000 g, and our 14 unaffected control samples, and prioritized according to putative functionality; splice site and coding nonsilent variants were given the highest priority.
Table 1.
Clinical Features, Origin, Inheritance, and Mutations in KIF22 in All Individuals with Lepto-SEMDJL
| Family | Subject | Origin | Short Stature | Skeletal Dysplasia | Laryngeal Stenosis | Joint Laxity | KIF22 Mutation (cDNA) | KIF22 Mutation (Protein) | Inheritance or De Novo |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1,2,3,4 | Italy | + | + | – | + | c.443C>T | p.Pro148Leu | autosomal dominant |
| 2 | 5,6 | UK | + | + | – | + | c.443C>T | p.Pro148Leu | autosomal dominant |
| 3 | 7,8,9 | USA | + | + | – | + | c.446G>A | p.Arg149Gln | autosomal dominant |
| 4 | 10,11 | Italy | + | + | – | + | c.446G>A | p.Arg149Gln | autosomal dominant |
| 5 | 12,13,14 | Japan | + | + | – | + | c.446G>A | p.Arg149Gln | autosomal dominant |
| 6 | 15,16 | UK | + | + | – | + | c.446G>A | p.Arg149Gln | autosomal dominant |
| 7 | 17,18 | Belgium | + | + | + | + | c.446G>A | p.Arg149Gln | autosomal dominant |
| 8 | 19,20 | USA | + | + | not known | + | c.446G>A | p.Arg149Gln | autosomal dominant |
| 9 | 21 | Lebanon | + | + | not known | + | c.446G>A | p.Arg149Gln | de novo |
| 10 | 22 | France | + | + | + | + | c.446G>A | p.Arg149Gln | de novo |
| 11 | 23 | Greece | + | + | – | + | c.446G>A | p.Arg149Gln | de novo |
| 12 | 24 | UK | + | + | + | + | c.446G>A | p.Arg149Gln | de novo |
| 13 | 25 | Germany | + | + | not known | + | c.446G>A | p.Arg149Gln | de novo |
| 14 | 26 | USA | + | + | not known | + | c.446G>A | p.Arg149Gln | de novo |
| 15 | 27 | USA | + | + | not known | + | c.446G>T | p.Arg149Leu | de novo |
| 16 | 28 | USA | + | + | not known | + | c.443C>T | p.Pro148Leu | de novo |
| 17 | 29 | Brazil | + | + | + | + | c.443C>T | p.Pro148Leu | de novo |
| 18 | 30 | Germany | + | + | + | + | c.443C>T | p.Pro148Leu | de novo |
| 19 | 31 | Japan | + | + | + | + | c.443C>T | p.Pro148Leu | de novo |
| 20 | 32 | Italy | + | + | not known | + | c.443C>T | p.Pro148Leu | de novo |
We identified one gene, KIF22, in which one of two heterozygous missense mutations (p.Pro148Leu [c.443C>T] or p.Arg149Gln [c.446G>A]) was present in all five lepto-SEMDJL cases and absent in 2,500 exomes from the National Heart Lung and Blood Institute Exome Sequencing Project (accessed August 2011). The mutations alter highly conserved residues within the KIF22 motor domain near an ATP-binding site (Figure 2). Sanger sequencing of KIF22 exon 4 in all 32 lepto-SEMDJL affected individuals and available relatives revealed that all affected persons were heterozygous for either p.Pro148Leu, p.Arg149Gln, or p.Arg149Leu [c.446G>T] allele (Table 1) and that these mutations were not present in unaffected relatives. We tested both parents (and unaffected siblings when available) of each affected individual and complete cosegregation of mutations with the phenotype was identified. Sequencing of exon 4 in 480 unrelated control samples of European descent (Sigma-Aldrich) revealed no mutations (data not shown).
Figure 2.
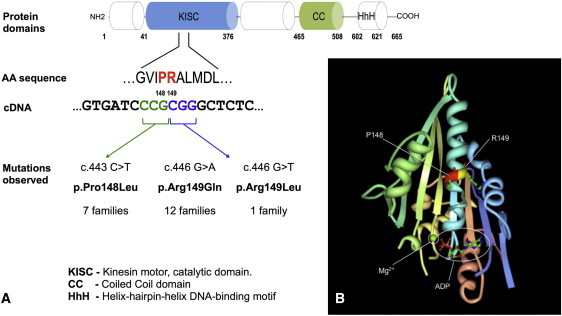
Schematic Representation of KIF22 Protein and Mutations Found in Individuals with Lepto-SEMDJL
(A) Functional domains of KIF22; the mutations found in lepto-SEMDJL patients are located on two adjacent amino acid residues in the motor/catalytic domain.
(B) Crystal structure of the motor domain of KIF22, generated with Research Collaboratory for Structural Bioinformatics-Protein Data Bank (RCSB-PDB), viewer Protein Workshop (see Web Resources; deposition: 2007-11-22; release: 2007-12-04; last modified 2011-07-13). The two contiguous residues mutated in lepto-SEMDJL (Pro148 and Arg149) are located close to the ATP/ADP binding site.
We next tested whether the lepto-SEMDJL mutations affect protein abundance, posttranslational processing, or cytoskeletal architecture by using skin fibroblast cell lines from three unrelated affected individuals (subjects 1, 10, and 32 in Table 1) and controls as well as in tendon derived fibroblast-like cells of subject 6 and a control cell line. We observed no differences from control in KIF22 localization or cytoskeletal architecture as determined by anti-KIF22 or anti-tubulin immunofluorescence (data not shown). Because immunoblot performed with a commercial antibody against human KIF22 failed to reveal expression in both skin fibroblasts and tendon fibroblast-like cells from lepto-SEMDJL affected individuals and controls (data not shown), we looked for specific expression in cartilage growth plates of wild-type mouse tibia. Quantitative RT-PCR in microdissected mouse growth plates, performed as previously described17 and with the housekeeping gene Mrps16 as a reference, showed strong upregulation of Kif22 in the proliferative zone of the growth plate (Figure 3 and Table S1, available online).
Figure 3.
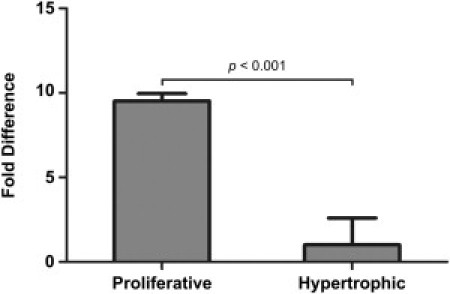
Quantitative RT-PCR Analysis of Kif22 Expression in Wild-Type Mouse Growth Plate Cartilage Zones
qPCR was performed for Kif22 on cDNA derived from proliferative and hypertrophic zones microdissected from 2-week-old wild-type mouse tibial growth plates (n = 3). qPCR was conducted with three technical replicates. Kif22 expression was calculated relative to the housekeeper gene Mrps16 and expressed as the fold difference in expression in the proliferative zone compared to the hypertrophic zone. Statistical significance was calculated with the Student's t test. Error bars represent standard deviation. Detailed quantitative expression data are reported in Table S1.
The observation that KIF22 mutations were restricted to two adjacent codons in all examined lepto-SEMDJL individuals confirms the genetic homogeneity of this disorder and the specificity of the diagnostic criteria as outlined by Hall et al.6 and Kim et al.18 The findings also raise questions on the possible molecular mechanisms. Haploinsufficiency seems unlikely, because it would be extremely unusual to have independent occurrence in a large number of unrelated pedigrees clustering on two adjacent amino acids; instead, the two residues must have a functional role that is hitherto unknown. Previously ascribed functions for KIF22, based upon knockout and knockdown studies, involved spindle formation,19 chromosomal movement,19 microtubule stabilization,5, 20 genomic stability, and cellular replication.21 However, the phenotype of lepto-SEMDJL does not have any feature that would seem related to these functions. Several kinesins play important roles in the transport of morphogens (KIF3B),22 cell surface receptors (KIF17),23 and matrix metalloproteinases (KIF5B, KIF3A, and KIF3B).24, 25, 26 By analogy, KIF22 might also have an important trafficking role in chondrocytes or the motor domain missense mutations might cause KIF22 to interfere with other motor domain kinesins that function in cartilage. As an alternative explanation, the mutations at residues 148 and 149 might trans-specify the protein, conferring to it some properties normally reserved to other members of the kinesin family. This latter hypothesis is supported by the observation that some kinesins do have Leu or Gln at position 149 (Supplemental Data). Finally, the mutations might produce a dominant negative effect if KIF22 heterodimerizes with other kinesins and/or interacts with other partner proteins.
To date, few human monogenic diseases have been associated with mutations in any of the 45 currently annotated human kinesin genes. Recessive mutations in kinesin genes have been associated with acrocallosal syndromes, Joubert syndrome (KIF7),27, 28 and hereditary sensory and autonomic neuropathy type 2 (KIF1A).29 Dominant, recurrent missense mutations in a methylated CpG dinucleotide in the C-terminal domain of KIF21A cause congenital fibrosis of extraocular muscles,30, 31 and a dominant missense mutation in the motor domain of KIF1B (p.Gln98Leu [c.293A>T]) has been associated with Charcot-Marie-Tooth type 2 disease in a single pedigree.32 Similar to the p.Gln98Leu (c.293A>T) KIF1B mutation, the KIF22 mutations found in lepto-SEMDJL affect the motor domain of the kinesin and might therefore result in a loss of its motor activity.32 However, as outlined above, simple loss of function is unlikely to explain the clustering of mutations on the two adjacent amino acids. Because kinesins have not been associated with human cartilage biology so far, our findings also raise the possibility that KIF22 might be implicated in intracellular transport (and possibly, secretion) of an extracellular matrix protein and/or in cilia-associated transport mechanisms, two mechanisms that have been evoked in Kif3a and Kif5b conditional chondrocytic knockouts.33, 34 This hypothesis is supported by our expression data in mouse growth plates. Strong expression of Kif22 in the proliferative zone of the growth plate with downregulation in hypertrophic chondrocytes is compatible with a broader role of KIF22 in the synthesis of extracellular matrix rather than with a more restricted role in the hypertrophic zone, that would be a prelude to calcification and vascular invasion. Alternatively, this finding might be explained by higher expression of KIF22 in proliferating cells because of its likely involvement in chromosomal movement during cell division.
The elucidation of the mechanism by which the specific mutations at codons 148 and 149 of KIF22 result in a phenotype restricted to bone and connective tissue will require the design of knockin experiments with appropriate cellular or animal models. Given the relatively high frequency of lepto-SEMDJL, its dominant inheritance, and the diagnostic difficulty in infancy and early childhood, we communicate our findings in order to allow the clinical community and the affected families to benefit from them and to inform basic scientists involved in the study of KIF22 and other kinesins about these unexpected aspects of KIF22 physiology.
Acknowledgments
This work was supported by the Howard Hughes Medical Institute, by the Swiss National Research Foundation, grant 310030_132940 to L.B., by the National Institutes of Health grant HD22657 to D.H.C., and by the National Health and Medical Research Council of Australia, Project grant 607398 to J.F.B. and the Victorian Government's Operational Infrastructure Support Program. A.S.F. is supported by the Leenaards Foundation (Lausanne, Switzerland) and by the Faculty of Biology and Medicine of the Lausanne University (Fonds de Recherche en Pédiatrie). We thank the staff of the Harvard Medical School Biopolymers Facility for assistance in exome sequencing. We are grateful to S. Miyagawa, Department of Pediatrics, Kure Medical Center, Japan, for collaboration in sample collection.
Published online: December 8, 2011
Footnotes
Supplemental Data include one figure and one table and can be found with this article online at http://www.cell.com/AJHG/.
Web Resources
The URLs for data presented herein are as follows:
Ensembl, http://www.ensembl.org/index.html
Integrative Genomics Viewer, http://www.broadinstitute.org/igv/
National Center for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov/
National Heart Lung and Blood Institute (NHLBI) Exome Sequencing Project, http://snp.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
RCSB-PDB, http://www.rcsb.org/
UCSC Genome Browser, http://genome.ucsc.edu/
Supplemental Data
References
- 1.Superti-Furga A., Bonafé L., Rimoin D.L. Molecular-pathogenetic classification of genetic disorders of the skeleton. Am. J. Med. Genet. 2001;106:282–293. [PubMed] [Google Scholar]
- 2.Warman M.L., Cormier-Daire V., Hall C., Krakow D., Lachman R., LeMerrer M., Mortier G., Mundlos S., Nishimura G., Rimoin D.L., et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am. J. Med. Genet. A. 2011;155A:943–968. doi: 10.1002/ajmg.a.33909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miki H., Setou M., Kaneshiro K., Hirokawa N. All kinesin superfamily protein, KIF, genes in mouse and human. Proc. Natl. Acad. Sci. USA. 2001;98:7004–7011. doi: 10.1073/pnas.111145398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yajima J., Edamatsu M., Watai-Nishii J., Tokai-Nishizumi N., Yamamoto T., Toyoshima Y.Y. The human chromokinesin Kid is a plus end-directed microtubule-based motor. EMBO J. 2003;22:1067–1074. doi: 10.1093/emboj/cdg102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shiroguchi K., Ohsugi M., Edamatsu M., Yamamoto T., Toyoshima Y.Y. The second microtubule-binding site of monomeric kid enhances the microtubule affinity. J. Biol. Chem. 2003;278:22460–22465. doi: 10.1074/jbc.M212274200. [DOI] [PubMed] [Google Scholar]
- 6.Hall C.M., Elçioglu N.H., Shaw D.G. A distinct form of spondyloepimetaphyseal dysplasia with multiple dislocations. J. Med. Genet. 1998;35:566–572. doi: 10.1136/jmg.35.7.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holder-Espinasse M., Fayoux P., Morillon S., Fourier C., Dieux-Coeslier A., Manouvrier-Hanu S., Le Merrer M., Hall C.M. Spondyloepimetaphyseal dysplasia (Hall type) with laryngeal stenosis: A new diagnostic feature? Clin. Dysmorphol. 2004;13:133–135. doi: 10.1097/01.mcd.0000130236.02981.c5. [DOI] [PubMed] [Google Scholar]
- 8.Nishimura G., Honma T., Shiihara T., Manabe N., Nakajima E., Adachi M., Mikawa M., Fukushima Y., Ikegawa S. Spondyloepimetaphyseal dysplasia with joint laxity leptodactylic form: Clinical course and phenotypic variations in four patients. Am. J. Med. Genet. A. 2003;117A:147–153. doi: 10.1002/ajmg.a.10927. [DOI] [PubMed] [Google Scholar]
- 9.Park S.M., Hall C.M., Gray R., Firth H.V. Persistent upper airway obstruction is a diagnostic feature of spondyloepimetaphyseal dysplasia with multiple dislocations (Hall type) with further evidence for dominant inheritance. Am. J. Med. Genet. A. 2007;143A:2024–2028. doi: 10.1002/ajmg.a.31857. [DOI] [PubMed] [Google Scholar]
- 10.Hall C.M., Elcioglu N.H., MacDermot K.D., Offiah A.C., Winter R.M. Spondyloepimetaphyseal dysplasia with multiple dislocations (Hall type): Three further cases and evidence of autosomal dominant inheritance. J. Med. Genet. 2002;39:666–670. doi: 10.1136/jmg.39.9.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rossi M., De Brasi D., Hall C.M., Battagliese A., Melis D., Sebastio G., Andria G. A new familial case of spondylo-epi-metaphyseal dysplasia with multiple dislocations Hall type (leptodactylic form) Clin. Dysmorphol. 2005;14:13–18. [PubMed] [Google Scholar]
- 12.Mégarbané A., Ghanem I., Le Merrer M. Spondyloepimetaphyseal dysplasia with multiple dislocations, leptodactylic type: Report of a new patient and review of the literature. Am. J. Med. Genet. A. 2003;122A:252–256. doi: 10.1002/ajmg.a.20262. [DOI] [PubMed] [Google Scholar]
- 13.Bowen M.E., Boyden E.D., Holm I.A., Campos-Xavier B., Bonafé L., Superti-Furga A., Ikegawa S., Cormier-Daire V., Bovée J.V., Pansuriya T.C., et al. Loss-of-function mutations in PTPN11 cause metachondromatosis, but not Ollier disease or Maffucci syndrome. PLoS Genet. 2011;7:e1002050. doi: 10.1371/journal.pgen.1002050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang K., Li M., Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cameron T.L., Bell K.M., Tatarczuch L., Mackie E.J., Rajpar M.H., McDermott B.T., Boot-Handford R.P., Bateman J.F. Transcriptional profiling of chondrodysplasia growth plate cartilage reveals adaptive ER-stress networks that allow survival but disrupt hypertrophy. PLoS ONE. 2011;6:e24600. doi: 10.1371/journal.pone.0024600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim O.H., Cho T.J., Song H.R., Chung C.Y., Miyagawa S., Nishimura G., Superti-Furga A., Unger S. A distinct form of spondyloepimetaphyseal dysplasia with joint laxity (SEMDJL)-leptodactylic type: Radiological characteristics in seven new patients. Skeletal Radiol. 2009;38:803–811. doi: 10.1007/s00256-009-0671-4. [DOI] [PubMed] [Google Scholar]
- 19.Levesque A.A., Howard L., Gordon M.B., Compton D.A. A functional relationship between NuMA and kid is involved in both spindle organization and chromosome alignment in vertebrate cells. Mol. Biol. Cell. 2003;14:3541–3552. doi: 10.1091/mbc.E03-02-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tokai-Nishizumi N., Ohsugi M., Suzuki E., Yamamoto T. The chromokinesin Kid is required for maintenance of proper metaphase spindle size. Mol. Biol. Cell. 2005;16:5455–5463. doi: 10.1091/mbc.E05-03-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maddika S., Sy S.M., Chen J. Functional interaction between Chfr and Kif22 controls genomic stability. J. Biol. Chem. 2009;284:12998–13003. doi: 10.1074/jbc.M900333200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nonaka S., Tanaka Y., Okada Y., Takeda S., Harada A., Kanai Y., Kido M., Hirokawa N. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 1998;95:829–837. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- 23.Yin X., Takei Y., Kido M.A., Hirokawa N. Molecular motor KIF17 is fundamental for memory and learning via differential support of synaptic NR2A/2B levels. Neuron. 2011;70:310–325. doi: 10.1016/j.neuron.2011.02.049. [DOI] [PubMed] [Google Scholar]
- 24.Wiesner C., Faix J., Himmel M., Bentzien F., Linder S. KIF5B and KIF3A/KIF3B kinesins drive MT1-MMP surface exposure, CD44 shedding, and extracellular matrix degradation in primary macrophages. Blood. 2010;116:1559–1569. doi: 10.1182/blood-2009-12-257089. [DOI] [PubMed] [Google Scholar]
- 25.Schnaeker E.M., Ossig R., Ludwig T., Dreier R., Oberleithner H., Wilhelmi M., Schneider S.W. Microtubule-dependent matrix metalloproteinase-2/matrix metalloproteinase-9 exocytosis: Prerequisite in human melanoma cell invasion. Cancer Res. 2004;64:8924–8931. doi: 10.1158/0008-5472.CAN-04-0324. [DOI] [PubMed] [Google Scholar]
- 26.Gueye Y., Ferhat L., Sbai O., Bianco J., Ould-Yahoui A., Bernard A., Charrat E., Chauvin J.P., Risso J.J., Féron F., et al. Trafficking and secretion of matrix metalloproteinase-2 in olfactory ensheathing glial cells: A role in cell migration? Glia. 2011;59:750–770. doi: 10.1002/glia.21146. [DOI] [PubMed] [Google Scholar]
- 27.Putoux A., Thomas S., Coene K.L., Davis E.E., Alanay Y., Ogur G., Uz E., Buzas D., Gomes C., Patrier S., et al. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat. Genet. 2011;43:601–606. doi: 10.1038/ng.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dafinger C., Liebau M.C., Elsayed S.M., Hellenbroich Y., Boltshauser E., Korenke G.C., Fabretti F., Janecke A.R., Ebermann I., Nürnberg G., et al. Mutations in KIF7 link Joubert syndrome with Sonic Hedgehog signaling and microtubule dynamics. J. Clin. Invest. 2011;121:2662–2667. doi: 10.1172/JCI43639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rivière J.B., Ramalingam S., Lavastre V., Shekarabi M., Holbert S., Lafontaine J., Srour M., Merner N., Rochefort D., Hince P., et al. KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am. J. Hum. Genet. 2011;89:219–230. doi: 10.1016/j.ajhg.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ali M., Venkatesh C., Ragunath A., Kumar A. Mutation analysis of the KIF21A gene in an Indian family with CFEOM1: Implication of CpG methylation for most frequent mutations. Ophthalmic Genet. 2004;25:247–255. doi: 10.1080/13816810490498198. [DOI] [PubMed] [Google Scholar]
- 31.Wang P., Li S., Xiao X., Guo X., Zhang Q. KIF21A novel deletion and recurrent mutation in patients with congenital fibrosis of the extraocular muscles-1. Int. J. Mol. Med. 2011;28:973–975. doi: 10.3892/ijmm.2011.759. [DOI] [PubMed] [Google Scholar]
- 32.Zhao C., Takita J., Tanaka Y., Setou M., Nakagawa T., Takeda S., Yang H.W., Terada S., Nakata T., Takei Y., et al. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell. 2001;105:587–597. doi: 10.1016/s0092-8674(01)00363-4. [DOI] [PubMed] [Google Scholar]
- 33.Koyama E., Young B., Nagayama M., Shibukawa Y., Enomoto-Iwamoto M., Iwamoto M., Maeda Y., Lanske B., Song B., Serra R., Pacifici M. Conditional Kif3a ablation causes abnormal hedgehog signaling topography, growth plate dysfunction, and excessive bone and cartilage formation during mouse skeletogenesis. Development. 2007;134:2159–2169. doi: 10.1242/dev.001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu, G. (2009). Study of the function of Kinesin-1 (KIF5B) in long bone development. PhD thesis, University of Hong Kong, Hong Kong.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.