

Abstract
Thiamine pyrophosphate (TPP) is an essential cofactor of the cytosolic transketolase and of three mitochondrial enzymes involved in the oxidative decarboxylation of either pyruvate, α-ketoglutarate or branched chain amino acids. Thiamine is taken up by specific transporters into the cell and converted to the active TPP by thiamine pyrophosphokinase (TPK) in the cytosol from where it can be transported into mitochondria. Here, we report five individuals from three families presenting with variable degrees of ataxia, psychomotor retardation, progressive dystonia, and lactic acidosis. Investigation of the mitochondrial energy metabolism showed reduced oxidation of pyruvate but normal pyruvate dehydrogenase complex activity in the presence of excess TPP. A reduced concentration of TPP was found in the muscle and blood. Mutation analysis of TPK1 uncovered three missense, one splice-site, and one frameshift mutation resulting in decreased TPK protein levels.
Main Text
Thiamine or vitamin B1, which was originally termed aneurin, is a water-soluble aromatic substance that has to be taken up from nutrition by many eukaryotes. Vitamin B1 is active as cofactor in the form of thiamine pyrophosphate (TPP) but is absorbed in form of either thiamine or thiamine monophosphate (Figure 1) in the small intestine. TPP is virtually absent from plasma and cerebrospinal fluid.1 In humans there are two isoforms of thiamine transporters in the plasma membrane encoded by SLC19A2 and SLC19A3. As soon as thiamine enters the cell it is pyrophosphorylated in the cytosol by the thiamine pyrophosphokinase (TPK, Enzyme Commission number EC 2.7.4.15) to form the enzymatically active TPP. TPP is either bound to the cytosolic thiamine-dependent enzyme transketolase from the pentose phosphate cycle or transported into mitochondria by means of the mitochondrial thiamine pyrophosphate carrier encoded by SLC25A19. There it serves as a cofactor of three distinct ketoacid dehydrogenases, namely pyruvate dehydrogenase complex (PDHC, EC 1.2.4.1), α-ketoglutarate dehydrogenase (α-KGDH, EC 1.2.4.2), and branched-chain α-keto acid dehydrogenase (BCKDH, EC 1.2.4.4).
Figure 1.
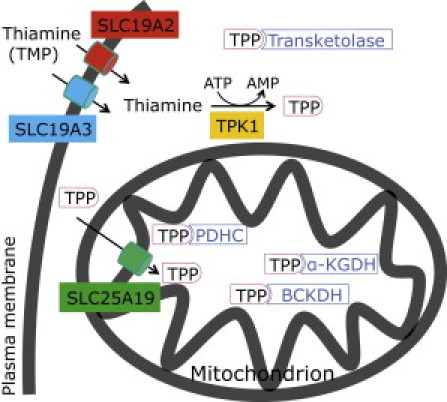
Thiamine Metabolism in Mammalian Cells
The following abbreviations are used: TPP, thiamine pyrophosphate; PDHC, pyruvate dehydrogenase complex; α-KGDH, α-ketoglutarate dehydrogenase; BCKDH, branched chain α-keto acid dehydrogenase.
Mutations in SLC19A2 have been identified as a cause of Rogers syndrome with megaloblastic anemia, thrombocytopenia, diabetes mellitus, and sensorineural deafness (MIM 249270).2 Mutations in SLC19A3 cause biotin responsive basal ganglia disease (MIM 607483), a subacute encephalopathy initially presenting with confusion, dysarthria, and occasional supranuclear facial nerve palsy or external ophthalmoplegia progressing to severe cogwheel rigidity, dystonia, and quadriparesis.3, 4 The different clinical manifestation of these disorders correlates with the different expression pattern of the two thiamine transporter within the organism.5
Defects of the mitochondrial thiamine pyrophosphate transporter SLC25A19, originally described as a deoxynucleotide carrier, result in severe encephalopathy with microcephaly, which was originally discovered in the Amish population in North America,6 or in a milder clinical presentation with episodes of flaccid paralysis and encephalopathy associated with bilateral striatal necrosis and chronic progressive polyneuropathy7 (MIM 607196).
Besides the genetic defects within the thiamine metabolism, a deficiency of this coenzyme is well known from nutritional deficits where it causes beriberi in case of depletion in the food, Wernicke encephalopathy (MIM 277730) in chronic alcohol abuse8 or in disorders with insufficient resorption of thiamine as in severe, chronic vomiting,9 prolonged fasting,10 anorexia nervosa11 gastric surgery,12 and peptic ulcer disease.13 All these forms of thiamine deficiency lead to neurological symptoms that are also found in inborn pyruvate oxidation deficiencies.
Here we describe five individuals from three different families with a disorder of the thiamine metabolism.
Individual P1 (family A II-1 in Figure 2), a girl, was born at term after an uneventful pregnancy from nonconsanguineous parents. From the first year of life she showed a developmental delay. At the age of 15 months, during an infectious episode with dehydration, she was lethargic and hypotonic and lost the ability to walk. Lactate was 3.5 mmol/l in plasma (normal is 0.5–2.2 mmol/l) and 2.7 mmol/l in cerebrospinal fluid (normal is 1.1–2.4 mmol/l). A cranial magnetic resonance image (MRI) was reported as normal. She started to walk again at 3 years of age but continued to show muscular hypotonia and remained delayed in her psychomotor development. A muscle biopsy was performed at the age of 3. Cardiac function documented during the first 3 years showed no abnormalities. She had two further crises with encephalopathy and lactic acidosis triggered by viral infections and died during the last crises at the age of 8 1/2. Urinary organic-acids analysis showed repeated but not always elevated α-ketoglutaric acid (up to 1,468 mmol/mol creatinine, normal < 190).
Figure 2.
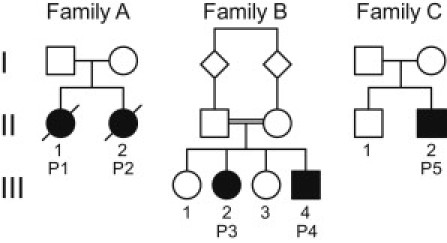
Pedigrees of the Five Affected Individuals from Three Families
Her sister (individual P2, family A II-2 in Figure 2) showed normal development when she was first seen at the age of 7 months. Her plasma lactate was normal (1.3 mmol/l). At 18 months she had an episode of ataxia and disturbed gait from which she only partially recovered. CSF lactate was 2.4 mmol/l. At the age of 2 years she presented with truncal ataxia, was unable to walk, and showed slight dystonia of the upper limbs. She could speak several words. Her head circumference was at the 3rd percentile. The clinical examination was otherwise normal. Echocardiography and ECG did not reveal any abnormalities. MRI was not performed as the parents refused. A muscle biopsy was taken at 2 years. She died at the age of 3 1/2 years during a viral infection. Like her sister, she showed elevated α-ketoglutaric acid in urine on several occasions (up to 1,911 mmol/mol creatinine).
Individual P3 (family B III-2 in Figure 2), a girl, and individual P4 (family B III-4 in Figure 2), a boy, are two of four children from consanguineous parents (first cousins) of a Christian minority from Iraq. Both showed normal development during the first 3 years of life. At the age of 4 spasticity of the lower extremity appeared and worsened during the next two years accompanied by a progressive dystonic movement disorder of all extremities and a loss of gait.
The girl lost the ability to speak and developed a symptomatic epilepsy. On clinical examination at the age of 11, she was wheelchair bound and showed severe dystonia of all extremities. She understood well but was not able to speak. She was treated with a baclofen pump, which slightly decreased her spasticity. Her lactate was between 1.5 and 4 mmol/l; during an intercurrent gastrointestinal infection, she had acidosis with lactate levels up to 17 mmol/l. Organic acids in urine showed elevated α-ketoglutaric acid and 3-hydroxyisovaleric acid. A cranial MRI at the age of 6 performed in Iraq was reported normal; an MRI at age of 11 revealed global brain atrophy and increased signal intensities in the globus pallidus. Magnetic resonance (MR) spectroscopy showed a lactate peak in the basal ganglia.
Echocardiography demonstrated mild left ventricular hypertrophy that moderately increased during the following 2 years. Biopsies of muscle and skin were taken at the age of 11. The spasticity increased during the following 2 years; her cognitive functions remained stable.
Her younger brother also showed progressive dystonia and had severe difficulties in walking. He was mildly microcephalic (49.5 cm at 7 years). He was still able to speak and had an adequate cognitive development at the age of 7. His blood lactate was between 2.3 and 4.6 mmol/l. An MRI at the age of 5 was normal, and he had normal lactate levels in MR spectroscopy. Organic-acids analysis in urine showed increased α-ketoglutaric acid (2,082 mmol/mol creatinine). Individual P3 and P4 are both supplemented with thiamine (100 mg/day initially, then 200 mg/day) and have a fat-rich diet (70% of daily caloric intake).
Individual P5 (family C II-2 in Figure 2), a boy, was born after uneventful pregnancy at term. Parents are nonconsanguineous; an older brother is healthy. At the age of 2 the boy became dizzy and vertiginous and developed a gait disturbance, but there was no nausea or vomiting. In the following period there were several episodes of dizziness and vertigo as well as intermittent gait ataxia. In all cases there was spontaneous remission without therapy. An MRI was normal. At the age of 10 1/2 years, he was admitted to the hospital with a loss of speech; he was weepy and also crying because of frontal headaches, and he had a seizure with clonic jerks of his arms and gaze. The Babinski reflex was positive on both sides. Emergency cranial computerized tomography was normal. Three days later there were clear signs of cerebellar and bulbar affection with dysarthria, intention tremor, confusion, and episodic ataxia. One day later ophthalmoplegia and nystagmus were noted, and there was right accented spasticity. Lactate was elevated up to 4.4 mmol/l in plasma and 3.3 mmol/l in the cerebrospinal fluid. MRI showed changes in the white matter of the cerebellum, increased signal intensities in the corticospinal tract at the medulla oblongata, and slightly increased intensities in the dorsal pons, a result that explains the ataxia. MR spectroscopy of the cerebellum revealed a soft decrease of N-acetylaspartate and an increase of choline; lactate was normal.
The Glasgow coma scale dropped down to 3, and he displayed respiratory insufficiency (pO2 was 63 mm Hg; pCO2 was 16 mm Hg), hence he had to be intubated and needed artificial ventilation for 6 days. There was a slow regression of clinical signs in the following months. Mental development was normal, the boy attends high school at his present age of 17. There is a mild paresis of the soft palate and dysphonia. A muscle biopsy was taken at the age of 11.
For metabolic flux and mitochondrial enzyme analysis fresh muscle biopsies were taken from subjects P2, P3, and P5 and homogenates containing intact mitochondria were prepared.14 Measurement of single enzymes of the mitochondrial energy metabolism15, 16 showed normal or only mildly affected activities for all respiratory chain complexes and the pyruvate dehydrogenase complex (Table 1). The investigation of oxidation rates of mitochondrial substrates revealed normal metabolization of acetylcarnitine-containing substrates, whereas pyruvate-containing substrates were significantly reduced (Table 1). This result indicated a defect within the mitochondrial pyruvate oxidation route. Immunoblot analysis17 with an antibody cocktail against PDHC subunits (MSP02, Mitosciences) showed no changes of the protein content of either subunit E1 α, E1 β, E2, or E3 binding protein (Figure S1, available online). Likewise, expression-level analysis by real-time PCR of the PDHC subunits (PDHA1 [MIM 300502], PDHB [MIM 179060], DLAT [MIM 608770], DLD [MIM 238331], and PDHX [MIM 608769]) and PDHC phosphatases (PPAPDC2 [MIM 605993], PDP2, and PDPR) revealed no abnormalities. Finally, mutation analysis of all these genes by Sanger sequencing14 discovered no abnormalities. Also the analysis of the thiamine transporter genes (SLC19A2 [MIM 603941], SLC19A3 [MIM 606152], and SLC25A19 [MIM 606521]) revealed no mutation.
Table 1.
Enzymatic Investigations in Muscle Tissue
| P2 | P3 | P5 | Controls | |
|---|---|---|---|---|
| Substrate Oxidation Rates (nmol/h/mUnit Citrate synthase) | ||||
| [1-14C]pyruvate+malate+ADP | 0.44 | 0.55 | 0.75 | 1.54–3.55 |
| [1-14C]pyruvate+carnitine+ADP | 0.42 | 0.54 | 0.80 | 1.65–3.66 |
| [U-14C]malate+pyruvate+malonate+ADP | 0.58 | 0.67 | 0.96 | 1.56–3.87 |
| [U-14C]malate+acetylcarn.+malonate+ADP | 1.13 | 1.51 | 1.70 | 1.16–2.82 |
| [U-14C]malate+acetylcarn.+arsenite+ADP | 0.68 | 0.94 | 0.92 | 0.57–1.52 |
| [1,4-14C]succinate+acetylcarnitine+ADP | 0.85 | 1.20 | 1.38 | 0.90–2.06 |
| Enzyme Activities (mUnit/mUnit citrate synthase) | ||||
| Complex I | 0.16 | 0.06 | 0.16 | 0.14–0.35 |
| Complex I+III | 0.51 | 0.21 | 0.42 | 0.24–0.81 |
| Complex II | 0.21 | 0.14 | 0.35 | 0.18–0.41 |
| Complex II+III | 0.42 | 0.31 | 0.67 | 0.30–0.67 |
| Complex III | 1.82 | 0.80 | 1.40 | 1.45–3.76 |
| Cytochrome c oxidase | 1.51 | 1.11 | 2.21 | 0.91–2.24 |
| Oligomycin sensitive ATPase (complex V) | 0.30 | 0.51 | 0.70 | 0.42–1.26 |
| Pyruvate dehydrogenase complex | 0.031 | 0.062 | 0.034 | 0.026–0.079 |
| Citrate synthase (mUnit/mg protein) | 154 | 294 | 244 | 150–338 |
Functional investigation of post nuclear supernatant prepared from native, unfrozen muscle showed reduced activities in all pyruvate containing oxidation reactions in the affected individuals (P2, family A II-2; P3, family B III-2; P5, family C II-2 in Figure 2). Analysis of respiratory chain enzymes, ATPase and pyruvate dehydrogenase complex revealed no significant reductions in muscle.
In order to test for a cofactor deficiency, we measured PDHC activity either under standard conditions15, 16 in the presence of 0.8 mmol/l TPP or without any addition of the cofactor. As shown in Figure 3, in the absence of TPP the PDHC activity was decreased in muscle extracts from individuals P2, P3, and P5 versus controls (p value 0.022, Student's unpaired t test). This difference became even more obvious when the ratio of TPP-unstimulated versus TPP-stimulated PDHC activity was calculated (p = 0.0012). The response to TPP indicated a deficiency within the thiamine metabolism (Figure 1).
Figure 3.
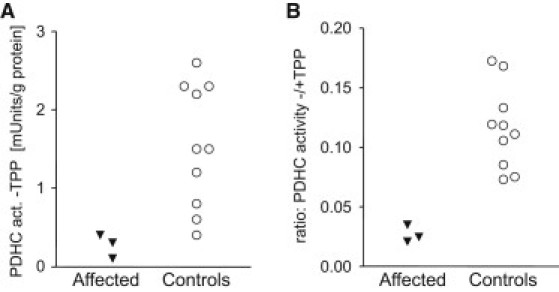
Investigations of Cofactor Dependency of Pyruvate Dehydrogenase Complex Activity
Pyruvate dehydrogenase complex was measured in the in the absence of thiamine pyrophosphate and showed a decrease in the affected individuals compared to controls (A). An even more pronounced decrease was found in the ratio of PDHC activities under TPP-unsupplemented versus TPP-supplemented (0.8 mmol/l) assay conditions (B).
Therefore, the thiamine content was quantified in muscle, blood, and fibroblast samples by an high-pressure liquid chromatography (HPLC) procedure,18 which was adapted to small sample sizes. Briefly, either 50 μl of muscle 600 g supernatant (protein content 2–6 mg/ml) or 50 μl of fibroblast cytosolic fraction was deproteinized by the addition of 5.5 μl of 50% trichloroacetic acid (TCA); 100 μl of whole EDTA-blood were deproteinised by the addition 100 μl of 10% TCA. After incubation for 15 min on ice the samples were centrifuged 5 min at 10,000 g, and TCA was removed from the supernatant by extracting with the two-fold volume (related to the original enzyme solution) of diethylether twice. Prior to HPLC analysis 45 μl of the thiamine-containing solutions were derivatized to the corresponding thiochromes by the addition of 5 μl of a freshly prepared solution of 10 mM potassium hexacyanoferrate (III) in 15% NaOH and immediate mixing. The samples were put in an autosampler protected from light, and 20 μl were applied with a flow rate of 1.0 ml/min to an HPLC system equipped with a reversed phase analytical column (Agilent, Eclipse Plus C18, 5 μm, 4.6 × 150 mm) and a C18 guard column (Supelco, 581372-U). The elution condition were 0–1.5 min: 0% B, 1.5–3.5 min: 0%–12% B, 3.5–5.0 min: 12%–50% B, 5.0–8.0 min 50% B, 8.0–9.5 min 50%–0% B, 9.5–12.1 min: 0% B, with linear gradients. Puffer A was 10% methanol, 90% 25 mmol/l sodium phosphate (pH 7.0); puffer B was 70% methanol, 30% 25 mmol/l sodium phosphate (pH 7.0). Application of the sample was at 1.5 min in this cycle. The fluorimetric detector was set to excitation wavelength 375 nm and emission wavelength 435 nm. TPP, TMP, and thiamine eluted at 5.0, 5.6, and 7.7 min, respectively. Standard solutions of these three thiamine species at a concentration of 10 and 100 nmol/l each were processed in the same way and used for the calculation of concentrations. As shown in Table 2, TPP was significantly (p = 0.000031) reduced in the muscle of the investigated individuals from all families with the pyruvate oxidation disorders. Also the investigation of the concentration of thiamine, TMP, and TPP in blood from individuals P3, P4, and P5 showed a significant (p = 0.00035) decrease (Table 2). Remarkably these three individuals were under oral supplementation of thiamine (100–200 mg/day) because of the previously discovered deficiency in the oxidation of pyruvate. In fibroblasts there was a reduction of the TPP concentration in the cells of individual P3, whereas the cells of individual P5 showed a normal concentration of TPP. These cells were grown in a thiamine-rich standard growth medium (DMEM, Sigma D5648, thiamine concentration 10.7 μmol/l).
Table 2.
Concentration of Thiamine, TMP, and TPP Muscle and Fibroblasts and Blood Samples of Affected Individuals and Controls
| TPP | TMP | Thiamine | |
|---|---|---|---|
| Muscle 600 g Supernatant (nmol/g protein) | |||
| Individual P2 | 8.8 | 0.1 | 1.5 |
| Individual P3 | 9.5 | 0.1 | 3.0 |
| Individual P5 | 9.3 | 0.2 | 1.9 |
| Controlsa: mean ± SD | 58.7 ± 12.6 | 1.1 ± 0.4 | 0.9 ± 1.4 |
| Controlsa: range | 41.6–81.6 | 0.4–1.7 | 0.2–4.4 |
| Fibroblast Cytosol (nmol/g protein) | |||
| Individual P3 (n = 2) | 11.2 | 0.1 | 129.9 |
| Individual P5 (n = 2) | 74.5 | 1.5 | 297.2 |
| Controlsb: mean ± SD | 92.6 ± 37.3 | 6.8 ± 4.2 | 160.2 ± 73.6 |
| Controlsb: range | 56.0–158.2 | 2.7–15.1 | 78.7–262.7 |
| Blood (nmol/l) | |||
| Individual P3 | 68.0 | 2.2 | 21.4 |
| Individual P4 | 50.4 | 1.2 | 5.3 |
| Individual P5 | 96.9 | 18.1 | 67.5 |
| Controlsc: mean ± SD | 190.9 ± 41.5 | 6.2 ± 1.3 | 10.7 ± 6.1 |
| Controlsc: Range | 132.2–271.2 | 4.1–8.8 | 5.0–26.4 |
Affected individuals P3–P5 (Figure 2) were under supplementation of thiamine (100–200 mg/day) when the blood sample was taken.
n = 9.
n = 8.
n = 10.
Although mutations in the coding region of genes associated with defects in PDHC and the thiamine metabolism have been excluded, the TPP-dependent activity of PDHC and the reduced levels of TPP still pointed to a defect within the thiamine metabolism in the investigated families. Finally, the analysis of TPK1 (RefSeq NM_022445.3), the gene of the thiamine pyrophosphokinase, led to the identification of mutations in the affected individuals from all three families (Figure 4). Individual P1 and P2 were compound heterozygous for the mutations c.[148A>C]+[501+4A>T] (p.[Asn50His]+[Val119_Pro167del]), individuals P3 and P4 were homozygous for the mutation c.119T>C (p.Leu40Pro), and individual P5 was compound heterozygous for the mutations c.[179_182delGAGA]+[656A>G] (p.[Arg60LysfsX52]+[Asn219Ser]). The missense mutation in individuals 1 and 2 affects an amino acid that is phylogenetically conserved within eukaryotes (Figure 5). The missense mutation of individual P5 is even conserved within eukaryots and prokaryots. The missense mutation of individuals P3 and P4 changes a less conserved amino acid to a proline, an exchange between the first alpha helix and a following beta sheet,19 which is not found in other organisms. The location of the mutations is illustrated in the crystalline structure of the human TPK (Figure S2), which was recently published in the Protein Data Bank, accession number 3S4Y. The splice-site mutation in individuals 1 and 2 dramatically decreases the splice efficiency of exon 7; the resulting full-length cDNA contained only the second allele with the missense mutation (Figure 4 and Figure S4). The frameshift mutation in individual P5 leads to an early termination and most likely no functional protein.
Figure 4.
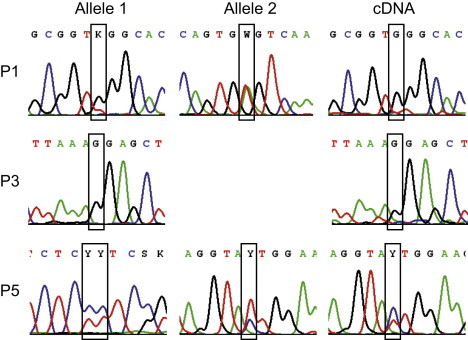
Sequence Analysis of TPK1
Sequence analysis revealed compound heterozygous mutations c.[148A>C]+[501+4A>T] p.[Asn50His]+[Val119_Pro167del] in the individuals P1 and P2 (Figure 2) in TPK1 (RefSeq NM_022445.3). In the individuals P3 and P4 a homozygous mutation c.119T>C p.Leu40Pro was found. In the individual P5 compound heterozygous mutations c.[179_182delGAGA]+[656A>G] p.[Arg60LysfsX52]+[Asn219Ser] were found. The expression of the missense mutations is shown in the cDNA of the affected individuals.
Figure 5.

Phylogenetic Conservation of the Identified Thiamine Phyrophosphokinase Mutations
Alignment by ClustalW24 shows phylogenetic conservation of missense mutations found in the thiamine pyrophosphokinase in the affected individuals (P1–P5, cf. Figure 2).
In order to analyze the effect of the mutations on the TPK protein amount an immunoblot analysis of cell extracts was performed with a rabbit anti-TPK1 antibody (HPA021849, Sigma-Aldrich). As shown in Figure 6, a clear decrease of the full-length (27.3 kDa) thiamine pyrophosphokinase was found in the samples of the affected individuals from all three families. The antibody also recognizes a second protein of lower molecular weight, which might represent the shorter splice variant b of the TPK enzyme (RefSeq NM_001042482.1). Splice variant b lacks exon 7. The corresponding protein has a deletion of 49 amino acids (5.4 kDa) compared to the full-length variant a. The physiological relevance of the short transcript b is not known. Because it lacks a conserved portion of the protein, a functional role as thiamine pyrophosphokinase is unlikely.
Figure 6.
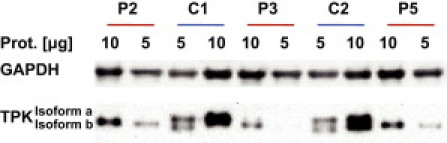
Immunoblot Analysis of Muscle Extracts
A decrease in the amount of the active isoform a (27.3 kDa) was found in all affected individuals with a TPK antibody (A). An antibody against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as loading control for cytosolic protein (B).
We found a deficiency of the TPK in five affected individuals from three families presenting with a variable degree of psychomotor retardation, progressive dystonia, and lactic acidosis. There was a delay of one to several years until the onset of first symptoms. All affected individuals had several crises, triggered by infections, and in four of five affected individuals there was a clearly progressive course of the disease. Two siblings, from the most severely affected family, died in childhood. Two siblings from the family with intermediate clinical affection have a milder but progressive course with onset in the fourth year of life with several crises and development of a severe dystonia. Individual P5 (family C, II-2 in Figure 2) with the best clinical outcome had several, in some cases severe, metabolic crises with onset in infancy, but in contrast to the others he always recovered more or less completely. Remarkably, this individual was compound heterozygous for a frameshift mutation and a missense mutation, p.Asn219Ser, which is highly conserved, even in prokaryotes. This p.Asn219Ser mutation, in contrast to the other missense mutations, is located in the C-terminal part of the protein that is close to the binding site for thiamine, whereas the other point mutations that result in a more severe phenotype are located close to the conserved Asp46 that is involved in magnesium binding.19
Deficiency of the cofactor TPP is known to be caused either by alimentary deficiency, by the metabolic defects in plasma membrane transport of thiamine (SLC19A2 and SLC19A3 isoforms), or by the transport of TPP across the inner mitochondrial membrane (SLC25A19). With the exception of the SLC19A2 defect, which results in megaloblastic anemia, thrombocytopenia, diabetes mellitus and sensorineural deafness (if not treated or if treated too late individuals also develop mental retardation), the other two metabolic defects and the alimentary deficiency of thiamine clinically resemble each other and the novel TPK defect. All of these diseases primarily result in neurologic symptoms, including ataxia and dystonia. An MRI usually shows symmetric affection of the basal ganglia and sometimes progressive brain atrophy. In severe forms of nutritional thiamine deficiency, cardiac affection has been reported,20, 21 as has also been found in one of our individuals with TPK deficiency (individual P3, family B, III-2 in Figure 2). The neurological symptoms in thiamine deficiency are similar to defects of the pyruvate dehydrogenase complex, which most frequently present as Leigh syndrome with basal ganglia involvement. Therefore, the nervous system, which is highly specialized in the use of glucose for energy generation, seems to be most vulnerable to PDHC deficiency due to TPP depletion. The phenotype of α-ketoglutarate dehydrogenase deficiency, the other TPP-dependent enzyme of the mitochondrial energy metabolism, is not known because no isolated defects of this enzyme have been reported so far. It can be assumed that α-KGDH defects would affect the energy generation from fatty acids to the same extent as glucose is. Therefore, an effect on the highly energy-dependent cardiac muscle can be anticipated. In fact, two of the three individuals with a mutation in the dihydrolipoamide dehydrogenase (DLD, E3 subunit, MIM 238331) and predominant deficiency of the α-KGDH suffered from hypertrophic cardiomyopathy.22 Furthermore, the elevated excretion of α-ketoglutaric acid, which was found several times in four of the five affected individuals in our study, might reflect the deficiency in the α-KGDH, and excretion of α-ketoglutaric acid was also found in individuals with Amish microcephaly due to defects in the mitochondrial TPP carrier SLC25A196 but not in the milder variant of this disease.7 Consequences of the deficiency in the other two TPP-dependent enzymes, transketolase and branched chain keto acid dehydrogenase, have not been observed in the affected individuals with TPK deficiency. Megaloblastic anemia, which is found because of transketolase deficiency in the SLC19A2 defect, is an unusual condition that is also found in Wernicke encephalopathy.11 Elevation of branched chain amino acids has not been reported in any condition of thiamine deficiency.
The therapeutic intervention in TPK deficiency was not the focus of this work. However, it has to be noted that the surviving individuals P3, P4, and P5 (Figure 2) were initially put on supplementation with thiamine (100–200 mg/day) because of the biochemically diagnosed defect in pyruvate oxidation and because a hidden PDHC deficiency was suspected. Individuals P4 and P5 stabilized and even improved to some extent after thiamine substitution was initiated; in individual P3 no clear improvement could be observed within 2 years of treatment. We have several considerations for modifying the therapy of this disease in the future: on the one hand the dosage of thiamine could be further increased in order to increase the substrate concentration for the residual TPK and prevent depletion of this vitamin. A positive effect of high thiamine concentration can be delineated, at least in case of individual P5, because the level of TPP reached the normal range in fibroblasts of this individual when in the presence of 10.7 μmol/l thiamine in the growth medium (Table 2). Because the synthesis of the active cofactor TPP from thiamine is affected, a direct supplementation of TPP might be considered. Usually thiamine and TMP but not TPP is taken up from the intestine. However, some transport activity for TPP has been reported for the folate transporter SLC19A1.23 Because TPP is not a licensed drug, the effectivity and safety need to be demonstrated. Furthermore, it is unclear whether TPP is able to cross the blood-brain barrier.
Our results suggest that TPK deficiency should be considered in individuals with suspect symptoms like ataxia and dystonia as well as lactate elevation during metabolic crises. If a muscle biopsy is performed, we advise the functional analysis of intact mitochondria from unfrozen samples in order to identify the pyruvate oxidation defect (Table 1). In frozen biopsy samples the activity of the pyruvate dehydrogenase complex should be measured in the presence and absence of TPP, the ratio of these activities should be indicative for TPP deficiency (Figure 3). Furthermore, the TPP concentration could be quantified in the tissue or tissue extracts.
In summary, we show a deficiency of the thiamine pyrophosphokinase in five individuals that results in a decrease of the essential cofactor thiamine pyrophosphate and a predominantly neurologic disease with onset in childhood. These results enlarge the spectrum of inborn errors of thiamine metabolism that usually have a severe neurologic involvement in common.
Acknowledgments
Supported by the Vereinigung zur Förderung der pädiatrischen Forschung und Fortbildung Salzburg (J.A.M, F.A.Z., J.K., and W.S.), the Jubiläumsfonds of the Oesterreichische Nationalbank (12568 to W.S.), by the Impulse and Networking Fund of the Helmholtz Association in the framework of the Helmholtz Alliance for Mental Health in an Ageing Society (HA-215 to H.P.), by the Bundesministerium für Bildung und Forschung (BMBF) funded German Network for Mitochondrial Disorders (mitoNET 01GM0867 to H.P., P.F., B.R. and U.A.) and Systems Biology of Metabotypes (SysMBo 0315494A to H.P.).
Published online: December 8, 2011
Footnotes
Supplemental Data include three figures and can be found with this article online at http://www.cell.com/AJHG/.
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Protein Data Bank, http://www.pdb.org
Supplemental Data
References
- 1.Gangolf M., Czerniecki J., Radermecker M., Detry O., Nisolle M., Jouan C., Martin D., Chantraine F., Lakaye B., Wins P., et al. Thiamine status in humans and content of phosphorylated thiamine derivatives in biopsies and cultured cells. PLoS ONE. 2010;5:e13616. doi: 10.1371/journal.pone.0013616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Labay V., Raz T., Baron D., Mandel H., Williams H., Barrett T., Szargel R., McDonald L., Shalata A., Nosaka K., et al. Mutations in SLC19A2 cause thiamine-responsive megaloblastic anaemia associated with diabetes mellitus and deafness. Nat. Genet. 1999;22:300–304. doi: 10.1038/10372. [DOI] [PubMed] [Google Scholar]
- 3.Ozand P.T., Gascon G.G., Al Essa M., Joshi S., Al Jishi E., Bakheet S., Al Watban J., Al-Kawi M.Z., Dabbagh O. Biotin-responsive basal ganglia disease: A novel entity. Brain. 1998;121:1267–1279. doi: 10.1093/brain/121.7.1267. [DOI] [PubMed] [Google Scholar]
- 4.Zeng W.Q., Al-Yamani E., Acierno J.S., Jr., Slaugenhaupt S., Gillis T., MacDonald M.E., Ozand P.T., Gusella J.F. Biotin-responsive basal ganglia disease maps to 2q36.3 and is due to mutations in SLC19A3. Am. J. Hum. Genet. 2005;77:16–26. doi: 10.1086/431216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Subramanian V.S., Marchant J.S., Said H.M. Biotin-responsive basal ganglia disease-linked mutations inhibit thiamine transport via hTHTR2: Biotin is not a substrate for hTHTR2. Am. J. Physiol. Cell Physiol. 2006;291:C851–C859. doi: 10.1152/ajpcell.00105.2006. [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg M.J., Agarwala R., Bouffard G., Davis J., Fiermonte G., Hilliard M.S., Koch T., Kalikin L.M., Makalowska I., Morton D.H., et al. Mutant deoxynucleotide carrier is associated with congenital microcephaly. Nat. Genet. 2002;32:175–179. doi: 10.1038/ng948. [DOI] [PubMed] [Google Scholar]
- 7.Spiegel R., Shaag A., Edvardson S., Mandel H., Stepensky P., Shalev S.A., Horovitz Y., Pines O., Elpeleg O. SLC25A19 mutation as a cause of neuropathy and bilateral striatal necrosis. Ann. Neurol. 2009;66:419–424. doi: 10.1002/ana.21752. [DOI] [PubMed] [Google Scholar]
- 8.Kril J.J. Neuropathology of thiamine deficiency disorders. Metab. Brain Dis. 1996;11:9–17. doi: 10.1007/BF02080928. [DOI] [PubMed] [Google Scholar]
- 9.Ogershok P.R., Rahman A., Nestor S., Brick J. Wernicke encephalopathy in nonalcoholic patients. Am. J. Med. Sci. 2002;323:107–111. doi: 10.1097/00000441-200202000-00010. [DOI] [PubMed] [Google Scholar]
- 10.Devathasan G., Koh C. Wernicke's encephalopathy in prolonged fasting. Lancet. 1982;2:1108–1109. doi: 10.1016/s0140-6736(82)90039-3. [DOI] [PubMed] [Google Scholar]
- 11.Saad L., Silva L.F., Banzato C.E., Dantas C.R., Garcia C., Jr. Anorexia nervosa and Wernicke-Korsakoff syndrome: A case report. J. Med. Case Reports. 2010;4:217. doi: 10.1186/1752-1947-4-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aasheim E.T. Wernicke encephalopathy after bariatric surgery: A systematic review. Ann. Surg. 2008;248:714–720. doi: 10.1097/SLA.0b013e3181884308. [DOI] [PubMed] [Google Scholar]
- 13.Uruha A., Shimizu T., Katoh T., Yamasaki Y., Matsubara S. Wernicke's Encephalopathy in a Patient with Peptic Ulcer Disease. Case Report Med. 2011;2011:156104. doi: 10.1155/2011/156104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mayr J.A., Merkel O., Kohlwein S.D., Gebhardt B.R., Böhles H., Fötschl U., Koch J., Jaksch M., Lochmüller H., Horváth R., et al. Mitochondrial phosphate-carrier deficiency: A novel disorder of oxidative phosphorylation. Am. J. Hum. Genet. 2007;80:478–484. doi: 10.1086/511788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mayr J.A., Paul J., Pecina P., Kurnik P., Förster H., Fötschl U., Sperl W., Houstek J. Reduced respiratory control with ADP and changed pattern of respiratory chain enzymes as a result of selective deficiency of the mitochondrial ATP synthase. Pediatr. Res. 2004;55:988–994. doi: 10.1203/01.pdr.0000127016.67809.6b. [DOI] [PubMed] [Google Scholar]
- 16.Strassburg H.M., Koch J., Mayr J., Sperl W., Boltshauser E. Acute flaccid paralysis as initial symptom in 4 patients with novel E1alpha mutations of the pyruvate dehydrogenase complex. Neuropediatrics. 2006;37:137–141. doi: 10.1055/s-2006-924555. [DOI] [PubMed] [Google Scholar]
- 17.Feichtinger R.G., Zimmermann F., Mayr J.A., Neureiter D., Hauser-Kronberger C., Schilling F.H., Jones N., Sperl W., Kofler B. Low aerobic mitochondrial energy metabolism in poorly- or undifferentiated neuroblastoma. BMC Cancer. 2010;10:149. doi: 10.1186/1471-2407-10-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu J., Frank E.L. Rapid HPLC measurement of thiamine and its phosphate esters in whole blood. Clin. Chem. 2008;54:901–906. doi: 10.1373/clinchem.2007.099077. [DOI] [PubMed] [Google Scholar]
- 19.Santini S., Monchois V., Mouz N., Sigoillot C., Rousselle T., Claverie J.M., Abergel C. Structural characterization of CA1462, the Candida albicans thiamine pyrophosphokinase. BMC Struct. Biol. 2008;8:33. doi: 10.1186/1472-6807-8-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sica D.A. Loop diuretic therapy, thiamine balance, and heart failure. Congest. Heart Fail. 2007;13:244–247. doi: 10.1111/j.1527-5299.2007.06260.x. [DOI] [PubMed] [Google Scholar]
- 21.Rao S.N., Chandak G.R. Cardiac beriberi: Often a missed diagnosis. J. Trop. Pediatr. 2010;56:284–285. doi: 10.1093/tropej/fmp108. [DOI] [PubMed] [Google Scholar]
- 22.Odièvre M.H., Chretien D., Munnich A., Robinson B.H., Dumoulin R., Masmoudi S., Kadhom N., Rötig A., Rustin P., Bonnefont J.P. A novel mutation in the dihydrolipoamide dehydrogenase E3 subunit gene (DLD) resulting in an atypical form of alpha-ketoglutarate dehydrogenase deficiency. Hum. Mutat. 2005;25:323–324. doi: 10.1002/humu.9319. [DOI] [PubMed] [Google Scholar]
- 23.Zhao R., Gao F., Wang Y., Diaz G.A., Gelb B.D., Goldman I.D. Impact of the reduced folate carrier on the accumulation of active thiamin metabolites in murine leukemia cells. J. Biol. Chem. 2001;276:1114–1118. doi: 10.1074/jbc.M007919200. [DOI] [PubMed] [Google Scholar]
- 24.Higgins D.G., Thompson J.D., Gibson T.J. Using CLUSTAL for multiple sequence alignments. Methods Enzymol. 1996;266:383–402. doi: 10.1016/s0076-6879(96)66024-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.