

Abstract
Joubert syndrome related disorders (JSRDs) have broad but variable phenotypic overlap with other ciliopathies. The molecular etiology of this overlap is unclear but probably arises from disrupting common functional module components within primary cilia. To identify additional module elements associated with JSRDs, we performed homozygosity mapping followed by next-generation sequencing (NGS) and uncovered mutations in TMEM237 (previously known as ALS2CR4). We show that loss of the mammalian TMEM237, which localizes to the ciliary transition zone (TZ), results in defective ciliogenesis and deregulation of Wnt signaling. Furthermore, disruption of Danio rerio (zebrafish) tmem237 expression produces gastrulation defects consistent with ciliary dysfunction, and Caenorhabditis elegans jbts-14 genetically interacts with nphp-4, encoding another TZ protein, to control basal body-TZ anchoring to the membrane and ciliogenesis. Both mammalian and C. elegans TMEM237/JBTS-14 require RPGRIP1L/MKS5 for proper TZ localization, and we demonstrate additional functional interactions between C. elegans JBTS-14 and MKS-2/TMEM216, MKSR-1/B9D1, and MKSR-2/B9D2. Collectively, our findings integrate TMEM237/JBTS-14 in a complex interaction network of TZ-associated proteins and reveal a growing contribution of a TZ functional module to the spectrum of ciliopathy phenotypes.
Introduction
Ciliopathies, including the Joubert syndrome related disorders (JSRDs [MIM 213300]), represent an expanding group of physiological and developmental disorders caused by dysfunction of primary cilia.1, 2, 3, 4 These microtubule-based structures are present on nearly every cell type in mammals and perform critical functions associated with sensory perception (e.g., vision, hearing, smell, and mechanosensation) and developmental signaling (including Hedgehog, Wnt, and cyclic nucleotide signaling).5, 6, 7 JSRDs are predominantly autosomal-recessive developmental syndromes that are characterized by a specific midhindbrain malformation (hypoplasia of the cerebellar vermis; abnormally deep interpeduncular fossa at the level of the isthmus and upper pons; and horizontalized, thickened and elongated superior cerebellar peduncles), visualized as the molar tooth sign (MTS) on brain imaging. Additional findings can include polydactyly, ocular coloboma, retinal dystrophy, renal disease, and hepatic fibrosis.8 The JSRDs are genetically heterogeneous with mutations in 14 genes identified to date,9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 although these only account for approximately 50% of mutations in affected individuals.8 The typically autosomal-recessive and genetically heterogeneous ciliopathies Meckel-Gruber syndrome (MKS [MIM 249000]), nephronophthisis (NPHP [MIM 256100]), and Bardet-Biedl syndrome (BBS [MIM 209900]) phenotypically overlap with the JSRDs. MKS is a more severe group of disorders characterized by posterior fossa defects including occipital encephalocele as well as cystic dysplastic kidneys, hepatic bile duct proliferation, and polydactyly.25 NPHP is characterized primarily by renal cysts and is the most frequent cause of end-stage kidney disease in the first three decades of life.26 Primary features of BBS include rod-cone dystrophy, polydactyly, truncal obesity, learning disabilities, hypogonadism and/or genital anomalies, and renal manifestations including NPHP.2, 3
The ciliary basal body is a modified centriolar structure that is anchored to the plasma membrane and is responsible for nucleating the ciliary axoneme, whose formation depends on intraflagellar transport (IFT). The transition zone (TZ), termed connecting cilium in photoreceptor cells, is the most proximal region of the axoneme and is characterized by Y-shaped structures of unknown composition that join the axonemal doublet microtubules to the ciliary membrane. Recently, many JSRDs/MKS/NPHP proteins have been found to localize either at the basal body, or more specifically, at the ciliary TZ just distal to the basal body.24, 27 These include the evolutionarily conserved B9 domain-containing proteins (MKS1 [MIM 609883], MKSR1/B9D1 [MIM 614144], and MKSR2/B9D2 [MIM 611951]) as well as MKS3/Meckelin/TMEM67 [MIM 609884], MKS5/RPGRIP1L [MIM 610937], MKS6/CC2D2A [MIM 612013], NPHP1 [MIM 607100], and NPHP4 [MIM 607215]. Studies in Caenorhabditis elegans have provided evidence that TZ-associated proteins collectively participate in basal body-TZ anchoring to the membrane and establishing a functional ciliary gate during ciliogenesis.27, 28, 29, 30 Several related roles for many of the above-mentioned proteins in mammalian cells have been ascribed, including basal body migration, docking, ciliogenesis, ciliary gate function, and cellular signaling.24, 25, 26, 31, 32 Whether there are additional TZ components relevant to these cellular processes, some of which could be implicated in human diseases, remains unclear.
Here, we identified mutations in TMEM237 in individuals affected with a JSRD and studied this largely uncharacterized gene, which encodes a predicted tetraspan transmembrane protein (TMEM) TMEM237/JBTS-14. Our findings reveal that TMEM237 is important for ciliogenesis in mammalian cells, Danio rerio (zebrafish), and C. elegans. Importantly, we show that TMEM237 interacts functionally with NPHP4, MKS2, MKSR1/B9D1, MKSR2/B9D2, and MKS5/RPGRIP1L at the transition zone. Together, our studies expand the interaction network of TZ proteins and reveal a growing contribution of the TZ in ciliogenesis, signaling, and the JBTS-MKS-NPHP ciliopathy spectrum.
Material and Methods
Research Subjects
We used standard methods to isolate genomic DNA from peripheral blood of the affected persons and family members or from frozen fetal tissue or amniocytes. Informed consent was obtained from all participating individuals, and the studies were approved by the Ethics Boards of the Children's Hospital of Eastern Ontario, Leeds (East) Local Research Ethics Committee, University of Calgary, University of Washington, the University Hospital of Cologne, Duke University, and according to Austrian legislation.
Homozygosity Mapping and Haplotype Analysis
Hutterite Families
A genome scan was initiated with DNA samples from four affected individuals. The samples were amplified individually with a set of 400 polymorphic microsatellite markers (ABI PrismTM Linkage Mapping Set Version 2) with an average spacing of 10 cM and heterozygosity of 0.79. Criteria for regions of interest included homozygosity in at least two out of the four affected individuals and allele sharing in the remaining affected individual(s). For candidate regions, DNA from nine affected individuals and their family members were analyzed with the markers of interest as well as additional microsatellite markers from the 5 cM Linkage Mapping Set (Applied Biosystems) and/or from the Généthon, deCode, or Marshfield maps. Haplotypes were constructed manually, and phase was assigned on the basis of the minimum number of recombinants. LOD scores were not calculated because they were estimated to be below significance for each nuclear family.
Tyrol Families
The 10K Affymetrix SNP array was used to perform genetic mapping with DNA sample from one affected individual. For candidate regions, DNA from all three affected individuals and their family members were analyzed with additional dense microsatellite markers. Haplotypes were constructed manually, and phase was assigned on the basis of the minimum number of recombinants.
Next-Generation Sequencing and Analysis
We used an array capture approach followed by high-throughput Roche 454 sequencing in one affected Hutterite child and his carrier mother. A custom array (NimbleGen) was designed with oligonucleotide probes targeting the minimal region of overlap between the Hutterite and Austrian families. The results from 454 sequencing were analyzed with NextGENe software (SoftGenetics). Sequence reads that passed the quality filtering were aligned to the reference sequences of the human genome with the Sequence Alignment function. Homozygous single nucleotide variants, microinsertions, and microdeletions were called, and variants were filtered out when they were present in dbSNP132.
Cells
Mouse inner medullary collecting duct (IMCD3) cells were grown in Dulbecco's modified Eagle's medium (DMEM)/Ham's F12 supplemented with 10% fetal calf serum at 37°C/5% CO2. Fibroblasts from a normal control were maintained in Fibroblast Growth Medium (Genlantis) supplemented with 0.2 mg/ml geneticin. Fibroblast lines derived from the affected individuals were grown in DMEM/F12 medium.
Antibodies
The following primary antibodies were used: rabbit anti-Tmem237, FLJ-FM, and FLJ-LG, have been described previously;33 mouse anti-polyglutamylated tubulin clone GT-335 (Enzo Life Sciences); rabbit anti-GFP (Living Colors A.v. Peptide Antibody, Clontech); rabbit-anti-γ-tubulin, mouse anti-β-actin clone AC-15 (Abcam Ltd.); and mouse anti-RhoA (Cytoskeleton). Rabbit anti-meckelin C terminus, raised against amino acids 982-995, has been described previously.34 Rabbit anti-TMEM216 has also been described.19 Secondary antibodies were Alexa-Fluor 488-, Alexa-Fluor 568-conjugated goat anti-mouse IgG, and goat anti-rabbit IgG (Molecular Probes). Alexa-Fluor 488 phalloidin conjugate (Molecular Probes) was used to visualize F-actin and 4′,6-diamidino-2-phenylindole (DAPI) to visualize nuclei.
Whole-Cell Extract Preparation and Immunoblotting
Whole-cell extracts (WCE) containing total soluble proteins were prepared from confluent IMCD3 cells that had been transiently transfected with 1.0 μg plasmid constructs in 90 mm tissue culture dishes, or scaled down as appropriate. Proteins were analyzed by SDS-PAGE (with 4%–12% acrylamide gradient gels) and immunoblot analysis according to standard protocols with either rabbit polyclonal antisera (final dilutions of ×200–1,000) or mouse monoclonal antibodies (MAbs) (×1,000–5,000). Appropriate HRP-conjugated secondary antibodies (Dako) were used (final dilutions of ×10,000–25,000) for detection by the enhanced chemiluminescence Femto West immunoblot detection system (Pierce). Full scans of immunoblots are shown in Figure S10 (available online).
RhoA Activation Assays
The activated GTP-bound isoform of RhoA was specifically assayed in pull-down assays with a GST fusion protein of the Rho effector rhotekin (Cytoskeleton) with conditions recommended by the manufacturers. WCEs were processed as rapidly as possible at 4°C and snap-frozen in liquid nitrogen. Total RhoA (in WCEs) and pull-down protein was immunodetected on immunoblot with a proprietary RhoA monoclonal antibody (Cytoskeleton). Immunoblot analysis for total RhoA and β-actin were used as loading controls.
Canonical Wnt Activity (Topflash) Luciferase Assays
For luciferase assays of canonical Wnt activity, fibroblasts were grown in 48-well plates and cotransfected with 0.25 μg Topflash firefly luciferase construct (or Fopflash as a negative control); 0.25 μg of expression constructs (pCMV HA-meckelin, pCMV GFP-TMEM216, pEGFPN1-TMEM237, or empty pCMV-HA/pEGFPN1 vector); and 0.05 μg of pRL-TK (Promega; Renilla luciferase construct used as an internal control reporter). Cells were treated with Wnt3A- [MIM 606359] or Wnt5A- [MIM 164975] conditioned media to stimulate or inhibit the canonical Wnt pathway. We obtained Wnt3A- or Wnt5A-conditioned media from stably-transfected L cells with Wnt3A or Wnt5A expression vectors.35 Control media were obtained from untransfected L cells. Activities from firefly and Renilla luciferases were assayed with the Dual-Luciferase Reporter Assay system (Promega) on a Mithras LB940 (Berthold Technologies) luminometer. Minimal responses were noted with coexpression of the Fopflash negative control reporter construct (data not shown). Raw readings were normalized with Renilla luciferase values. Results reported are from at least three independent biological replicates.
Transfection and siRNA
For transfection with plasmids, cells at 80% confluency were transfected with TransIT-LK (Mirus) according to the manufacturer's instructions and as described previously.34 For RNAi knockdown, siRNA duplexes were designed against mouse Tmem237 sequence (NC_000067.5) with Ambion's custom Silencer Select siRNA service. Antisense sequences were as follows: Tmem237: duplex 1 5′-GGAUCUUAGUGAAGAGUUATT and duplex 2 5′-GAACGAAAACGGCAUUGAUTT. The medium or low GC nontargeting scrambled siRNA duplexes (Invitrogen) were used as negative controls. IMCD3 cells were transfected with 100 pmol of each siRNA at 60%–80% confluency with Lipofectamine RNAiMax (Invitrogen) and as described previously.19
Immunofluorescence and Confocal Microscopy
Cells were seeded at 2 × 104 cells/well on glass coverslips in six-well plates and fixed in ice-cold methanol (5 min at −20°C) or 2% paraformaldehyde (20 min at room temperature). Permeabilization, blocking methods and immunofluorescence staining were essentially as described previously.34 Primary antibodies were used at final dilutions of ×200–1,000. Secondary antibodies and phalloidin conjugate were diluted ×200. Confocal images were obtained with a Nikon Eclipse TE2000-E system controlled and processed by EZ-C1 3.50 (Nikon) software. Images were assembled with Adobe Illustrator CS2.
Whole-Mount In Situ Hybridization on Zebrafish Embryos
Whole-mount in situ hybridization was performed as described previously.36 Briefly, full-length cDNAs of zebrafish tmem237a (XM_680251.3), tmem237b (NM_001004636.1), krox20 (NM_130997.2), and myoD (NM_131262.2) were obtained by RT-PCR amplification from total RNA extracted from 24 hpf embryos. The cDNAs were cloned into pGEM-T vector (Promega) and used as templates for in vitro synthesis of antisense RNA probes. Embryos were fixed in 4% paraformaldehyde in PBS, hybridized with DIG-labeled riboprobes at 65°C, and followed by incubation with anti-DIG antibody conjugated with alkaline phosphatase (AP) (Roche). NBT and BICP were used as the substrates of AP to generate the purple coloration.
Morpholino Knockdown and Rescue
Two splice-blocking morpholino antisense oligonucleotides (MO) (zf.tmem237a MO: 5′-TTGTCTGTGTGAAAGGCAGAAATCA-3′ and zf.tmem237b MO: 5′- TGGAAACCTACACTTAACAATATGT-3′) (Gene Tools) were used to knockdown tmem237a (NC_007120.5) and tmem237b (NC_007117.5), respectively, in zebrafish. Gene-specific MOs or control MO (2 ng for rescue and 1 ng for interaction studies) were microinjected into one-two cell stage embryos. Full-length cDNA of human TMEM237 (NM_001044385.1) was obtained by RT-PCR amplification of total RNA extracted from a human fibroblast cell line, which was then subcloned into pCS2+ vector. The human TMEM216 and human MKS3 cDNAs subcloned into pCS2+ vector were described previously.37, 38 In vitro mRNA synthesis was carried out with the mMessagemMachine kit (Ambion). The synthetic mRNA was coinjected with MOs where indicated. For gastrulation phenotypes, embryos were scored according to previously established objective criteria37, 38, 39 at 8–10 somites with masked scoring, n = 76–112 embryos/injection batch. The efficacy of the morpholinos is shown in Figure S11.
C. elegans Strain Construction and Imaging
All strains were maintained and cultured at 20°C. Those carrying mutations in C. elegans mksr-1(ok2092), mksr-2(tm2452), mks-5(tm3100), mks-3(tm2547), mks-6(gk674), and nphp-4(tm925), were obtained from the C. elegans Gene Knockout Consortium and the National Bioresource Project and outcrossed to wild-type (N2) at least four times. Standard mating procedures were used to introduce GFP-tagged protein constructs for JBTS-14 and MKS-2 into different genetic backgrounds. Single-worm PCR reactions were used to genotype the various mutants. GFP-tagged variants of C. elegans JBTS-14 and MKS-2 were generated by producing translational constructs containing each native promoter and all the exons and introns fused in-frame to EGFP; transgenic lines for these constructs were generated as reported previously.40 Subcellular localization was assessed by fluorescence microscopy for both the wild-type (N2) or the indicated TZ mutant backgrounds. Mislocalization phenotypes were confirmed by analyzing at least 50 individuals for each strain.
C. elegans Phenotypic Assays for Ciliary Structure and Chemosensation
Assays for defective osmotic avoidance were performed as described with a ring of 8 M glycerol and five individuals per ring for 10 min.41 The filling of environmentally exposed ciliated sensory neurons with DiI was completed as previously described.40 Dye-stained worms were imaged with fluorescence microscopy and intensities were quantitated with ImageJ.
Transmission Electron Microscopy
Transmission Electron Microscopy (TEM) was performed on staged young adult worms exactly as described previously.42
Identification of jbts-14 Mutant Strain
A nonsense allele (gk287997) of jbts-14 (F53A9.4) was identified by genome sequencing of an EMS-mutagenized C. elegans strain.
Generation of mks-2 Mutant Strain
The mutagenesis protocol for generating an mks-2 (C30B5.9) allele was modified from the protocol described previously.43 Strain EG1642 (genotype lin-15B (n765)X; oxEx166[HSP::MosTransposase + cc::gfp]) carrying the extrachromosomal Mos transposase under a heat-shock promoter was crossed into strain ttTi38815, which contains a Mos1 insertion in an intergenic region ∼2.4 kb away from C30B5.9 (mks-2). Two hundred and fifty young adult (ttTi38815;nxEx166[HSP::MosTransposase + cc::gfp]) worms (P0) were placed on one plate and heat shocked (33°C in a water bath for 1 hr; 1 hr in a 20°C room; 33°C in the water bath for 1 hr again). After 2 hr recovery at 20°C, the P0 worms were placed onto individual plates and allowed to lay eggs (F1) for 40 hr at 20°C. The P0 worms were removed and the plates were placed at 20°C until the worms were grown to the F2 generation. Then, ∼50% of worms (a population of F1 and F2) from each plate were recovered, and crude DNA lysates (200 μl) were prepared as described at Protocol Online. To identify lysates containing an mks-2 deletion, we performed PCR by using a primer set (GTACTATAGCGGTGCATTCCAAC and AAAAAACCAACAGAACCAGGCTGC) flanking the gene and Mos1 transposon. One lysate containing an ∼3.5 kb deletion was identified. We cloned 400 worms from this plate and screened them by using the same PCR strategy. Using this scheme, we isolated one allele, mks-2(nx111), containing a 3573 bp deletion that removes the entire mks-2 coding region.
Results
Mutations in TMEM237 Cause a JSRD
To uncover additional genes mutated in ciliopathies, we investigated a group of ten related Canadian Hutterite individuals with a severe JSRD44 that was previously reported as MKS45 (Table 1). The known loci for JSRDs and MKS were excluded by homozygosity mapping, which suggested that this condition represented a distinct JSRD locus.44 Genome-wide homozygosity mapping with microsatellite markers with the DNA from four affected individuals localized the gene to the JBTS-14 locus on 2q33. Genotyping additional affected and healthy family members refined the region to a 7.5 Mb interval between D2S2327 and D2S1384 (Figure 1A and Figure S1A). A JSRD in a consanguineous family from Tyrol, Austria46 (Table 1), was mapped to an overlapping 9.3 Mb region between D2S115 and D2S325 (Figure 1A and Figure S1B). The minimal gene-rich region (D2S115-D2S1384) contains more than 70 genes; we excluded over 30 of these by using Sanger sequencing.
Table 1.
Clinical and Molecular Data from Families with TMEM237 Mutations
| Individual | Age at Last Assessment | Sex | Living or Deceased | Current Age or Age at Death | Origin | CNS | Kidney | Eye | Other | Nucleotide Changes | Protein Alterations |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family A | |||||||||||
| 1 | 3.5 years | M | L | 8 years | Hutterite | MTS, PF, H | CK | Nys, Str, OA | – | c.52C>T | p.Arg18∗ |
| 2 | 2.5 years | F | D | 7 years | Hutterite | MTS, PF, H, En | CK | Nys, Str | – | c.52C>T | p.Arg18∗ |
| Family B | |||||||||||
| 3 | 27 years | F | D | 28 years | Hutterite | CVH | – | Nys, Str | – | c.52C>T | p.Arg18∗ |
| 4 | 6 years | F | D | 12 years | Hutterite | CVH | – | Nys, Str | – | c.52C>T | p.Arg18∗ |
| Family C | |||||||||||
| 5 | 10 years | M | D | 12 years | Hutterite | MTS | – | Nys, Str, C | IB | c.52C>T | p.Arg18∗ |
| Family D | |||||||||||
| 6 | 0 days | F | D | SB | Hutterite | En, H | CK | ND | CP | c.52C>T | p.Arg18∗ |
| 7 | 2 days | F | D | 2 days | Hutterite | H | CK | ND | P | c.52C>T | p.Arg18∗ |
| Family E | |||||||||||
| 8 | 5 years | F | L | 9 years | Hutterite | MTS, En | CK | Nys, Str, C | VSD | c.52C>T | p.Arg18∗ |
| Family F | |||||||||||
| 9 | 4 months | F | D | 13 months | Hutterite | En, H, DWV | CK | Nys, Str | – | c.52C>T | p.Arg18∗ |
| Family G | |||||||||||
| 10 | 4 years | M | L | 8 years | Hutterite | MTS, PF | CK | Nys, Str | – | c.52C>T | p.Arg18∗ |
| Family H | |||||||||||
| 11 | 3 weeks | F | D | 3 weeks | Hutterite | MTS, DWV, H, En | CK | ND | – | c.52C>T | p.Arg18∗ |
| 12 | 3 months | F | D | 3 months | Hutterite | MTS, DWV, H, En | CK | Nys | – | c.52C>T | p.Arg18∗ |
| Family I | |||||||||||
| 13 | 3 years | M | L | 3 years | Hutterite | MTS, DWV, H | CK | Nys | – | c.52C>T | p.Arg18∗ |
| Family J | |||||||||||
| 14 | 1 days | F | D | 2 days | Hutterite | En | CK | ND | – | c.52C>T | p.Arg18∗ |
| Family K | |||||||||||
| VIII-2 | 3 years | F | D | 4 years | Austrian | CVA | CK | MGDA | – | c.677+1G>T | p.Phe187_Ala251 delinsSer; p.Arg186∗ |
| VIII-4 | Fetus, 22 weeks | ND | D | fetus, 22 weeks | Austrian | H, CVA, CCH, MC | CK | MGDA | no LF | c.677+1G>T | p.Phe187_Ala251 delinsSer; p.Arg186∗ |
| VIII-5 | 2 years | F | L | 11 years | Austrian | CVA, BH | CK | Nys, MGDA | – | c.677+1G>T | p.Phe187_Ala251 delinsSer; p.Arg186∗ |
| Family L | |||||||||||
| 1 | 1.5 years | M | ND | ND | Jordanian | MYC, CVI | ND | ND | – | c.1066dupC | p.Gln356Profs∗23 |
| Family M | |||||||||||
| 1 | 4 years | F | L | 4 years | Hispanic, European | MTS, H, PF, CVI | CK | Nys, Str | – | c.[943+1G>T]+ [76C>T] | p.[IIe291_Trp346 del]+[Gln26∗] |
The following abbreviations are used: –, not affected; BH, brain stem hypoplasia; C, coloboma; CCH, corpus callosum hypoplasia; CK,cystic kidney; CP, cleft palate; CVA, cerebellar vermis aplasia; CVH, cerebellar vermis hypoplasia; CVI, cortical visual impairment; D, deceased; DWV, Dandy–Walker variant; En, encephalocele; H, hydrocephalus; IB, intracranial bleed; L, living; LF, liver fibrosis; MGDA, morning glory disc anomaly; MC, meningocele; MYC, meningomyelocele; MTS, molar tooth sign; ND, no data; Nys, nystagmus; OA, optic atrophy; P, postaxial polydactyly; PF, posterior fossa abnormality; SB, stillborn; Str, strabismus; VSD, ventricular septal defect.
Figure 1.
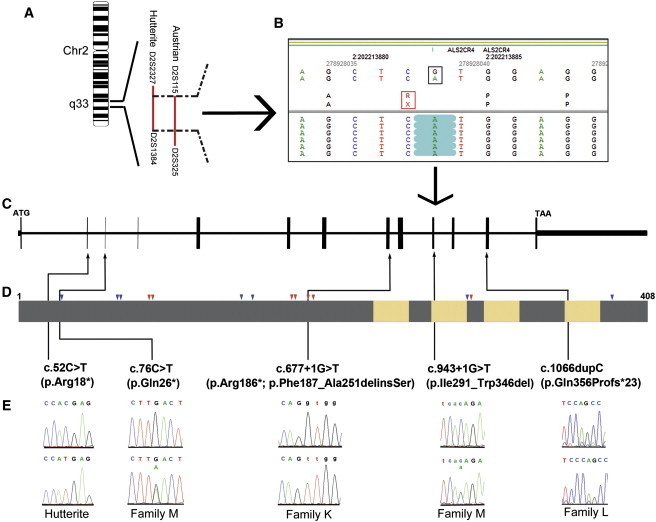
Mutations in TMEM237 Cause a Joubert Syndrome Related Disorder
(A) Chromosomal location of the overlapping JSRD loci of the Hutterite families and the Austrian family.
(B) Targeted sequence capture followed by massively parallel sequencing and data analysis identified a homozygous mutation in TMEM237 (ALS2CR4) in an affected Hutterite individual. The black box shows the G>A change at the genomic level. The red box shows the resultant amino acid change (R>X). The green highlights show that all the sequence reads from the affected child contain an A.
(C) TMEM237 genomic organization.
(D) Domain structure of TMEM237 protein. Yellow indicates the transmembrane domains. Grey indicates the extramembrane region. Red arrows and blue arrows indicate the highly conserved short motifs of basic and acidic amino acids, respectively. Black arrows indicate the positions of identified mutations.
(E) Sequence traces of normal controls (top) and affected individuals (bottom) are shown.
We next employed an array capture approach followed by high-throughput Roche 454 sequencing in one affected Hutterite child and his carrier mother and identified a homozygous C to T transition in TMEM237 ([NM_001044385.1] c.52C>T) leading to a premature stop codon ([NP_001037850.1] p.Arg18∗) (Figures 1B–1E). Screening of normal Hutterite controls (N = 1758) revealed a carrier frequency of 6% for this mutation, but none of the Hutterite controls carried the mutation in the homozygous state. Furthermore, the mutation was not identified in over 105 normal Northern European controls. We next sequenced all the coding and exon/intron boundaries of TMEM237 in the Austrian JSRD family and identified a homozygous change (c.677+1G>T) at the splice donor site of intron 9 (Figures 1C and 1E, and Table S1), which was not present in the Northern European controls. Subsequently, we performed RT-PCR on RNA from a fibroblast cell line derived from an affected individual; two abnormal transcripts were obtained with primers bordering exons 8 and 11. Sequencing of the products revealed: (1) the usage of a cryptic splice donor site at the beginning of exon 9 and a cryptic splice acceptor site in exon 10, leading to a deletion of 65 amino acids and an insertion of a Serine residue (p.Phe187_Ala251delinsSer) and (2) the skipping of exon 9, resulting in a premature stop codon (p.Arg186∗) (Figure 1D and Figure S2).
We screened an additional 201 individuals with JSRD and 90 individuals with MKS/JSRD and identified two additional families with mutations in TMEM237. In a Jordanian child born to consanguineous parents (Table 1), a homozygous TMEM237 frameshift mutation in exon 13 (c.1066dupC) was identified (Figures 1C and 1E). The frameshift mutation is predicted to result in a truncated protein (p.Gln356Profs∗23) (Figure 1D) and was absent in over 105 normal Jordanian controls. Compound heterozygous mutations c.[943+1G>T]+[76C>T] were identified (Figures 1C and 1E) in an affected child from a nonconsanguineous JSRD family of European and Spanish descent (Table 1); her mother carries the c.76C>T mutation, and the father carries the c.943+1G>T mutation. c.76C>T is a nonsense mutation resulting in a premature stop codon (p.Gln26∗). We performed RT-PCR on RNA from a lymphoblast cell line derived from the affected individual; one abnormal transcript was obtained with primers bordering exons 8 and 13 (Figure S3). Sequencing of the product revealed deletion of exons 11 and 12, leading to a deletion of 56 amino acids (p.IIe291_Trp346del) (Figure 1D). These mutations were absent in over 105 normal controls. Together, these data demonstrate that mutations in TMEM237 are causative for JSRD and implicate a largely uncharacterized member of the tetraspan transmembrane protein family, TMEM237, in normal functions of the primary cilium.
TMEM237 spans 23 kb on 2q33 and contains 14 exons. Two alternative TMEM237 transcripts (1 [NM_001044385.1] and 2 [NM_152388.2]), translating into two protein isoforms, a (NP_001037850.1) and b (NP_689601.2), have been proposed in humans. Each transcript utilizes one of the two alternative exons, 1 and 2, which are spliced in a mutually exclusive manner. All positional information in this paper refers to transcript 1 and isoform a. In two mouse proteomic studies, Tmem237 was found in the photoreceptor connecting cilium complex and outer segments.47, 48 TMEM237 is a predicted tetraspan transmembrane protein with both amino and carboxyl termini directed to the cytoplasmic side.33 Alignment of metazoan orthologs with the human protein revealed strong conservation of repetitive short motifs of both basic (R and/or K) and acidic (D and/or E) amino acid residues throughout the intracellular N-terminal region and predicted intracellular loops between transmembrane helices (Figure 1D and Figure S4). Members of the TMEM family, TMEM216/MKS2 [MIM 613277] and meckelin/MKS3/TMEM67, have both been implicated in JSRDs and MKS14, 19, 20, 49 and both localize to the ciliary basal body or TZ region. Because other tetraspan transmembrane proteins function through the formation of complexes with each other and other membrane proteins such as Frizzled receptors,50 it suggests that TMEM237, TMEM216, and MKS3 might cooperate in maintaining normal ciliary functions.
Loss of TMEM237 Causes Ciliogenesis Defects in Mammalian Cells
To elucidate potential roles for TMEM237, we first determined the subcellular localization of the encoded protein. We used two affinity-purified polyclonal antibodies raised against mouse Tmem237 amino acids 76-88 (FLJ-FM) and 390-403 (FLJ-LG) in the amino- and carboxy-terminal domains, respectively.33 These antibodies were used for immunocytochemical staining of a polarized monolayer of ciliated mouse inner medullary collecting duct (IMCD3) cells. Discrete signals at the proximal region of primary cilia were observed, consistent with localization to the TZ (Figure 2A).
Figure 2.
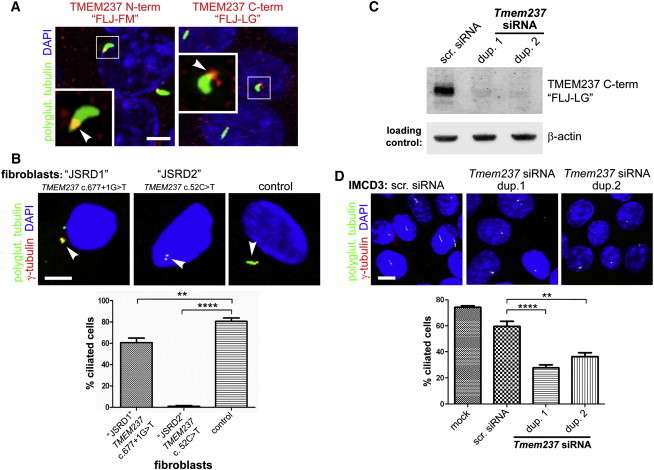
TMEM237 Is Localized to the Ciliary Transition Zone Region and Mutation or Knockdown Results in Defective Ciliogenesis
(A) Overlapping localization of endogenous N-terminal (N-term) or C-terminal (C-term) mouse Tmem237 (red) and polyglutamylated (polyglut.) tubulin (green) at the base of primary cilia and/or mother centriole (arrowheads in magnified insets) of IMCD-3 cells. The scale bar represents 5 μm.
(B) Two different TMEM237-mutated fibroblasts lines (JSRD1 with homozygous splice-site mutation c.677+1G>T and JSRD2 with homozygous nonsense mutation c.52C>T as indicated) show defective ciliogenesis and pairing of centrioles (marked by γ-tubulin, red) compared to a normal control line. The scale bar represents 5 μm. Ciliogenesis is quantitated in the lower panel (n = 500 to 600 for each line; statistical significance of comparisons are shown (∗∗p < 0.01, ∗∗∗∗p < 0.0001 Student t test); error bars indicate standard error of the mean (SEM).
(C) Tmem237 antisera against the C-terminal mouse protein detects a specific 45 kD band33 in immunoblot of control cells transfected with scrambled (scr.) siRNA that is lost after transfection with Tmem237 siRNA duplexes (dup.) 1 or 2. Equal loading is indicated by immunoblot analysis for β-actin.
(D) Defective ciliogenesis in IMCD-3 cells after Tmem237 siRNA knockdown with duplexes (dup.) 1 or 2. Cells stained and ciliogenesis quantified as in (B); the scale bar represents 10 μm.
We next explored the role of TMEM237 function in ciliogenesis. Quantitative real-time (qRT) PCR was used to measure TMEM237 transcript expression in fibroblasts derived from the affected individual with the splice-site mutation (c.677+1G>T, line JSRD1), and the nonsense mutation (c.52C>T, p.Arg18∗, line JSRD2), because existing antibodies were not specific for human TMEM237. Total TMEM237 transcript levels in the JSRD1 and JSRD2 lines were reduced by 99.6% and 98.4%, respectively (Figure S5), although low levels of an exon-skipped mutant transcript were detectable by RT-PCR in JSRD1 with the c.677+1G>T splice-site mutation (Figure S2A). A failure in ciliogenesis was observed in these fibroblasts, after 48 hr of serum starvation (Figure 2B) but not in controls; similar results were seen previously in cells deficient for MKS3 and TMEM216.19, 31 These data were quantified by analyzing the percentage of cells with evident cilia (defined as >1 μm in length) versus those without cilia (defined as <1 μm in length) (Figure 2B). It is possible that the low levels of the exon-skipped mutant transcript in JSRD1 (c.677+1G>T) were sufficient to maintain some ciliogenesis in those fibroblasts. For independent confirmation of these results, we also tested for ciliogenesis defects in IMCD3 cells disrupted for TMEM237 by transient siRNA knockdown. We first confirmed that the expected 45 kDa protein recognized by immunoblotting with the FLJ-LG antibody33 in immunoblotting experiments was lost after transient siRNA knockdown (Figure 2C and Figure S6). Consistent with the results obtained from fibroblasts from affected individuals, transfection with two separate Tmem237 siRNA duplexes impaired ciliogenesis in polarized cells (Figure 2D and Figure S7).
TMEM237 Disruption Results in Deregulation of Wnt Signaling
Many aspects of actin-dependent polarized cell behavior, including morphogenetic cell movements51 and ciliogenesis,52 are mediated by the planar cell polarity (PCP) pathway of noncanonical Wnt signaling.53 Perturbation of noncanonical Wnt signaling is implicated in the pathogenesis of MKS19, 34, 37 and deregulation of the canonical β-catenin pathway is implicated in the ciliopathy disease state.54, 55 To assess whether these pathways were perturbed after TMEM237 mutation or loss, we first determined levels of key mediators in fibroblasts from affected and control individuals. Immunoblot analysis of protein extract from affected individuals demonstrated constitutive phosphorylation and hyperactivation of Dishevelled-1 (Dvl1 [MIM 601365]), a core Wnt signaling protein,56 and an increase in both soluble and total levels of β-catenin [MIM 116806] (Figure 3A). Levels of downstream effectors for both canonical and noncanonical Wnt signaling, cyclin-D1 [MIM 168461] and phosphorylated myosinIIB [MIM 160776] were also assessed with immunoblot analysis. We observed a slight increase in phosphorylated myosinIIB levels and a striking decrease in the amount of cyclin-D1 in JSRD2 cells (Figure 3A). We next assayed RhoA [MIM 165390] activation because the Rho family of small GTPases are key PCP mediators.57 Consistent with previous results after MKS3 or TMEM216 mutation or knockdown,19, 34 we found that RhoA signaling was hyperactivated in TMEM237-mutated fibroblasts (Figure 3A) or after Tmem237 knockdown (Figure 3B) despite normal total amounts of RhoA in these cells. RhoA localized to the basal body in confluent IMCD3 cells (Figure 3C), supporting a role in mediating centrosome docking at the apical cell surface prior to ciliogenesis.19, 56 However, after Tmem237 knockdown, RhoA was mislocalized to peripheral regions of the basal body and to basolateral cell-cell contacts (Figure 3C), consistent with translocation of ectopically-activated RhoA to the cytosol.58 Because RhoA modulates the actin cytoskeleton in the PCP pathway, we evaluated the cytoskeletal phenotype of TMEM237 fibroblast lines from affected individuals; strikingly, we uncovered prominent actin stress fibers in these but not in control cells (Figure 3C).
Figure 3.
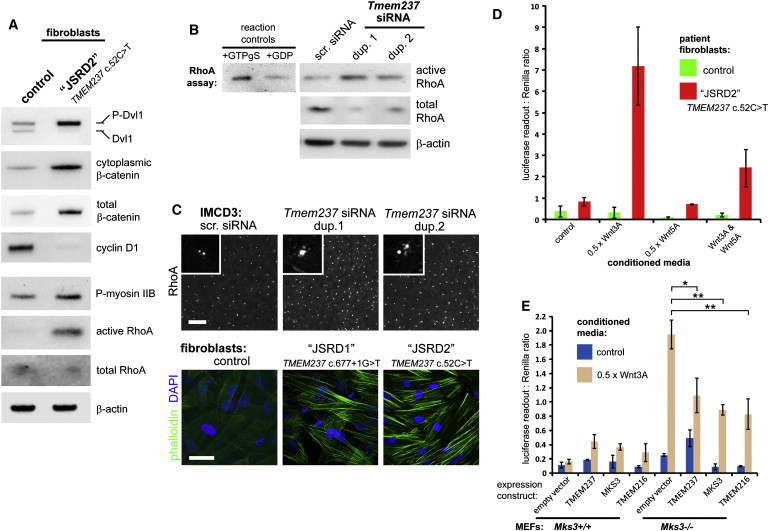
Deregulation of Wnt Signaling Following Mutation or Loss of TMEM237
(A) Immunoblots showing the relative levels of the indicated endogenous mediators of both canonical and noncanonical-PCP Wnt signaling in normal control fibroblasts compared to the TMEM237-mutated fibroblast line (JSRD2 c.52C>T line). Identical results were seen for the JSRD1 c.677+1G>T line (data not shown).
(B) Increase in levels of activated RhoA-GTP after Tmem237 siRNA knockdown with duplexes (dup.) 1 or 2, compared to scrambled (scr.) siRNA. Levels of β-actin are shown as loading controls. Positive control for the assay (loading with nonhydrolyzable GTPγS) and a negative control (loading with GDP) are shown on the left.
(C) Pericentriolar and basolateral subcellular localization of RhoA (top panels) and increase of actin stress fibers (bottom panels; visualized by phalloidin staining, green) in TMEM237-mutated fibroblast lines; the scale bars represent 10 μm.
(D) TOP Flash assays of canonical Wnt signaling, showing deregulation in affected fibroblasts (JSRD2 c.52C>T, red) compared to normal control cells (green) after treatment with 0.5 × L cell control-conditioned media (control), and conditioned media containing expressed Wnt3A and/or Wnt5A, as indicated. Activity is expressed as ratios of luciferase reporter construct expression, normalized for loading by measurement of a Renilla construct expression. Values shown are means of three independent replicates; error bars indicate SEM.
(E) TOP Flash assays of canonical Wnt signaling after cotransfection of Mks3+/+ and Mks3−/− MEFs with reporter constructs and empty vector control, wild-type GFP-TMEM237, HA-MKS3, or GFP-TMEM216 as indicated. The empty vector results combine the data from cotransfections with pCMV-HA and pGFPN1. Responses are shown to L cell control-conditioned media (control, blue bars, values are means of three independent replicates) and conditioned media containing expressed Wnt3A (0.5 × Wnt3A, brown bars, values are means of four independent replicates). Error bars indicate SEM. Statistical significance of comparisons are shown (∗p < 0.05, ∗∗p < 0.01, Student t test).
TMEM237-mutated fibroblasts carrying the putative null mutation c.52C>T (line JSRD2) also demonstrated dysregulated canonical Wnt signaling (over 5-fold basal levels) compared to control fibroblasts upon stimulation with Wnt3A-conditioned media (Figure 3D). Treatment with Wnt5A had no effect on activation; however, as expected, Wnt5A suppressed the activation by Wnt3A (Figure 3D). Deregulated Wnt signaling activation by Wnt3A was also apparent in mouse embryonic fibroblasts (MEFs) derived from wild-type Mks3+/+ and knockout mutant Mks3−/− E18 embryos (Figure 3E). Interestingly, responses were attenuated by expression of TMEM237, MKS3, and TMEM216, indicating either partial complementation of MKS3 loss by other TZ-localized members of the TMEM family or Wnt signal compensation from another cellular site (or process).
Knockdown of tmem237 Causes Developmental Defects in Zebrafish
To provide complementary evidence of a role for TMEM237 in ciliary function, signaling, and development, we turned to zebrafish, an established vertebrate model system for ciliopathies.37, 38, 39, 59 Two zebrafish paralogs of TMEM237, tmem237a and tmem237b, have been annotated. They share over 50% protein sequence identity with human TMEM237 (Figure S4) and demonstrate a significant overlap in expression pattern in the developing central nervous system, the eyes, pharyngeal arch cartilage, and the pectoral fin buds (Figure S8). The PCP pathway is known to play an essential role in polarized convergent extension (CE) movements during gastrulation and neurulation.60 To investigate the possible role of TMEM237 in the PCP pathway and embryonic development, we examined morphant phenotypes after knockdown of tmem237. When disrupting both tmem237a and tmem237b simultaneously, we observed defects in midsomitic embryos that are consistent with other ciliary morphants,19, 37, 38, 39, 59 including shortening of the anterior-posterior axis and small anterior structures, kinking of the notochord, and broadening and thinning of the somites (Figure 4A). To quantify these defects, we measured the widening of the 5th rhombomere and the shortening of the notochord as labeled by krox20 and myoD riboprobes, respectively, on whole-embryo flatmounts; results suggest altered convergence to the midline and extension along the anterior-posterior axis (Figures 4B and C). These defects could be rescued significantly by coinjection of human TMEM237 mRNA (Figure 4D). Notably, coinjection with either human MKS3 or TMEM216 mRNA also led to significant improvement of tmem237 morphant phenotypes that were indistinguishable from rescue with human TMEM237 transcript (Figure 4D). Furthermore, reciprocal injections (TMEM237 mRNA coinjected with either mks3 or tmem216 MOs) also resulted in significant improvement (Figures 4E and 4F). Together, these data suggest a functional complementation of TMEM237, TMEM216, and MKS3, consistent with the in vitro functional attenuation of canonical Wnt signaling in mammalian cells (Figure 3E).
Figure 4.
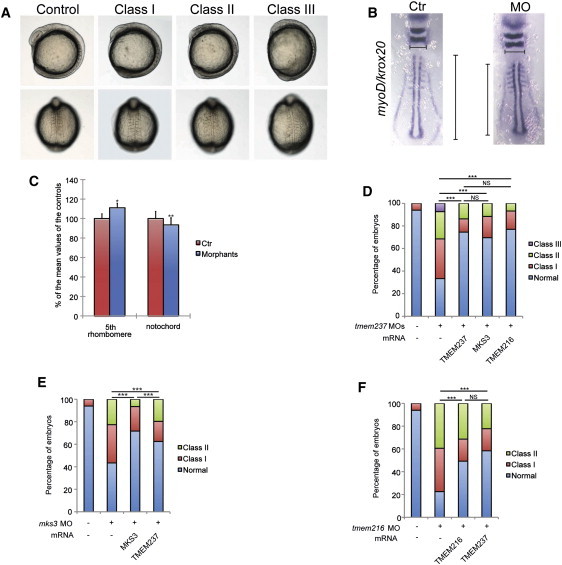
Zebrafish Morphant Phenotypes and Functional Complementation of TMEM Proteins
(A) Representative live embryo images of tmem237 morphants at 8–10 somites (top, lateral view; bottom, dorsal view). Class I and II embryos were scored on the basis of previously established objective criteria.37, 38, 59 Class I, short anterior-posterior axis, small anterior structures, and mild somitic defects; Class II, severe defects in head and eye development, shortening of the body axis, kinked notochords, and broadening of the somites; Class III, severe thinning and shortening of the body axis, curved notochord, broadened and thin somites, and an ovoid embryonic shape suggestive of dorsalization—embryos were severely delayed in development and typically did not survive past the ten somite stage.
(B) Zebrafish embryo flatmounts at the six somite stage hybridized with krox20 and myoD riboprobe cocktails. Representative embryos are shown: control MO (Ctr) injected (left); tmem237 MOs (MO) injected (right). Gastrulation defects were measured in two dimensions (medial-lateral and anterior-posterior) by the length of the 5th rhombomere (horizontal lines) and notochord (vertical lines), respectively.
(C) Quantification of the mean length of the 5th rhombomere or notochord of morphants (n = 29) are shown as the percentage of the mean length of the controls (n = 22). t test, ∗p < 0.01; ∗∗p < 0.001; error bars indicate SEM.
(D) Coinjection of tmem237 MOs with human TMEM237 mRNA, MKS3 mRNA, or TMEM216 mRNA significantly rescued morphant phenotypes at eight to ten somites.
(E) TMEM237 can partially ameliorate mks3 suppression defects of midsomitic embryos. Comparison of rescue efficiency of human MKS3 versus TMEM237 mRNA is shown.
(F) TMEM237 mRNA rescues tmem216 suppression phenotypes in a manner indistinguishable from TMEM216 mRNA coinjection in midsomitic embryos (∗∗∗p < 0.0001; NS, not significant for D and F).
TMEM237 Functionally Interacts with Other Transition Zone Proteins to Control Basal Body-Transition Zone Anchoring to the Membrane
To ascertain whether TMEM237 functions together with other ciliopathy proteins localized at the ciliary transition zone, we turned to the model organism C. elegans, where a functional network of TZ-associated proteins (MKS-1, MKSR-1/B9D1, MKSR-2/B9D2, MKS-3, MKS-5, MKS-6, NPHP-1, and NPHP-4) has been established and shown to be important for cilium formation and function.27, 28, 29, 30, 61, 62, 63, 64 We first confirmed that, as with mammalian TMEM237 (Figures 2A and 5A), the C. elegans ortholog, which we name JBTS-14, localizes to the ciliary TZ. In IMCD3 cells, Tmem237 localized to proximal regions of the cilium (Figure 2A) but just distal to the basal body (Figures 5A and 5B, marked with γ-tubulin and Dvl-1, respectively). In C. elegans, GFP-tagged JBTS-14 also concentrated to this ciliary region (marked by the tdTomato-tagged IFT dynein subunit XBX-1) within the cilia of head (amphid) and tail (phasmid) sensory neurons (Figure 6A). This localization is identical to that of other established C. elegans TZ proteins.27 Next, we identified a likely null mutant in the C. elegans jbts-14 from a deep-sequenced EMS-mutagenized animal. Similar to other C. elegans single TZ gene mutants,27 the jbts-14 mutant displayed no gross structural or functional ciliary defects, as judged by its ability to take up fluorescent dye through environmentally-exposed ciliary endings (Figure 6B), and its ability to sense its chemical environment (an avoidance of a high osmolarity solution; Figure 6C). Importantly, we identified a functional (genetic) interaction between jbts-14 and another ciliopathy-associated gene, nphp-4. Compared to the single jbts-14 or nphp-4 single mutants, the jbts-14;nphp-4 double mutant displays prominent dye-filling (dyf) and osmolarity avoidance (osm) phenotypes (Figures 6B and C). Such dyf and osm phenotypes occur with essentially all known ciliary mutants in the worm.65
Figure 5.
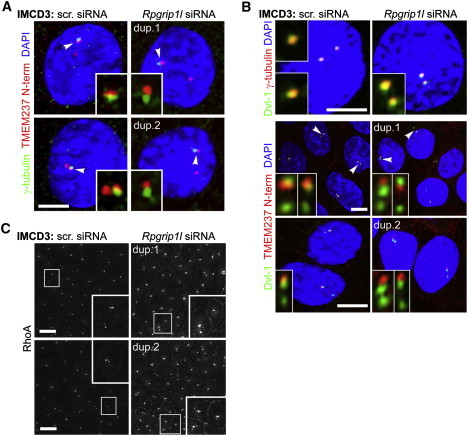
Ciliopathy TMEM Proteins Are Anchored at the Transition Zone by MKS5/RPGRIP1L
(A) In scrambled (scr.) siRNA-treated IMCD-3 cells, Tmem237 (red channel) is at proximal regions of the cilium and partially colocalizes with γ-tubulin (green channel) at the basal body, consistent with Tmem237 localization to the TZ. Colocalization with γ-tubulin is disrupted after Rpgrip1l siRNA knockdown with duplexes (dup.) 1 or 2. Insets show magnified regions (indicated by arrowheads on the main image) for the red and green channels only; the scale bars represent 5 μm.
(B) Top panels: Dvl-1 (green channel) is at the basal body that colocalizes with γ-tubulin (red channel) in IMCD-3 cells, as described previously.19Rpgrip1l siRNA knockdown does not disrupt overall basal body architecture. Bottom panels: in scrambled (scr.) siRNA-treated IMCD-3 cells, Tmem237 partially colocalizes with Dvl-1, but this is disrupted after Rpgrip1l siRNA knockdown.
(C) As above, but with RhoA staining; magnified insets are indicated by white frames.
Figure 6.
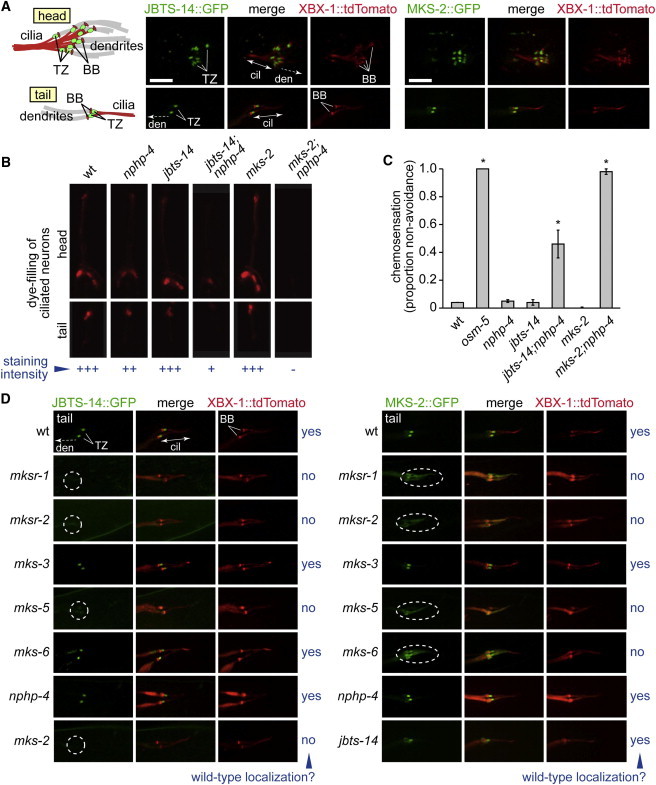
C. elegans JBTS-14 Functionally Interacts with NPHP-4 to Promote Ciliogenesis and Forms Part of a Functional Assembly of Proteins at the Transition Zone that Includes MKSR-1/B9D1, MKSR-2/B9D2, MKS-2, and MKS-5/RPGRIP1L
(A) C. elegans JBTS-14 and MKS-2 (GFP-tagged) localize specifically to the transition zone (TZ) in amphid (head) and phasmid (tail) cilia. The TZ is immediately distal to the basal body (BB) on the ciliary axoneme, both of which are marked by the tdTomato-tagged XBX-1 intraflagellar transport protein.
(B) Disruption of jbts-14 or mks-2 alone does not cause overt ciliogenesis defects, as judged by the ability of worms to take up fluorescent dye through environmentally-exposed cilia. In contrast, the jbts-14;nphp-4, or mks-2;nphp-4 double-mutant animals are dye-filling defective, indicative of cilia structure defects. The osm-5 ciliary mutant, which lacks cilia altogether, is included as a control.
(C) jbts-14 or mks-2 single mutants can avoid a high osmolarity solution, which depends on functional cilia, whereas the jbts-14;nphp-4 or mks-2;nphp-4 double mutants are defective in this chemosensory behavior. Chi-square test, ∗p<0.0001; error bars indicate SEM.
(D) The localization of JBTS-14 (left panels) and MKS-2 (right panels) depends on the presence of several other TZ proteins but not others, as indicated by yes (properly localized) or no and a dotted ellipse for a mislocalized protein. The following abbreviations are used: BB, basal body; cil, cilium; TZ, transition zone; den, dendrite. The scale bar represents 5 μm.
To provide a more detailed look at the ciliary anomalies in the jbts-14;nphp-4 double mutant, we performed TEM on this strain. In wild-type amphid channel neurons, ciliary axonemes extend from a basal body region that contains transition fibers and is found at the distal dendrite tip; the axonemes are defined at their proximal ends by the TZ compartment that makes axoneme-to-membrane connections via Y-links and are followed by microtubule doublets in the middle segment compartment and singlets in the distal segment compartment (wild-type TEMs and corresponding schematics shown in Figure 7). As expected, the single jbts-14 mutant displayed an essentially wild-type ciliary ultrastructure (data not shown). In contrast, the jbts-14;nphp-4 double mutant displays distinct cilium formation anomalies, such as axonemes that are either truncated or absent (Figure 7A). Strikingly, improper associations between the basal body-TZ region and the ciliary membrane are almost always observed, coincident with the loss or reduction of Y-link connectors (Figure 7B). In extreme cases, the TZ is fully disconnected (unanchored) from the ciliary membrane, existing instead as an ectopic structure within the distal dendrite region (Figure 7B; +10 image). In addition, many vesicle or membrane accumulations are found in proximity to the TZ, something never observed in wild-type animals. Overall, these ciliary phenotypes are very similar to those observed in other TZ gene mutant combinations with nphp-4.27 This strongly suggests that JBTS-14 (TMEM237) has a ciliogenesis function that overlaps with that of at least five other TZ proteins: namely, MKS-1, MKSR-1/B9D1, MKS-3/Meckelin/TMEM67, MKS-5/RPGRIP1L, and MKS-6/CC2D2A. Such a shared function is consistent with our finding that the localization of C. elegans JBTS-14 to the TZ depends on multiple other TZ proteins (see below), all of which are implicated in JSRDs, MKS, and NPHP ciliopathies.
Figure 7.
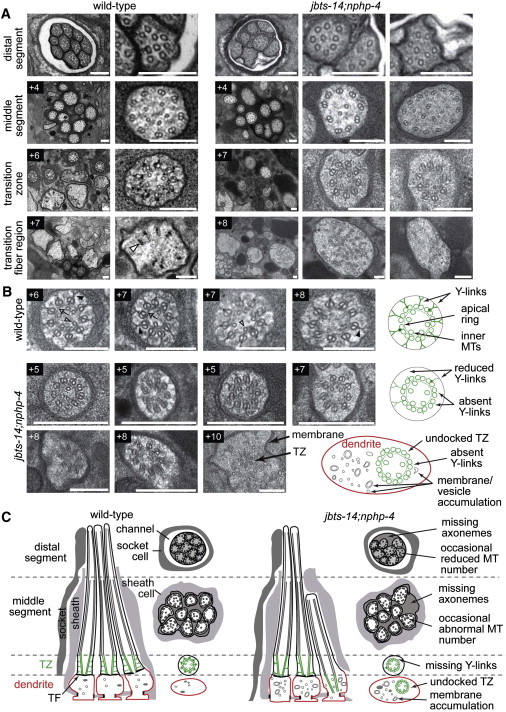
Basal Body-Transition Zone Attachment to the Ciliary Membrane Is Disrupted in Mutant Strain Lacking JBTS-14 and NPHP-4
(A) Low and high-magnification images of C. elegans amphid channel sensory cilia from TEM serial cross sections of the amphidal pore. Top row: images from the distal pore region. Boxed numbers denote proximal positioning of sections relative to the top row sections. In wild-type (N2) worms, the amphid pore contains ten axonemes, each consisting of a distal segment (top row: nine singlet microtubules), a middle segment (second row: doublet microtubules) and a transition zone (TZ) (third row: ring of doublet microtubules drawn together by the apical ring and connected to the ciliary membrane via Y-links). At the base of the cilia, within the far distal dendrite region, are transition fibers and occasional vesicles. In jbts-14;nphp-4 mutants, two to three axonemes are typically missing from the distal pore (top row, left image) and occasionally from more proximal regions (+4; left image), microtubule number is sometimes elevated (+4; right image), and Y-links are consistently missing or reduced from the transition zone compartment (+7; middle and right images). In distal dendrite regions, vesicles and membranes are often abnormally accumulated (+8; middle image) and incomplete, ectopically positioned, ciliary axonemes are occasionally observed (+8; right image). The open arrowhead indicates a transition fiber. The scale bars represent 200 nm.
(B) High-magnification images of transition zone phenotypes in the jbts-14;nphp-4 double mutant presenting multiple examples of the phenotypes described above, as well as an image showing an undocked TZ in the distal dendrite region (+10). The arrow indicates an apical ring; the closed arrowhead, Y-links; and the open arrowhead, inner microtubules. Images taken from three independently sectioned worms. Boxed numbers denote proximal positioning of sections relative to the distal end of the amphidal pore. Schematics to the right of each row depict normal and abrogated ciliary structures in wild-type and jbts-14;nphp-4 mutant worms, respectively. The scale bars represent 200 nm.
(C) Schematics of amphid channel cilia (longitudinal and transverse views) showing the major ultrastructural defects observed, including the loss of TZ connections to the ciliary membrane via Y-links. TZ subcompartment (microtubules and Y-links) is highlighted in green, dendritic membrane is highlighted in red.
To determine whether these ciliary phenotypes are specific for JBTS-14 or are general to other ciliopathy-associated TMEMs, we studied MKS-2/TMEM216. The mammalian TMEM216 is another tetraspan transmembrane protein localizing to the base of cilia,19 and it can partially complement TMEM237 loss in vitro and in vivo (Figures 3E and 4D). As expected, the C. elegans MKS-2/TMEM216 ortholog specifically localizes to the TZ (Figure 6A) and functions in a manner similar to JBTS-14. We used transposon-mediated mutagenesis to obtain a likely null allele of mks-2 and found, as with jbts-14, that the single mutant displays no ciliary or chemotaxis defects but functionally interacts with nphp-4 (Figures 6B and 6C). To further delineate the functional and modular or hierarchical relationships between ciliopathy-associated TMEMs and other established TZ proteins, we determined whether GFP-tagged JBTS-14 or MKS-2 proteins localized correctly in different TZ gene mutant backgrounds. In mks-5 mutants, both JBTS-14 and MKS-2 failed to localize to the TZ and displayed a diffuse staining pattern within the dendrite and cilium (Figure 6D), indicating that MKS-5/RPGRIP1L is required for proper TZ localization of these two TMEM proteins, similar to that reported for C. elegans MKS-3 as well.27 To confirm this result in mammalian cells, we performed knockdowns of Mks5/Rpgrip1l in IMCD3 cells and stained for Tmem237 and the basal body markers γ-tubulin (Figure 5A) or Dvl1 (Figure 5B), and RhoA (Figure 5C). We observed that after scrambled siRNA transfection, basal body architecture was intact (Figure 5B) and that Tmem237 was just distal to the basal body (Figures 5A and 5B; marked by either γ-tubulin or Dvl-1, respectively). This localization to the TZ was disrupted after knockdown of Mks5/Rpgrip1l. RhoA was also mislocalized to peripheral regions of the basal body and to basolateral cell-cell contacts (Figure 5C), a pattern identical to that in the cellular phenotype after Tmem237 knockdown (Figure 3C) as well as Tmem216/Mks2 knockdown.19
We then further assessed the functional requirements for C. elegans JBTS-14 and MKS-2 localization to the TZ. We uncovered two additional TZ components, the B9 domain proteins MKSR-1/B9D1 and MKSR-2/B9D2, whose functions are required for the proper TZ localization of JBTS-14 and MKS-2 (Figure 6D). Furthermore, C. elegans MKS-2 is at least partially delocalized in the mks-6 mutant. In contrast, both JBTS-14 and MKS-2 localized normally in two other mutants, mks-3 and nphp-4 (Figure 6D). Interestingly, although C. elegans MKS-2 localizes properly in the jbts-14 mutant, the reciprocal experiment shows that JBTS-14 is mislocalized in the mks-2 mutant. Thus, in a hierarchical or modular interaction network, JBTS-14 could potentially be considered peripheral with respect to MKS-2, whereas MKS-2 connects JBTS-14 to other TZ proteins. Together, these data demonstrate that both JBTS-14 and MKS-2 are likely to be part of a hierarchical or modular assembly that includes other established TZ proteins. Because MKS5/RPGRIP1L is required for the correct localization of JBTS-14/TMEM237 at the TZ in both C. elegans and mammalian cells, such functional modules are likely to be evolutionarily conserved.
Assessing the Possible Contribution of TMEM237 to BBS
The potential functional overlap of TMEM237 with other bona fide ciliopathy causal loci (both in vitro and in vivo), as well as the documented genetic overlap across ciliopathies, raised the question of whether loss of function in this gene transcript could contribute to other overlapping clinical phenotypes. To begin to address this question, we sequenced TMEM237 in 90 individuals of Northern European origin who had been diagnosed with BBS but were unselected for known mutations. We found one heterozygous change (p.Asp155Ala) in a BBS family with a heterozygous, functionally null BBS6 [MIM 604896] Thr57Ala mutation,38, 66 which was not present in HapMap, the 1000 Genomes Project, or an additional 200 controls. When tested by our established in vivo zebrafish-based complementation assay,37, 38, 39, 59 this allele was shown to be hypomorphic, because it could only partially rescue tmem237 MO phenotypes (Figure S9A). Digenic inheritance is unlikely because of segregation of the alleles in the family and the fact that we observed no synergistic effect when subeffective doses of tmem237 and bbs6 MOs were coinjected in zebrafish embryos (Figure S9B).
Discussion
Ciliopathies are a unique group of genetically heterogeneous human developmental disorders that collectively affect nearly every tissue and organ in the body, presenting with a wide array of physiological and developmental phenotypes.1, 2, 3, 4, 5, 6, 67 Remarkably, although discrete clinical entities such as Joubert syndrome, Meckel-Gruber syndrome, Bardet-Biedl syndrome, and nephronophthisis are apparent, there is considerable phenotypic overlap between these conditions and even allelism at a number of loci.6, 8 A key question is therefore whether the phenotypic overlap—and variability—of these cilium-associated disorders can be explained by a common molecular and cellular etiology.
Many of the known JBTS/MKS/BBS/NPHP proteins from vertebrates or other species such as C. elegans have now been ascribed roles in cilium biogenesis and signaling at the ciliary transition zone.27, 28, 29, 30 To gain further insight into the genetic causes of the MKS/JBTS group of ciliopathies, we first mapped a locus for JSRD type 14 (JBTS-14) in the Canadian Hutterites, leading to the identification of mutations in TMEM237. The mutations identified in this study are all predicted to be null, suggesting that complete loss or near-absence of TMEM237 causes the JSRD type 14 phenotype. Despite this common mechanism, it is interesting to note that the large subgroup of the affected individuals with cystic kidneys also have posterior fossa findings in addition to a MTS, suggesting that these developmental pathways are affected by additional, probably genetic, modifying factors. Although mutations in TMEM237 only account for <1% cases in our JSRDs-MKS cohort, the high carrier frequency of the c.52C>T mutation in the Hutterite population (approximately 1:15) means that carrier testing, cascade testing in families, and prenatal diagnosis will be of high clinical utility. Families with JSRDs will also benefit from improved diagnosis and accurate genetic counseling.
To further elucidate the molecular etiology of TMEM237-associated JSRD, we characterized the function of the TZ-localized TMEM237 protein. Loss of human TMEM237 results in failure of ciliogenesis and deregulation of both canonical and noncanonical/PCP Wnt signaling pathways. These findings are strikingly similar to previous studies of both TMEM216 and MKS3,19, 34 two additional transmembrane proteins associated with JSRDs and MKS. MKS3 and TMEM216 have been suggested to be noncanonical Wnt receptors that regulate the RhoA pathway and thus mediate the cytoskeleton rearrangements required for basal body docking at the apical region of the cells prior to ciliogenesis.34 Importantly, we demonstrate that in a complementary zebrafish model system, disruption of TMEM237 causes developmental (convergent extension) phenotypes comparable to those obtained upon abrogation of TZ-localized proteins, including TMEM216 and MKS3.19 Furthermore, we now show by both an in vitro (Figure 3E) and an in vivo (Figures 4D–4F) assay that there is the probable functional complementation of TMEM237, TMEM216, and MKS3. This finding, and the similarities between the three TMEMs in cellular localization, protein structure, role in cilium formation and function, and clinical phenotypes caused by mutations indicate that TMEM237, TMEM216, and MKS3 probably function in the same pathway or module to regulate ciliogenesis and signaling. This conclusion is supported by the functional interaction of C. elegans TZ-localized proteins belonging to the MKS module in mediating basal body-TZ attachment to the membrane and cilium formation.27 We speculate that TMEM237, TMEM216, MKS3, and perhaps other TMEMs form a receptor-coreceptor complex at the ciliary TZ to mediate and integrate signaling pathways impacting the primary cilium.
One of the central questions remaining concerning the molecular etiology and underlying phenotypic variability of the JBTS-MKS-NPHP ciliopathy spectrum is whether the ciliary defects arise from disruption in one or more macromolecular assemblies, or modules, comparable to that of the BBSome, an oligomeric protein complex containing proteins associated with BBS.68, 69 Support for this model comes from the study of C. elegans, which has suggested the existence of two genetically defined modules: one, being an MKS module (consisting of MKS-1, MKSR-1/B9D1, MKSR-2/B9D2, MKS-3, and MKS-6), and the other, an NPHP module, consisting of NPHP-1 and NPHP-4.27 Specifically, disruption of any single mutant, or combination of mutants, in gene(s) within either module abrogates a ciliary gate function but does not significantly impair the structure of most cilia; in contrast, disrupting any combination of two genes, one from each module, causes transition zone structure anomalies that are concomitant with loss of basal body-TZ anchoring to the membrane and ciliary axoneme structure defects. Importantly, these modules are largely consistent with a physical interaction network recently uncovered in mammalian cells.22 For example, NPHP1 and NPHP4 are ascribed to a distinct protein interaction module, coincident with the genetically-defined C. elegans NPHP module, and MKS1 and MKS6 are part of a physical interaction network that is consistent with the two respective genes being components of the C. elegans MKS genetic module. Additionally, disruption of individual mammalian TZ proteins (such as MKS1 and MKS3) in mouse models is on the whole phenotypically less severe than that observed for disruption of IFT, which causes global defects in ciliogenesis;27 this suggests a functional redundancy in mammals that might be similar to that observed in C. elegans and could be exposed by analysis of mouse double knockouts. In this study, we show that C. elegans JBTS-14 (TMEM237) functionally interacts with NPHP-4 to promote basal body-TZ attachment to the membrane and cilium formation. Such functions parallel those reported for the other C. elegans TZ-localized proteins belonging to the MKS module and thus implicate JBTS-14 in the same molecular pathway.
Overall, our analyses of TMEM237 in mammalian cells, zebrafish, and C. elegans strongly support a role for this tetraspan transmembrane protein within an expansive genetic and physical interaction network of proteins localized to the TZ, where it participates in a functional module that regulates cilium biogenesis and signaling. Our findings therefore extend the interaction network of ciliopathy proteins at the TZ, revealing additional complexity at this ciliary region, and underline the importance of an evolutionary conserved JSRD-MKS-NPHP functional module for cilium biogenesis and signaling.
Acknowledgments
We thank the families for participation in this study, L. Racacho (D.E.B.) and J. Zhang (M.-A.A.) for technical support, R.T. Moon at University of Washington for the Topflash and Fopflash constructs, and D.G. Moerman (University of British Columbia; funded by Genome Canada, Genome British Columbia, and Canadian Insitutes of Health Research [CIHR]) and R. Waterston (University of Washington; funded by American Recovery and Reinvestment Act grant HG 005921 from the National Human Genome Research Institute) for the C. elegans jbts-14 mutant strain. V.L.J. was supported by a postdoctoral fellowship from the Michael Smith Foundation for Health Research (MSFHR). A.R.J. received funding from the Austrian Science Foundation (FWF grant P18470) and the Dr.-Legerlotz foundation Austria. E.E.D. is supported by National Institutes of Health (NIH) grant R01EY021872. P.F. and C.M.L. were supported by the Canadian Insitutes of Health Research (CIHR) Training Program in Genetics, Child Development, and Health. T.M. received funding from the Tiroler Wissenschaftsfonds. C.M.C. is the Mary D. Allen Chair and acknowledges funding from NIH EY015851, EY03040, 1F31GM079910, and Research to Prevent Blindness. H.J.B. received funding from the Gertrud-Kusen-Stiftung. N.K. acknowledges funding from NIH R01HD04260, R01DK072301, and R01DK075972 and is the Distinguished George W. Brumley Professor. O.E.B. acknowledges a Science Foundation Ireland President of Ireland Young Researcher Award (06/Y12/B928). D.D. is funded by the NIH grants KL2RR025015 and R01NS064077. J.S.P acknowledges funding from the Alberta Children's Hospital Foundation. M.R.L. is funded by the March of Dimes and a senior scholar award from the MSFHR. C.A.J. acknowledges funding from a Sir Jules Thorn Charitable Trust Biomedical Research Award (09/JTA). C.A.J. and N.K. are supported by the European Community's Seventh Framework Programme FP7/2009 under grant agreement 241955, SYSCILIA. K.M.B. is funded by the SickKids Foundation/CIHR IHDCYH (NI10-008), and a Clinical Investigatorship Award from the CIHR Institute of Genetics.
Published online: December 8, 2011
Footnotes
Supplemental Data include 11 figures and 1 table and can be found with this article online at http://www.cell.com/AJHG/.
Contributor Information
Michel R. Leroux, Email: leroux@sfu.ca.
Kym M. Boycott, Email: kboycott@cheo.on.ca.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://browser.1000genomes.org/index.html
C. elegans Gene Knockout Consortium, http://celeganskoconsortium.omrf.org/
Entrez Gene, http://www.ncbi.nlm.nih.gov/gene
National Bioresource Project, http://shigen.lab.nig.ac.jp/c.elegans/index.jsp
National Center for Biotechnology Information (NCBI) dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/
NCBI Nucleotide database, http://www.ncbi.nlm.nih.gov/nuccore
NCBI protein database, http://www.ncbi.nlm.nih.gov/protein
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Protocol Online, http://www.protocol-online.org/
SYSCILIA, http://syscilia.org/
Zebrafish Model Organism Database, http://zfin.org
Supplemental Data
References
- 1.Brancati F., Dallapiccola B., Valente E.M. Joubert Syndrome and related disorders. Orphanet J. Rare Dis. 2010;5:20. doi: 10.1186/1750-1172-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker K., Beales P.L. Making sense of cilia in disease: The human ciliopathies. Am. J. Med. Genet. C. Semin. Med. Genet. 2009;151C:281–295. doi: 10.1002/ajmg.c.30231. [DOI] [PubMed] [Google Scholar]
- 3.Sharma N., Berbari N.F., Yoder B.K. Ciliary dysfunction in developmental abnormalities and diseases. Curr. Top. Dev. Biol. 2008;85:371–427. doi: 10.1016/S0070-2153(08)00813-2. [DOI] [PubMed] [Google Scholar]
- 4.Lee J.E., Gleeson J.G. Cilia in the nervous system: Linking cilia function and neurodevelopmental disorders. Curr. Opin. Neurol. 2011;24:98–105. doi: 10.1097/WCO.0b013e3283444d05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cardenas-Rodriguez M., Badano J.L. Ciliary biology: Understanding the cellular and genetic basis of human ciliopathies. Am. J. Med. Genet. C. Semin. Med. Genet. 2009;151C:263–280. doi: 10.1002/ajmg.c.30227. [DOI] [PubMed] [Google Scholar]
- 6.Hildebrandt F., Benzing T., Katsanis N. Ciliopathies. N. Engl. J. Med. 2011;364:1533–1543. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson J.L., Leroux M.R. cAMP and cGMP signaling: Sensory systems with prokaryotic roots adopted by eukaryotic cilia. Trends Cell Biol. 2010;20:435–444. doi: 10.1016/j.tcb.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 8.Parisi M.A. Clinical and molecular features of Joubert syndrome and related disorders. Am. J. Med. Genet. C. Semin. Med. Genet. 2009;151C:326–340. doi: 10.1002/ajmg.c.30229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bielas S.L., Silhavy J.L., Brancati F., Kisseleva M.V., Al-Gazali L., Sztriha L., Bayoumi R.A., Zaki M.S., Abdel-Aleem A., Rosti R.O., et al. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat. Genet. 2009;41:1032–1036. doi: 10.1038/ng.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valente E.M., Brancati F., Silhavy J.L., Castori M., Marsh S.E., Barrano G., Bertini E., Boltshauser E., Zaki M.S., Abdel-Aleem A., et al. International JSRD Study Group AHI1 gene mutations cause specific forms of Joubert syndrome-related disorders. Ann. Neurol. 2006;59:527–534. doi: 10.1002/ana.20749. [DOI] [PubMed] [Google Scholar]
- 11.Parisi M.A., Bennett C.L., Eckert M.L., Dobyns W.B., Gleeson J.G., Shaw D.W., McDonald R., Eddy A., Chance P.F., Glass I.A. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am. J. Hum. Genet. 2004;75:82–91. doi: 10.1086/421846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valente E.M., Silhavy J.L., Brancati F., Barrano G., Krishnaswami S.R., Castori M., Lancaster M.A., Boltshauser E., Boccone L., Al-Gazali L., et al. International Joubert Syndrome Related Disorders Study Group Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat. Genet. 2006;38:623–625. doi: 10.1038/ng1805. [DOI] [PubMed] [Google Scholar]
- 13.Sayer J.A., Otto E.A., O'Toole J.F., Nurnberg G., Kennedy M.A., Becker C., Hennies H.C., Helou J., Attanasio M., Fausett B.V., et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat. Genet. 2006;38:674–681. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- 14.Baala L., Romano S., Khaddour R., Saunier S., Smith U.M., Audollent S., Ozilou C., Faivre L., Laurent N., Foliguet B., et al. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am. J. Hum. Genet. 2007;80:186–194. doi: 10.1086/510499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delous M., Baala L., Salomon R., Laclef C., Vierkotten J., Tory K., Golzio C., Lacoste T., Besse L., Ozilou C., et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet. 2007;39:875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- 16.Arts H.H., Doherty D., van Beersum S.E., Parisi M.A., Letteboer S.J., Gorden N.T., Peters T.A., Märker T., Voesenek K., Kartono A., et al. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat. Genet. 2007;39:882–888. doi: 10.1038/ng2069. [DOI] [PubMed] [Google Scholar]
- 17.Cantagrel V., Silhavy J.L., Bielas S.L., Swistun D., Marsh S.E., Bertrand J.Y., Audollent S., Attié-Bitach T., Holden K.R., Dobyns W.B., et al. International Joubert Syndrome Related Disorders Study Group Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am. J. Hum. Genet. 2008;83:170–179. doi: 10.1016/j.ajhg.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorden N.T., Arts H.H., Parisi M.A., Coene K.L., Letteboer S.J., van Beersum S.E., Mans D.A., Hikida A., Eckert M., Knutzen D., et al. CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am. J. Hum. Genet. 2008;83:559–571. doi: 10.1016/j.ajhg.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valente E.M., Logan C.V., Mougou-Zerelli S., Lee J.H., Silhavy J.L., Brancati F., Iannicelli M., Travaglini L., Romani S., Illi B., et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat. Genet. 2010;42:619–625. doi: 10.1038/ng.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edvardson S., Shaag A., Zenvirt S., Erlich Y., Hannon G.J., Shanske A.L., Gomori J.M., Ekstein J., Elpeleg O. Joubert syndrome 2 (JBTS2) in Ashkenazi Jews is associated with a TMEM216 mutation. Am. J. Hum. Genet. 2010;86:93–97. doi: 10.1016/j.ajhg.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coene K.L., Roepman R., Doherty D., Afroze B., Kroes H.Y., Letteboer S.J., Ngu L.H., Budny B., van Wijk E., Gorden N.T., et al. OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin. Am. J. Hum. Genet. 2009;85:465–481. doi: 10.1016/j.ajhg.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sang L., Miller J.J., Corbit K.C., Giles R.H., Brauer M.J., Otto E.A., Baye L.M., Wen X., Scales S.J., Kwong M., et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell. 2011;145:513–528. doi: 10.1016/j.cell.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dafinger C., Liebau M.C., Elsayed S.M., Hellenbroich Y., Boltshauser E., Korenke G.C., Fabretti F., Janecke A.R., Ebermann I., Nürnberg G., et al. Mutations in KIF7 link Joubert syndrome with Sonic Hedgehog signaling and microtubule dynamics. J. Clin. Invest. 2011;121:2662–2667. doi: 10.1172/JCI43639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia-Gonzalo F.R., Corbit K.C., Sirerol-Piquer M.S., Ramaswami G., Otto E.A., Noriega T.R., Seol A.D., Robinson J.F., Bennett C.L., Josifova D.J., et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Genet. 2011;43:776–784. doi: 10.1038/ng.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Logan C.V., Abdel-Hamed Z., Johnson C.A. Molecular genetics and pathogenic mechanisms for the severe ciliopathies: Insights into neurodevelopment and pathogenesis of neural tube defects. Mol. Neurobiol. 2011;43:12–26. doi: 10.1007/s12035-010-8154-0. [DOI] [PubMed] [Google Scholar]
- 26.Hildebrandt F., Attanasio M., Otto E. Nephronophthisis: Disease mechanisms of a ciliopathy. J. Am. Soc. Nephrol. 2009;20:23–35. doi: 10.1681/ASN.2008050456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williams C.L., Li C., Kida K., Inglis P.N., Mohan S., Semenec L., Bialas N.J., Stupay R.M., Chen N., Blacque O.E., et al. MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J. Cell Biol. 2011;192:1023–1041. doi: 10.1083/jcb.201012116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bialas N.J., Inglis P.N., Li C., Robinson J.F., Parker J.D., Healey M.P., Davis E.E., Inglis C.D., Toivonen T., Cottell D.C., et al. Functional interactions between the ciliopathy-associated Meckel syndrome 1 (MKS1) protein and two novel MKS1-related (MKSR) proteins. J. Cell Sci. 2009;122:611–624. doi: 10.1242/jcs.028621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams C.L., Masyukova S.V., Yoder B.K. Normal ciliogenesis requires synergy between the cystic kidney disease genes MKS-3 and NPHP-4. J. Am. Soc. Nephrol. 2010;21:782–793. doi: 10.1681/ASN.2009060597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Williams C.L., Winkelbauer M.E., Schafer J.C., Michaud E.J., Yoder B.K. Functional redundancy of the B9 proteins and nephrocystins in Caenorhabditis elegans ciliogenesis. Mol. Biol. Cell. 2008;19:2154–2168. doi: 10.1091/mbc.E07-10-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dawe H.R., Smith U.M., Cullinane A.R., Gerrelli D., Cox P., Badano J.L., Blair-Reid S., Sriram N., Katsanis N., Attie-Bitach T., et al. The Meckel-Gruber Syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum. Mol. Genet. 2007;16:173–186. doi: 10.1093/hmg/ddl459. [DOI] [PubMed] [Google Scholar]
- 32.Vierkotten J., Dildrop R., Peters T., Wang B., Rüther U. Ftm is a novel basal body protein of cilia involved in Shh signalling. Development. 2007;134:2569–2577. doi: 10.1242/dev.003715. [DOI] [PubMed] [Google Scholar]
- 33.Zuniga F.I., Craft C.M. Deciphering the structure and function of Als2cr4 in the mouse retina. Invest. Ophthalmol. Vis. Sci. 2010;51:4407–4415. doi: 10.1167/iovs.10-5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dawe H.R., Adams M., Wheway G., Szymanska K., Logan C.V., Noegel A.A., Gull K., Johnson C.A. Nesprin-2 interacts with meckelin and mediates ciliogenesis via remodelling of the actin cytoskeleton. J. Cell Sci. 2009;122:2716–2726. doi: 10.1242/jcs.043794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Willert J., Epping M., Pollack J.R., Brown P.O., Nusse R. A transcriptional response to Wnt protein in human embryonic carcinoma cells. BMC Dev. Biol. 2002;2:8. doi: 10.1186/1471-213x-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thisse C., Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 2008;3:59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- 37.Leitch C.C., Zaghloul N.A., Davis E.E., Stoetzel C., Diaz-Font A., Rix S., Alfadhel M., Lewis R.A., Eyaid W., Banin E., et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 2008;40:443–448. doi: 10.1038/ng.97. [DOI] [PubMed] [Google Scholar]
- 38.Zaghloul N.A., Liu Y., Gerdes J.M., Gascue C., Oh E.C., Leitch C.C., Bromberg Y., Binkley J., Leibel R.L., Sidow A., et al. Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet-Biedl syndrome. Proc. Natl. Acad. Sci. USA. 2010;107:10602–10607. doi: 10.1073/pnas.1000219107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khanna H., Davis E.E., Murga-Zamalloa C.A., Estrada-Cuzcano A., Lopez I., den Hollander A.I., Zonneveld M.N., Othman M.I., Waseem N., Chakarova C.F., et al. A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat. Genet. 2009;41:739–745. doi: 10.1038/ng.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blacque O.E., Reardon M.J., Li C., McCarthy J., Mahjoub M.R., Ansley S.J., Badano J.L., Mah A.K., Beales P.L., Davidson W.S., et al. Loss of C. elegans BBS-7 and BBS-8 protein function results in cilia defects and compromised intraflagellar transport. Genes Dev. 2004;18:1630–1642. doi: 10.1101/gad.1194004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Culotti J.G., Russell R.L. Osmotic avoidance defective mutants of the nematode Caenorhabditis elegans. Genetics. 1978;90:243–256. doi: 10.1093/genetics/90.2.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cevik S., Hori Y., Kaplan O.I., Kida K., Toivenon T., Foley-Fisher C., Cottell D., Katada T., Kontani K., Blacque O.E. Joubert syndrome Arl13b functions at ciliary membranes and stabilizes protein transport in Caenorhabditis elegans. J. Cell Biol. 2010;188:953–969. doi: 10.1083/jcb.200908133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boulin T., Bessereau J.L. Mos1-mediated insertional mutagenesis in Caenorhabditis elegans. Nat. Protoc. 2007;2:1276–1287. doi: 10.1038/nprot.2007.192. [DOI] [PubMed] [Google Scholar]
- 44.Boycott K.M., Parboosingh J.S., Scott J.N., McLeod D.R., Greenberg C.R., Fujiwara T.M., Mah J.K., Midgley J., Wade A., Bernier F.P., et al. Meckel syndrome in the Hutterite population is actually a Joubert-related cerebello-oculo-renal syndrome. Am. J. Med. Genet. A. 2007;143A:1715–1725. doi: 10.1002/ajmg.a.31832. [DOI] [PubMed] [Google Scholar]
- 45.Schurig V., Bowen P., Harley F., Schiff D. The Meckel syndrome in the Hutterites. Am. J. Med. Genet. 1980;5:373–381. doi: 10.1002/ajmg.1320050408. [DOI] [PubMed] [Google Scholar]
- 46.Janecke A.R., Müller T., Gassner I., Kreczy A., Schmid E., Kronenberg F., Utermann B., Utermann G. Joubert-like syndrome unlinked to known candidate loci. J. Pediatr. 2004;144:264–269. doi: 10.1016/j.jpeds.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 47.Liu Q., Tan G., Levenkova N., Li T., Pugh E.N., Jr., Rux J.J., Speicher D.W., Pierce E.A. The proteome of the mouse photoreceptor sensory cilium complex. Mol. Cell. Proteomics. 2007;6:1299–1317. doi: 10.1074/mcp.M700054-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kwok M.C., Holopainen J.M., Molday L.L., Foster L.J., Molday R.S. Proteomics of photoreceptor outer segments identifies a subset of SNARE and Rab proteins implicated in membrane vesicle trafficking and fusion. Mol. Cell. Proteomics. 2008;7:1053–1066. doi: 10.1074/mcp.M700571-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith U.M., Consugar M., Tee L.J., McKee B.M., Maina E.N., Whelan S., Morgan N.V., Goranson E., Gissen P., Lilliquist S., et al. The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat. Genet. 2006;38:191–196. doi: 10.1038/ng1713. [DOI] [PubMed] [Google Scholar]
- 50.Junge H.J., Yang S., Burton J.B., Paes K., Shu X., French D.M., Costa M., Rice D.S., Ye W. TSPAN12 regulates retinal vascular development by promoting Norrin- but not Wnt-induced FZD4/beta-catenin signaling. Cell. 2009;139:299–311. doi: 10.1016/j.cell.2009.07.048. [DOI] [PubMed] [Google Scholar]
- 51.Wallingford J.B., Rowning B.A., Vogeli K.M., Rothbächer U., Fraser S.E., Harland R.M. Dishevelled controls cell polarity during Xenopus gastrulation. Nature. 2000;405:81–85. doi: 10.1038/35011077. [DOI] [PubMed] [Google Scholar]
- 52.Park T.J., Haigo S.L., Wallingford J.B. Ciliogenesis defects in embryos lacking inturned or fuzzy function are associated with failure of planar cell polarity and Hedgehog signaling. Nat. Genet. 2006;38:303–311. doi: 10.1038/ng1753. [DOI] [PubMed] [Google Scholar]
- 53.Veeman M.T., Axelrod J.D., Moon R.T. A second canon. Functions and mechanisms of beta-catenin-independent Wnt signaling. Dev. Cell. 2003;5:367–377. doi: 10.1016/s1534-5807(03)00266-1. [DOI] [PubMed] [Google Scholar]
- 54.Gerdes J.M., Liu Y., Zaghloul N.A., Leitch C.C., Lawson S.S., Kato M., Beachy P.A., Beales P.L., DeMartino G.N., Fisher S., et al. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet. 2007;39:1350–1360. doi: 10.1038/ng.2007.12. [DOI] [PubMed] [Google Scholar]
- 55.Lancaster M.A., Louie C.M., Silhavy J.L., Sintasath L., Decambre M., Nigam S.K., Willert K., Gleeson J.G. Impaired Wnt-beta-catenin signaling disrupts adult renal homeostasis and leads to cystic kidney ciliopathy. Nat. Med. 2009;15:1046–1054. doi: 10.1038/nm.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Park T.J., Mitchell B.J., Abitua P.B., Kintner C., Wallingford J.B. Dishevelled controls apical docking and planar polarization of basal bodies in ciliated epithelial cells. Nat. Genet. 2008;40:871–879. doi: 10.1038/ng.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Winter C.G., Wang B., Ballew A., Royou A., Karess R., Axelrod J.D., Luo L. Drosophila Rho-associated kinase (Drok) links Frizzled-mediated planar cell polarity signaling to the actin cytoskeleton. Cell. 2001;105:81–91. doi: 10.1016/s0092-8674(01)00298-7. [DOI] [PubMed] [Google Scholar]
- 58.Lang P., Gesbert F., Delespine-Carmagnat M., Stancou R., Pouchelet M., Bertoglio J. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J. 1996;15:510–519. [PMC free article] [PubMed] [Google Scholar]
- 59.Merveille A.C., Davis E.E., Becker-Heck A., Legendre M., Amirav I., Bataille G., Belmont J., Beydon N., Billen F., Clément A., et al. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 2011;43:72–78. doi: 10.1038/ng.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Keller R. Shaping the vertebrate body plan by polarized embryonic cell movements. Science. 2002;298:1950–1954. doi: 10.1126/science.1079478. [DOI] [PubMed] [Google Scholar]
- 61.Jauregui A.R., Barr M.M. Functional characterization of the C. elegans nephrocystins NPHP-1 and NPHP-4 and their role in cilia and male sensory behaviors. Exp. Cell Res. 2005;305:333–342. doi: 10.1016/j.yexcr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 62.Winkelbauer M.E., Schafer J.C., Haycraft C.J., Swoboda P., Yoder B.K. The C. elegans homologs of nephrocystin-1 and nephrocystin-4 are cilia transition zone proteins involved in chemosensory perception. J. Cell Sci. 2005;118:5575–5587. doi: 10.1242/jcs.02665. [DOI] [PubMed] [Google Scholar]
- 63.Hopp K., Heyer C.M., Hommerding C.J., Henke S.A., Sundsbak J.L., Patel S., Patel P., Consugar M.B., Czarnecki P.G., Gliem T.J., et al. B9D1 is revealed as a novel Meckel syndrome (MKS) gene by targeted exon-enriched next-generation sequencing and deletion analysis. Hum. Mol. Genet. 2011;20:2524–2534. doi: 10.1093/hmg/ddr151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dowdle W.E., Robinson J.F., Kneist A., Sirerol-Piquer M.S., Frints S.G., Corbit K.C., Zaghloul N.A., van Lijnschoten G., Mulders L., Verver D.E., et al. Disruption of a ciliary B9 protein complex causes Meckel syndrome. Am. J. Hum. Genet. 2011;89:94–110. doi: 10.1016/j.ajhg.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Inglis, P.N., Ou, G., Leroux, M.R., and Scholey, J.M. (2007). The sensory cilia of Caenorhabditis. In WormBook, The C. elegans Research Community, ed. 10.1895/wormbook.1.126.2, http://www.wormbook.org. [DOI] [PMC free article] [PubMed]
- 66.Katsanis N., Beales P.L., Woods M.O., Lewis R.A., Green J.S., Parfrey P.S., Ansley S.J., Davidson W.S., Lupski J.R. Mutations in MKKS cause obesity, retinal dystrophy and renal malformations associated with Bardet-Biedl syndrome. Nat. Genet. 2000;26:67–70. doi: 10.1038/79201. [DOI] [PubMed] [Google Scholar]
- 67.Wolf M.T., Hildebrandt F. Nephronophthisis. Pediatr. Nephrol. 2011;26:181–194. doi: 10.1007/s00467-010-1585-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nachury M.V., Loktev A.V., Zhang Q., Westlake C.J., Peränen J., Merdes A., Slusarski D.C., Scheller R.H., Bazan J.F., Sheffield V.C., Jackson P.K. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129:1201–1213. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- 69.Jin H., White S.R., Shida T., Schulz S., Aguiar M., Gygi S.P., Bazan J.F., Nachury M.V. The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell. 2010;141:1208–1219. doi: 10.1016/j.cell.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.