

Abstract
Flecked-retina syndromes, including fundus flavimaculatus, fundus albipunctatus, and benign fleck retina, comprise a group of disorders with widespread or limited distribution of yellow-white retinal lesions of various sizes and configurations. Three siblings who have benign fleck retina and were born to consanguineous parents are the basis of this report. A combination of homozygosity mapping and exome sequencing helped to identify a homozygous missense mutation, c.133G>T (p.Gly45Cys), in PLA2G5, a gene encoding a secreted phospholipase (group V phospholipase A2). A screen of a further four unrelated individuals with benign fleck retina detected biallelic variants in the same gene in three patients. In contrast, no loss of function or common (minor-allele frequency>0.05%) nonsynonymous PLA2G5 variants have been previously reported (EVS, dbSNP, 1000 Genomes Project) or were detected in an internal database of 224 exomes (from subjects with adult onset neurodegenerative disease and without a diagnosis of ophthalmic disease). All seven affected individuals had fundoscopic features compatible with those previously described in benign fleck retina and no visual or electrophysiological deficits. No medical history of major illness was reported. Levels of low-density lipoprotein were mildly elevated in two patients. Optical coherence tomography and fundus autofluorescence findings suggest that group V phospholipase A2 plays a role in the phagocytosis of photoreceptor outer-segment discs by the retinal pigment epithelium. Surprisingly, immunohistochemical staining of human retinal tissue revealed localization of the protein predominantly in the inner and outer plexiform layers.
Main Text
Benign fleck retina (MIM 228980) refers to an autosomal-recessive condition associated with a distinctive retinal appearance and no apparent visual or electrophysiological deficits.1 Affected individuals are asymptomatic, but fundus examination reveals a striking pattern of diffuse, yellow-white, fleck-like lesions extending to the far periphery of the retina but sparing the foveal region.2–5 The phenotype associated with benign fleck retina was first described in 1980 in seven affected siblings born to consanguineous parents.2 A similar clinical appearance was subsequently reported in three unrelated individuals originating from diverse ethnic backgrounds.3–5 Elucidating the genetic basis of human ocular phenotypes such as that of benign fleck retina remains a major goal because it will provide important insights into the complex biochemistry and cellular physiology of the human eye.
In order to characterize the clinical consequences of mutations in genes previously associated with abnormal retinal function and/or structure, as well as to identify novel disease-associated genes, as part of an ongoing study we recruited families that presented themselves to the inherited eye disease clinics at Moorfields Eye Hospital and that showed evidence of parental consanguinity. One such family (family J, Figure 1) of South Asian origin is the basis of this report. The study was approved by the local research ethics committee, and all investigations were conducted in accordance with the principles of the Declaration of Helsinki; informed consent was obtained from all participating individuals. Initially, subject J-4, a healthy, asymptomatic 10-year-old girl (IV-2, family J in Figure 1), was referred after abnormal retinal appearance was noticed on a routine eye test. No family history of retinal disease was reported. Visual acuity was normal. Fundus examination revealed multiple, discrete, polymorphous, yellow-white flecks at the level of the retinal pigment epithelium (RPE). The flecks affected both fundi in a symmetrical pattern, spread peripherally beyond the major vascular arcades, and spared the maculae (Figure 2). Other family members, including three siblings and both parents, were also examined. Findings similar to those for the proband were obtained in subjects J-5 (aged 9; IV-3, family J in Figure 1) and J-6 (aged 7; IV-4, family J in Figure 1); normal retinal appearance was observed in subject J-3 (IV-1, family J in Figure 1) and the parents, J-1 (III-6, family J in Figure 1) and J-2 (III-7, family J in Figure 1). Electrophysiological assessment was performed. Full-field and pattern electroretinograms (ERGs) as well as electrooculograms (EOGs) were normal in all three affected subjects, and a diagnosis of benign fleck retina was confirmed. The clinical findings are summarized in Table 1.
Figure 1.
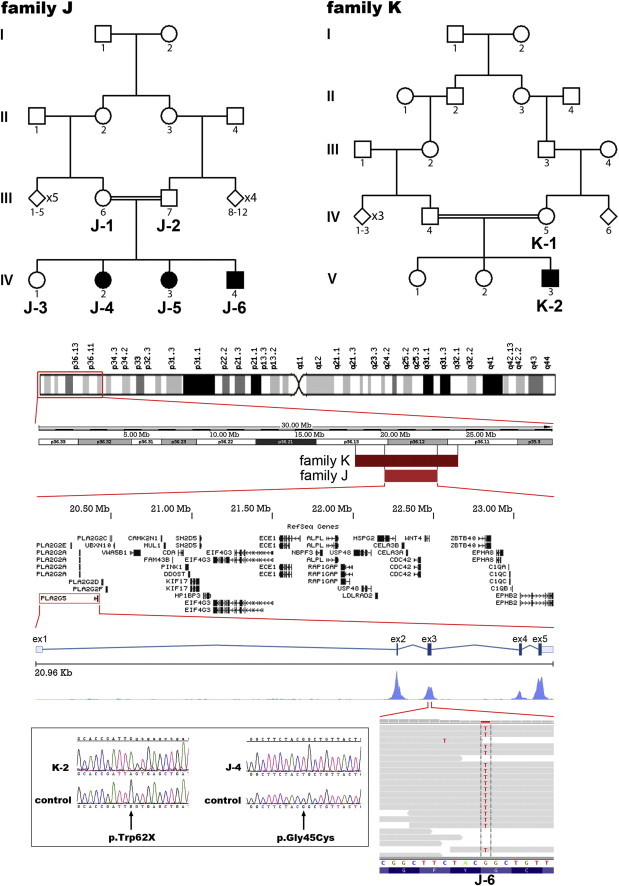
Identification of PLA2G5 Mutations in Individuals from Two Families with Benign Fleck Retina
Pedigrees of families J and K are shown. Homozygosity mapping with DNA from subject K-2 revealed a 12 cM region on 1p (flanked by rs10796459 and rs12407356). DNA samples from subjects J-1, J-2, J-3, J-4, J-5, and J-6 were also genotyped, and a 5 cM region (flanked by rs3738122 and rs1832047) was found to be homozygous in all affected individuals and was found to be consistent with disease segregation. RefSeq genes contained in this shared region between families K and J are shown. Exome sequencing with DNA from subject J-6 revealed a homozygous missense change, c.133G>T (p.Gly45Cys) in PLA2G5. Gene structure of PLA2G5, coverage depth distribution of the mapped reads along its five exons (Savant Genome Browser), and sequencing reads corresponding to this variant are presented (IGV viewer; 34 reads total: 10 forward and 24 reverse,100% thymine). Subsequently, bidirectional Sanger sequencing confirmed segregation of the p.Gly45Cys change in family J and identified a homozygous nonsense mutation (c.185G>A [p.Trp62X]) in individual K-2. Electropherograms of DNA sequences surrounding these two variants are shown. Both sequences are displayed in the forward orientation.
Figure 2.
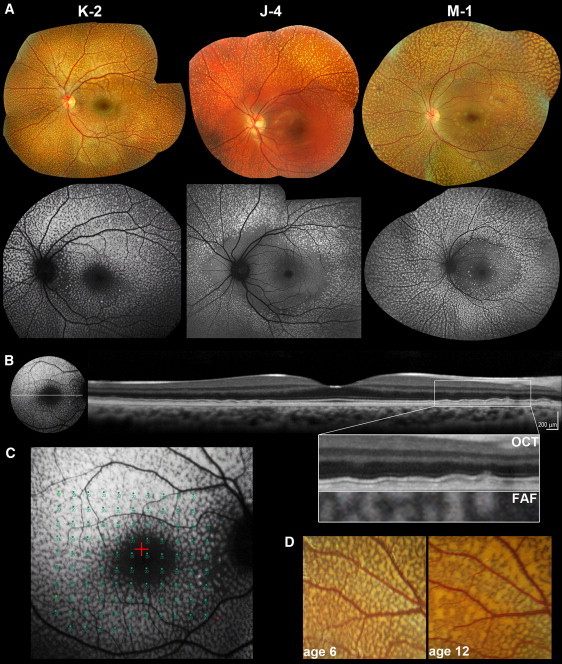
Retinal Imaging of Individuals with Benign Fleck Retina and PLA2G5 Mutations
(A) Color photographs and corresponding fundus autofluorescence (FAF) images of the left fundi of subjects K-2 (aged 12), J-4 (aged 12), and M-1 (aged 39). On fundus photography, multiple yellow-white flecks of various sizes are observed. FAF reveals hyperautofluorecent lesions corresponding in location with the flecks. The macula is relatively spared in subjects J-4 and M-1 but not in K-2, in whom only the fovea appears to be unaffected. This might reflect a more detrimental effect of the c.185G>A (p.Trp62X) mutation in the homozygous state (subject K-2) as opposed to homozygous missense (c.133G>T [p.Gly45Cys] in subject J-4) or compound heterozygous (p.Gly45Cys and c.383delA, p.Gln128ArgfsX88 in M-1) mutations.
(B) FAF imaging and linear spectral domain optical coherence tomography (OCT) scan of the left retina of subject K-2. Deep, discrete, hyper-reflective deposits, more obvious at the edge of the foveal scan, are observed. The panel with an enlarged image of the boxed region shows the outer retina and RPE in detail. The lesions are located posterior to the hyperreflective band corresponding to the photoreceptor inner/outer segment junction and do not disrupt it. An overlay of OCT with FAF is also presented. Deposits are spatially associated with the hyperautofluorescent lesions and thus correspond to the flecks.
(C) Functional assessment of the central retina in subject K-2. Static-perimetry testing (threshold sensitivities from 0 to 20 dB, test spot size Goldmann III) results overlaid with FAF are presented. Retinal sensitivity was normal.
(D) Longitudinal data showing evolution of fleck-like lesions over time. Magnified view of fundus photographs from the left eye (vascular arcades) of subject K-2 at ages 6 and 12. Flecks increase in number and size and become more confluent.
Table 1.
Clinical Characteristics and Molecular Pathology of Subjects with Benign Fleck Retina
| Subject | Gender | Visual Functiona[age at examination] | Lipid Levelsb | Other Systemic Findings | Molecular Diagnosis, Amino Acid Changes in PLA2G5 |
|---|---|---|---|---|---|
| J-4 | female | normal ERG, pERG, EOG, DA [12] | not tested | p.[Gly45Cys];[Gly45Cys] | |
| J-5 | female | normal ERG, pERG, EOG [12] | not tested | p.[Gly45Cys];[Gly45Cys] | |
| J-6 | male | normal ERG, pERG, EOG [10] | not tested | p.[Gly45Cys];[Gly45Cys] | |
| K-2 | male | normal ERG, pERG [6],4 and MP [12] | LDL 3.6 mmol/liter, chol 5.5 mmol/liter | high BMI (31), allergic rhinitis | p.[Trp62X];[Trp62X] |
| L-1 | female | normal ERG, EOG [12]3 | not tested | high BMI | p.[Gly49Ser];[Arg53X] |
| M-1 | female | normal ERG, pERG [37] | LDL 3.9 mmol/liter chol 6.3 mmol/liter |
high BMI (26) | p.[Gly45Cys];[Gln128ArgfsX45] |
| N-1 | female | normal ERG, pERG [10] | normal LDL, chol | normal BMI | no mutation identified |
Subjects J-4, J-5, J-6, and K-2 are of South Asian origin and were born to consanguineous parents; subject L-1 is of mixed Australian aboriginal and white descent; subject M-1 is of South Asian origin; subject N-1 is of white British origin. All affected individuals presented with abnormal retinal appearance on a routine eye test, were asymptomatic, reported no night blindness, and had visual acuities of 0.2 logMAR (logarithm of the minimal angle of resolution) or better. Color vision was normal in all eyes (evaluated with the Farnsworth D-15 test[(L-2],3 Hardy-Rand-Rittler test [HRR; K-2, M-1 and N-1], or Ishihara test plates [J-4, J-5, J-6, L-2,3 M-1 and N-1}. Subjects K-2 and L-1 had mild myopic astigmatism, and subject M-1 is a high myope. Abbreviations are as follows: ERG, electroretinogram; pERG, pattern electroretinogram; EOG, electrooculogram; DA, dark adaptometry; chol, cholesterol; and BMI, body mass index.
Subjects K-2 and M-1 had mild eosinophilia (0.45 × 109 and 0.64 × 109 eosinophils/liter respectively; normal levels are from 0.0 × 109 to 0.4 × 109 eosinophils/liter).
Visual function was evaluated via electrophysiology or fundus-controlled perimetry (Nidek MP1, Goldmann III stimulus size).
Normal levels are from 2.3 to 4.9 mmol/liter for cholesterol and from 0.0 to 3.0 mmol/liter for LDL.
DNA samples from the three affected siblings (subjects J-4, J-5, and J-6) and their unaffected sister (subject J-3) and parents (subjects J-1 and J-2) were genotyped with the use of single-nucleotide polymorphism (SNP) chip arrays (GeneChip Human Mapping 50K Xba Array, Affymetrix, Santa Clara, CA, USA) according to the manufacturer's recommendations. The Bayesian Robust Linear Model with Mahalanobis distance classifier (BRLMM) genotype-calling algorithm was used;6 CEL files were input, and the threshold was set at 0.01. The pedigree was consistent with the propagation of a single mutant allele from a recent ancestor such that affected individuals were autozygous for this allele and the unaffected sibling was not. We wrote a python script interacting with a MySQL database to detect regions obeying this rule and rank them by genetic distance; the Marshfield linkage map was used. Three chromosomal segments of more than 1 cM were identified (Table S1 available online): two regions on 1p (19 cM and 5 cM) and one region on 2q (14 cM).
Exon capture and high-throughput sequencing of DNA from subject J-6 was undertaken. The solution-phase Agilent SureSelect 38 Mb exome capture (SureSelect Human All Exon Kit, Agilent, Santa Clara, CA, USA) and the Illumina HiSeq2000 sequencer (Illumina, San Diego, CA, USA) were used. Reads were aligned to the hg19 human reference sequence; average sequencing depth on target was 72, and 87% of the targeted region was covered with a minimum read depth of 10. Overall, we identified 15,611 exonic sequence alterations with respect to the reference sequence (Table 2). Given the level of consanguinity in this family, we hypothesized that the trait is recessive and focused on homozygous variants. On the basis of the prior belief that benign-fleck-retina-associated mutations are rare, calls with minor-allele frequency of more than 0.5% in the 1000 Genomes dataset (May 2011 release) or an internal set of 224 exomes (from individuals with adult-onset neurodegenerative disease) were filtered. Subsequently, we focused on the three homozygous regions found by SNP arrays to be shared among affected family members; no loss-of-function variants were identified, and three homozygous rare missense changes were detected: c.133G>T (p.Gly45Cys) in PLA2G5 (MIM 601192), c.1154A>G (p.Asn385Ser) in ECE1 (MIM 600423), and c.722G>A (p.Arg241Gln) in NEU2 (MIM 605528) (Table S1).
Table 2.
Prioritization of Variants Identified by Exome Sequencing of DNA from Subject J-6
| Total | Within Regions of Homozygositya | Within Regions of Homozygosity Shared among Affected but Not Unaffected Siblingsb | |
|---|---|---|---|
| All variants | 15,611 | 1,223 | 81 |
| Only NS/SS/I, | 7,247 | 588 | 40 |
| AND ≤ 0.5% MAF in 1000 genomes, | 648 | 41 | 3 |
| AND ≤ 0.5% MAF in internal database | 580 | 36 | 3 |
| AND are predicted to be loss of function | 80 | 7 | 0 |
Variants presented were sequentially filtered on the basis of effect on protein sequence (synonymous or intronic variants were excluded), presence in the 1000 Genomes Project dataset (with ≤ 0.5% MAF; the 20101123 sequence and alignment release including 1094 individuals was used), presence in exomes from an internal database (with ≤ 0.5% MAF; DNA from 224 samples processed with the same tools as J-6), and being presumed to cause loss of protein function (nonsense, splice site variants and frameshifting insertions-deletions).
Abbreviations are as follows: SNP, single-nucleotide polymorphism; NS/SS/I, nonsynonymous, splice site or coding insertion-deletion variants; and MAF, minor-allele frequency.
Based on exome sequencing data.
based on SNP genotyping data.
Simultaneously, a DNA sample was obtained from a previously reported case of benign fleck retina (K-2; V-3, family K in Figure 1).4 There was evidence of parental consanguinity, and homozygosity mapping via the Affymetrix SNP Array 6.0 (performed as previously described7) yielded four homozygous regions that were more than 10 cM (Table S1). The third-largest segment (12 cM) encompassed one of the loci detected in family J. Thus, we focused on the PLA2G5:p.Gly45Cys and ECE1:p.Asn385Ser variants found within this shared region. On the basis of physiological relevance (Unigene and OMIM), the PLA2G5 change appeared to be more likely to cause disease, and Sanger sequencing of the open-reading frame (exons 2 to 5, 138 amino acids, Ensembl transcript ENST00000375108) and intron-exon boundaries was undertaken in four unrelated individuals with benign fleck retina (primer details are listed in Table S2). Clinical and electrophysiological characteristics of two of these cases (K-2 and L-1) have been detailed in previous reports.3,4
Biallelic PLA2G5 variants were identified in three of four cases; all changes were novel (Figure 3 and Table 1). Notably, seven nonsynonymous sequence alterations (all with minor-allele frequency < 0.05%) and no-loss-of function PLA2G5 variants have been previously reported (EVS, dbSNP, 1000 Genomes) or were identified in an internal set of 224 exomes (Table S3). Subject L-1, a 28-year-old female,3 was found to carry two changes in a heterozygous state (c.145G>A [p.Gly49Ser] and c.157C>T [p.Arg53X]). PCR amplification and subsequent TA cloning of exon 3 (pGEM-T Easy Vector, Promega, Madison, WI, USA) demonstrated that these variants were present on different alleles. Two heterozygous changes (c.133G>T [p.Gly45Cys] and c.383delA [p.Gln128ArgfsX88]) were also identified in subject M-1, a 39-year-old female; these variants were also shown to be biallelic by a similar approach (long-range PCR and TA cloning of the 5 kb of DNA encompassing exons 3–5). Interestingly, the p.Gly45Cys variant was detected in a homozygous state in the three affected members of family J. Both missense changes identified in benign fleck retina patients (p.Gly45Cys and p.Gly49Ser) were highly conserved among orthologs and paralogs (Figures S1 and S2).
Figure 3.
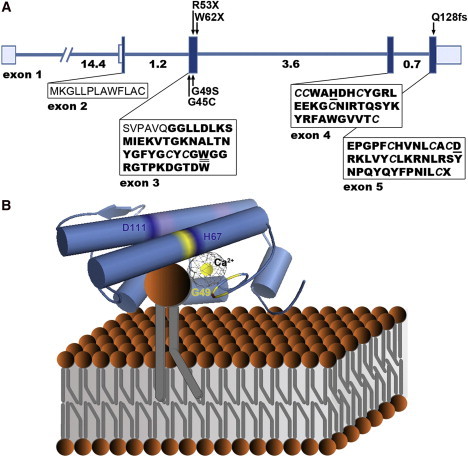
Structure of PLA2G5 and Hypothetical Model of Human Group V Phospholipase A2 Binding to a Phospholipid Membrane Surface
(A) Exons are depicted with boxes in which the shaded areas denote the coding sequence and the unshaded areas denote the 5′ and 3′ untranslated regions. Numbers under the line correspond to intron size (kb), and arrows indicate the position of mutations identified in this study. The amino acid sequence of the signal peptide is shown in normal font; the sequence of the 118 amino acid mature enzyme after cleavage of the prepeptide is shown in bold font (Uniprot8). Cystine residues forming the six disulfide bridges maintaining the enzyme's rigid three-dimensional structure are italicized (Uniprot8). Amino acids responsible for interfacial binding (tryptophan 50)43 and catalytic activity (histidine 67 and aspartic acid 111)12 are underlined.
(B) A homology model of human group V phospholipase A2 (Protein Data Bank accession code 2ghn)44 after hypothetical association with a phospholipid membrane is presented. Structural features of the active site, conserved among secreted phospholipase A2s, are highlighted; these features include a catalytic Ca2+ ion bound by a peptide loop (yellow) and a catalytic dyad formed by amino acids His67 and Asp111 (dark blue).12 The Ca2+ coordination includes carbonyl backbone interactions from Tyr47, Gly49, and Gly51, as well as a shared bidentate interaction from Asp68 (amino acids colored in yellow; Uniprot). Trp50, a key amino acid in the enzyme's interfacial binding surface (distinct from the active site) is highlighted in red; its indole chain contributes to the characteristic ability of group V phospholipase A2 to bind to both zwitterionic and anionic phospholipid vesicles.43 Cationic residues that are also responsible for membrane binding at the carboxyl end of the protein are colored in purple.45
PyMOL (Delano Scientific, Portland, OR) was used for viewing the human group V phospholipase A2 three-dimensional molecular structure (orthoscopic view, cartoon setting, cylindrical helices).
Subject K-2, a 12-year-old boy,4 was found to be homozygous for a c.185G>A (p.Trp62X) change, altering the last base of exon 3. Using patient-derived leukocyte mRNA, we investigated how this variant affects pre-mRNA splicing of the PLA2G5 transcript in vivo. To do this, we performed a series of reverse transcriptase PCR (RT-PCR) experiments (Figure S3 and Table S4). Two amplimers of different size were detected for each of the control and patient-derived samples. Both were confirmed by direct sequencing to represent distinct PLA2G5 transcripts: (1) the expected segment of the protein-coding PLA2G5 transcript (Ensembl transcript ENST00000375108); and (2). a segment of a transcript containing an additional alternatively spliced 77 bp exon, between exons 3 and 4, previously observed in a non-coding PLA2G5 transcript (Ensembl transcript ENST00000478803). Interestingly, the relative abundance of these two amplimers was different in the control versus the patient sample; the alternatively spliced exon was present at a higher level in the latter. This finding indicates that this c.185G>A variant alters the relative expression of different PLA2G5 transcript levels in vivo. Importantly, with or without the addition of the alternately spliced exon, the c.185G>A variant leads to the production of transcripts containing a premature termination codon. Therefore, if the p.Trp62X mutant mRNA did not succumb to nonsense-mediated decay and was translated, the encoded protein would be severely truncated.
PLA2G5 encodes group V phospholipase A2 (PLA2), a secreted PLA2 first described in 1994.8 The PLA2 superfamily includes a broad range of enzymes defined by their ability to catalyze the hydrolysis of the middle (sn-2) ester bond of glycerophospholipids and thus release potentially bioactive lipids, namely lysophospholipids and free fatty acids (arachidonic acid and others).9,10 PLA2s have been subdivided into several classes, including secreted PLA2s.11 These are water-soluble, Ca2+-requiring enzymes that contain Histidine- and Aspartic-acid-catalytic dyads and have the ability to function during secretion (in the secretory compartment or in the extracellular space, in an autocrine or paracrine manner) or after internalization.12 On the basis of selected structural determinants, secreted PLA2s have been classified into six groups. Individual secreted PLA2s exhibit unique enzymatic properties and show diverse tissue and cellular localizations; thus, distinct physiological roles and nonredundant functions are likely.12 PLA2G5 is highly expressed in the eye and heart and is present in other tissues as well, including placenta, lung and brain (eyeSAGE, Unigene,8,13–17). A number of human cells, including macrophages, neutrophils, bronchial and renal tubular epithelium, subendocardial cells (cardiomyocytes), and interstitial fibroblasts of gastric submucosa, have been shown to express PLA2G5.16,18–22
A variety of biological functions have been attributed to group V PLA2. These functions are often related to the enzyme's ability to provide arachidonic acid for eicosanoid (prostaglandins, leukotrienes, and others) generation.20,23 Additional functions not directly related to lipid-mediator biosynthesis have also been demonstrated; these include regulation of phagocytosis, foam cell formation, and anti-bacterial activities.21,24,25 This combination of pro- and anti-inflammatory properties, as well as the presence of cell-type-specific functions, suggests that group V PLA2 has distinct anatomical and context-dependent roles.18,25 Studies employing transgenic26 and knockout27 mice have provided important insights into the role of group V PLA2 in various pathophysiological events. Enzyme deficiency in Pla2g5-null mice leads to marked attenuation of airway inflammation (asthma28,29 and acute respiratory distress syndrome30) and reduced atherosclerosis.31,32 Conversely, because group V PLA2 modulates immune complex clearance by stimulating phagocytosis, knockout mice demonstrate exacerbation of autoantibody-induced arthritis.25 Pla2g5-transgenic mice overexpressing PLA2G5 die soon after birth as a result of aberrant hydrolysis of lung surfactant phospholipids.26 Despite the growing body of research focusing on animal model studies, definite evidence for an in vivo role of group V PLA2 in human tissues is lacking, and it is likely that some biological functions are not conserved from mice to humans.12
None of the affected individuals in this study reported a medical history of major or chronic illness (Table 1). Subject K-2 experiences symptoms of mild seasonal allergic rhinitis and infrequently receives antihistamine tablets. A high body mass index was recorded in three patients. In both mutation-positive individuals tested (subjects K-2 and M-1), a blood test revealed slight eosinophilia and mildly elevated low-density lipoprotein (LDL) and total cholesterol levels (Table 1). Notably, an association of human PLA2G5 haplotypes with total and LDL cholesterol has been previously reported.33 Although there is strong in vitro evidence that group V PLA2 is enzymatically active in serum and hydrolyses LDL,34 no effect on plasma lipoproteins was observed in mice with enzyme deficiency.31 It is possible that the raised LDL levels are unrelated to the PLA2G5 mutations, and further studies would be of interest. It is, however, noteworthy that a phase II trial of varsepladib, an inhibitor of secreted PLA2s (with selectivity against group IIA, V and X PLA2s) has demonstrated efficacy in reducing the concentrations of LDL cholesterol.35
To determine the consequences of reduced levels or the absence of group V PLA2 on retinal structure and function, we performed clinical investigations of individuals with mutations in PLA2G5. First, in vivo cross-sectional imaging via spectral domain optical coherence tomography36 (SD-OCT; Spectralis HRA+OCT, Heidelberg Engineering, Heidelberg, Germany) was undertaken. Deposit accumulation within the RPE monolayer and/or the area between the RPE and photoreceptor cells was observed (subjects K-2 and M-1; Figure 2). Second, we used fundus autofluorescence imaging37 (HRA2, Heidelberg Engineering) to gain insight into the molecular composition of the fleck-like lesions; hyperautofluorescent material, i.e., material rich in lipofuscin or other fluorophores, was observed (subjects J-5, J-6, K-2 and M-1; Figure 2). Lipofuscin accumulation is a hallmark of aging in metabolically active cells, including cardiac myocytes, neurons, and the RPE.38 In the latter, the main source of lipofuscin is the undegradable components of phagocytosed photoreceptor outer-segment disks.37,39 Excessive build-up has been associated with various forms of photoreceptor degeneration, namely retinal dystrophies and age-related macular degeneration.37 In order to assess the functional significance of abnormalities detected by fundus autofluorescence and SD-OCT in benign fleck retina patients, we performed fundus-controlled perimetry (MP1 Microperimeter, Nidek Technologies, Padova, Italy). Fundus-controlled perimetry provides a method for accurate functional assessment of the central retina with high spatial resolution.40 Retinal sensitivity was normal in a 10-year-old individual (subject K-2) even when areas corresponding to large flecks were stimulated (Figure 2). This suggests that the compounds of lipofuscin accumulating in this condition have no or minimal functional consequences; this observation is supported by normal electrophysiological findings (Table 1). Finally, fundus photography in subject K-2 at 6 and 12 years of age has documented an increase in number and size of retinal flecks (Figure 2). This is not evident from cross-sectional analysis across four decades, and a genotype-phenotype correlation cannot be excluded (Figure 2).
Despite the fact that Kolko et al. previously demonstrated high levels of PLA2G5 mRNA expression within the rat retina,14 the precise protein localization is currently unknown. To determine the precise localization, we performed immunohistochemical staining of human retinal tissue obtained from an 87-year-old male donor eye (Figure 4). Immunoreactivity was predominantly detected in the outer and inner plexiform layers (Figure 4). This result is unexpected because imaging data (SD-OCT, fundus autofluorescence imaging) indicate that the primary defect in individuals with mutated group V PLA2 is in close proximity to the RPE. More specifically, abnormal RPE phagocytosis could explain the level and autofluorescent nature of the fleck-like lesions (Figure 2); this hypothesis would be supported by previous reports that demonstrated the capacity of the protein to promote phagosome maturation in other tissues.24 The inconsistency between protein localization in donor retina and the site of structural change in patients is difficult to explain, and future studies investigating group V PLA2 staining in younger retinae should provide further insight.
Figure 4.
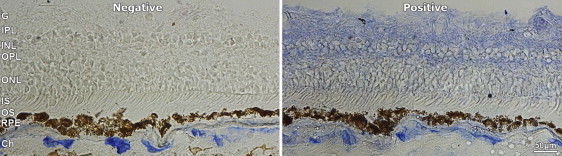
Localization of Group V Phospholipase A2 within a Control Human Retinal Tissue
Human retinal tissue from an 87-year-old male donor's eye was obtained from the eye bank at Moorfields Eye Hospital with the approval of Moorfields and Whittington Research Ethics Committee (06/Q0504/78) and embedded in an optimal-cutting-temperature compound. Cryostat sections were cut at 10 μm and thaw-mounted onto charged slides. Immunohistochemistry was performed at room temperature to reveal group V phospholipase A2 localization via mouse anti-human PLA2G5 monoclonal antibody (LS-C11702, clone MCL-3G1, Lifespan Bioscience, Seattle, WA, USA)30 at a dilution factor of 1/20. An alkaline phosphatase-conjugated avidin-biotin complex kit (Vectastain ABC-AP Mouse IgG kit, Vector Laboratories, Burlingame, CA, USA) was used as a secondary detection method according to the manufacturer's guidelines. An additional quenching step was performed with 1% Levamisole for 30 min so that autofluorescence would be reduced.
Abbreviations are as follows: Ch, Choroid; RPE, retinal pigment epithelium; OS, photoreceptor outer segments; IS photoreceptor inner segment; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; an dG ganglion cell layer. The scale bar represents 50 μm.
No mutation was detected in the PLA2G5 coding region or intron-exon boundaries of subject N-1, a 10-year-old girl with a typical benign fleck retina phenotype (rs2020887, a previously reported SNP, was found in heterozygous state). This finding suggests that benign fleck retina might be a genetically heterogeneous condition. Interestingly, group IB PLA2 (MIM 172410), another conventional secretory phospholipase, has been shown to be expressed at similar levels and to have a comparable localization to group V PLA2 within the rat retina.14 We therefore selected PLA2G1B as a candidate gene and screened its coding region and intron-exon boundaries; no variants were identified in subject N-1.
Retinal disease due to mutations in PLA2G5 adds to a small group of human Mendelian disorders associated with genes encoding PLA2s; these diseases involve neurodegeneration (mutations in PLA2G6 [MIM 603604] and PNPLA6 [MIM 612020]), abnormal lipid storage (mutations in PNPLA2 [MIM 609059]) or platelet dysfunction (mutations in PLA2G4A [MIM 600522] and PLA2G7 [MIM 601690]). Notably, PLA2G7 encodes a lipoprotein-associated PLA2, and its natural deficiency (due to a functionally validated Val279Phe-null allele; allele frequency is from 4% to 18% in East Asian and around 0.03% in European populations) is not detrimental to human health; carriers have a low risk for coronary artery disease.41,42
In this study biallelic nonsense and missense PLA2G5 variants are identified in four families with benign fleck retina. This finding facilitates differential diagnosis of this benign condition from other fleck retina syndromes associated with abnormal retinal function. A role of group V PLA2 in RPE phagocytosis through phagosome maturation can be speculated.24 Affected individuals reported here have reduced levels or an absence of functional group V PLA2 and remain systemically well; this suggests that pharmacological abrogation of group V PLA2 function, as a strategy for treating systemic disease, would be unlikely to have deleterious consequences on the patient. Future studies on older subjects with benign fleck retina as well as detailed investigations aimed at delineating the effect of mutant PLA2G5 alleles in other tissues will provide important insights.
Acknowledgements
We acknowledge the cooperation and help provided by the family members in this study. We thank Thomas Daskalakis for his key contribution in developing the python program and Jill Urquhart and Sarah Daly at the National Institute for Health Research Manchester Biomedical Research Centre for their technical assistance with Affymetrix SNP Array 6.0 genotyping. We are grateful to colleagues who referred individuals from Moorfields Eye Hospital to us, as well as to those who contributed to the assembly of the Benign Fleck Retina panel; we are particularly grateful to Naushin Waseem, Bev Scott, Genevieve Wright, Sophie Devery, Michel Michaelides, and Mandeep Sagoo. We thank Kaoru Fujinami, Professor Alan Bird, Professor Philippa Talmud, and Professor Yozo Miyake for their insightful comments. We acknowledge the following sources of funding: British Retinitis Pigmentosa Society, Fight for Sight, Moorfields Eye Hospital Special Trustees, National Institute for Health Research UK (Moorfields Eye Hospital and Institute of Ophthalmology, London, UK), the Foundation Fighting Blindness (USA), the Medical Research Council UK, the Wellcome Trust, and the University College London Hospitals Biomedical Research Centre.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data/
Blossum62, http://www.ncbi.nlm.nih.gov/Class/FieldGuide/BLOSUM62.txt
Exome Variant Server (EVS), NHLBI Exome Sequencing Project (ESP), http://snp.gs.washington.edu/EVS/
Interactive Genomics Viewer (IGV), http://www.broadinstitute.org/software/igv/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Polymorphism Phenotyping (PolyPhen) version 2, http://genetics.bwh.harvard.edu/pph2/
Protein Data Bank, http://www.pdb.org/pdb/home/home.do
PyMOL, http://www.pymol.org/
Savant Genome Browser, http://genomesavant.com/
Sorting intolerant from tolerant (SIFT), http://sift.bii.a-star.edu.sg/
Swiss-Model, http://swissmodel.expasy.org/
Unigene, http://www.ncbi.nlm.nih.gov/UniGene/
UniProt, http://www.uniprot.org/
References
- 1.Gass J.D. Mosby; St Louis: 1997. Stereoscopic Atlas of Macular Disease: Diagnosis and Treatment. [Google Scholar]
- 2.Sabel Aish S.F., Dajani B. Benign familial fleck retina. Br. J. Ophthalmol. 1980;64:652–659. doi: 10.1136/bjo.64.9.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Isaacs T.W., McAllister I.L., Wade M.S. Benign fleck retina. Br. J. Ophthalmol. 1996;80:267–268. doi: 10.1136/bjo.80.3.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Audo I., Tsang S.H., Fu A.D., Barnes J.A., Holder G.E., Moore A.T. Autofluorescence imaging in a case of benign familial fleck retina. Arch. Ophthalmol. 2007;125:714–715. doi: 10.1001/archopht.125.5.714. [DOI] [PubMed] [Google Scholar]
- 5.Galindo-Ferreiro A., Sanabria M.R., Garcia E.P., Coco-Martin R.M., Galindo-Alonso J., Palencia-Ercilla J. Benign fleck retinal findings on multifocal ERG, microperimetry, and OCT. Ophthalmic Surg. Lasers Imaging. 2010;41:e1–e5. doi: 10.3928/15428877-20101025-05. [DOI] [PubMed] [Google Scholar]
- 6.Affymetrix. (2006). BRLMM: An improved genotype calling method for the GeneChip Mapping 500K Array Set. http://media.affymetrix.com/support/technical/whitepapers/brlmm_whitepaper.pdf
- 7.Sergouniotis P.I., Davidson A.E., Mackay D.S., Li Z., Yang X., Plagnol V., Moore A.T., Webster A.R. Recessive mutations in KCNJ13, encoding an inwardly rectifying potassium channel subunit, cause leber congenital amaurosis. Am. J. Hum. Genet. 2011;89:183–190. doi: 10.1016/j.ajhg.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen J., Engle S.J., Seilhamer J.J., Tischfield J.A. Cloning and recombinant expression of a novel human low molecular weight Ca(2+)-dependent phospholipase A2. J. Biol. Chem. 1994;269:2365–2368. [PubMed] [Google Scholar]
- 9.Six D.A., Dennis E.A. The expanding superfamily of phospholipase A(2) enzymes: classification and characterization. Biochim. Biophys. Acta. 2000;1488:1–19. doi: 10.1016/s1388-1981(00)00105-0. [DOI] [PubMed] [Google Scholar]
- 10.Dennis E.A. Diversity of group types, regulation, and function of phospholipase A2. J. Biol. Chem. 1994;269:13057–13060. [PubMed] [Google Scholar]
- 11.Murakami M., Taketomi Y., Miki Y., Sato H., Hirabayashi T., Yamamoto K. Recent progress in phospholipase A2 research: from cells to animals to humans. Prog. Lipid Res. 2011;50:152–192. doi: 10.1016/j.plipres.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Lambeau G., Gelb M.H. Biochemistry and physiology of mammalian secreted phospholipases A2. Annu. Rev. Biochem. 2008;77:495–520. doi: 10.1146/annurev.biochem.76.062405.154007. [DOI] [PubMed] [Google Scholar]
- 13.Valentin E., Singer A.G., Ghomashchi F., Lazdunski M., Gelb M.H., Lambeau G. Cloning and recombinant expression of human group IIF-secreted phospholipase A(2) Biochem. Biophys. Res. Commun. 2000;279:223–228. doi: 10.1006/bbrc.2000.3908. [DOI] [PubMed] [Google Scholar]
- 14.Kolko M., Christoffersen N.R., Barreiro S.G., Bazan N.G. Expression and location of mRNAs encoding multiple forms of secretory phospholipase A2 in the rat retina. J. Neurosci. Res. 2004;77:517–524. doi: 10.1002/jnr.20187. [DOI] [PubMed] [Google Scholar]
- 15.Cupillard L., Koumanov K., Mattéi M.G., Lazdunski M., Lambeau G. Cloning, chromosomal mapping, and expression of a novel human secretory phospholipase A2. J. Biol. Chem. 1997;272:15745–15752. doi: 10.1074/jbc.272.25.15745. [DOI] [PubMed] [Google Scholar]
- 16.Seeds M.C., Jones K.A., Duncan Hite R., Willingham M.C., Borgerink H.M., Woodruff R.D., Bowton D.L., Bass D.A. Cell-specific expression of group X and group V secretory phospholipases A(2) in human lung airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2000;23:37–44. doi: 10.1165/ajrcmb.23.1.4034. [DOI] [PubMed] [Google Scholar]
- 17.Suzuki N., Ishizaki J., Yokota Y., Higashino K., Ono T., Ikeda M., Fujii N., Kawamoto K., Hanasaki K. Structures, enzymatic properties, and expression of novel human and mouse secretory phospholipase A(2)s. J. Biol. Chem. 2000;275:5785–5793. doi: 10.1074/jbc.275.8.5785. [DOI] [PubMed] [Google Scholar]
- 18.Masuda S., Murakami M., Ishikawa Y., Ishii T., Kudo I. Diverse cellular localizations of secretory phospholipase A2 enzymes in several human tissues. Biochim. Biophys. Acta. 2005;1736:200–210. doi: 10.1016/j.bbalip.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 19.Masuda S., Murakami M., Mitsuishi M., Komiyama K., Ishikawa Y., Ishii T., Kudo I. Expression of secretory phospholipase A2 enzymes in lungs of humans with pneumonia and their potential prostaglandin-synthetic function in human lung-derived cells. Biochem. J. 2005;387:27–38. doi: 10.1042/BJ20041307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim K.P., Rafter J.D., Bittova L., Han S.K., Snitko Y., Munoz N.M., Leff A.R., Cho W. Mechanism of human group V phospholipase A2 (PLA2)-induced leukotriene biosynthesis in human neutrophils. A potential role of heparan sulfate binding in PLA2 internalization and degradation. J. Biol. Chem. 2001;276:11126–11134. doi: 10.1074/jbc.M004604200. [DOI] [PubMed] [Google Scholar]
- 21.Balestrieri B., Arm J.P. Group V sPLA2: Classical and novel functions. Biochim. Biophys. Acta. 2006;1761:1280–1288. doi: 10.1016/j.bbalip.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 22.Murakami M., Masuda S., Shimbara S., Ishikawa Y., Ishii T., Kudo I. Cellular distribution, post-translational modification, and tumorigenic potential of human group III secreted phospholipase A(2) J. Biol. Chem. 2005;280:24987–24998. doi: 10.1074/jbc.M502088200. [DOI] [PubMed] [Google Scholar]
- 23.Muñoz N.M., Kim Y.J., Meliton A.Y., Kim K.P., Han S.K., Boetticher E., O'Leary E., Myou S., Zhu X., Bonventre J.V. Human group V phospholipase A2 induces group IVA phospholipase A2-independent cysteinyl leukotriene synthesis in human eosinophils. J. Biol. Chem. 2003;278:38813–38820. doi: 10.1074/jbc.M302476200. [DOI] [PubMed] [Google Scholar]
- 24.Balestrieri B., Maekawa A., Xing W., Gelb M.H., Katz H.R., Arm J.P. Group V secretory phospholipase A2 modulates phagosome maturation and regulates the innate immune response against Candida albicans. J. Immunol. 2009;182:4891–4898. doi: 10.4049/jimmunol.0803776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boilard E., Lai Y., Larabee K., Balestrieri B., Ghomashchi F., Fujioka D., Gobezie R., Coblyn J.S., Weinblatt M.E., Massarotti E.M. A novel anti-inflammatory role for secretory phospholipase A2 in immune complex-mediated arthritis. EMBO Mol Med. 2010;2:172–187. doi: 10.1002/emmm.201000072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohtsuki M., Taketomi Y., Arata S., Masuda S., Ishikawa Y., Ishii T., Takanezawa Y., Aoki J., Arai H., Yamamoto K. Transgenic expression of group V, but not group X, secreted phospholipase A2 in mice leads to neonatal lethality because of lung dysfunction. J. Biol. Chem. 2006;281:36420–36433. doi: 10.1074/jbc.M607975200. [DOI] [PubMed] [Google Scholar]
- 27.Satake Y., Diaz B.L., Balestrieri B., Lam B.K., Kanaoka Y., Grusby M.J., Arm J.P. Role of group V phospholipase A2 in zymosan-induced eicosanoid generation and vascular permeability revealed by targeted gene disruption. J. Biol. Chem. 2004;279:16488–16494. doi: 10.1074/jbc.M313748200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muñoz N.M., Meliton A.Y., Arm J.P., Bonventre J.V., Cho W., Leff A.R. Deletion of secretory group V phospholipase A2 attenuates cell migration and airway hyperresponsiveness in immunosensitized mice. J. Immunol. 2007;179:4800–4807. doi: 10.4049/jimmunol.179.7.4800. [DOI] [PubMed] [Google Scholar]
- 29.Giannattasio G., Fujioka D., Xing W., Katz H.R., Boyce J.A., Balestrieri B. Group V secretory phospholipase A2 reveals its role in house dust mite-induced allergic pulmonary inflammation by regulation of dendritic cell function. J. Immunol. 2010;185:4430–4438. doi: 10.4049/jimmunol.1001384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muñoz N.M., Meliton A.Y., Meliton L.N., Dudek S.M., Leff A.R. Secretory group V phospholipase A2 regulates acute lung injury and neutrophilic inflammation caused by LPS in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009;296:L879–L887. doi: 10.1152/ajplung.90580.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bostrom M.A., Boyanovsky B.B., Jordan C.T., Wadsworth M.P., Taatjes D.J., de Beer F.C., Webb N.R. Group V secretory phospholipase A2 promotes atherosclerosis: evidence from genetically altered mice. Arterioscler. Thromb. Vasc. Biol. 2007;27:600–606. doi: 10.1161/01.ATV.0000257133.60884.44. [DOI] [PubMed] [Google Scholar]
- 32.Boyanovsky B., Zack M., Forrest K., Webb N.R. The capacity of group V sPLA2 to increase atherogenicity of ApoE-/- and LDLR-/- mouse LDL in vitro predicts its atherogenic role in vivo. Arterioscler. Thromb. Vasc. Biol. 2009;29:532–538. doi: 10.1161/ATVBAHA.108.183038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wootton P.T., Arora N.L., Drenos F., Thompson S.R., Cooper J.A., Stephens J.W., Hurel S.J., Hurt-Camejo E., Wiklund O., Humphries S.E., Talmud P.J. Tagging SNP haplotype analysis of the secretory PLA2-V gene, PLA2G5, shows strong association with LDL and oxLDL levels, suggesting functional distinction from sPLA2-IIA: results from the UDACS study. Hum. Mol. Genet. 2007;16:1437–1444. doi: 10.1093/hmg/ddm094. [DOI] [PubMed] [Google Scholar]
- 34.Rosengren B., Jönsson-Rylander A.C., Peilot H., Camejo G., Hurt-Camejo E. Distinctiveness of secretory phospholipase A2 group IIA and V suggesting unique roles in atherosclerosis. Biochim. Biophys. Acta. 2006;1761:1301–1308. doi: 10.1016/j.bbalip.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 35.Rosenson R.S., Hislop C., McConnell D., Elliott M., Stasiv Y., Wang N., Waters D.D., PLASMA Investigators Effects of 1-H-indole-3-glyoxamide (A-002) on concentration of secretory phospholipase A2 (PLASMA study): a phase II double-blind, randomised, placebo-controlled trial. Lancet. 2009;373:649–658. doi: 10.1016/S0140-6736(09)60403-7. [DOI] [PubMed] [Google Scholar]
- 36.Drexler W., Fujimoto J.G. State-of-the-art retinal optical coherence tomography. Prog. Retin. Eye Res. 2008;27:45–88. doi: 10.1016/j.preteyeres.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 37.Schmitz-Valckenberg S., Holz F.G., Bird A.C., Spaide R.F. Fundus autofluorescence imaging: Review and perspectives. Retina. 2008;28:385–409. doi: 10.1097/IAE.0b013e318164a907. [DOI] [PubMed] [Google Scholar]
- 38.Terman A., Kurz T., Navratil M., Arriaga E.A., Brunk U.T. Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid. Redox Signal. 2010;12:503–535. doi: 10.1089/ars.2009.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wing G.L., Blanchard G.C., Weiter J.J. The topography and age relationship of lipofuscin concentration in the retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci. 1978;17:601–607. [PubMed] [Google Scholar]
- 40.Rohrschneider K., Bültmann S., Springer C. Use of fundus perimetry (microperimetry) to quantify macular sensitivity. Prog. Retin. Eye Res. 2008;27:536–548. doi: 10.1016/j.preteyeres.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 41.Jang Y., Waterworth D., Lee J.E., Song K., Kim S., Kim H.S., Park K.W., Cho H.J., Oh I.Y., Park J.E. Carriage of the V279F null allele within the gene encoding Lp-PLA2 is protective from coronary artery disease in South Korean males. PLoS ONE. 2011;6:e18208. doi: 10.1371/journal.pone.0018208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song K., Nelson M.R., Aponte J., Manas E.S., Bacanu S.A., Yuan X., Kong X., Cardon L., Mooser V.E., Whittaker J.C. Sequencing of Lp-PLA2-encoding PLA2G7 gene in 2000 Europeans reveals several rare loss-of-function mutations. Pharmacogenomics J. 2011 doi: 10.1038/tpj.2011.20. in press. Published online May 24, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han S.K., Kim K.P., Koduri R., Bittova L., Munoz N.M., Leff A.R., Wilton D.C., Gelb M.H., Cho W. Roles of Trp31 in high membrane binding and proinflammatory activity of human group V phospholipase A2. J. Biol. Chem. 1999;274:11881–11888. doi: 10.1074/jbc.274.17.11881. [DOI] [PubMed] [Google Scholar]
- 44.Winget J.M., Pan Y.H., Bahnson B.J. The interfacial binding surface of phospholipase A2s. Biochim. Biophys. Acta. 2006;1761:1260–1269. doi: 10.1016/j.bbalip.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 45.Murakami M., Shimbara S., Kambe T., Kuwata H., Winstead M.V., Tischfield J.A., Kudo I. The functions of five distinct mammalian phospholipase A2S in regulating arachidonic acid release. Type IIa and type V secretory phospholipase A2S are functionally redundant and act in concert with cytosolic phospholipase A2. J. Biol. Chem. 1998;273:14411–14423. doi: 10.1074/jbc.273.23.14411. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.