

Abstract
Very-long-chain fatty acids (VLCFAs) play important roles in membrane structure and cellular signaling, and their contribution to human health is increasingly recognized. Fatty acid elongases catalyze the first and rate-limiting step in VLCFA synthesis. Heterozygous mutations in ELOVL4, the gene encoding one of the elongases, are known to cause macular degeneration in humans and retinal abnormalities in mice. However, biallelic ELOVL4 mutations have not been observed in humans, and murine models with homozygous mutations die within hours of birth as a result of a defective epidermal water barrier. Here, we report on two human individuals with recessive ELOVL4 mutations revealed by a combination of autozygome analysis and exome sequencing. These individuals exhibit clinical features of ichthyosis, seizures, mental retardation, and spasticity—a constellation that resembles Sjögren-Larsson syndrome (SLS) but presents a more severe neurologic phenotype. Our findings identify recessive mutations in ELOVL4 as the cause of a neuro-ichthyotic disease and emphasize the importance of VLCFA synthesis in brain and cutaneous development.
Main Text
Very-long-chain fatty acids (VLCFAs) with chain length ≥24 carbon atoms exist in both saturated and unsaturated forms; the latter are defined by the presence of one (monounsaturated) or more (polyunsaturated) double bonds. VLCFAs are largely components of more complex lipids, such as sphingolipids and phospholipids, that participate in a wide array of physiological functions. These functions include nuclear membrane stabilization, cell signaling, lipid and protein trafficking, modulation of inflammatory responses through the formation of eicosanoids, myelin production, normal photoreceptor function, and contribution to the epidermal water barrier.1, 2, 3, 4, 5, 6
VLCFAs are synthesized from shorter fatty acids by the sequential addition of 2-carbons. The first and rate-limiting step of this process is a condensation reaction with malonyl-CoA; this reaction is catalyzed by a family of enzymes collectively known as fatty acid elongases.7 The diversity of elongases seems to correlate with the complexity of organisms. In humans, seven elongases designated as ELOVL1-7 are known to exist, and these possess different substrate preferences, tissue expressions, and developmental patterns.8, 9, 10
Despite the critical role of elongases in the production of VLCFAs, the specific contribution of each elongase to tissue function is unclear. Mutations in only one elongase gene, ELOVL4 (MIM 605512), are known to cause disease in humans. Patients with heterozygous ELOVL4 mutations display a juvenile form of macular degeneration known as Stargardt disease type 3 (STGD3, MIM 600110).11 Interestingly, only three ELOVL4 mutations have been reported to date and all have segregated in an autosomal-dominant pattern, which has experimentally been shown to be dominant-negative in nature.11, 12, 13, 14 These ELOVL4-deficient patients have no apparent phenotype beyond their macular degeneration, which is probably related to a deficiency of VLCFA-containing lipids in the retina.15 The phenotypic effects of biallelic ELOVL4 mutations in humans are not known. However, homozygous Elovl4 knockout in mice results in a lethal cutaneous disease arising from a defective epidermal water barrier.6
Here, we report on two human individuals with recessive ELOVL4 mutations who display congenital ichthyosis, as seen in Elovl4 −/− mice, along with a profound neurological phenotype. This neuro-ichthyotic disease is characterized by a constellation of clinical features resembling Sjögren-Larsson syndrome (SLS) (MIM 270200).16 These ELOVL4-deficient “pseudo-SLS” individuals provide researchers with a unique opportunity to study the clinical effects of severe ELOVL4 deficiency and showcase the power of exome sequencing, even in simplex cases, to reveal the genetics of rare Mendelian disorders.
The index person (person 1), now almost 6 years old, was born to healthy first-cousin Saudi Arabian parents after an uncomplicated pregnancy. He was born by normal vaginal delivery at 38 weeks gestation and had a birth weight of 2.81 kg. A collodion membrane covering the skin was noted immediately after birth, which necessitated admission to the neonatal intensive-care unit for fluid management. The collodion membrane resolved and was replaced with dry ichthyotic skin. After he was discharged, his ichthyosis was noted to wax and wane periodically despite meticulous topical emollient therapy by the parents. Although no body part was fully spared, significant predilection to the neck and diaper areas was noted, and later the focus shifted to the hands and feet. Profound developmental delay had been evident since early infancy. Recalcitrant seizures started at the age of 4 months and progressively increased in frequency to approximately one every 1–2 hr, and there was no clear response to antiepileptic medications. Past medical history is also significant for recurrent and severe bronchial asthmatic episodes often requiring hospital admissions. He underwent surgical repair for bilateral inguinal hernias as an infant. Physical examination at 5 yr, 10 months of age was notable for head circumference at the fifth percentile, length <fifth percentile, and weight at the 25th percentile. He showed very little interest in his surroundings. No clear dysmorphia was noted. His oral exam was significant for gingivitis and loss of most of his teeth. His skin was remarkably dry, and scaling was particularly noticeable on his hands and feet. Severe hypertonia in the upper and lower extremities was evident, and he was unable to roll over or turn his head toward his spoken name. He could not bring his hands to a midline position and was generally immobile. Testicular size was small. Examination of his chest, heart, and abdomen was within normal limits. Ophthalmologic examination was limited by a lack of cooperation and parental refusal to sedate the patient. Nevertheless, the macula could be visualized and had a normal appearance. Limited electroretinograms (ERGs) revealed a normal flicker response. High myopia (−11 D) was diagnosed, and amblyopia was also suspected. Interestingly, although the mother's eye examination was completely normal, the father's eye examination revealed some drusen-like flecks in the macula and moderate myopia.
Laboratory investigations included a normal karyotype and normal plasma acylcarnitines and amino acids, liver function tests, electrolytes, thyroid function tests, blood lactate, ammonia, and urine organic acids. Brain magnetic resonance imaging (MRI) was performed at 6 months of age and revealed severely delayed myelination and evidence of brain atrophy (Figure 1). The combination of ichthyosis, intellectual disability, and spastic quadriplegia in person 1 raised the possibility of SLS,16 so biochemical analysis of fibroblast fatty aldehyde dehydrogenase (FALDH) activity was pursued because FALDH deficiency is the hallmark of SLS.17 However, FALDH activity was found to be normal, which suggested that person 1 had “pseudo-SLS.” Therefore, we enrolled him in a study approved by the institutional review board of the King Faisal Specialist Hospital and Research Center. Our hypothesis was that, in view of parental consanguinity, his condition was caused by autozygosity for an ancestral haplotype that harbors an autosomal-recessive mutation.18
Figure 1.
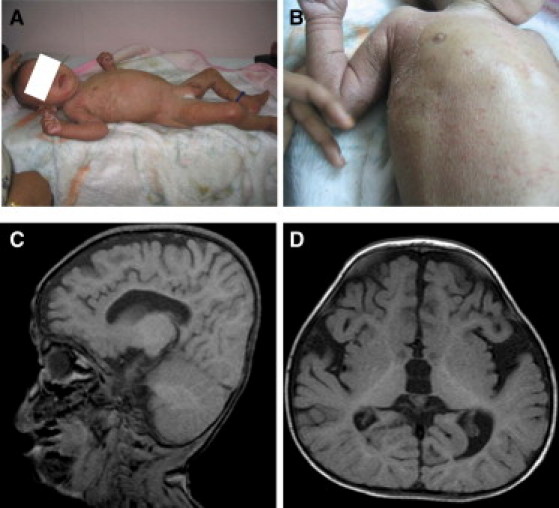
Clinical Images of Two Individuals with ELOVL4-Related Pseudo-SLS
(A) A photograph of person 2 shows the hypertonic posture and ichthyotic skin.
(B) A close-up image of the skin demonstrates the erythematous ichthyosis and a well-demarcated but less-affected region on the upper chest.
(C and D) A brain MRI of person 1 (sagittal and coronal views, respectively) shows delayed myelination and evidence of brain atrophy.
A QIAGEN kit was used for extracting genomic DNA from EDTA-treated whole blood from the index patient and his two parents. Genotyping was performed with the Axiom platform (Affymetrix), and autozygome analysis was run on autoSNPs as previously described.19 Several runs of homozygosity (ROH, cutoff 2 Mb) that were not shared by the parents were evident (Figure 2 and Table S1). Because ALDH3A2 (MIM 609523), the only known gene mutated in SLS, was present in one ROH, it was sequenced both at the genomic and cDNA levels as described,17 but no mutation was identified. Therefore, we proceeded with the sequencing of patient 1's exome at 30× coverage by using Illumina's HiSeq 2000 platform.
Figure 2.
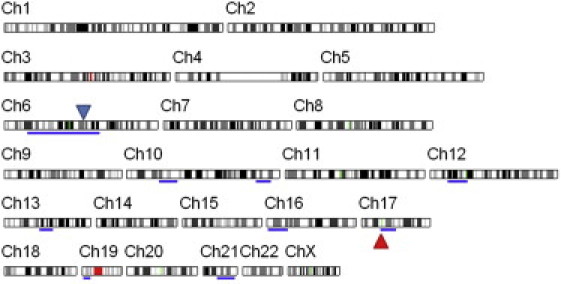
Autozygome Analysis of Person 1
Blue bars indicate ROH. The locations of ALDH3A2 and ELOVL4 are indicated by red and blue triangles, respectively.
Figure 3 summarizes the filtration strategy used in this study. We used dbSNP132 and the 1000 Genomes Project to highlight novel variants. We also used 72 in-house Saudi exomes to exclude variants, even when present once in the heterozygous state, because we reasoned that a 1/144 allele frequency is too high for a disease this rare. We have previously demonstrated the efficiency of using the autozygome as a filter of the variants generated by exome sequencing.20, 21 Indeed, by applying the autozygome among other filters (PolyPhen for the probability of pathogenicity and Tissue-specific Gene Expression and Regulation [TiGER] and UniGene expression databases for skin expression), we narrowed the search to just three novel coding variants: TBCC (MIM 602971)(NM_003192.2: c.258G>T [p.Glu86Asp]), ELOVL4 (NM_022726.2:c.646C>T [p.Arg216X]), and TIAM1 (MIM 600687) (NM_003253.2:c.3020G>A [p.Arg1007His]) (Figure 3, Figure 4 and Table S2). Of these, the truncating nonsense mutation in exon 5 of ELOVL4 was the most plausible candidate for causing the disease phenotype. This mutation was identified in 86 reads, which suggested high reliability of the call. Indeed, Sanger sequencing confirmed the homoallelic state of this mutation in the patient and the carrier state of the parents, and a screen of 192 Saudi controls was negative.
Figure 3.
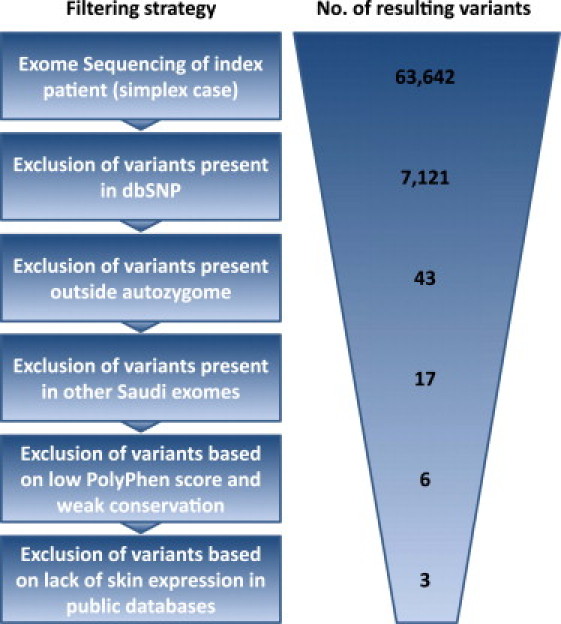
Filtering Strategy Used for Identification of a Causative Recessive Mutation in a Simplex Case with Pseudo-SLS
Figure 4.
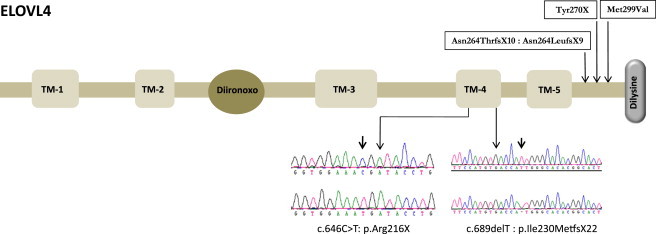
Cartoon of ELOVL4
Five transmembrane (TM) domains are indicated by boxes 1–5. The diiron-oxo binding motif (His158-His162) is circled, and Carboxy-terminal dilysine is columned. The previously reported mutations are shown on top of the cartoon, and the two mutations in this work with the DNA chromatogram are shown below the cartoon in the TM-4 domain.
In view of this finding, we screened 15 additional pseudo-SLS individuals (defined as having an SLS phenotype with normal FALDH activity) for ELOVL4 mutations using Sanger sequencing (primer sequences and PCR conditions for ELOVL4 are shown in Table S3). These individuals were recruited by one of the authors (W.B.R.) as part of an IRB-approved protocol at the University of Nebraska Medical Center. All had ichthyosis, psychomotor retardation, and spasticity, along with normal fibroblast FALDH activity and/or sequencing of ALDH3A2. One individual was found to harbor a novel homozygous truncating mutation in exon 6 of ELOVL4 (c.690del [p.Ile230Metfs∗22]) (Figure 4). DNA analysis of the person's parents showed that they were heterozygous for this mutation.
This person (person 2) was born at 35 weeks gestation to consanguineous Asian Indian parents. He was noted to have dry skin shortly after birth but exhibited no collodion membrane. At 2 months of age, myoclonic seizures began, and he had profound motor delay over the ensuing first year. He underwent surgery for a left-sided inguinal hernia. At 11 months of age, physical examination showed a height of 63 cm (<third percentile), a weight of 4.5 kg (<third percentile), and a head circumference of 40 cm (<third percentile). There were no dysmorphic features. His hair and nails appeared normal. No teeth had erupted. The ophthalmologic exam showed no cataract or macular abnormalities, but he did have temporal pallor in both eyes and exhibited photophobia. The skin had generalized erythematous ichthyosis with fine scales over most of the body but sparing the face (Figure 1). There were well-demarcated regions of skin that appeared less erythematous and hyperkeratotic. The neurologic exam showed increased tone in all four limbs and scissoring and contractures of the lower extremities.
An EEG showed bilateral posterior and generalized epileptogenic activity consistent with myoclonic epilepsy. Studies showed abnormal visual evoked potentials. No fibroblasts were available for study. Person 2 was bedridden and never gained the ability to sit, speak, or feed himself. He died from aspiration at 2 years of age. A similarly affected sibling died at 6 months of age.
The identification of ELOVL4 mutations in patients with the atypical juvenile form of macular degeneration (SGDT3) sparked interest in the role of VLCFAs in the physiology of the retina.22, 23 Using several mouse models in which the heterozygous-dominant SGDT3 mutation was introduced by genetic knockin made it possible to recapitulate a retinal phenotype and confirm the deficiency of phosphatidylcholine-containing fatty acids >28 carbons long.24 Interestingly, this finding was true both in heterozygous mice carrying the human dominant SGDT3 5 bp deletion as well as homozygous-knockout mice with a null allele. However, mice heterozygous for the null allele appeared to have no phenotypic consequences, suggesting that the heterozygous 5 bp deletion in humans is a dominant-negative mutation.25 This discovery is further supported by functional studies that convincingly showed that the mutant ELOVL4 failed to be targeted to the endoplasmic reticulum and caused mislocalization of the wild-type protein.26, 27, 28, 29 The loss of the ER targeting signal in the C terminus of ELOVL4, which characterizes all three dominant mutations reported in humans, is a likely explanation of this observation. We ruled out nonsense-mediated decay as a result of the truncating mutation in fibroblasts from person 1 (Figure S1), so it seemed likely that a truncated version of ELOVL4 was being made by this individual. However, immunoblot analysis of his cells consistently showed that he produced normal-sized ELOVL4, albeit at about a 45% reduced level (Figure S2). This is surprising because his nonsense mutation predicted a 24 kDa protein compared to the normal 37 kDa ELOVL4. We considered the possibility that this might represent a nonspecific 37 kDa band, but two independent anti-ELOVL4 antibodies gave the exact same result. The possibility that this protein was a product of an alternatively spliced transcript that skipped exon 5 and did not contain the mutation was not supported by RT-PCR analysis, which failed to identify any RNA transcript other than the mutant RNA that harbored the nonsense mutation (Figure S1). Therefore, we were left with the possibility of a nonsense readthrough. Such readthroughs have previously been detected under experimental conditions such as aminoglycoside treatment, suppressor tRNA, and depletion of eRF1 and eRF3. We did a 2D gel electrophoresis to examine the effect of the presumed readthrough on the protein, and other than the difference in abundance, there was no apparent difference in charge (Figure S3). It is not known whether the apparent nonsense readthrough is seen in keratinocytes and neural cells, which are probably more directly responsible for the cutaneous and neurologic phenotypes. Thus, it remains unclear whether the mutational mechanism in person 1 is mediated by a defective or deficient ELOVL4. Given that person 2 has a similar phenotype and harbors a homozygous frameshift mutation, it is possible that the overall mutational mechanism is loss of function.
The cutaneous phenotype of the two affected children is comparable to that of homozygous mouse models of Elovl4 mutations. These mice die within hours after birth as a result of severe ichthyosis, disruption of the epidermal water barrier, and consequent dehydration.25 Consistent with the role of ELOVL4 in VLCFA synthesis and its localization in skin, the epidermis of these mice completely lacks acylceramide, an essential VLCFA-containing component of the water barrier.25 The apparent lack of a cutaneous phenotype in heterozygous mice therefore suggests that the retinal phenotype is more sensitive to ELOVL4 dosage than is the epidermal phenotype.
In addition to the ichthyosis phenotype, the study individuals have suffered from severe brain damage likely caused by a deficiency of VLCFAs that are >28 carbons long, which are essential components of the sphingolipids that make up 30% of the myelin sheath of the brain.30 Unfortunately, early lethality of the homozygous mouse model precludes observation of a comparable neurologic phenotype.
We are intrigued by the apparent lack of a macular phenotype in person 1. Obviously, we cannot fully rule out the presence of a macular phenotype given the limited eye exam, including our inability to perform a full ERG or more detailed optical coherence tomography. However, the lack of macular flecks indicative of lipofuscin accumulation, which is typical of STGD3, might be explained by his young age. We also note that despite the recessive nature of person 1's mutation, the very mild macular changes in the father might be related to his heavy smoking habit. Several studies have shown the interaction between smoking as an environmental factor and the genetic predisposition to macular degeneration.31, 32 It would be of interest to observe the father's macula over time after smoking cessation, which he promised to consider after the revelation of his carrier status.
Although much remains to be learned about the biochemical changes in ELOVL4-deficient individuals, the deficiency in VLCFA synthesis suggests a therapeutic approach to repletion of these fatty acids. The parents of person 1 have independently tried to bypass the metabolic defect in their child by administering oral fish oil supplements, and we also recommended ceramide-based skin lotions. Whether this intervention will modify the natural history of the disease remains unknown. Feeding the patient a diet enriched with VLCFAs, i.e., supplementation with peanut oil, seems a rational approach, but entry of these exogenous fatty acids into the brain is less likely. On the other hand, topical application of VLCFAs to the skin might be considered as a potential therapy for the ichthyosis.
In summary, we show the power of combining homozygosity mapping with next-generation sequencing for identifying the cause of autosomal-recessive diseases even in simplex cases.18, 20 Our findings highlight the critical role of ELOVL4 and VLCFAs in early brain development and in the integrity of the epidermal water barrier.
Acknowledgments
We thank the two families for their participation, the Genotyping and Sequencing Core Facilities at King Faisal Specialist Hospital and Research Center for their help, and Kristen Jackson and Dana S'Aulis for their technical assistance. This work was funded in part by the King Abdulaziz City for Science and Technology grant 08-MED497-20 (F.S.A.), Dubai Harvard Foundation for Medical Research Collaborative grant (F.S.A.), and National Institutes of Health grant R01 AR 044552 (W.B.R.).
Published online: November 17, 2011
Footnotes
Supplemental data include three figures and three tables and can be found with this article online at http://www.cell.com/AJHG/.
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Tissue-specific Gene Expression and Regulation (TiGER), http://bioinfo.wilmer.jhu.edu/tiger/
UniGene, http://www.ncbi.nlm.nih.gov/unigene
Mutalyzer, https://mutalyzer.nl/
Supplemental Data
References
- 1.Schneiter R., Brügger B., Amann C.M., Prestwich G.D., Epand R.F., Zellnig G., Wieland F.T., Epand R.M. Identification and biophysical characterization of a very-long-chain-fatty-acid-substituted phosphatidylinositol in yeast subcellular membranes. Biochem. J. 2004;381:941–949. doi: 10.1042/BJ20040320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Toulmay A., Schneiter R. Lipid-dependent surface transport of the proton pumping ATPase: A model to study plasma membrane biogenesis in yeast. Biochimie. 2007;89:249–254. doi: 10.1016/j.biochi.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 3.Leonard A.E., Kelder B., Bobik E.G., Chuang L.T., Lewis C.J., Kopchick J.J., Mukerji P., Huang Y.S. Identification and expression of mammalian long-chain PUFA elongation enzymes. Lipids. 2002;37:733–740. doi: 10.1007/s11745-002-0955-6. [DOI] [PubMed] [Google Scholar]
- 4.Poulos A., Beckman K., Johnson D.W., Paton B.C., Robinson B.S., Sharp P., Usher S., Singh H. Very long-chain fatty acids in peroxisomal disease. Adv. Exp. Med. Biol. 1992;318:331–340. doi: 10.1007/978-1-4615-3426-6_30. [DOI] [PubMed] [Google Scholar]
- 5.McMahon A., Butovich I.A., Mata N.L., Klein M., Ritter R., 3rd, Richardson J., Birch D.G., Edwards A.O., Kedzierski W. Retinal pathology and skin barrier defect in mice carrying a Stargardt disease-3 mutation in elongase of very long chain fatty acids-4. Mol. Vis. 2007;13:258–272. [PMC free article] [PubMed] [Google Scholar]
- 6.Vasireddy V., Uchida Y., Salem N., Jr., Kim S.Y., Mandal M.N., Reddy G.B., Bodepudi R., Alderson N.L., Brown J.C., Hama H., et al. Loss of functional ELOVL4 depletes very long-chain fatty acids (> or =C28) and the unique omega-O-acylceramides in skin leading to neonatal death. Hum. Mol. Genet. 2007;16:471–482. doi: 10.1093/hmg/ddl480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nugteren D.H. The enzymic chain elongation of fatty acids by rat-liver microsomes. Biochim. Biophys. Acta. 1965;106:280–290. doi: 10.1016/0005-2760(65)90036-6. [DOI] [PubMed] [Google Scholar]
- 8.Jakobsson A., Westerberg R., Jacobsson A. Fatty acid elongases in mammals: Their regulation and roles in metabolism. Prog. Lipid Res. 2006;45:237–249. doi: 10.1016/j.plipres.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Tamura K., Makino A., Hullin-Matsuda F., Kobayashi T., Furihata M., Chung S., Ashida S., Miki T., Fujioka T., Shuin T., et al. Novel lipogenic enzyme ELOVL7 is involved in prostate cancer growth through saturated long-chain fatty acid metabolism. Cancer Res. 2009;69:8133–8140. doi: 10.1158/0008-5472.CAN-09-0775. [DOI] [PubMed] [Google Scholar]
- 10.Leonard A.E., Pereira S.L., Sprecher H., Huang Y.S. Elongation of long-chain fatty acids. Prog. Lipid Res. 2004;43:36–54. doi: 10.1016/s0163-7827(03)00040-7. [DOI] [PubMed] [Google Scholar]
- 11.Zhang K., Kniazeva M., Han M., Li W., Yu Z., Yang Z., Li Y., Metzker M.L., Allikmets R., Zack D.J., et al. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat. Genet. 2001;27:89–93. doi: 10.1038/83817. [DOI] [PubMed] [Google Scholar]
- 12.Edwards A.O., Donoso L.A., Ritter R., 3rd A novel gene for autosomal dominant Stargardt-like macular dystrophy with homology to the SUR4 protein family. Invest. Ophthalmol. Vis. Sci. 2001;42:2652–2663. [PubMed] [Google Scholar]
- 13.Bernstein P.S., Tammur J., Singh N., Hutchinson A., Dixon M., Pappas C.M., Zabriskie N.A., Zhang K., Petrukhin K., Leppert M., Allikmets R. Diverse macular dystrophy phenotype caused by a novel complex mutation in the ELOVL4 gene. Invest. Ophthalmol. Vis. Sci. 2001;42:3331–3336. [PubMed] [Google Scholar]
- 14.Maugeri A., Meire F., Hoyng C.B., Vink C., Van Regemorter N., Karan G., Yang Z., Cremers F.P., Zhang K. A novel mutation in the ELOVL4 gene causes autosomal dominant Stargardt-like macular dystrophy. Invest. Ophthalmol. Vis. Sci. 2004;45:4263–4267. doi: 10.1167/iovs.04-0078. [DOI] [PubMed] [Google Scholar]
- 15.Vasireddy V., Wong P., Ayyagari R. Genetics and molecular pathology of Stargardt-like macular degeneration. Prog. Retin. Eye Res. 2010;29:191–207. doi: 10.1016/j.preteyeres.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rizzo W.B. Sjögren-Larsson syndrome: Molecular genetics and biochemical pathogenesis of fatty aldehyde dehydrogenase deficiency. Mol. Genet. Metab. 2007;90:1–9. doi: 10.1016/j.ymgme.2006.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rizzo W.B., Carney G., Lin Z. The molecular basis of Sjögren-Larsson syndrome: Mutation analysis of the fatty aldehyde dehydrogenase gene. Am. J. Hum. Genet. 1999;65:1547–1560. doi: 10.1086/302681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alkuraya F.S. Autozygome decoded. Genet. Med. 2010;12:765–771. doi: 10.1097/GIM.0b013e3181fbfcc4. [DOI] [PubMed] [Google Scholar]
- 19.Shaheen R., Faqeih E., Seidahmed M.Z., Sunker A., Alali F.E., Alkuraya F.S. A TCTN2 mutation defines a novel meckel gruber syndrome locus. Hum. Mutat. 2011;36:573–578. doi: 10.1002/humu.21507. [DOI] [PubMed] [Google Scholar]
- 20.Shaheen R., Faqeih E., Sunker A., Morsy H., Al-Sheddi T., Shamseldin H.E., Adly N., Hashem M., Alkuraya F.S. Recessive mutations in DOCK6, encoding the guanidine nucleotide exchange factor DOCK6, lead to abnormal actin cytoskeleton organization and Adams-Oliver syndrome. Am. J. Hum. Genet. 2011;89:328–333. doi: 10.1016/j.ajhg.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aldahmesh M.A., Khan A.O., Mohamed J.Y., Alkuraya H., Ahmed H., Bobis S., Al-Mesfer S., Alkuraya F.S. Identification of ADAMTS18 as a gene mutated in Knobloch syndrome. J. Med. Genet. 2011;48:597–601. doi: 10.1136/jmedgenet-2011-100306. [DOI] [PubMed] [Google Scholar]
- 22.Liu A., Chang J., Lin Y., Shen Z., Bernstein P.S. Long-chain and very long-chain polyunsaturated fatty acids in ocular aging and age-related macular degeneration. J. Lipid Res. 2010;51:3217–3229. doi: 10.1194/jlr.M007518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Molday R.S., Zhang K. Defective lipid transport and biosynthesis in recessive and dominant Stargardt macular degeneration. Prog. Lipid Res. 2010;49:476–492. doi: 10.1016/j.plipres.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McMahon A., Jackson S.N., Woods A.S., Kedzierski W. A Stargardt disease-3 mutation in the mouse Elovl4 gene causes retinal deficiency of C32-C36 acyl phosphatidylcholines. FEBS Lett. 2007;581:5459–5463. doi: 10.1016/j.febslet.2007.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McMahon A., Butovich I.A., Kedzierski W. Epidermal expression of an Elovl4 transgene rescues neonatal lethality of homozygous Stargardt disease-3 mice. J. Lipid Res. 2011;52:1128–1138. doi: 10.1194/jlr.M014415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raz-Prag D., Ayyagari R., Fariss R.N., Mandal M.N., Vasireddy V., Majchrzak S., Webber A.L., Bush R.A., Salem N., Jr., Petrukhin K., Sieving P.A. Haploinsufficiency is not the key mechanism of pathogenesis in a heterozygous Elovl4 knockout mouse model of STGD3 disease. Invest. Ophthalmol. Vis. Sci. 2006;47:3603–3611. doi: 10.1167/iovs.05-1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vasireddy V., Vijayasarathy C., Huang J., Wang X.F., Jablonski M.M., Petty H.R., Sieving P.A., Ayyagari R. Stargardt-like macular dystrophy protein ELOVL4 exerts a dominant negative effect by recruiting wild-type protein into aggresomes. Mol. Vis. 2005;11:665–676. [PubMed] [Google Scholar]
- 28.Karan G., Yang Z., Howes K., Zhao Y., Chen Y., Cameron D.J., Lin Y., Pearson E., Zhang K. Loss of ER retention and sequestration of the wild-type ELOVL4 by Stargardt disease dominant negative mutants. Mol. Vis. 2005;11:657–664. [PubMed] [Google Scholar]
- 29.Grayson C., Molday R.S. Dominant negative mechanism underlies autosomal dominant Stargardt-like macular dystrophy linked to mutations in ELOVL4. J. Biol. Chem. 2005;280:32521–32530. doi: 10.1074/jbc.M503411200. [DOI] [PubMed] [Google Scholar]
- 30.Brush R.S., Tran J.T., Henry K.R., McClellan M.E., Elliott M.H., Mandal M.N. Retinal sphingolipids and their very-long-chain fatty acid-containing species. Invest. Ophthalmol. Vis. Sci. 2010;51:4422–4431. doi: 10.1167/iovs.09-5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thornton J., Edwards R., Mitchell P., Harrison R.A., Buchan I., Kelly S.P. Smoking and age-related macular degeneration: A review of association. Eye (Lond.) 2005;19:935–944. doi: 10.1038/sj.eye.6701978. [DOI] [PubMed] [Google Scholar]
- 32.Conley Y.P., Thalamuthu A., Jakobsdottir J., Weeks D.E., Mah T., Ferrell R.E., Gorin M.B. Candidate gene analysis suggests a role for fatty acid biosynthesis and regulation of the complement system in the etiology of age-related maculopathy. Hum. Mol. Genet. 2005;14:1991–2002. doi: 10.1093/hmg/ddi204. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.