

1. DISEASE CHARACTERISTICS
1.1 Name of the disease (synonyms)
Multiple endocrine neoplasia 2A (MEN 2A), multiple endocrine neoplasia 2B (MEN 2B), familial medullary thyroid carcinoma (FMTC).
1.2 OMIM# of the disease
MEN 2A: 171400; MEN 2B: 162300; FMTC: 155240.
1.3 Name of the analysed genes or DNA/chromosome segments
RET proto-oncogene/10q11.2.
1.4 OMIM# of the gene(s)
164761.
1.5 Mutational spectrum
Mostly point mutations, insertions of few nucleotides, small indels.
Presently mutations are described in exons 5, 8, 10, 11, 13, 14, 15 and 16.1, 2, 3, 4
1.6 Analytical methods
Bidirectional sequencing of coding regions.
1.7 Analytical validation
Direct sequencing of both strands, verification of sequence results with genomic DNA from the second blood sample. External validation through exchange of DNA control samples with other diagnostic institutions.
1.8 Estimated frequency of the disease (incidence at birth (‘birth prevalence') or population prevalence)
1 per 30 000.
1.9 If applicable, prevalence in the ethnic group of investigated person
Not available.
1.10 Diagnostic setting

Comment: Pre-implantation/prenatal diagnostics is rarely done because adequate prevention/therapy of the associated diseases is available. Theoretically, we would recommend pre-implantation diagnosis in MEN 2B families, but these cases are very rare.
2. TEST CHARACTERISTICS
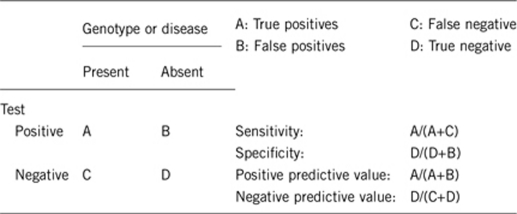
2.1 Analytical sensitivity (proportion of positive tests if the genotype is present)
About 98%.
2.2 Analytical specificity (proportion of negative tests if the genotype is not present)
About 98%.
2.3 Clinical sensitivity (proportion of positive tests if the disease is present)
The clinical sensitivity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
About 97%.5
2.4 Clinical specificity (proportion of negative tests if the disease is not present)
The clinical specificity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
About 98%.
2.5 Positive clinical predictive value (lifetime risk of developing the disease if the test is positive)
Depending on the specific codon mutated:
MEN 2A: frequency of MTC 90–95%, pheochromocytoma 40–50%, hyperparathyroidism 10–20%.
MEN 2B: frequency of MTC 100%, pheochromocytoma 50%, hyperparathyroidism 0%.
FMTC: frequency of MTC 80%, rarely pheochromocytoma/parathyroid disease (below 5%).6, 7, 8
2.6 Negative clinical predictive value (probability of not developing the disease if the test is negative)
Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
About 98%.
Index case in that family had not been tested:
Depending on age, phenotype and number of family members under investigation.
3. CLINICAL UTILITY
In MEN 2, DNA analysis has obtained a central role in diagnosis and management. For this reason it is inconceivable that in MEN 2 families patients may refuse early DNA analysis for their children at risk. Delay in treatment may have serious consequences for survival and quality of life.
3.1 (Differential) diagnosis: The tested person is clinically affected
(To be answered if in 1.10 ‘A' was marked)
If a person is clinically affected with MTC, about 70–75% of cases are sporadic, the remaining 25–30% belong to the hereditary variety (MEN 2/FMTC). In all cases of MTC genetic testing for mutations in the RET proto-oncogene is recommended.1 The probability that an individual with apparent sporadic MTC will be found to have a RET mutation is 1–7%.1, 9, 10
In rare families, both HSCR and MEN 2 appear to segregate with germline RET mutations in codon 609, 611, 618, or 620.11, 12
3.1.1 Can a diagnosis be made other than through a genetic test?

3.1.2 Describe the burden of alternative diagnostic methods to the patient?
In the past, pentagastrin and calcium infusion tests were used to stimulate calcitonin secretion by thyroid C-cells, but nowadays RET-mutation screening is available for familial MTC and thus these tests have lost their clinical significance with respect to diagnosis. The widespread availability of RET gene mutation analysis and prophylactic thyroidectomy in MEN 2 disease-gene carriers has made periodical C-cell stimulation tests largely obsolete for detection of MTC in MEN 2 families.
For detection of MTC, C-cell stimulation tests are useful for family members who refuse DNA analysis. In addition, preoperatively it may facilitate a decision with regard to the extensiveness of the surgical procedure.
Occasionally, MTCs do not secrete calcitonin,13, 14 but this has not been observed in MEN 2 gene carriers.
Tests for pheochromocytoma and HPT significantly add to the burden.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
After 3–4 appointments with calcitonin stimulation test the costs are equal to the molecular diagnostic test. Savings through unnecessary testing for pheochromocytoma and HPT further increase the cost effectiveness.
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.10 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive (please describe):
Positive test result: Depending on the detected mutation the patient should undergo prophylactic thyroidectomy or yearly calcitonin measurement. Likewise, also depending on the detected mutation the patient should undergo biochemical testing for pheochromocytoma and HPT.
If the test result is negative (please describe):
Negative test result: Person can be excluded from further evaluations.
3.2.2 Which options in view of lifestyle and prevention does a person at risk have if no genetic test has been done (please describe)?
Calcitonin test and clinical evaluation are necessary every year, with an increasing risk of MTC development and development of lymph node and distant metastases. There is an increasing risk of developing pheochromocytoma or hyperparathyroidism during one's lifetime.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.10 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Yes, autosomal dominant inheritance.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
Early DNA analysis (already at birth) may be considered as a safe clinical test with clinical consequences and makes periodical measurement of calcitonin levels and ultrasound redundant in non-carriers.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes.
3.4 Prenatal diagnosis
(To be answered if in 2.10 ‘D' was marked)
This is rarely necessary, because the disease can be managed properly (prophylactic thyroidectomy at an adequate age determined by the respective RET mutation, adrenalectomy in pre-symptomatic stage of pheochromocytoma, parathyroidectomy in asymptomatic stage of hyperparathyroidism).
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnostic?
Yes, but this is rarely done, because postnatal therapy is available.
4. IF APPLICABLE, FURTHER CONSEQUENCES OF TESTING
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe)
Molecular diagnosis of the proband allows for a precise recommendation of further therapy (prophylactic thyroidectomy at the appropriate age, depending on the respective RET mutation) and also for recommendation concerning the commencement and frequency of biochemical testing for pheochromocytoma and hyperparathyroidism. Cure of MTC is only possible as long as MTC is limited to the thyroid gland; therefore diagnosis at an early stage is the prerequisite for cure. Genetic test allows for regular biochemical screening using epinephrine and nor-epinephrine analysis to prevent hypertensive crisis induced by pheochromocytoma,16 and regular serum calcium and parathyroid hormone measurement to diagnose hyperparathyroidism at an early stage.15
Molecular diagnosis of the proband allows for an accurate and faster diagnosis in first-degree relatives.
Acknowledgments
This work was supported by EuroGentest, an EU-FP6-supported NoE, contract number 512148 (EuroGentest Unit 3: ‘Clinical genetics, community genetics and public health', Workpackage 3.2).
The authors declare no conflict of interest.
References
- Kloos RT, Eng C, Evans DB, et al. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid. 2009;19:565–612. doi: 10.1089/thy.2008.0403. [DOI] [PubMed] [Google Scholar]
- Frank-Raue K, Rondot S, Raue F. Molecular genetics and phenomics of RET mutations: impact on prognosis of MTC. Mol Cell Endocrinol. 2010;322:2–7. doi: 10.1016/j.mce.2010.01.012. [DOI] [PubMed] [Google Scholar]
- Elisei R, Romei C, Cosci B, et al. RET genetic screening in patients with medullary thyroid cancer and their relatives: experience with 807 individuals at one center. J Clin Endocrinol Metab. 2007;92:4725–4729. doi: 10.1210/jc.2007-1005. [DOI] [PubMed] [Google Scholar]
- de Groot JW, Links TP, Plukker JT, Lips CJ, Hofstra RM. RET as a diagnostic and therapeutic target in sporadic and hereditary endocrine tumors. Endocr Rev. 2006;27:535–560. doi: 10.1210/er.2006-0017. [DOI] [PubMed] [Google Scholar]
- Romei C, Mariotti S, Fugazzola L, et al. Multiple endocrine neoplasia type 2 syndromes (MEN 2): results from the ItaMEN network analysis on the prevalence of different genotypes and phenotypes. Eur J Endocrinol. 2010;163:301–308. doi: 10.1530/EJE-10-0333. [DOI] [PubMed] [Google Scholar]
- Machens A, Niccoli-Sire P, Hoegel J, et al. Early malignant progression of hereditary medullary thyroid cancer. N Engl J Med. 2003;349:1517–1525. doi: 10.1056/NEJMoa012915. [DOI] [PubMed] [Google Scholar]
- Frank-Raue K, Rybicki LA, Erlic Z, et al. Risk profiles and penetrance estimations in multiple endocrine neoplasia type 2A caused by germline RET mutations located in exon 10. Hum Mutat. 2010;32:51–58. doi: 10.1002/humu.21385. [DOI] [PubMed] [Google Scholar]
- Raue F, Frank-Raue K. Update multiple endocrine neoplasia type 2. Fam Cancer. 2010;9:449–457. doi: 10.1007/s10689-010-9320-2. [DOI] [PubMed] [Google Scholar]
- Brandi ML, Gagel RF, Angeli A, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab. 2001;86:5658–5671. doi: 10.1210/jcem.86.12.8070. [DOI] [PubMed] [Google Scholar]
- Wiench M, Wygoda Z, Gubala E, et al. Estimation of risk of inherited medullary thyroid carcinoma in apparent sporadic patients. J Clin Oncol. 2001;19:1374–1380. doi: 10.1200/JCO.2001.19.5.1374. [DOI] [PubMed] [Google Scholar]
- Hansford JR, Mulligan LM. Multiple endocrine neoplasia type 2 and RET: from neoplasia to neurogenesis. J Med Genet. 2000;37:817–827. doi: 10.1136/jmg.37.11.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks AS, Oostra BA, Hofstra RM. Studying the genetics of Hirschsprung's disease: unraveling an oligogenic disorder. Clin Genet. 2005;67:6–14. doi: 10.1111/j.1399-0004.2004.00319.x. [DOI] [PubMed] [Google Scholar]
- Wang TS, Ocal IT, Sosa JA, Cox H, Roman S. Medullary thyroid carcinoma without marked elevation of calcitonin: a diagnostic and surveillance dilemma. Thyroid. 2008;18:889–894. doi: 10.1089/thy.2007.0413. [DOI] [PubMed] [Google Scholar]
- Dora JM, Canalli MH, Capp C, Punales MK, Vieira JG, Maia AL. Normal perioperative serum calcitonin levels in patients with advanced medullary thyroid carcinoma: case report and review of the literature. Thyroid. 2008;18:895–899. doi: 10.1089/thy.2007.0231. [DOI] [PubMed] [Google Scholar]
- Raue F, Kraimps JL, Dralle H, et al. Primary hyperparathyroidism in multiple endocrine neoplasia type 2A. J Intern Med. 1995;238:369–373. doi: 10.1111/j.1365-2796.1995.tb01212.x. [DOI] [PubMed] [Google Scholar]
- Pacak K, Eisenhofer G, Ilias I. Diagnosis of pheochromocytoma with special emphasis on MEN2 syndrome. Hormones (Athens) 2009;8:111–116. doi: 10.14310/horm.2002.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]