

Abstract
Organophosphates such as methamidophos, usually used in the agricultural field, have harmful effects on humans. Exposures to insecticides has been associated with many disorders, including damage to the central and peripheral nervous system. Chronic exposure to organophosphates may lead to persistent neurological and neurobehavioral effects. This study was conducted to determine the effect of methamidophos on [3H]-dopamine (DA) and gamma aminobutyric acid (GABA) release from different brain regions after chronic exposure to it for 3, 6 or 9 months. After a six-month methamidophos treatment, the mice showed high susceptibility to convulsive seizures and a reduction in stimulated gamma aminobutyric acid release from the cerebral cortex and hippocampal slices, whereas stimulated (DA) release was slightly decreased from the striatum after three months of methamidophos exposure. The results indicate changes in gamma aminobutyric acid and dopamine neurotransmission, suggesting a specific neuronal damage.
Keywords: brain, neurotransmitter, GABA, dopamine, chronic toxicity, methamidophos.
Introduction
Insecticides are essential to the human population in terms of food security and disease prevention. There has been an increase in the use of insecticides, but the pests themselves have become more resistant and, consequently, the tolerated minimum dose has increased (it can range from a few grams), causing health problems. The mechanism of action is by inhibition of the enzyme acetylcholinesterase (AChE) and formation of acetylcholine (ACh) with its consequent alterations of the central nervous system, affecting the cholinergic neurotransmission in the peripheral and central nervous system1,2. The spectrum of effects caused by excess ACh depends upon the distribution of the insecticide in the body and the receptor type with which ACh interacts3.
On the other hand, neurotoxic and schizophrenic reactions, depression, and damage to memory with thought difficulties have been observed in groups of workers exposed to different insecticides4. That damage can persist for some months with neuropathologic injuries and cognoscitive and neuromuscular function damage with symptoms of ataxia, convulsions, parestesia and defects in speech, lack of memory, insomnia and mental disorder5. Exposure to organophosphates may produce neuropsychiatric symptoms and biochemical alterations in humans, such as anxiety, depression, impairment of concentration and learning, fatigue, postural instability and rigidity of face muscles, bradykynesia and transient parkinsonism6. Exposure to phosphorated insecticides for a long time results in convulsions, respiratory damage and cardiac arrhythmia that can lead to cerebral anoxia7. Comparative studies have been performed on the action of the receptor for gamma aminobutyric acid (GABA), N-methyl-D-aspartate (NMDA), and dopamine (DA) release. With kainic acid administration by perfusion and microdialysis, there were toxic signs of convulsions with greater DA, GABA and aspartate release at the striatum8. Gabaergic power stations and neurons, and the number of positive cells for GABA were reduced in the brain, causing abnormal movements of the eyes in humans9. Only a few studies on chronic poisoning by phosphorated insecticides exist. This work therefore aimed to study the effects of GABA and DA release on different brain regions of mice after chronic exposure to a methamidophos device.
Materials and Methods
Animals
Forty adult male Balb/c mice at 3 months (12 weeks) of age and weighing 22–25 g were used. They were kept under standard conditions with 12-h light:12-h dark cycles and free access to water and food in polypropylene cages with controlled temperature of 23 ± 2 °C and relative humidity of 85 ± 5 percent.
The dose of insecticide to which the experimental animals were exposed was determined by a dose-response curve. We used several groups of 10 mice to find the median lethal dose (LD50) of methamidophos with different concentrations following the method of Miller and Tainter10 and determined the subacute or chronic exposure dose, 2.6 mg/kg.
The animals were divided into two groups, one exposed to the insecticide methamidophos at a dose of 2.6 mg/kg administered subcutaneously every third day throughout the duration of this study (3, 6 and 9 months) and the other constituting the control group, which was treated with physiological saline as the vehicle solution. At the end of the exposure period, the animals were sacrificed by decapitation. Their brains were removed, and slices of the different brain regions (cerebral cortex, striatum, and hippocampus) were dissected to perform the GABA and DA release experiments. Tissue slices were immediately washed with a 0.32 M sucrose solution at temperatures ranging from 0 to 4 °C.
The experimental methods applied to laboratory animals used in this investigation 22 were carried out under the guidelines of the Mexican Official Standard NOM-062-ZOO- 23 1999 backed by the Coordination of Research and Ethics Committee of the Faculty of 24 Medicine of the National Autonomous University of Mexico (UNAM).
Materials
Methamidophos (Tamaron 600®) formulated as a soluble liquid base of methamidophos (O,S-dimethyl phosphoramidothioate) for agricultural use was purchased from Bayer Chemical Corporation in Mexico.
Dihydroxyphenylethylamine, 3,4-[7-3H(N)]-, or tritiated [3H]-dopamine ([3H]-DA) (31 Ci/mmol) and γ -[2,3-3H(N)]-aminobutyric acid (-[3H]-GABA) (36.8 Ci/mmol) were purchased from New England Nuclear, Wilmington, DE, USA. All other reagents were from Sigma-Aldrich (St. Louis, MO, USA).
[3H]-DA and [3H]-GABA
Release experiments were conducted in accordance with the following protocol: to measure the [3H]-DA baseline flow, a Krebs-bicarbonate medium was made with 115 mM NaCl, 3 mM KCl, 1.2 mM Mg2SO4, 1.2 mM NaH2PO4, 25 mM NaHCO3, 1.5 mM CaCl2, 10 mM glucose, 0.1 mM ascorbic acid and 25 μM Iproniazide and adjusted to pH 7.4 with a CO2-O2 mixture (5–95%). For [3H]-GABA, release experiments were carried out as [3H]-DA release using only the incubation medium containing 1 mM aminooxyacetic acid to avoid GABA metabolism11.
Slices of different brain regions (cerebral cortex, hippocampus and striatal) were incubated for 20 min in 1 mL of incubation medium for GABA at 37 °C together with [3H]-DA. The samples were then transferred to a filter perfusion Whatman GF/B system. Both filters and tissue samples were washed an SP-06 Brandel perfusion system at a 250-μl flow for 30 min. Three consecutive 10 min fractions were then collected and taken as the baseline flow. Spontaneous release was conducted with a solution containing drugs for the following four fractions (40 min) with a 10 µM concentration in all cases. Finally, three additional fractions were collected with a baseline medium. Every fraction was collected in vials with radioactive fluid, and radioactivity was measured with a model LS6000 Beckman scintiscan.
Spontaneous [3H]-DA release was calculated as the amount of DA released in the presence of drugs over the baseline flow (read as 0%). Results were expressed as the overflow in percentage of the total radioactivity present in the samples, with fraction number 6 having the greatest release value.
Statistical analysis
Statistical significance between group means was found by one-way ANOVA analysis using the SPSS computer program with significance denoted by p<0.001–p <0.05.
Results
Collateral effects of methamidophos treatment
Treatment of the animals with methamidophos produced a significant body weight reduction at the end of the 6-month period, and a loss of 25% was observed within 6 and 9 months of treatment (data not shown). All animals showed tonic convulsive seizures characterized by a generalized body tremor; the latency time was observed to be between 7 and 15 min after receipt of the methamidophos treatment. The convulsive duration was approximately 20 min, and some aggressive behavior was also observed in some animals after 5–6 months of treatment. During acute poisoning, it was also observed that the mice had white eyes, spiky hair and sometimes nosebleed.
Cortical and hippocampal [3H]-GABA release
Methamidophos treatment produced a reduction in stimulated [3H]-GABA release in slices obtained from the cerebral cortex compared with the control (Fig. 1). This effect was observed after methamidophos exposure for 3 and 6 months (Fig. 1a–b). Basal [3H]-GABA release was not modified in any of the groups studied at this time. However, after 9 months, stimulated [3H]-GABA release was significantly increased in slices from the cerebral cortex compared with the control group. At this time, basal [3H]-GABA release did not differ between the groups studied (Fig. 1c).
Fig. 1.

[3H]-GABA release from cerebral cortex slices of mice exposed to
methamidophos. Statistics are presented as the mean ± SEM of six–seven experiments
(n=1 per experiment) with determinations duplicated. Statistically different with
respect to the control group ( and
and 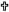 : p<0.001 and p<0.05, respectively).
: p<0.001 and p<0.05, respectively).
On the other hand, stimulated [3H]-GABA release in hippocampal slices was also decreased by methamidophos exposure during the initial 6 months of this study (Fig. 2a–b). However, at the end of 9 months of methamidophos treatment, changes in stimulated [3H]-GABA release were observed at the hippocampal region (Fig. 2c). No differences in basal [3H]-GABA release were observed at any time studied.
Fig. 2.

[3H]-GABA release from hippocampal slices of mice exposed to
methamidophos. Statistics are presented as the mean ± SEM of six–seven experiments
(n=1 per experiment) with determinations duplicated. Statistically different with
respect to the control group ( and
and 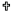 : p<0.001 and p<0.05, respectively).
: p<0.001 and p<0.05, respectively).
Striatal [3H]-DA release
Methamidophos exposure for 3 months resulted in an insignificant light reduction in stimulated [3H]-DA release in slices obtained from the striatal region compared with the control group (Fig. 3a), whereas, no difference was observed after 6 months of methamidophos exposure in stimulated [3H]-DA release at the striatum compared with the control (Fig. 3b). Basal [3H]-DA release was not modified in any of the groups during the study period.
Fig. 3.

[3H]-DA release from striatal slices of mice exposed to methamidophos. Statistics are presented as the mean ± SEM of six–seven experiments (n=1 per experiment) with determinations duplicated.
Discussion
Organophosphates represent a group of compounds sharing a similar chemical structure with resulting inhibition of cholinesterase enzymes. They are also well absorbed by all routes of exposure, including the dermal, respiratory and gastrointestinal routes. Their primary route of toxicity is irreversible binding of phosphate radicals to the active site of the enzyme AChE. This results in excess cholinergic stimulation and subsequently the acute clinical effects seen in poisoning. Clinical effects from acute poisoning can be separated into autonomic nervous system, neuromuscular junction, and central nervous system (CNS) categories3.
Postnatal exposure of mice to insecticide not only led to a permanent and selective loss of dopaminergic neurons but also enhanced the impact of these insecticides during adulthood relative to developmental-only or adult-only treatment8.
Various parameters of neurotransmission can be directly affected by neurotoxicants; these include the enzyme(s) of neurotransmitter synthesis, the release and uptake processes, the enzyme(s) that metabolizes the neurotransmitter, the receptors and postsynaptic events associated with receptor activation. Some neurotoxicants can interfere with neurotransmission indirectly by interacting, for example, with energy metabolism, sodium channels or ATPases. Furthermore, measured alterations of any neurotransmission parameter can be the result of neuronal death caused by a cytotoxic effect of the neurotoxicants. Thus, the decrease in stimulated GABA and DA release observed in this work was associated with convulsive seizures shown by the mice after methamidophos administration, indicating reduced neurotransmission as a result of neuronal damage in the region studied. A possible decrease in release of GABA and concomitant increase in seizure susceptibility have been demostrated by blocking the GABA-gated chloride channel12. A similar work13 showed that treatment with aldrin, an organochlorine insecticide, made rats more susceptible to sound-induced seizures, suggesting the involvement of both noradrenergic and alteration excitability of the CNS with the observed increments in seizure susceptibility. Furthermore, organophosphoruses owe their acute toxicity to inhibition of AChE and accumulation of ACh at cholinergic receptors. Chronic exposure to these compounds results in the development of tolerance for their toxicity, which was associated with a decrease in the density of muscarinic and nicotinic receptors in both the central and peripheral nervous systems14.
These works may suggest that developmental exposure to insecticides elicits long-lasting alterations in cell-signaling cascades that are shared by multiple neurotransmitters and hormonal inputs; the resultant abnormalities of synaptic communication are thus likely to occur in widespread neural circuits and induce corresponding behaviors9.
The toxic actions of insecticides are due primarily to their ability to inhibit AChE, an enzyme responsible for the breakdown of the neurotransmitter ACh. The result is excessive ACh accumulation at synapses, where only small quantities of ACh are needed for transmission. Agent-induced ACh accumulation generates side effects that involve action on other CNS neurotransmitter systems (e.g., norepinephrine, dopamine, and γ-amino butyric acid). The interplay of various neurotransmitters within the nervous system probably results in these varied side effects15, although nerve agents may exert direct effects on these same noncholinergic systems16.
Developmental neurotoxicity is concerned with adverse health effects of exogenous agents acting on neurodevelopment. Because human brain development is a delicate process involving many cellular events, the developing fetus is rather susceptible to compounds that can alter the structure and function of the brain. Today, there is clear evidence that early exposure to many neurotoxicants can severely damage the developing nervous system. Although, in recent years, there has been much attention given to model development and risk assessment procedures for developmental toxicants, the area of developmental neurotoxicity has been largely ignored. Here, we consider the problem of risk estimation for developmental neurotoxicants from animal bioassay data. Since most responses from developmental neurotoxicity experiments are nonquantal in nature, an adverse health effect will be defined as a response that occurs with very small probability in unexposed animals1.
Acknowledgements
The authors thank the Consejo Nacional de Ciencia y Tecnología (CONACYT) de México, Centro de Investigación Biomédica de Occidente (CIBO) del Instituto Mexicano del Seguro Social (IMSS), Centro Universitario de Ciencias de la Salud (CUCS) de la Universidad de Guadalajara (U de G), Centro Universitario de Ciencias Biológicas y Agropecuarias (CUCBA) de la Universidad de Guadalajara (U de G) and Facultad de Medicina de la Universidad Autónoma de Sinaloa (UAS) for their support for this work.
References
- 1.Razzaghi M, Kodell R. Quantitative risk assessment for developmental neurotoxic effects. Risk Anal. 24: 1673–1681 2004. [DOI] [PubMed] [Google Scholar]
- 2.Costa LG. Current issues in organophosphate toxicology. Clin Chim Acta. 366: 1–13 2006. [DOI] [PubMed] [Google Scholar]
- 3.Mileson BE, Chambers JE, Chen WL, Dettbarn W, Ehrich M, Eldefrawi AT. Common mechanism of toxicity: a case study of organophosphorus pesticides. Toxicol Sci. 41: 8–20 1998. [DOI] [PubMed] [Google Scholar]
- 4.Kamel F, Engel LS, Gladen BC, Hoppin JA, Alavanja MC. Neurologic symptoms in licensed private pesticide applicators in the agricultural health study. Environ Health Perspect. 113: 877–882 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stallones L, Beseler C. Pesticide illness, farm practices, and neurological symptoms among farm residents in Colorado. Environ Res. 90: 89–97 2002. [DOI] [PubMed] [Google Scholar]
- 6.Salvi RM, Lara DR, Ghisolfi ES, Portela LV, Dias RD, Souza DO. Neuropsychiatric evaluation in subjects chronically exposed to organophosphate pesticides. Toxicol Sci. 72: 267–271 2003. [DOI] [PubMed] [Google Scholar]
- 7.Wu SY, Casida JE. Subacute neurotoxicity induced in mice by potent organophosphorus neuropathy target esterase inhibitors. Toxicol Appl Pharmacol. 139: 195–202 1996. [DOI] [PubMed] [Google Scholar]
- 8.Jacobsson SO, Casseñ GE, Karlsson BM, Sellstrom A, Persson SA. Release of dopamine, GABA and EAA in rats during intrastriatal perfusion with kainic acid, NMDA and soman: a comparative microdialysis study. Arch Toxicol. 71: 756–765 1997. [DOI] [PubMed] [Google Scholar]
- 9.Kojima Y, Sexiya H, Ishikawa S. The effect of organophosphorus pesticide (Fenthion) on Gaba-cell in the central nuclei. Nippon Ganka Gakkai Zasshi. 97: 50–57 1993. [PubMed] [Google Scholar]
- 10.Miller LC, Tainter ML. Graphical method for determination of LD50. Proc Soc Exp Biol Med. 57: 261–271 1944. [Google Scholar]
- 11.Beas-Zárate C, del Ángel Meza AR, Morales-Villagrán A, Feria Velasco A. Monosodium L-glutamate induced convulsions: changes in uptake and release of catecholamines in cerebral cortex and caudate nucleus of adult rats. Epilepsy Res. 4: 20–27 1989. [DOI] [PubMed] [Google Scholar]
- 12.Lutz B. On-demand activation of the endocannabinoid system in the control of neuronal excitability and epileptiform seizures. Biochem Pharmacol. 68: 1691–1698 2004. [DOI] [PubMed] [Google Scholar]
- 13.Castro VL, Palermo-Neto J. Effects of long-term aldrin administration on seizure susceptibility of rats. Pharmacol Toxicol. 65: 204–208 1989. [DOI] [PubMed] [Google Scholar]
- 14.Costa LG. Interactions of neurotoxicants with neurotransmitter systems. Toxicology. 49: 359–366 1988. [DOI] [PubMed] [Google Scholar]
- 15.Camera AL, Braga MF, Rocha ES, Santos MD, Cortes WS. Methamidophos: anticholinesterase without significant effects on postsynaptic receptors or transmitter release. Neurotoxicol. 18: 589–602 1997 [PubMed] [Google Scholar]
- 16.Bertolazzi M, Caroldi S, Moretto A. Interaction of methamidophos with hen and human acetylcholinesterase and neuropathy target esterase. Arch Toxicol. 65: 580–585 1991. [DOI] [PubMed] [Google Scholar]