

Abstract
Signal Transducer and Activator of Transcription 4 (STAT4) is a transcription factor that is activated by IL-12 signaling and promotes Th1-cell differentiation and IFN-γ production. Defective IFN-γ production because of STAT4 mRNA and protein deficiency occurs after autologous stem cell transplantation for lymphoma. In the present study, we investigated the mechanisms of STAT4 deficiency in lymphoma patients. The tumor-bearing state is not responsible, because STAT4 levels were not significantly different in PBMCs obtained from healthy control subjects compared with those from lymphoma patients before treatment. STAT4 protein levels were significantly decreased in PBMCs and T cells obtained from lymphoma patients after standard-dose chemotherapy. Furthermore, treatment of control PBMC cultures or a natural killer cell line with chemotherapy drugs in vitro also resulted in reduced STAT4 protein and diminished, IL-12–induced IFN-γ production. Translation of STAT4 protein was not impaired in chemotherapy-treated cells, whereas the STAT4 protein half-life was significantly reduced. Chemotherapy drugs promoted the ubiquitination and proteasomal degradation of STAT4. Treatment with the proteasome inhibitor bortezomib reversed chemotherapy-induced STAT4 deficiency and defective IFN-γ production. We conclude that acquired STAT4 deficiency in lymphoma patients is a consequence of treatment with chemotherapy, results that have important implications for the design of optimal immunotherapy for lymphoma.
Introduction
Signal Transducer and Activator of Transcription 4 (STAT4) is required for the biologic functions of IL-12, including the differentiation of Th1 cells and optimal IFN-γ production.1–3 IL-12 showed potent antitumor activity in preclinical models.4–7 IL-12–mediated antitumor effects are dependent on the production of IFN-γ.8,9 In a Th1-mediated inflammatory environment, IFN-γ has pleiotropic effects such as promoting antitumor immunity and antimicrobial activity. IFN-γ induces apoptosis in tumor cells and bacterially infected monocytes,10,11 enhances major histocompatibility class I and II antigen expression, and augments cytotoxic T lymphocyte and natural killer (NK)–cell cytotoxicity.12,13
In the context of IL-12–based immunotherapy, it was observed that IFN-γ production in vivo was markedly defective in patients with lymphoma who had undergone autologous peripheral blood stem cell transplantation (PBSCT). Moreover, PBMCs obtained from patients after PBSCT were profoundly deficient in IFN-γ production after direct stimulation with IL-12 in vitro.14 We subsequently showed that defective IFN-γ production in this setting is because of a profound and selective deficiency in STAT4.1,15 STAT4 deficiency may impair not only IL-12–based immunotherapy, but any therapeutic approach that requires Th1 immunity or optimal production of IFN-γ. The molecular mechanisms responsible for the observed deficiency of STAT4 are not known.
Unlike several other STAT proteins (eg, STAT1 and STAT3), which appear to be constitutively expressed in many tissues, STAT4 expression is restricted to hematopoietic cells.16,17 STAT4 is weakly expressed by resting T cells and is up-regulated after T-cell activation.18 STAT4 expression is maintained during the development of Th1 cells, but is down-regulated during the development of Th2 cells.19 Human NK cells constitutively express STAT4, but STAT4 protein levels can be increased or decreased in NK cells after cytokine stimulation.20,21 Despite abundant evidence that STAT4 expression is subject to tight regulation, the mechanisms that control STAT4 expression in physiologic or pathologic conditions have not been well characterized. Previous studies indicate that transcriptional silencing of the STAT4 gene due to hypermethylation of its promoter region and proteasome-dependent degradation of STAT4 protein can decrease STAT4 expression in human lymphocytes.22,23 We have undertaken studies to elucidate the mechanisms of STAT4 deficiency in patients with lymphoma.
Methods
Cytokines, Abs, chemotherapy drugs, and other reagents
Recombinant human IL-2 was obtained from Chiron and recombinant human IL-12 from PeproTech. Anti-STAT4 mAb for immunoblot analysis, allophycocyanin-conjugated annexin V, 7-amino-actinomycin, Alexa Fluor 647–conjugated streptavidin, and fluorochrome-conjugated mAbs recognizing human CD3, CD4, CD8, CD20, and CD56 were obtained from BD Biosciences. Biotin-labeled anti-STAT4 Ab, affinity purified goat anti–STAT4 Abs, and PE-conjugated donkey anti–goat Abs were from R&D Systems. Anti-STAT4 polyclonal Abs (SC-486), anti-STAT3 polyclonal Abs (SC-482), and anti–β-actin mAbs (SC-47 778) were from Santa Cruz Biotechnology. Bortezomib was from Millennium Pharmaceuticals. Phytohemagglutinin, propidium iodide, actinomycin D, cycloheximide, MG-132, 5-aza-2′-deoxycytidine (5-Aza-dC), carmustine, and etoposide were from Sigma-Aldrich. Ficoll-Paque PLUS was from GE Healthcare Bio-Sciences.
Blood samples, cell cultures, and cell lines
Collection of blood samples was approved by the Institutional Review Board at Indiana University Medical Center and written informed consent was obtained from each study subject in accordance with the Declaration of Helsinki. Blood samples were obtained from patients with Hodgkin or non-Hodgkin lymphoma before and after treatment with standard chemotherapy or high-dose chemotherapy and autologous PBSCT. Standard-dose chemotherapy regimens included rituximab, cyclophosphamide, vincristine, and prednisone with (R-CHOP) or without (R-CVP) doxorubicin. Data are derived from experiments done using samples obtained from 17 patients with non-Hodgkin lymphoma (10 diffuse large B-cell lymphoma, 5 follicular lymphoma, and 2 mantle-cell lymphoma) who received standard-dose chemotherapy; 13 patients received R-CHOP and 4 received R-CVP. High-dose chemotherapy regimens included cyclophosphamide, carmustine, and etoposide (CBV) and carmustine, etoposide, cytarabine, and melphalan (BEAM). Control PBMCs were obtained from healthy volunteer donors. Aliquots of PBMCs were cryopreserved in liquid nitrogen. NKL, a human NK cell line, was grown in culture as described previously.24 Activated PBMCs were obtained by culturing PBMCs in medium containing PHA (2.5 μg/mL) and IL-2 (50 U/mL) for 3 days in a 5% CO2 incubator at 37°C. Activated PBMCs and NKL cells were incubated in medium with or without chemotherapy drugs for 2-3 days. For some experiments, NKL cells were incubated with or without 5.2nM bortezomib simultaneously with the addition of corresponding chemotherapy drugs for 2 days. In other experiments, activated PBMCs were incubated with or without 5-Aza-dC (2.5 ng/mL) for 1 day after incubation with or without chemotherapy drugs.
STAT4 protein levels after etoposide treatment in a mouse tumor model
C56BL/6 mice (7-9 weeks old) were inoculated subcutaneously with 0.5 × 106 B16F10 murine melanoma cells into the right flank (day 0). At day 6, these mice were treated without or with etoposide at 35 mg/kg body weight. Etoposide treatment continued every 3 days until day 21 (on days 6, 9, 12, 15, 18, and 21). The control group without etoposide received vehicle solution. Mice were killed on day 29.
Analysis of STAT4 protein and RNA levels and flow cytometry
STAT4 mRNA and protein levels were analyzed using real-time PCR and immunoblotting analysis, respectively.1,15 In some experiments, STAT4 protein expression was analyzed from isolated subsets using positive selection with corresponding magnetic beads (Miltenyi Biotech).
For flow cytometric analysis, aliquots of PBMCs were stained with allophycocyanin-conjugated CD56 or CD20 and FITC-conjugated CD3, washed, and fixed with 2% paraformaldehyde. For detection of intracellular STAT4, fixed cells were permeabilized in 90% methanol, incubated with goat anti–STAT4 Abs or medium alone, washed, and incubated with PE-conjugated donkey anti–goat Abs, and analyzed by flow cytometry. In some experiments, a biotin-conjugated anti-STAT4 Ab followed by Alexa Fluor 647–conjugated streptavidin was used. During analysis, forward- and side-scattering properties were used to create a live lymphocyte gate. Thresholds for discriminating levels of staining above background were established by analysis of cells stained with fluorochrome-conjugated control mAbs. Mean fluorescence intensity (MFI) of intracellular STAT4 staining was obtained from the flow cytometer after electronic gating on NK cells, T cells, and B cells.
Assessment of STAT4 mRNA and protein half-life
Patient or control subject PBMCs were incubated with or without actinomycin D at 1 μg/mL for 0, 2, 4, and 6 hours in a 5% CO2 incubator at 37°C. RNA was extracted, first-strand cDNA was synthesized, and real-time PCR was performed.25 The half-life of STAT4 mRNA from each sample was calculated accordingly.25
NKL cells were treated without or with carmustine or etoposide for 2-3 days. Dead cells were removed by prior centrifugation on Ficoll-Paque PLUS gradient, and the collected viable cells were subsequently incubated with cycloheximide at 80 μg/mL for 0, 2, 4, 6, 8, 10, 12, and 24 hours. The amount of STAT4 and β-actin protein was determined using Western blot analysis. STAT4 protein levels were normalized to β-actin, and the half-life was calculated.26
Analysis of translation in NKL cells using metabolic labeling
NKL cells treated without or with carmustine or etoposide for 2 day were used for [35S] methionine and cysteine labeling (10 μCi/μL; Perkin Elmer). Dead cells were removed as described in “Assessment of STAT4 mRNA and protein half-life,” and the live cells were pulsed for various time points with [35S] methionine/cysteine. The cell extract (1%) was separated by SDS-PAGE followed by exposure to X-ray film after drying on a gel dryer. [35S]-labeled STAT4 and STAT3 after labeling were immunoprecipitated using polyclonal Abs against STAT4 and STAT3, respectively. Preimmunized rabbit serum was used in immunoprecipitation as a negative background control. The immunoprecipitated proteins were separated by SDS-PAGE followed by exposure to X-ray film after drying.
Analysis of ubiquitin-conjugated STAT4 protein
NKL cells were incubated without or with carmustine or etoposide for 2 days. Total protein lysates were extracted as described previously.27 MG-132 at concentration of 20μM was added to the cell lysis buffer to inhibit the activity of proteasome and to prevent the further degradation of protein.28 Ubiquitin-conjugated protein was first enriched using the Ubiquitin Enrichment Kit (Thermo Scientific) with a specialized affinity resin binding to polyubiquitinylated proteins from cell lysates. The negative control was the resin slurry lacking affinity to polyubiquitinylated proteins. The enriched proteins were subjected to immunoblot analysis. The levels of ubiquitin-conjugated STAT4 protein were analyzed using an anti-STAT4 mAb.
Evaluation of IFN-γ production
NKL cells treated without or with carmustine, etoposide, and bortezomib were incubated for 24 hours in medium alone or medium containing IL-12. Supernatant IFN-γ protein levels were measured using ELISA as described previously.1,15
Statistical analysis
Mean, SE, SD, and P values were determined using PASW Statistics software (IBM-SPSS 18.0) with a 2-sided test. P ≤ .05 was considered statistically significant.
Results
STAT4 deficiency is a consequence of chemotherapy treatment and is not because of lymphoma tumor burden
We demonstrated previously that STAT4 protein levels are decreased in PBMCs obtained from lymphoma patients after PBSCT.15 The observed STAT4 deficiency could be because of the tumor-bearing state per se or could occur as a consequence of prior therapy for lymphoma. To address this question, STAT4 protein levels were analyzed in PBMCs obtained from patients with active lymphoma who had not received any therapy. Levels of STAT4 protein, as assessed by immunoblot analysis, in PBMCs of untreated lymphoma patients were not significantly different (P > .05) from those in PBMCs of healthy controls (Figure 1A-C). Results of intracellular staining for STAT4 protein by flow cytometry (data not shown) were consistent with the results obtained by immunoblotting. The STAT4 protein level in gated lymphocytes from 10 samples of control PBMCs (MFI 50 ± 4; mean ± SE) was not significantly different (P = .728) than the STAT4 level (MFI 52 ± 6) in gated lymphocytes from 5 samples of PBMCs obtained from untreated lymphoma patients. Similarly, there were no significant differences in STAT4 levels in gated T cells (P = .295), NK cells (P = .118), or B cells (P = .33) when control PBMCs were compared with PBMCs from untreated lymphoma patients. Therefore, the presence of tumor burden does not result in STAT4 deficiency in patients with lymphoma.
Figure 1.

Expression of STAT4 in cells obtained before and after chemotherapy. (A) STAT4 protein expression was analyzed by immunoblotting of PBMCs from 4 healthy controls (C1-C4) and 6 untreated lymphoma patients (P1-P6). Samples from controls and patients were run in separate gels and exposure was done at the same time. The indicated upper STAT4 band detected with anti-STAT4 mAb was confirmed with anti-STAT4 polyclonal Ab.15 (B) Immunoblot analysis of STAT4 protein levels in control (C1-C3 and C5-C15) and lymphoma patient (P4, P7-P10, P12, P19-P23) PBMCs. Lymphoma patient PBMCs were obtained before (lanes labeled “U” for untreated) and 3 weeks after (lanes labeled “S” for standard dose) their first cycle of standard-dose chemotherapy plus rituximab. The chemotherapy regimen was R-CVP for patients P1, P6, P10, and P20 and R-CHOP for all other patients. A vertical line has been inserted to indicate the repositioned gel lane. (C) The levels of STAT4 protein in panel B were quantified by densitometry of the corresponding bands using the National Institutes of Health ImageJ program, and each sample was normalized to endogenous control β-actin as the ratio. Results are presented as means ± SD from 14 control and 11 patient samples. Blots in panel B were run in different gels but exposed to the same extent. *P < .05 relative to controls (C) or untreated lymphoma patients (U); **P > .05 relative to controls (C). (D) Changes in STAT4 and STAT3 protein levels in PBMCs obtained from each patient after standard-dose chemotherapy (S) are presented as the percentage of reduction compared with PBMCs obtained before chemotherapy treatment (U). The average percentage of reduction was obtained from the same 11 patients as in panel B and shown as the horizontal line in the graph. (E) Analysis of STAT4 protein in mice treated without or with etoposide. Tumor-bearing mice were treated with vehicle or etoposide as described in “Methods.” Spleens were harvested from 2 mice killed on day 29 of study. CD4+ T cells were isolated from each spleen using positive selection with CD4 magnetic beads (Miltenyi Biotec). Total protein extracts from isolated cells were subjected to immunoblotting analysis. The level of STAT4 protein in each mouse was normalized to internal control β-actin and is presented as the ratio indicated below. Nondetectable STAT4 protein is presented as (-). Results shown are representative from 2 independent studies. A vertical line has been inserted to indicate the repositioned gel lane.
We hypothesized that STAT4 deficiency is caused by the chemotherapy used to treat lymphoma. To test this hypothesis, STAT4 levels were analyzed by immunoblotting in PBMCs collected from lymphoma patients before and after they had received the initial chemotherapy used to treat their lymphoma. STAT4 protein levels were significantly decreased in PBMCs obtained from patients after standard-dose chemotherapy treatment compared with their pretreatment levels (Figure 1B-C). This finding is compatible with our hypothesis that chemotherapy treatment can cause relative STAT4 deficiency in lymphoma patients. However, interpretation of the immunoblotting results could be confounded if baseline STAT4 protein levels differ significantly in the cell subsets present in PBMCs and if the distribution of these subsets is altered after chemotherapy. To address this issue, we analyzed STAT4 protein levels in various PBMC subsets by both immunoblotting and flow cytometric analysis (Figure 2).
Figure 2.

Expression of STAT4 in different hematopoietic-cell subsets. (A) Immunoblot analysis of STAT4 protein levels. Various subsets of cells as indicated were isolated from control PBMCs using positive selection with magnetic beads (Miltenyi Biotec). The level of STAT4 protein in each subset was determined as described in Figure 1E. Nondetectable STAT4 protein is presented as (-). Results shown are representative of 3 different control samples that were tested. (B) Flow cytometric analysis of STAT4 protein levels. Control PBMCs were stained with mAbs specific for lineage-associated antigens, fixed, permeabilized, and stained with anti-STAT4 Abs as described in “Methods.” Histograms represent data obtained by electronic gating on CD3−CD56+ cells (NK-cell subset; top panel), CD3+CD56− cells (T-cell subset; middle panel), and CD3−CD20+ cells (B-cell subset; bottom panel). Logarithm of red fluorescence is displayed on the abscissa and relative cell number on the ordinate. STAT4 Ab staining is indicated by shaded histograms and control staining by open histograms. Results shown are representative of 10 different control samples that were tested. Summary data and statistics are presented in the text.
Among control PBMCs, STAT4 levels as assessed by immunoblotting were highest in NK cells, intermediate in CD4+ and CD8+ T cells, and barely detectable or undetectable in B cells and monocytes (Figure 2A and data not shown). Results of intracellular staining for STAT4 by flow cytometry (Figure 2B) were consistent with the results obtained by immunoblotting. Analysis of 10 samples of control PBMCs indicated that the expression of STAT4 was significantly higher (P = .002) in NK cells (MFI 65 ± 5) than in T cells (MFI 47 ± 5). Furthermore, the expression of STAT4 was significantly higher (P < .001) in T cells than in B cells (MFI 30 ± 3). We could not reliably measure STAT4 protein expression in monocytes by flow cytometry because of relatively high levels of nonspecific staining (data not shown).
STAT4 protein levels were relatively higher in NK cells and T cells and relatively lower in B cells and monocytes, and a change in the distribution of these subsets after chemotherapy could alter the STAT4 levels in total PBMCs even if there is no change in the STAT4 levels of individual cells. Nevertheless, we detected no significant change in the percentage of lymphocytes (P = .252), monocytes (P = .263), T cells (P = .453), or NK cells (P = .073) when we compared PBMCs obtained from 10 lymphoma patients before chemotherapy with samples obtained from the same patients after chemotherapy. In contrast, the percentage of B cells before chemotherapy was 17% ± 6%, whereas B cells were undetectable after treatment (P = .029). This result is to be expected for chemotherapy regimens that include rituximab. However, because B cells express significantly lower levels of STAT4 than T cells or NK cells (Figure 2), their depletion after chemotherapy would be expected to bias results toward higher STAT4 levels in analyses of unfractionated PBMCs. Therefore, our immunoblot analyses of total PBMCs may actually underestimate the degree of STAT4 deficiency induced by standard-dose chemotherapy. Nevertheless, it would appear that standard-dose chemotherapy is associated with a less severe STAT4 deficiency (overall, an approximately 65% reduction; Figure 1D) compared with high-dose chemotherapy and PBSCT (overall, an ∼ 97% reduction15).
Flow cytometric analysis of PBMCs obtained from 5 lymphoma patients indicated that STAT4 protein levels in T cells decreased significantly (P = .011) from MFI 52 ± 4 before chemotherapy to MFI 42 ± 2 after chemotherapy (data not shown). STAT4 protein levels were also decreased in NK cells from 3 of these 5 patients after chemotherapy, although the difference was not statistically significant when all 5 paired samples were analyzed (data not shown). STAT4 protein levels were also significantly lower in splenic T cells of tumor-bearing mice treated with etoposide compared with control mice treated with vehicle alone (Figure 1E). These data indicate that chemotherapy treatment is associated with an acquired STAT4 deficiency in the lymphocytes of tumor-bearing humans and mice.
The STAT4 deficiency that we described previously as occurring after high-dose therapy and PBSCT is relatively selective, because levels of JAK2, Tyk2, STAT3, and STAT1 were not significantly different in PBMCs obtained after PBSCT compared with control PBMCs (Robertson et al15 and our unpublished data). We have also analyzed STAT3 protein levels in lymphoma patient PBMCs obtained before and after standard-dose chemotherapy (Figure 1B,D). Although STAT3 levels did decline in the PBMCs of some patients after standard-dose chemotherapy, this effect was less consistent and usually more modest than the reduction in STAT4 levels (Figure 1D).
Acquired STAT4 deficiency of control PBMCs treated in vitro with chemotherapy drugs
To test directly our hypothesis that chemotherapy drugs cause STAT4 deficiency, PBMCs obtained from healthy control subjects were incubated in vitro with chemotherapeutic agents. Carmustine and etoposide were used in these experiments, because these cytotoxic drugs are commonly included in the high-dose chemotherapy regimens (CBV and BEAM) that are associated with profound STAT4 deficiency after PBSCT. The levels of STAT4 protein detected in activated PBMCs incubated with etoposide or carmustine were significantly decreased compared with the levels detected in cells cultured in medium alone (Figure 3A). In contrast, the level of STAT3 protein was not significantly decreased after in vitro treatment with chemotherapy drugs. The levels of STAT4 mRNA were also diminished in activated PBMCs treated in vitro with etoposide or carmustine (Figure 3B).
Figure 3.

Expression of STAT4 in cells treated in vitro with chemotherapy drugs. PBMCs obtained from controls were activated with PHA and IL-2 for 3 days and then cultured for 3 more days with the indicated concentrations of etoposide (Eto) and carmustine (Car). RNA was extracted and the first-strand cDNA was synthesized from the cells. STAT4 expression was analyzed using immunoblot (A) and real-time PCR (B). (C) STAT4 protein levels in CD4+ and CD8+ cells from control activated PBMCs treated without (-D) or with carmustine (Car) or etoposide (Eto) were analyzed using flow cytometry as described in “Methods.” Histograms represent the STAT4 expression gated on 5000 events of live CD4+ or CD8+ cells using WinMDI software. (D) NKL cells were treated without (-D) or with 50μM carmustine (Car) or 2μM etoposide (Eto) for 2-3 days. STAT4 protein levels were analyzed using Western blotting. (E) NKL cells treated as described in panel D were incubated with medium alone (unfilled bars) or medium containing 2 ng/mL of IL-12 (filled bars) for 1 day. The cell-free supernatants were analyzed for IFN-γ production using ELISA. The data are presented as means ± SD from 3 independent experiments.
The reduced STAT4 protein levels detected after chemotherapy exposure were measured by immunoblotting of whole-cell lysates from unfractionated PBMCs. These studies cannot elucidate whether STAT4 deficiency is more or less severe in particular subsets of lymphocytes. To address this question, the effect of chemotherapy drugs on STAT4 expression in different cell types was analyzed by flow cytometry. STAT4 protein levels were found to be reduced in both CD4+ and CD8+ T-cell subsets after chemotherapy exposure (Figure 3C). The relative paucity of NK cells in activated PBMCs generated under these culture conditions hindered flow cytometric assessment of STAT4 levels in the NK-cell subset (data not shown). Therefore, the effect of chemotherapeutic agents on STAT4 expression by NK cells was examined using a human NK cell line, NKL.24 Immunoblot analysis demonstrated reduced STAT4 protein levels in NKL cells treated with carmustine or etoposide for 2 days (Figure 3D), and reduction of STAT4 protein was seen as early as 24 hours after chemotherapy exposure (data not shown).
To determine the potential functional consequences of chemotherapy-induced STAT4 deficiency, we measured IFN-γ production by NK cells incubated with and without chemotherapy drugs. The levels of IFN-γ secreted by IL-12–stimulated NKL cells were significantly lower in the presence of either carmustine or etoposide (Figure 3E). Therefore, the partial reduction of STAT4 protein levels in NK cells exposed to chemotherapy drugs in vitro is associated with impaired IFN-γ production after cytokine stimulation.
STAT4 mRNA stability is not affected by chemotherapy
Our results indicate that STAT4 mRNA levels are diminished in cells exposed to chemotherapy drugs in vitro (Figure 3B) or high-dose chemotherapy in vivo (Figure 4A). The reduction in STAT4 mRNA levels could be because of decreased transcription of the STAT4 gene and/or decreased stability of the STAT4 mRNA. We measured the half-life of STAT4 mRNA in PBMCs obtained from control subjects or patients after high-dose chemotherapy and PBSCT (Figure 4B). The mean half-life of STAT4 mRNA from patient PBMCs obtained after PBSCT (3.967 hours) was not significantly different (P = .13) than that of control subject PBMCs (2.797 hours); therefore, although steady-state levels of STAT4 mRNA are decreased, STAT4 mRNA stability is not affected by high-dose chemotherapy. These results suggest that transcription of the STAT4 gene may be impaired after high-dose chemotherapy. Therefore, we evaluated the transcriptional regulation of STAT4 in chemotherapy-treated cells. STAT4 promoter activity was measured using a RenSP luciferase reporter construct (SwitchGear Genomics) in NKL cells treated in vitro without or with carmustine or etoposide. The expression of reporter driven by the STAT4 promoter was moderately reduced (18% ± 10% averaged from 3 independent experiments) in cells treated with carmustine or etoposide compared with untreated cells (data not shown), suggesting that transcriptional regulation is involved in reduction of STAT4 gene expression after chemotherapy.
Figure 4.
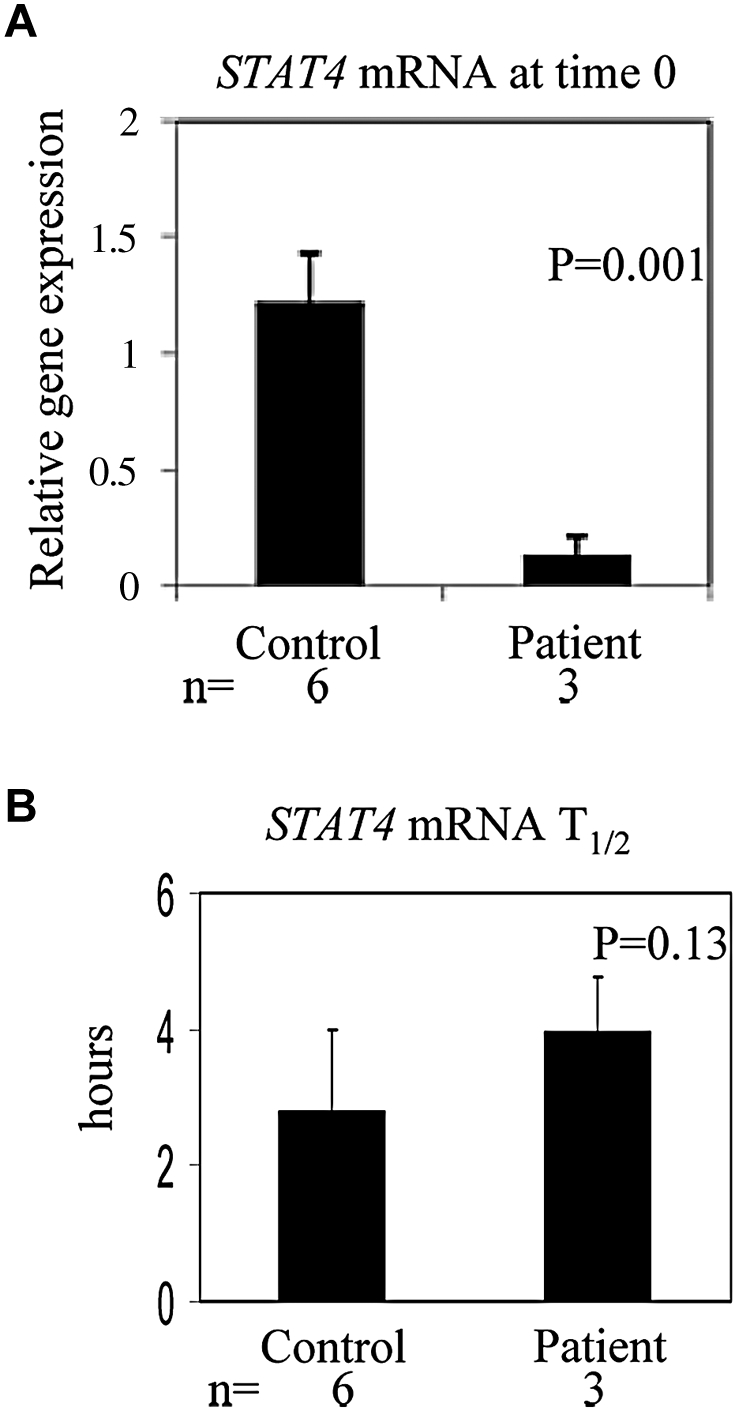
STAT4 mRNA levels and half-life of STAT4 mRNA in PBMCs. PBMCs obtained from 6 healthy control subjects and 3 patients after high-dose chemotherapy and PBSCT were treated with actinomycin D at 1 μg/mL for 0, 2, 4, and 6 hours in a 5% CO2 incubator at 37°C. RNA was extracted and first-strand cDNA was synthesized, followed by the real-time PCR.25 The half-life of STAT4 mRNA from each sample was calculated accordingly.25 Results shown are means ± SD.
It has been shown that DNA methylation in the proximal promoter region plays a direct role in the regulation of STAT4 transcriptional activity.22 Furthermore, primary T cells and cell lines with relatively low STAT4 expression exhibit increased STAT4 mRNA and protein levels after treatment with the DNA methyltransferase inhibitor 5-Aza-dC.22 We therefore used 5-Aza-dC to inhibit de novo DNA methylation in both control PBMCs and NKL cells treated in vitro with or without chemotherapy drugs. Despite increases in STAT4 gene expression after 5-Aza-dC treatment of cells incubated with carmustine or etoposide, the total level of STAT4 protein was minimally increased after 5-Aza-dC treatment (data not shown).
Effect of chemotherapy drugs on the efficiency of protein translation
Our data indicate that an intervention that can increase STAT4 mRNA levels in chemotherapy-treated cells was not sufficient to restore STAT4 protein levels. These results suggest that inefficient protein translation and/or reduced stability of the translated STAT4 protein may be more important mechanisms of chemotherapy-induced STAT4 deficiency. We therefore evaluated the efficiency of protein translation in cells exposed to chemotherapy drugs. Experiments using metabolic labeling with [35S] methionine/cysteine did indeed reveal a reduction in overall translation products after 15 to 60 minutes of pause in cells treated with carmustine or etoposide (Figure 5A).
Figure 5.

Analysis of translational regulation of STAT4 protein. (A) NKL cells treated without (-D) or with 50μM carmustine (Car) or 2μM etoposide (Eto) as described in Figure 3D were used for [35S] methionine and cysteine labeling (10 μCi/μL; Perkin Elmer). Dead cells were removed from the culture using Ficoll centrifugation, and the collected viable cells at 10 × 106 cells/2 mL were subsequently pulsed for 15, 30, and 60 minutes with [35S] methionine/cysteine. The cell extract (1%) was separated by SDS-PAGE followed by exposure to X-ray film after drying on a Bio-Rad gel dryer. The molecular weight (MW) is accompanied on the side of the gel. (B) NKL cells treated as described in panel A were pulsed with [35S] methionine/cysteine for 24 hours. STAT4 and STAT3 proteins were immunoprecipitated from the total protein extracts using polyclonal anti-STAT4 and anti-STAT3 Abs, which were subjected to SDS-PAGE and then exposure to X-ray film after drying. A preimmunized rabbit serum (IgG) was used in immunoprecipitation as a negative background control. One percent of the total protein extracts from each sample was separated on the same gel as loading control (1% loading). The molecular weight (MW) at 75 kD is labeled.
To elucidate whether reduced protein translation is involved in STAT4 protein regulation, SDS-PAGE was performed on immunoprecipitates of [35S]-labeled proteins from cells incubated in the presence or absence of chemotherapy drugs. We failed to detect any mature [35S]-labeled STAT4 or STAT3 protein on gels of immunoprecipitates from cells after short-term labeling (up to 6 hours), suggesting that the STAT4 and STAT3 proteins are both synthesized at low rates. We then performed immunoprecipitation from cells after long-term labeling for 24 hours with [35S] to accumulate mature labeled STAT proteins. The production of STAT4 and STAT3 proteins was similar in cells treated without and with carmustine or etoposide (Figure 5B). The specificity of immunoprecipitated STAT4 or STAT3 protein was confirmed by the absence of the corresponding band when using the preimmunized rabbit serum (Figure 5B). These results indicate that translation of STAT4 protein is not reduced in chemotherapy-treated cells despite an overall reduction in translational efficiency.
Ubiquitin-dependent proteasomal degradation of STAT4 in chemotherapy-treated cells
Because STAT4 protein translation did not appear to be impaired, we hypothesized that the reduced stability of the STAT4 protein was the dominant mechanism of chemotherapy-induced STAT4 deficiency. To test this hypothesis, we measured the half-life of STAT4 protein in human NK cells cultured in the presence or absence of chemotherapy drugs. Consistent with our hypothesis, the half-life of STAT4 protein was found to be significantly reduced in NKL cells treated with carmustine or etoposide (Figures 6A-B).
Figure 6.

Chemotherapy drugs reduce STAT4 protein levels via ubiquitin-mediated proteasomal degradation. NKL cells were treated without (-D) or with 50μM carmustine (Car) or 2μM etoposide (Eto) as described in Figure 3D. Dead cells were removed by Ficoll centrifugation from the culture of each treatment, and the collected viable cells were subsequently incubated with cycloheximide (CHX) for 0, 2, 4, 6, 8, 10, 12, and 24 hours. STAT4 protein expression was analyzed by immunoblotting, and a vertical line has been inserted to indicate the repositioned gel lane (A). The levels of STAT4 protein were determined by the densitometry of the corresponding bands normalized to endogenous control β-actin using the National Institutes of Health ImageJ program. The half-life of STAT4 protein was calculated accordingly, and the results shown are means ± SD from a total of 3 independent experiments (B). *P < .05 relative to treatment without chemotherapy drug (-D). (C) NKL cells were treated without (-D) or with 50μM carmustine (Car) or 2μM etoposide (Eto) as in Figure 3D. Dead cells were removed as described in panel A, followed by total protein extraction. Ubiquitin-conjugated protein was first enriched using the Ubiquitin Enrichment Kit (Thermo Scientific) with a specialized affinity resin binding to polyubiquitinylated proteins from cell lysates (Ub-beads). The negative control is the resin slurry lacking affinity to polyubiquitinylated proteins (beads). The enriched proteins were subjected to immunoblotting. Ubiquitin-conjugated STAT4 protein levels were analyzed using an anti-STAT4 mAb (left panel). Total STAT4 protein levels were analyzed using immunoblotting from 10 μg of whole-cell lysates (right panel). The ratio of Ub-STAT4 to total STAT4 is indicated below. The molecular weight (MW) at 75 and 118 kD is labeled. The panel is representative of 3 independent experiments, and a vertical line has been inserted to indicate the repositioned gel lane. (D) NKL cells were incubated with the proteasome inhibitor bortezomib at 5.2nM simultaneously with 50μM carmustine (Car) or 2μM etoposide (Eto) for 2 days. STAT4 protein levels were determined by immunoblotting. The results are representative of 3 independent experiments. Ratio of total STAT4 to β-actin is indicated below. (E) Densitometric analysis of STAT4 protein levels in NKL cells treated with carmustine or etoposide in the presence or absence of bortezomib. The results are presented as the averaged ratio of STAT4 to β-actin from 2 independent experiments as means ± SD (F) NKL cell treated as described in panel D were stimulated with IL-12 at 2 ng/mL for 1 day. The IFN-γ levels in the cell supernatants were evaluated using ELISA. Results are averaged from 2 independent experiments as means ± SD.
Ubiquitin-dependent proteasomal degradation has been implicated in the regulation of levels of tyrosine phosphorylated29 and total STAT4.30 To evaluate the role of the ubiquitin-mediated degradation pathway in the regulation of STAT4 protein, the levels of ubiquitinated STAT4 protein were determined in NKL cells treated with or without carmustine and etoposide. The ratios between ubiquitin-conjugated STAT4 and total STAT4 were substantially higher in cells treated with chemotherapy drugs compared with those incubated in medium alone (Figure 6C).
To test the hypothesis that the observed chemotherapy-induced reduction in STAT4 protein stability was due to proteasomal degradation, human NK cells were incubated with or without the proteasome inhibitor bortezomib in the presence of either carmustine or etoposide. The magnitude of the decrease in STAT4 protein levels after chemotherapy exposure was greatly reduced in the presence compared with the absence of bortezomib (Figure 3D and Figure 6D-E). This result confirms that the proteasome pathway is involved in STAT4 protein degradation after chemotherapy exposure. In the absence of chemotherapy drugs, incubation of NKL cells or PBMCs with bortezomib caused modest increases in protein levels of STAT4 and granzyme B without any detectable effect on STAT3 and IL-12 receptor β2 (data not shown). These results suggest that the proteasome is involved in the baseline regulation of STAT4 levels in human lymphocytes.
We next evaluated whether the increased STAT4 protein levels in presence of bortezomib could circumvent the previously observed defective IFN-γ production by NK cells treated with carmustine or etoposide. In the presence of bortezomib, IL-12–induced IFN-γ production by chemotherapy-treated cells was equivalent to or superior to that of cells that had not been exposed to chemotherapy (Figure 6F). This further supports the importance of STAT4 in IL-12–mediated IFN-γ production. In addition, circumventing STAT4 deficiency by bortezomib has the potential for enhancing the efficacy of IL-12 immunotherapy and any other therapeutic regimen that requires Th1 immunity for the production of IFN-γ.
Discussion
We demonstrated previously that a selective STAT4 deficiency contributes to impaired IFN-γ production in lymphoma patients after autologous PBSCT.15 However, the mechanisms responsible for this STAT4 deficiency have not been elucidated previously. In the present study, we have shown that STAT4 protein levels are normal in PBMCs obtained from treatment-naive patients with active lymphoma. These results confirm that STAT4 deficiency in patients is acquired and is not an inherited condition that predisposes them to develop lymphoma. Furthermore, these data refute the possibility that acquired STAT4 deficiency is because of the lymphoma-bearing state. Therefore, the mechanism of STAT4 deficiency presumably differs from that of the acquired ζ chain deficiency previously identified in lymphocytes of patients with advanced cancer.31
Compared with the levels seen in PBMCs obtained from lymphoma patients before treatment, STAT4 protein levels were significantly diminished in PBMCs obtained after standard-dose chemotherapy. Our data have excluded the possibility that the observed reductions in STAT4 levels were because of chemotherapy-induced alterations in cell subsets within PBMCs. Moreover, STAT4 protein levels were significantly reduced in T cells obtained from lymphoma patients or tumor-bearing mice after chemotherapy. These results are consistent with the hypothesis that chemotherapy treatment causes an acquired STAT4 deficiency in tumor-bearing hosts. Our in vitro experiments have also demonstrated directly that exposure to chemotherapy drugs induces the down-regulation of the STAT4 protein in human lymphocytes. The severity of STAT4 deficiency appears to be greater after high-dose therapy compared with the deficiency observed after standard-dose therapy. We favor the hypothesis that the severity of the STAT4 deficiency is directly related to the intensity of chemotherapy exposure in vivo; however, we cannot exclude the possibility that other factors related to the autologous transplantation procedure could affect the magnitude of STAT4 deficiency after transplantation.
In contrast to STAT4, levels of STAT3 (Figure 3A) and STAT1 (data not shown) were not significantly decreased in cells exposed to chemotherapy drugs in vitro. Therefore, the chemotherapy-induced decrease in STAT4 expression is relatively selective and cannot be ascribed to nonspecific cellular damage by cytotoxic agents. Moreover, STAT4 deficiency was observed in viable cells obtained from patients after chemotherapy and in the viable cells that remained after chemotherapy exposure in vitro. Therefore, STAT4 deficiency induced by chemotherapy is likely to be physiologically important and is not simply an artifact related to cell death caused by chemotherapeutic agents.
Chemotherapy-induced STAT4 deficiency was seen in multiple lymphocyte populations, including CD4 T cells, CD8 T cells, and NK cells. These results suggest that anti-tumor immune responses may be impaired after treatment with systemic chemotherapy. STAT4 protein levels were significantly diminished but generally still detectable in cells exposed to chemotherapy drugs in vitro or in vivo. Nevertheless, the degree of STAT4 deficiency caused by chemotherapy was sufficient to profoundly inhibit IL-12–induced IFN-γ production (Figure 3E). Therefore, the partial STAT4 deficiency occurring after chemotherapy is likely to be functionally relevant.
Chemotherapy-induced STAT4 deficiency could be because of reduced transcription of the STAT4 gene, reduced stability of STAT4 mRNA, impaired translation of STAT4 mRNA into protein, or posttranslational modifications of STAT4 protein that reduce its stability. We found that STAT4 mRNA levels were reduced in the PBMCs of patients after PBSCT even though STAT4 mRNA stability did not appear to be affected. Moreover, expression driven by the STAT4 promoter was modestly reduced in NKL cells after chemotherapy exposure. This result prompted the hypothesis that chemotherapy causes epigenetic modifications of the STAT4 gene that inhibit its transcription. Hypermethylation of CpG islands located 3′ to the core promoter of the STAT4 gene have been shown to inhibit its promoter activity.22 We therefore examined the effect of the DNA methyltransferase inhibitor 5-Aza-dC on STAT4 expression in chemotherapy-treated cells. Compatible with our hypothesis, STAT4 mRNA levels were significantly higher in cells incubated with carmustine or etoposide plus 5-Aza-dC compared with levels in cells incubated in carmustine or etoposide alone. However, STAT4 protein levels were only minimally increased after 5-Aza-dC treatment. Furthermore, translation of the STAT4 protein was not impaired in chemotherapy-treated cells. Therefore, posttranslational regulation of STAT4 protein levels may be the dominant mechanism of STAT4 deficiency after chemotherapy treatment.
We found that the half-life of STAT4 protein was significantly reduced in cells exposed to chemotherapy drugs, suggesting that the degradation of STAT4 protein is enhanced by chemotherapy. Ubiquitin-mediated proteasomal degradation has been implicated in the regulation of tyrosine-phosphorylated STAT4 as well as total STAT4.29,30 Furthermore, the H2.0-like homeobox 1 protein (HLX1) promotes proteasome-dependent STAT4 regulation in NK cells after IL-12 stimulation.32 We observed increased ubiquitination of STAT4 in cells treated with carmustine or etoposide, suggesting that chemotherapy drugs reduce the stability of STAT4 protein at least in part by promoting ubiquitin-dependent proteasomal degradation of STAT4. Compatible with this hypothesis, we found that the proteasome inhibitor bortezomib can substantially rescue STAT4 protein in cells exposed to chemotherapy drugs. Bortezomib is used for the treatment of lymphoma patients33 and can be safely given after autologous PBSCT.34 Therefore, it is clinically feasible to administer bortezomib to lymphoma patients in an attempt to ameliorate chemotherapy-induced STAT4 deficiency.
The mechanisms by which chemotherapy drugs target STAT4 for ubiquitination and proteasomal degradation remain to be identified. It has been shown that STAT-interacting LIM protein (SLIM) promotes ubiquitination and degradation of STAT4 in mice.30 However, SLIM also promotes the proteasomal degradation of STAT1 and STAT4. In contrast to STAT4, we have found that STAT1 levels were not reduced in PBMCs obtained from patients after PBSCT or in control activated PBMCs treated with chemotherapy drugs in vitro. Therefore, it is not likely that SLIM mediates the acquired STAT4 deficiency that we have demonstrated.
Becknell et al have reported that cycloheximide treatment leads to increased STAT4 protein levels in the human NK cell line NK-92.32 Similarly, we observed that the levels of STAT4 protein are increased in PBMCs treated with cycloheximide for 4 hours (data not shown). These results suggest that a labile protein can suppress STAT4 protein levels in human cells. We speculate that chemotherapy drugs can induce the expression of a labile ubiquitin E3 ligase that participates in the selective ubiquitination of STAT4 protein, leading to its proteasomal degradation. Further investigation is necessary to test this hypothesis.
Systemic combination chemotherapy with or without rituximab is currently the mainstay of initial treatment for patients with lymphoma. Moreover, high-dose therapy and autologous PBSCT is the treatment of choice for eligible patients with relapsed or refractory lymphoma. We have shown both standard-dose and high-dose chemotherapy can cause a selective, acquired STAT4 deficiency leading to impaired IFN-γ production. Sufficient IFN-γ production has been shown to be required for effective cellular immunity to tumors in many experimental models. Optimal immunotherapy concurrent with or after chemotherapy for lymphoma will require strategies that can ameliorate or circumvent chemotherapy-induced STAT4 deficiency.
Acknowledgments
The authors thank Dr Menggang Yu for assistance with the statistical analysis, Dr Shreevrat Goenka for providing the β-galactosidase–containing plasmid, and Dr Mark Kaplan for his continued support and suggestions.
This work was supported in part by the Indiana University Pilot Funding for Research Use of Core Facilities (to H.-C.C.), by Showalter Trust Funds (to H.-C.C.), by a Indiana University Biomedical Research Pilot Grant through the Indiana University Melvin and Bren Simon Cancer Center Translational Research Acceleration Collaboration (to H.-C.C.), by the Oncological Sciences Center in Discovery Park at Purdue (to H.-C.C.), and by National Institutes of Health grant RO1 CA118118 (to M.J.R.).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: I.P.L. performed the experiments, analyzed the data, and wrote the manuscript; L.H., L.V., M.D.L.R., K.O., A.S., and D.P. performed the experiments; R.P.S. and J.B.T. designed and performed the experiments in the mouse tumor model; M.J.R. designed the research, analyzed the data, and wrote the manuscript; and H.-C.C. designed the research, performed the experiments, analyzed the data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Hua-Chen Chang, Department of Biology, Indiana University-Purdue University Indianapolis, 723 W Michigan St, SL310, Indianapolis, IN 46202; e-mail: huchang@iupui.edu; or Michael J. Robertson, Division of Hematology/Oncology, Indiana University Simon Cancer Center, 535 Barnhill Dr, Rm 473, Indianapolis, IN 46202; e-mail: mjrobert@iupui.edu.
References
- 1.Chang HC, Han L, Goswami R, et al. Impaired development of human Th1 cells in patients with deficient expression of STAT4. Blood. 2009;113(23):5887–5890. doi: 10.1182/blood-2008-09-179820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382(6587):174–177. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- 3.Thierfelder WE, van Deursen JM, Yamamoto K, et al. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature. 1996;382(6587):171–174. doi: 10.1038/382171a0. [DOI] [PubMed] [Google Scholar]
- 4.Kozar K, Kaminski R, Switaj T, et al. Interleukin 12-based immunotherapy improves the antitumor effectiveness of a low-dose 5-Aza-2′-deoxycitidine treatment in L1210 leukemia and B16F10 melanoma models in mice. Clin Cancer Res. 2003;9(8):3124–3133. [PubMed] [Google Scholar]
- 5.Kubin M, Kamoun M, Trinchieri G. Interleukin 12 synergizes with B7/CD28 interaction in inducing efficient proliferation and cytokine production of human T cells. J Exp Med. 1994;180(1):211–222. doi: 10.1084/jem.180.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robertson MJ, Ritz J. Interleukin 12: basic biology and potential applications in cancer treatment. Oncologist. 1996;1(1-2):88–97. [PubMed] [Google Scholar]
- 7.Smyth MJ, Taniguchi M, Street SE. The anti-tumor activity of IL-12: mechanisms of innate immunity that are model and dose dependent. J Immunol. 2000;165(5):2665–2670. doi: 10.4049/jimmunol.165.5.2665. [DOI] [PubMed] [Google Scholar]
- 8.Brunda MJ, Luistro L, Hendrzak JA, Fountoulakis M, Garotta G, Gately MK. Role of interferon-gamma in mediating the antitumor efficacy of interleukin-12. J Immunother Emphasis Tumor Immunol. 1995;17(2):71–77. doi: 10.1097/00002371-199502000-00001. [DOI] [PubMed] [Google Scholar]
- 9.Nastala CL, Edington HD, McKinney TG, et al. Recombinant IL-12 administration induces tumor regression in association with IFN-gamma production. J Immunol. 1994;153(4):1697–1706. [PubMed] [Google Scholar]
- 10.Dellacasagrande J, Ghigo E, Raoult D, Capo C, Mege JL. IFN-gamma-induced apoptosis and microbicidal activity in monocytes harboring the intracellular bacterium Coxiella burnetii require membrane TNF and homotypic cell adherence. J Immunol. 2002;169(11):6309–6315. doi: 10.4049/jimmunol.169.11.6309. [DOI] [PubMed] [Google Scholar]
- 11.Wang SH, Mezosi E, Wolf JM, et al. IFNgamma sensitization to TRAIL-induced apoptosis in human thyroid carcinoma cells by upregulating Bak expression. Oncogene. 2004;23(4):928–935. doi: 10.1038/sj.onc.1207213. [DOI] [PubMed] [Google Scholar]
- 12.Steimle V, Siegrist CA, Mottet A, Lisowska-Grospierre B, Mach B. Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Science. 1994;265(5168):106–109. doi: 10.1126/science.8016643. [DOI] [PubMed] [Google Scholar]
- 13.Wang H, Ruan Z, Wang Y, et al. MHC class I chain-related molecules induced on monocytes by IFN-gamma promote NK cell activation. Mol Immunol. 2008;45(6):1548–1556. doi: 10.1016/j.molimm.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 14.Robertson MJ, Pelloso D, Abonour R, et al. Interleukin 12 immunotherapy after autologous stem cell transplantation for hematological malignancies. Clin Cancer Res. 2002;8(11):3383–3393. [PubMed] [Google Scholar]
- 15.Robertson MJ, Chang HC, Pelloso D, Kaplan MH. Impaired interferon-gamma production as a consequence of STAT4 deficiency after autologous hematopoietic stem cell transplantation for lymphoma. Blood. 2005;106(3):963–970. doi: 10.1182/blood-2005-01-0201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamamoto K, Quelle FW, Thierfelder WE, et al. Stat4, a novel gamma interferon activation site-binding protein expressed in early myeloid differentiation. Mol Cell Biol. 1994;14(7):4342–4349. doi: 10.1128/mcb.14.7.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhong Z, Wen Z, Darnell JE., Jr Stat3 and Stat4: members of the family of signal transducers and activators of transcription. Proc Natl Acad Sci U S A. 1994;91(11):4806–4810. doi: 10.1073/pnas.91.11.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bacon CM, Petricoin EF, 3rd, Ortaldo JR, et al. Interleukin 12 induces tyrosine phosphorylation and activation of STAT4 in human lymphocytes. Proc Natl Acad Sci U S A. 1995;92(16):7307–7311. doi: 10.1073/pnas.92.16.7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Usui T, Nishikomori R, Kitani A, Strober W. GATA-3 suppresses Th1 development by downregulation of Stat4 and not through effects on IL-12Rbeta2 chain or T-bet. Immunity. 2003;18(3):415–428. doi: 10.1016/s1074-7613(03)00057-8. [DOI] [PubMed] [Google Scholar]
- 20.Wang KS, Frank DA, Ritz J. Interleukin-2 enhances the response of natural killer cells to interleukin-12 through up-regulation of the interleukin-12 receptor and STAT4. Blood. 2000;95(10):3183–3190. [PubMed] [Google Scholar]
- 21.Wang KS, Ritz J, Frank DA. IL-2 induces STAT4 activation in primary NK cells and NK cell lines, but not in T cells. J Immunol. 1999;162(1):299–304. [PubMed] [Google Scholar]
- 22.Shin HJ, Park HY, Jeong SJ, et al. STAT4 expression in human T cells is regulated by DNA methylation but not by promoter polymorphism. J Immunol. 2005;175(11):7143–7150. doi: 10.4049/jimmunol.175.11.7143. [DOI] [PubMed] [Google Scholar]
- 23.Wang KS, Zorn E, Ritz J. Specific down-regulation of interleukin-12 signaling through induction of phospho-STAT4 protein degradation. Blood. 2001;97(12):3860–3866. doi: 10.1182/blood.v97.12.3860. [DOI] [PubMed] [Google Scholar]
- 24.Robertson MJ, Cochran KJ, Cameron C, Le JM, Tantravahi R, Ritz J. Characterization of a cell line, NKL, derived from an aggressive human natural killer cell leukemia. Exp Hematol. 1996;24(3):406–415. [PubMed] [Google Scholar]
- 25.De SK, Enders GC, Andrews GK. Metallothionein mRNA stability in chicken and mouse cells. Biochim Biophys Acta. 1991;1090(2):223–229. doi: 10.1016/0167-4781(91)90105-u. [DOI] [PubMed] [Google Scholar]
- 26.Flavell JR, Baumforth KR, Wood VH, et al. Down-regulation of the TGF-beta target gene, PTPRK, by the Epstein-Barr virus encoded EBNA1 contributes to the growth and survival of Hodgkin lymphoma cells. Blood. 2008;111(1):292–301. doi: 10.1182/blood-2006-11-059881. [DOI] [PubMed] [Google Scholar]
- 27.Chang HC, Zhang S, Thieu VT, et al. PU. 1 expression delineates heterogeneity in primary Th2 cells. Immunity. 2005;22(6):693–703. doi: 10.1016/j.immuni.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 28.Cotter L, Ozcelik M, Jacob C, et al. Dlg1-PTEN interaction regulates myelin thickness to prevent damaging peripheral nerve overmyelination. Science. 2010;328(5984):1415–1418. doi: 10.1126/science.1187735. [DOI] [PubMed] [Google Scholar]
- 29.Hoey T, Zhang S, Schmidt N, et al. Distinct requirements for the naturally occurring splice forms Stat4alpha and Stat4beta in IL-12 responses. EMBO J. 2003;22(16):4237–4248. doi: 10.1093/emboj/cdg393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tanaka T, Soriano MA, Grusby MJ. SLIM is a nuclear ubiquitin E3 ligase that negatively regulates STAT signaling. Immunity. 2005;22(6):729–736. doi: 10.1016/j.immuni.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 31.Renner C, Ohnesorge S, Held G, et al. T cells from patients with Hodgkin's disease have a defective T-cell receptor zeta chain expression that is reversible by T-cell stimulation with CD3 and CD28. Blood. 1996;88(1):236–241. [PubMed] [Google Scholar]
- 32.Becknell B, Hughes TL, Freud AG, et al. Hlx homeobox transcription factor negatively regulates interferon-gamma production in monokine-activated natural killer cells. Blood. 2007;109(6):2481–2487. doi: 10.1182/blood-2006-10-050096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goy A, Bernstein SH, Kahl BS, et al. Bortezomib in patients with relapsed or refractory mantle cell lymphoma: updated time-to-event analyses of the multicenter phase 2 PINNACLE study. Ann Oncol. 2009;20(3):520–525. doi: 10.1093/annonc/mdn656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cavo M, Tacchetti P, Patriarca F, et al. Bortezomib with thalidomide plus dexamethasone compared with thalidomide plus dexamethasone as induction therapy before, and consolidation therapy after, double autologous stem-cell transplantation in newly diagnosed multiple myeloma: a randomised phase 3 study. Lancet. 2010;376(9758):2075–2085. doi: 10.1016/S0140-6736(10)61424-9. [DOI] [PubMed] [Google Scholar]