

Abstract
Cav1.2 Ca2+ channel activity diminishes in inside-out patches (run-down). Previously, we have found that with ATP, calpastatin domain L (CSL) and calmodulin (CaM) recover channel activity from the run-down in guinea pig cardiac myocytes. Because the potency of the CSL repriming effect was smaller than that of CaM, we hypothesized that CSL might act as a partial agonist of CaM in the channel-repriming effect. To examine this hypothesis, we investigated the effect of the competitions between CSL and CaM on channel activity and on binding in the channel. We found that CSL suppressed the channel-activating effect of CaM in a reversible and concentration-dependent manner. The channel-inactivating effect of CaM seen at high concentrations of CaM, however, did not seem to be affected by CSL. In the GST pull-down assay, CSL suppressed binding of CaM to GST fusion peptides derived from C-terminal regions in a competitive manner. The inhibition of CaM binding by CSL was observed with the IQ peptide but not the PreIQ peptide, which is the CaM-binding domain in the C terminus. The results are consistent with the hypothesis that CSL competes with CaM as a partial agonist for the site in the IQ domain in the C-terminal region of the Cav1.2 channel, which may be involved in activation of the channel.
Keywords: Calcium-binding Proteins, Calcium Channels, Calmodulin, Ion Channels, Protein-Protein Interactions, Calpastatin, Patch Clamp, Pull-down Assay
Introduction
Calpastatin (CS)2 is a specific endogenous inhibitor of calpain, a Ca2+-activated cysteine protease. The calpain and CS complex plays a pivotal role in cell adhesion, shape change, migration, and apoptosis. CS is composed of a unique leader domain (domain L) in the N-terminal and four repetitive homologous domains (domains 1–4), each of which has calpain-inhibiting activity. However, CS domain L (CSL) has no calpain-inhibiting activity (1), and its physiological role remains largely unknown. Previous studies have shown that CS or CSL reprimes activity of Cav1.2 Ca2+ channels, in a concentration-dependent manner, in cardiac myocytes using the inside-out patch clamp technique (2, 3). Subsequent studies have further shown that the effective region of CSL is confined to a region spanning 11 amino acid residues (amino acids 54–64 in exon 5) and that CSL can bind to a fragment derived from the intracellular C-terminal region of Cav1.2 channels (4, 5). However, it has not been clarified how CSL interacts with the channel and regulates its activity.
Calmodulin (CaM) is a small (18 kDa), ubiquitous, Ca2+-binding protein that mediates Ca2+-dependent regulation of various target proteins, such as enzymes, ion channels, and transporters (6). The activity of the Cav1.2 Ca2+ channel is controlled by Ca2+-dependent facilitation (CDF) and inactivation (CDI) (7–11). CaM has been suggested to serve as a Ca2+ sensor and a transducer to produce Ca2+-dependent conformation changes of the channel protein in both CDF and CDI. In the case of CDF, although another mechanism involving phosphorylation of the channels by Ca2+/CaM-dependent protein kinase has been suggested (12), the activation of the kinase is also mediated by CaM. In our previous studies, we have found that CaM enhances (facilitation) and then inhibits (inactivation) channel activity of the Cav1.2 channels (in a concentration-dependent manner) in the inside-out patch mode (13), in which the channel shows no activity without CaM (i.e. run-down). Although a central role for CaM in the Ca2+-dependent regulation of Cav1.2 channels is widely accepted, the molecular basis for these effects is yet to be established.
The effect of CSL on the activity of the Cav1.2 channel in the inside-out mode is small (∼30% of control activity seen in the preceding cell-attached mode), whereas that of CaM is much larger (>100% of control). This finding led us to hypothesize that CSL might act as a partial agonist of the CaM-binding site, which is involved in the repriming or facilitation of the channel. In general, a partial agonist can bind and activate a given receptor but with partial efficiency relative to a full agonist. However, the partial agonist acts as a competitive antagonist in the presence of a full agonist. To test this hypothesis, we compared the effects of CSL with those of CaM on Cav1.2 channels and investigated a functional relation between CSL and CaM in the inside-out patch mode in guinea pig ventricular myocytes. Then we explored the binding sites of CSL and CaM on the channel using a pull-down assay with GST fusion peptides of the α1C subunit of the channel. We found that CSL regulates the channel activity in a manner consistent with a partial agonist of CaM and that the binding site locates in the IQ region of the C-terminal tail of the Cav1.2 channel.
EXPERIMENTAL PROCEDURES
Preparation of Single Cardiac Myocytes
Ventricular cells were obtained from adult guinea pig hearts. Female guinea pigs (350–600 g) were anesthetized with sodium pentobarbital (30 mg/kg, intraperitoneally), and the aorta was cannulated under artificial respiration. The dissected heart was mounted on a Langendorff apparatus and perfused with Tyrode solution for 3 min at 37 °C, nominally Ca2+-free Tyrode solution for 5 min, Ca2+-free Tyrode solution containing collagenase (0.08 mg/ml; Yakult, Tokyo, Japan) for 9–15 min, and finally washed out with a high K+ and low Ca2+ storage solution. The left ventricular myocytes were dispersed and filtered through a stainless steel mesh (105 μm). Then isolated myocytes were treated with storage solution containing alkaline protease (Nagase NK-103 (0.03 mg/ml), Katayama (Osaka, Japan)) and DNase I (type IV (0.02 mg/ml), Sigma-Aldrich) for 3 min at 37 °C and washed twice with storage solution followed by centrifugation at 800 rpm for 3 min. The isolated cells were stored at 4 °C in storage solution until use in the experiments. Experiments were carried out after approval was received from the Committee of Animal Experimentation (Kagoshima University).
Solutions
Tyrode solution contained 135 mm NaCl, 5.4 mm KCl, 0.33 mm NaH2PO4, 1 mm MgCl2, 5.5 mm glucose, 1.8 mm CaCl2, and 10 mm Hepes-NaOH buffer, pH 7.4. Storage solution contained 70 mm KOH, 50 mm glutamic acid, 40 mm KCl, 20 mm KH2PO4, 20 mm taurin, 3 mm MgCl2, 10 mm glucose, 10 mm Hepes, and 0.5 mm EGTA buffer, pH 7.4 (with KOH). Pipette solution contained: 50 mm BaCl2, 70 mm tetraethylammonium-Cl, 0.5 mm EGTA, 0.003 mm Bay K 8644, and 10 mm Hepes-CsOH buffer, pH 7.4. The basic internal solution (I.O. solution) contained 120 mm potassium aspartate, 30 mm KCl, 1 mm EGTA, 0.5 mm MgCl2, 0.5 mm CaCl2 (free [Ca2+] = 80 nm), and 10 Hepes-KOH buffer, pH 7.4.
Patch Clamp and Data Analysis
Ca2+ channel activity was monitored using the patch clamp technique. The myocytes were superfused with I.O. solution. Channel activity was elicited by a depolarizing pulse from a holding potential of −70 to 0 mV for 200 ms at a rate of 0.5 Hz with a patch clamp amplifier (EPC-7; List, Darmstadt, Germany). The current signals were filtered at 1–1.5 Hz and fed to a computer at a sampling rate of 3.3 kHz, where the capacity and the leakage currents were subtracted digitally. The NPo value was used to represent the channel activity, where N is the number of channels in the patch and Po is the time-averaged open state probability of the channels at each depolarizing pulse. NPo was calculated based on the equation NPo = I/i, where I was the mean current during the 5–105 ms period after the onset of the test pulses, and i was the unitary current amplitude. In each experiment, basal activity was recorded in the cell-attached mode for 2 min and then in the inside-out patch mode. Test solutions were applied by moving the patch into a small inlet of the perfusion chamber, which was connected to a microinjection system. Data are presented as mean ± S.E. (number of observations). Student's t test was used to estimate the statistical significance.
Fusion Peptide Constructs
The coding sequence of CSL (amino acids 3–148) and calpastatin domain 1 (CSD1; amino acids 151–279) were subcloned from a human CS cDNA (sequence identical to GenBankTM accession number D16217) (14), and a human CaM sequence (amino acids 3–149) was cloned from HEK 293 cDNA. Eleven constructs encoding the cytoplasmic regions of α1C (see Fig. 3A) were generated by PCR based on guinea pig cDNA (GenBankTM accession number AB016287) (15) and inserted into pGEX6p-3 (GE Healthcare) as a GST fusion peptide. The identities of all constructs were confirmed by DNA sequencing prior to use in experiments.
FIGURE 3.

CaM binding to Cav1.2 channel peptides. A, schematic drawing of fragment peptides of Cav1.2 (α1C). Eleven peptides derived from the intracellular regions of the channel were constructed as GST fusion peptides, including NT, LI-II, LII-III, and LIII-IV. The C-terminal domain was divided into three parts as illustrated. CT1 contains regulatory regions (shown as boxes), a Ca2+-binding EF-hand-like motif (EF), and two CaM-binding domains (PreIQ and IQ). Fragments of CT1 (i.e. CT1A, CT1B, PreIQ, and IQ) were also prepared. B, GST pull-down assay for CaM-binding at 1 mm Ca2+. GST fusion peptides (∼1 μm) were mixed with 1 μm CaM, and bound CaM was separated by SDS-PAGE and stained with Coomassie Brilliant Blue. 2 μg of BSA (66 kDa) and 2 μg of CaM (18 kDa) were run as standards. C, summary of pull-down assay for CaM binding. Optical densities of bound CaM were calibrated and displayed in arbitrary units with mean ± S.E. (n = 3–8). Significant differences are marked by *** (p < 0.001; Dunnett's test). D, concentration-dependent binding of CaM to NT (circles), LI-II (triangles), and CT1 (squares) at 1 mm Ca2+ (upper graph) and at 80 nm Ca2+ (lower graph). Data in molar ratio (CaM/GST fusion peptide) were plotted against total [CaM] with mean ± S.E. (n = 4–10). Data were fitted to theoretical curves using Kell software with parameters shown in Table 2.
Purification of Recombinant GST Fusion Peptides
Vectors were transformed into the Escherichia coli BL21 (DE3) strain (Stratagene, La Jolla, CA), and bacteria were cultured to an A600 of 0.8. Expression of GST fusion peptides was induced by 1 mm isopropyl β-thiogalactopyranoside for 4 h at 37 °C. Next, bacteria were resuspended in Tris buffer (50 mm Tris, pH 8.0, and 150 mm NaCl). The suspension was treated with lysis buffer (Tris buffer containing 0.1 mg/ml lysozyme and 5 mm DTT) for 30 min. In the case of the α1C fragments, in particular CT1 due to its limited solubility, the bacterial precipitates were treated with 1.5% n-lauroylsarcosine (Sigma-Aldrich) for 30 min (16, 17). The mixture was sonicated at 50% output with an ultrasonic homogenizer, NR-50 M (Microtec Nition, Funabashi, Japan) for 1 min and 20-s rest, seven cycles. After treatment with 1% Triton X-100 for 30 min, the lysate was separated by centrifugation at 12,000 rpm for 20 min, and then the supernatant was incubated with glutathione-Sepharose 4B (GE Healthcare) for 4 h. Bound fusion proteins were extensively washed with nominally Ca2+-free I.O. solution (pH 8.0), and GST was removed by PreScission Protease (GE Healthcare). All steps were performed on ice or at 4 °C. The content of purified CSL and CSD1 was estimated by the Bradford method using a protein assay kit (Pierce) and BSA (Pierce) as a standard. CaM concentration was determined by extinction coefficient at 276 nm (E276) of 0.18 (mg/ml)−1cm−1 or the Bradford method using authentic CaM (Sigma-Aldrich) as a standard. The purity of yields was confirmed by densitometry on SDS-polyacrylamide gels.
GST Pull-down Assay
Fusion peptides corresponding to intracellular regions of α1C at saturating concentrations (2–4 μg) were immobilized on glutathione-Sepharose 4B. Each peptide was incubated with CSL, CaM, or both in 100 μl of I.O. solution with 80 nm or 1 mm Ca2+ for 3 h at 4 °C with agitation. Then these reaction mixtures were washed twice with two volumes of the same buffer and centrifuged at 5,000 rpm for 1 min at 4 °C. Bound CSL/CaM and α1C peptides were resuspended in SDS sample loading buffer at 50 °C for 5 min and separated on a 12% SDS-polyacrylamide gel. Proteins were visualized by Coomassie Brilliant Blue R staining. Protein amounts were quantified by scanning with the Photoshop software (Adobe, San Jose, CA), and optical density was analyzed by Image J software (National Institutes of Health, Bethesda, MD). The standard curves were generated with authentic BSA (E286 = 0.66 (mg/ml)−1 cm−1; Pierce), CaM (E276 = 0.18 (mg/ml)−1 cm−1; Sigma-Aldrich), and GST (E280 = 1.69 (mg/ml)−1 cm−1). We found that relative optical densities of the same amount of CaM and GST on the gel in reference to BSA were 0.59 and 0.80, respectively. The contents of GST fusion peptides were calculated using the standard curve for GST, assuming similar E values. Data were presented as mean ± S.E. (number of observations). Student's t test and Dunnett's test were used to estimate the statistical significance.
Curve Fitting and Analysis Derived from a Simplified Model
We have hypothesized that α1C has at least two CaM binding sites: one for the facilitatory (F) and the other for the inhibitory (I) effects (13). Thus, channel activity (A) including the overall effect of CaM would be as follows,
where Amax represents the maximum value of A at F = I = 1. The effects of CaM were fitted with a modified Hill's equation,
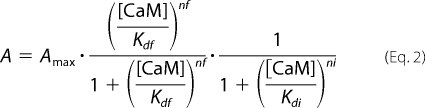 |
where Kdf and nf denote the concentration of CaM, [CaM], producing 50% of the maximum effect and an apparent Hill's coefficient, respectively, and Kdi and ni denote [CaM] producing 50% of the inhibitory effect and an apparent Hill's coefficient, respectively. The repriming effect of CSL on Cav1.2 channel activity (A) was fitted with the following Hill's equation,
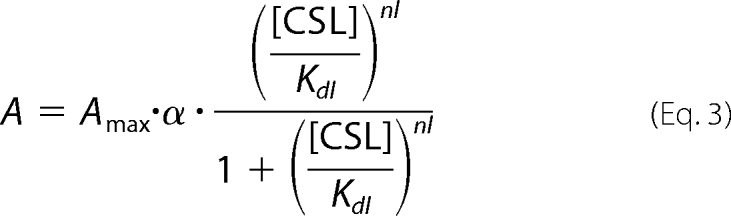 |
where α is the efficacy of CSL as a partial agonist of the CaM F site, and Kdl and nl denote [CSL] producing 50% of the maximum effect and an apparent Hill's coefficient, respectively. The channel activity in the presence of both CaM and CSL was fitted with the equation,
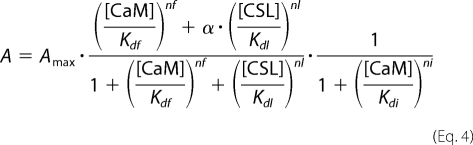 |
using DeltaGraph software (Red Rock Software, Salt Lake City, UT).
In the pull-down assay, bound ligand (CaM or CSL; denoted as Y) for the one-site model was expressed by the following Hill's equation,
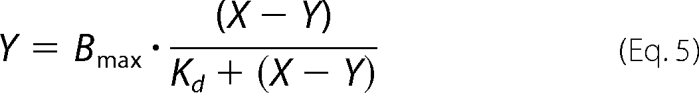 |
where Bmax is the maximum binding, X is total amount of ligand, and Kd is an apparent dissociation constant. In the case of the two-binding site model, a sum of two Hill's equations was used, assuming independent binding. The value of Y can be obtained from the solution to the cubic equation and was analyzed by the Kell Ligand program (Biosoft, Cambridge, UK) (18).
RESULTS
Calpastatin Domain L Suppresses Cav1.2 Channel Activity in the Presence of Calmodulin
To explore the functional regulation of the channel, we measured its activity using an electrophysiological technique (Fig. 1 shows a representative experiment). After inside-out configuration, Ca2+ channel activity was significantly diminished in 1 min (run-down; data not shown). In this experiment, test solution was applied within 1 min to prevent the complete run-down. CSL and CaM reprimed channel activity from the run-down state as described in previous studies (2, 4, 13, 19–21). We used 10 μm CSL and 1.22 μm CaM, which were submaximum concentrations for the channel-repriming effects, together with 3 mm ATP in all of the inside-out patch experiments. ATP was necessary to induce channel activity (19, 22, 23). In the previous studies, it has been reported that ATP supports channel activity by a mechanism independent of protein phosphorylation but not fully understood (22, 23). CSL recovered channel activity at the early transient phase, lasting for about 2 min after treatment, to 29.3% of that recorded in the cell-attached mode (control) and to 2.1% of the control at the late phase (Fig. 1A, a) (20). The addition of CaM to a channel preactivated by CSL increased the activity only to 5.4% of the control (Fig. 1A, b), which was markedly smaller than the effect expected with CaM alone (Fig. 1B, b). After CSL wash-out, the channel activity increased to 41.3% of the control (Fig. 1A, c), suggesting that CSL had an antagonistic effect on the CaM action to reprime channel activity. In another experiment, CaM was applied to the channel first, which recovered the channel activity to 117.0% of the control (Fig. 1B, a and b), and the effect was maintained stably for longer than 10 min (19). Then the addition of CSL to the CaM-preactivated channel conversely suppressed channel activity to 16.5% of the control, and this effect was repeatable (Fig. 1B, c). We confirmed that the inhibitory effect of CSL on the CaM-induced channel activity was seen even in the presence of protein kinase inhibitors KN-93 and K252a (supplemental Fig. S1), excluding a possible involvement of protein kinases in the CSL effect.
FIGURE 1.

CSL suppresses the effect of CaM on the Cav1.2 channel. A, the open state probability of the channels for each depolarization pulse (0.5 Hz) was measured as the NPo value and plotted against time. After NPo recording in the cell-attached mode, an inside-out patch (I.O) was initiated, as indicated by an arrow. Applications of 10 μm CSL and 1.22 μm CaM together with 3 mm ATP are indicated by boxes (black and white, respectively). The representative consequent current traces recorded at the time points indicated in the graph (a–c) are shown below, in the presence of CSL (a), CSL + CaM (b), and CaM (c). B, experiment similar to that shown in A, except that first CaM was applied, and CSL was applied in the presence of CaM. Representative current traces recorded at the indicated time points (a–c) in the graph are shown below for control activity in the cell-attached mode (a) and in the presence of CaM (b) and CaM + CSL (c). Scale bars for the current traces represent 50 ms and 1 pA.
These results suggest that there might be a competition between CaM and CSL for the binding to some region of the channel. CSL alone has a partial repriming effect on channel activity. On the other hand, it has an inhibitory effect on channel activity in the presence of CaM. Based on these findings, we hypothesized that CSL acts as a partial agonist of the CaM-binding repriming (facilitatory) site of the channel.
Concentration-dependent Effects of Calpastatin Domain L and Calmodulin
To examine our hypothesis, we investigated the concentration-dependent effects of CSL and CaM. First, we plotted the concentration-response curves of CSL and CaM. Channel activity was recorded in the inside-out patch mode and expressed as relative values normalized by the basal activity recorded in the preceding cell-attached mode. Fig. 2A, a, shows the [CSL]-activity relationship (open circles, n = 4–5), which was fitted by Equation 3. It was apparent that the maximum effect of CSL was 25% of the control. This result is consistent with our previous report (2). The [CaM]-activity relationship (Fig. 2B, a, open circles, n = 4–10) had a clear bell-shaped curve. The data were fitted by Equation 2. Channel activity appeared at 0.04 μm CaM, reached 100% at 0.6 μm, and peaked at around 3 μm (∼150% of control). Further increase in CaM decreased channel activity toward null activity. These results are consistent with our previous experiments (13, 19). From the maximum effects of CSL and CaM, the value of α was estimated as 0.14.
FIGURE 2.

Concentration-dependent effects of CSL and CaM on Cav1.2 channels. A, channel activity (NPo) induced by CSL in the absence (open circles) and presence (closed circles) of 1.22 μm CaM was normalized by control values (recorded in the cell-attached mode) and plotted against [CSL]. Data in the absence and presence of CaM were fitted by Equation 3 (a) and Equation 4 (b), respectively, with parameters listed in Table 1. B, normalized channel activity induced by CaM in the absence (open circles) and presence (closed circles) of 10 μm CSL was plotted against [CaM]. Data in the absence and presence of CSL were fitted by Equation 2 (a) and Equation 4 (b), respectively, with parameters listed in Table 1. Data are displayed as mean ± S.E. (n = 4–10). Significant differences compared at the same concentrations are marked by asterisks (*, p < 0.05; **, p < 0.01; Student's t test).
We then examined the concentration-dependent effect of CSL on channel activity in the presence of 1.22 μm CaM (Fig. 2A, b, filled circles; n = 4–10). CSL suppressed the repriming effect of 1.22 μm CaM in a concentration-dependent manner with an IC50 of ∼2 μm and a maximum inhibition to ∼20% of control at >5 μm. The relationship was fitted by Equation 4. These values were comparable with those obtained with either CSL or CaM alone. We also examined the concentration-dependent effect of CaM in the presence of 10 μm CSL (Fig. 2B, b, filled circles; n = 4–5). We noted that the facilitatory effect of CaM (<3 μm) was markedly suppressed, whereas the inhibitory effect did not seem to be affected. The data were fitted by Equation 4. Again, these values were not much different from those obtained with either CSL or CaM alone. The parameter values to fit the theoretical curves are summarized in Table 1. These results are consistent with the hypothesis that CSL interacts with CaM as a partial agonist at the CaM-repriming site, presumably located within the Cav1.2 channel. The inhibitory effect of CSL was also seen in the whole-cell Ca2+ current recorded from intact myocytes (supplemental Fig. S2).
TABLE 1.
Parameter values for [CSL]- and [CaM]-dependent activity of the Cav1.2 channel
Data shown in Fig. 2 were fitted by Equations 2–4 using DeltaGraph software. To reduce the number of variables in the curve fitting, values of Kdl, nl, and nf obtained in the fittings of CSL data and CaM data were used as fixed values in other curve fittings (marked with asterisks). The partial agonist efficacy (α) was calculated from a ratio of the effects of CSL (Amax) on CaM (Amax·α) as 0.14 and treated as a fixed value in further analysis. For further explanation, see “Experimental Procedures.”
| Test solution | Amax | Kdl | nl | Kdf | nf | Kdi | ni |
|---|---|---|---|---|---|---|---|
| % | μm | μm | μm | ||||
| CSL | 177 | 0.7 | 1.6 | ||||
| CSL with 1.22 μm CaM | 130 | 0.7* | 1.6* | 0.3 | 1.4* | 7.4 | 3.7 |
| CaM | 177 | 0.5 | 1.4 | 6.4 | 3.0 | ||
| CaM with 10 μm CSL | 211 | 0.7* | 1.6* | 0.5 | 1.4* | 6.4 | 2.8 |
Calmodulin Can Bind to the N-terminal and C-terminal Peptide of Cav1.2 Channel
As shown in Fig. 3A, we prepared seven constructs of GST fusion peptides of the α1C intracellular domains: N terminus (NT), cytosolic linker between channel domains I and II (LI-II), domain II to III linker (LII-III), domain III to IV linker (LIII-IV) and three C-terminal regions, proximal (CT1), middle (CT2), and distal (CT3). Because the CT1 region contains important regulatory domains, such as the EF-hand-like Ca2+-binding motif (EF) and two CaM-binding domains (PreIQ and IQ), four constructs related to CT1 were prepared: CT1A (EF + PreIQ), CT1B (PreIQ + IQ), PreIQ, and IQ. The GST fusion peptides immobilized to glutathione-Sepharose beads were mixed with CaM and 1 mm Ca2+, and then bound CaM was analyzed on SDS-PAGE (Fig. 3B). CaM bound markedly to NT and CT1 and weakly to LI-II and CT2, whereas its binding to LII-III, LIII-IV, and CT3 was minimal. Furthermore, CT1B, PreIQ, and IQ were also found to bind to CaM (example shown in Fig. 4A). Fig. 3C summarizes the pull-down assay results. CaM significantly bound to five peptides (NT, CT1, CT1B, PreIQ, and IQ; p < 0.001 compared with CaM-GST binding, n = 3–8) and weakly to three peptides (LI-II, CT2, and CT1A) although not statistically significant. On the other hand, CaM binding to LII-III, LIII-IV, and CT3 was negligible. We also confirmed that there was no binding between CSL and CaM using a GST fusion CSL peptide (Fig. 3, B and C). As reported previously (18), in the absence of Ca2+ (in the presence of 5 mm EGTA), it was difficult to detect CaM binding to any GST fusion peptide (data not shown).
FIGURE 4.

CSL competes with CaM for its site. A, GST pull-down assay for CaM binding to the peptides (NT, CT1, CT1A, CT1B, PreIQ, and IQ) at 1 μm CaM and 1 mm Ca2+. CaM binding was examined in the presence of 10 μm CSL (top) or 10 μm CSD1 (bottom). BSA (2 μg) and CaM (1 μg) were run as standards. B, summary of suppression of CaM binding by 10 μm CSL at 1 μm (open columns) and 10 μm CaM (filled columns). Data are displayed as mean ± S.E. (n = 5–9). Significant differences versus GST-NT (Dunnett's test) are marked by asterisks (**, p < 0.01; ***, p < 0.001). C–F, dose-dependent bindings of CaM to NT (C), CT1 (D), and IQ (E) at 1 mm Ca2+ and to CT1 at 80 nm Ca2+ (F) in the presence of 10 μm CSL. Data in molar ratio (CaM/GST peptide) were plotted against total [CaM] with mean ± S.E. (n = 4–10). Data were fitted to theoretical curves using Kell software with parameters shown in Table 2. For comparison, CaM binding curves in the absence of CSL were taken from Fig. 3D or measured (open symbols), and their fitted curves were superimposed by broken lines. Significant differences from the value without CSL at each point (Dunnett's test) are marked by asterisks (*, p < 0.05; **, p < 0.01).
In this study, we confirmed the report that three peptides, NT, LI-II, and CT1, interact with CaM (24), except that the binding to LI-II was weak and not statistically significant. We attempted to analyze the stoichiometry of CaM binding with these peptides in two [Ca2+] conditions, 80 nm (a resting [Ca2+] level in the myocytes) and 1 mm (a saturating [Ca2+] level). In 1 mm Ca2+, CaM binding to the three peptides was detected with a wide range of [CaM], as illustrated in Fig. 3D (upper graph). The curve-fitting analysis suggested that CaM bound to NT (open circles; n = 4–9), and to LI-II (open triangles; n = 4–5). CaM binding to CT1 was better fitted with a two-site model (open squares; n = 4–9). As a result, 1.94 mol/mol CaM (Bmax1 + Bmax2) was estimated to bind with CT1. On the other hand, as reported previously (25), CaM binding at 80 nm Ca2+ was much lower than at 1 mm Ca2+ (Fig. 3D, lower graph; n = 4–10). Nevertheless, we could estimate the ranking of the Kd values: CT1 < NT < LI-II. This tendency was similar to that found at the high [Ca2+] condition (18). The parameter values analyzed from the theoretical curves of CaM binding to NT, LI-II, CT1, and IQ are summarized in Table 2. These results supported the view that CaM bound to both N-terminal and proximal C-terminal regions of α1C and that more than one molecule of CaM could bind the C-terminal region, at least in high [Ca2+] conditions.
TABLE 2.
Parameter values for CaM binding to GST fusion peptides
Data from the GST pull-down assay shown in Figs. 3 and 4 were analyzed based on Equation 5 by Kell Ligand software. ND, no data.
| GST fusion peptide | Bmax |
Kd |
|
|---|---|---|---|
| Without CSL | With CSL | ||
| mol/mol | μm | ||
| 2+ | |||
| NT | 1.01 | 0.42 | 1.1 |
| LI-II | 0.34 | 0.54 | ND |
| CT1 | 1.14/0.8a | 0.23/17.84b | 1.17/29.12b |
| IQ | 0.76 | 1.72 | 5.81 |
| In 80 nm Ca2+ | |||
| NT | 0.28 | 4.58 | ND |
| LI-II | 0.16 | 4.74 | ND |
| CT1 | 0.37 | 0.66 | 5.22 |
a CaM binding to CT1 in 1 mm Ca2+ was fitted to a two-site model and expressed as Bmax1/Bmax2.
b CaM binding to CT1 in 1 mm Ca2+ was fitted to a two-site model and expressed as Kd1/Kd2.
Calpastatin Domain L Suppresses Calmodulin Binding to the C-terminal Peptides of the Cav1.2 Channel
Finally, we investigated the effects of CSL on the binding of CaM to the GST fusion peptides of α1C (i.e. NT, CT1, CT1B, PreIQ, and IQ). Binding of CaM (in 1 μm total [CaM]) to these peptides in the absence or presence of 10 μm CSL was compared using the pull-down assay. As shown in Fig. 4A (upper gel), whereas CaM binding to NT, CT1, CT1B, and PreIQ tended to be only slightly reduced, CaM binding to IQ was clearly reduced in the presence of CSL. The binding of CaM to CT1A, examined as control, was negligible. The densitometric analysis revealed that the reduction in CaM binding to IQ by CSL was about 40% (Fig. 4B). This effect was thought to be specific to CSL, because 10 μm CSD1 produced negligible or no effect (Fig. 4A, lower gel). We also performed similar experiments with 10 μm CaM and obtained essentially the same results. The summary of the densitometric analysis with 1 and 10 μm CaM is shown in Fig. 4B. CaM binding to IQ was significantly suppressed by CSL (p < 0.01 at 1 μm and p < 0.001 at 10 μm CaM versus GST-NT). Although 10 μm CSL reduced CaM binding to NT, CT1, CT1B, and PreIQ by 10–20%, this effect was considered as a nonspecific effect of CSL. We further analyzed the competition between CSL and CaM for the binding to NT, CT1, and IQ peptides at various [CaM]. Fig. 4C shows concentration-dependent CaM binding to NT at 1 mm Ca2+ in the presence of 10 μm CSL. CaM binding was not significantly affected by CSL compared with the binding in the absence of CSL (broken line) at total [CaM] higher than 1 μm. However, CaM binding to CT1 at 1 mm Ca2+ showed a small but significant rightward shift by CSL (Fig. 4D), suggesting the existence of a CaM-CSL competition to the CT1 peptide. This tendency was more pronounced with the IQ peptide (Fig. 4E); CSL shifted the concentration-binding curve, changing the apparent CaM Kd value from 1.72 to 5.81 μm. We also examined the competition between CSL and CaM for binding to the CT1 peptide at 80 nm Ca2+. The CaM binding curve for CT1 at 80 nm Ca2+ was largely shifted rightward by CSL, changing the apparent CaM Kd value from 0.66 to 5.22 μm (Fig. 4F). The reduction of CaM binding by 10 μm CSL was 67.2 ± 13.1% at 1.22 μm CaM (n = 5) and 29.2 ± 7.6% at 10 μm CaM (n = 10). It was difficult to examine the CaM-CSL competition for the IQ peptide at 80 nm Ca2+ in the pull-down assay, due to a low affinity between IQ and CaM. However, we confirmed obvious CaM binding to the IQ peptide at 80 nm Ca2+ and a concentration- dependent inhibition by CSL of the CaM binding to the IQ peptide in native PAGE (gel shift assay; supplemental Fig. S3). As expected, the inhibitory effect of CSL was much stronger at 80 nm than at 1 mm Ca2+ (supplemental Fig. S3C).
DISCUSSION
Dual CaM Effects on Channel Activity
CaM has been suggested to mediate both CDF and CDI of Cav1.2 channels (7–11). Our previous studies have revealed that CaM reprimes the channels in a run-down state in inside-out patches (13, 19, 21). Furthermore, an inhibitory effect of CaM on channel activity has been found at high [CaM], indicating that CaM can manifest both facilitatory and inhibitory effects in the inside-out mode. The fact that these CaM effects are Ca2+-dependent implies that the effects may reflect those seen in CDF and CDI in intact myocytes (13, 19, 21). In this study, we confirmed the dual CaM effects on channel activity and provided further support for the view that CaM interacts dynamically with the channel, thereby regulating basal activity and mediating CDF and CDI in cardiac myocytes.
CSL Is a Partial Agonist
CS has been known as the specific inhibitor of calpain. In addition, CSL can reprime the activity of Cav1.2 channels from run-down (3, 20, 26). This effect is specific to CSL, as demonstrated by the findings that a synthetic calpain inhibitor has no effect on the channel (26) and that CSL peptides, but not CSD1, reproduced the same effect (2, 4). In this study, we examined an interrelation between CaM and CSL in the regulation of Cav1.2 channels and obtained several new findings. First, although CaM and CSL showed similar effects in producing channel activity in the run-down channels, the maximum effect of CSL was only 14% of the CaM effect. The concentration-dependent effects of CSL could be fitted with a Hill's equation (Fig. 2, open circles) by introducing an efficacy factor of 0.14 (Equation 4). Second, the effects of CSL and CaM were not simply additive, but CSL suppressed the effect of CaM in a concentration-dependent manner (Fig. 2, filled circles). Third, unlike CaM, CSL by itself did not show an inhibitory effect on channel activity at high concentrations (>10 μm). These results strongly suggest that CSL may be a partial agonist of the CaM activation site in the channel and compete with CaM over this site.
CSL Binding Site in the Channel
Previously, we explored the possible CSL binding site in the channel using the pull-down method (5). It has been found that, in the 1 mm Ca2+ condition, CSL does not bind to NT, LI-II, LII-III, LIII-IV, CT2, or CT3 but only to the CT1 peptide. Further analysis has revealed that the binding is confined to the IQ but not to the PreIQ region. In this study, we confirmed that CaM binding to the IQ domain in the C-terminal proximal region was competed by CSL in the pull-down assay (Fig. 4E) and in the gel shift assay (supplemental Fig. S3). A direct binding between CaM and CS was excluded by the experiments, in which no specific binding between CaM and CS was detected with the SPR assay (27) and the GST pull-down assay (Fig. 3). Thus, these data are in accordance with the patch clamp experiments, which suggested that the C-terminal proximal region of α1C is a target site for a modulatory effect by endogenous CS (3). It has been suggested that CaM can bind to both PreIQ and IQ at the same time (18, 28–30), which is also supported by this study. It is interesting to note that CSL competes with CaM only for the IQ site.
In the patch clamp experiments of this study, Kd values of CSL and CaM for channel activation were estimated at 0.7 and 0.5 μm, respectively (Fig. 2), supporting the view that CaM had ∼1.5-fold higher affinity than CSL for the “putative” activation site. Amino acid sequence analysis showed that there is no homologues sequence between CSL and CaM. CSL probably has no α-helix region and no other secondary structure, as suggested by in silico analysis (4). Thus, it is difficult to speculate on the structural basis for CSL binding with the CaM site. Nevertheless, CSL provides a useful tool for investigating the putative CaM-activation site in the Cav1.2 channel.
CaM-mediated Regulation of Cav1.2 Channel
Although CaM has been known to interact with the α1C subunit during CDF and CDI, it is still unclear how and to which of the various CaM binding regions it associates. Many reports have identified important roles for the C-terminal regions in CDF and CDI. Three regions in the C terminus have been reported to be involved in CDF (31) and CDI (8, 11, 17, 32–34): the EF-hand motif-like region, the PreIQ, and the IQ region. Although the latter two regions can bind with CaM at high [Ca2+], a number of studies have failed to detect CaM binding under low [Ca2+] conditions (17, 28, 35). It has also been reported that a Ca2+-insensitive CaM mutant hardly binds to the C-terminal peptides (9, 31, 36) or to the IQ peptide (3, 9). On the other hand, some reports suggested that CaM binds to the Pre-IQ (3) and IQ peptides (10, 31) independently of Ca2+. Thus, it still remains to be determined whether or not CaM is tethered to or constitutively integrated with the C-terminal region. In the present study, we observed a weak but consistent binding of CaM (Kd ∼0.7 μm) to the proximal C-terminal region (CT1) at a low [Ca2+] compared with a relatively tight binding at a high [Ca2+] (Kd ∼0.2 μm). This result is consistent with the electrophysiological observation that the channel-repriming effect of CaM is Ca2+-dependent; the EC50 value was smaller at lower [Ca2+] (13). We hypothesized that CaM is not permanently tethered to the Cav1.2 channel but dynamically interacts with the channel, depending on CaM and Ca2+ concentrations (13, 18, 19, 21). The results of this study further support this hypothesis.
Deletion of the EF-hand region (8) or mutations in the PreIQ (33) or IQ regions (8, 10, 17, 24, 31, 32) eliminated CDI. Gel shift assay and FRET analysis have revealed that the interaction of the PreIQ and IQ region peptides with CaM is competitive (7), and peptides including the PreIQ and IQ regions bind to CaM with a 1:1 stoichiometry (7–9, 17, 25, 35, 37). Thus, it has been suggested that one molecule of CaM interacts with a site in the C-terminal region (either PreIQ or IQ) of the Cav1.2 channel during CDI (38). We have suggested, however, that more than one CaM molecule binds to a peptide containing both the PreIQ and IQ region, at least in the high [Ca2+] (18). In the present study, we obtained a similar result with the CT1 peptide. Binding of multiple CaM molecules to the C-terminal region was further supported by recent crystallographic studies (28, 29) and a thermodynamic analysis (30). Because CaM affinity for the IQ region seems to be higher than for the PreIQ region, we propose that the first molecule of CaM may interact with the IQ region to reprime the channel in a low [Ca2+], and then, when [Ca2+] is increased, a second molecule of CaM binds to the PreIQ region, triggering CDI. Although there is little evidence to support this idea, the finding that CSL interacts with the IQ peptide is consistent with this hypothesis. Thus, it would be helpful to pursue further studies on CaM-mediated regulation of Cav1.2 channels.
In addition to the C-terminal tail of the Cav1.2 channel, CaM has been reported to bind with the N-terminal and the LI-II peptide (24, 36). Although CaM binding to these regions was confirmed in this study, the affinities of these regions for CaM seemed lower than that of CT1, suggesting that these regions could not be the CaM-binding sites that produce the basal activity of the channel. Instead, it is possible that these regions, in particular the N-terminal region, serve as a CaM-interacting site for CDI (24, 36). Alternatively, both the N terminus and C terminus might collaborate with CaM to trigger CDI. The function of CaM binding to LI-II is largely unknown, although this region was also suggested to be involved in CDI (9, 11).
Ca2+ Dependence of Channel Activity
The location of the Ca2+-binding sites for CDF and CDI requires some discussion. Both the facilitatory and inhibitory actions of CaM on the Cav1.2 channel in cell-free patches are Ca2+-dependent, indicating that the responsible Ca2+-binding sites are in the EF-hand regions of CaM (13, 19). This is supported by studies showing that a Ca2+-insensitive CaM mutant loses Ca2+ dependence at its major component of action (13, 21). Ca2+-dependent binding of CaM to peptides containing PreIQ and/or IQ peptides has been reported in many studies (7, 9–11, 17, 31, 33, 35) and confirmed in this study. In addition, it has been suggested that Ca2+/CaM-dependent activation of CaM kinase II, and thereby phosphorylation of Cav1.2 channels, underlies CDF (12). We believe that both mechanisms, direct action of CaM on the channel and phosphorylation of the channel by CaM kinase II, take place during CDF in intact myocytes. On the other hand, Höfer et al. (39) showed that Cav1.2 channel activity was inhibited by Ca2+ with an IC50 of ∼4 μm and a Hill coefficient of ∼1, leading to the proposal that Ca2+ may act directly on the channel's inactivation site (16, 40), on a one-to-one molecular basis (33, 36, 39). We have also observed that channel activity induced by the Ca2+-insensitive CaM mutant still showed a Ca2+ dependence with a relatively low affinity for [Ca2+] (13). Whether this putative Ca2+-binding site is in the EF-hand region (17) or somewhere downstream of the EF-hand region (8) is not clear. In any case, it seems that there may be at least two Ca2+-sensing mechanisms for CDI.
CSL-CaM Competition Model
Based on previous studies (4, 5) and the present findings, we propose a hypothesis for the actions of CaM and CSL on the Cav1.2 channel (Fig. 5). At low [Ca2+], CaM mainly interacts with the IQ region due to its relatively high affinity for Ca2+-free CaM (apoCaM). CSL competes with apoCaM for this site as a partial agonist, thereby partially suppressing channel activity in the presence of apoCaM, in a concentration-dependent manner, as exemplified by a computer simulation (supplemental Fig. S4). Interactions of apoCaM with the N-terminal, LI-II, or PreIQ region may be minimal due to their relatively low affinity for apoCaM (10, 36). At high [Ca2+], because the affinity of the IQ region for Ca2+-bound CaM (Ca2+/CaM) is higher than for apoCaM, the IQ region occupancy with Ca2+/CaM is higher, leading to a further increase in channel activity (facilitation). At the same time, because the affinities of other sites, such as the N-terminal, LI-II, and PreIQ region, for Ca2+/CaM are also higher than for apoCaM, these sites may also interact with Ca2+/CaM, leading to the inactivation of the channel. Thus, the model explains CDF at a medium range of [Ca2+] and CDI at higher [Ca2+]. The model does not exclude the possibility, however, that CaM kinase II-mediated phosphorylation of the channel contributes to CDF in addition to the direct effect of Ca2+/CaM on the channel.
FIGURE 5.

A hypothetical model for competitions between CSL and CaM on the Cav1.2 channel. A, at low [Ca2+], CaM is mostly in the Ca2+-free form (apoCaM) and interacts with the IQ domain in the C terminus (C-term) of the Cav1.2 channel, producing basal channel activity. CSL competes with CaM for the site in the IQ domain. Interaction of CaM with NT, LI-II, or PreIQ is minimal in this condition. B, when [Ca2+] is increased, Ca2+-bound CaM (Ca2+/CaM) is formed and interacts with higher affinity with the IQ domain, resulting in Ca2+-dependent facilitation. Further increase in [Ca2+] promotes the interaction of Ca2+/CaM with NT and/or PreIQ and possibly with LI-II, leading to Ca2+-dependent inactivation. CSL does not interact with these regions and therefore does not affect the Ca2+-dependent inactivation.
Implications for the Physiological Significance
Although cardiac myocytes reportedly contain 0.7–1 μm CS (41), its concentration near the intracellular orifice of Cav1.2 channel is not known. However, because CS is localized around the Z-disk (42), where Cav1.2 channels in the T-tubular system are also localized, we speculate that the concentration of CS near the channels is higher than the estimated average value. If so, it may be reasonable to speculate further that the concentration of CS may be high enough to affect Cav1.2 channel activity by competing with CaM for its site, as predicted by a computer simulation (supplemental Fig. S4). The finding that CSL inhibits whole-cell Ca2+ current recorded from intact myocytes is in line with this idea (supplemental Fig. S2). CS is sensitive to proteolysis (43, 44) and degraded during ischemia and reperfusion in rat hearts (45). This may provide a possible mechanism by which Cav1.2 channel activity is affected during ischemic dysfunction of the heart.
In conclusion, we found that CSL inhibits the channel-activating effect of CaM. CSL competes with CaM for its binding site, located in the IQ region of the channel's C terminus. Thus, we suggest that CSL acts as a partial agonist of the CaM-binding site for channel activation.
Supplementary Material
Acknowledgment
We thank E. Iwasaki for secretarial work.
This work was supported by a grant-in-aid for young scientists from the Ministry of Education, Culture, Sports, Science, and Technology (to E. M.), a grant-in-aid for scientific research from the Japan Society for the Promotion of Science (to M. K.), and a grant from the Kodama Memorial Foundation for Medical Research (to E. M.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4.
- CS
- calpastatin
- CSL
- calpastatin domain L
- CaM
- calmodulin
- CDF
- Ca2+-dependent facilitation
- CDI
- Ca2+-dependent inactivation
- CSD1
- calpastatin domain 1
- NT
- N terminus
- LI-II
- LII-III, and LIII-IV, cytosolic linker between channel domains I and II, II and III, and III and IV, respectively
- CT1
- CT2, and CT3, C-terminal region 1 (proximal), 2 (middle), and 3 (distal), respectively.
REFERENCES
- 1. Maki M., Takano E., Mori H., Sato A., Murachi T., Hatanaka M. (1987) FEBS Lett. 223, 174–180 [DOI] [PubMed] [Google Scholar]
- 2. Hao L. Y., Kameyama A., Kuroki S., Takano J., Takano E., Maki M., Kameyama M. (2000) Biochem. Biophys. Res. Commun. 279, 756–761 [DOI] [PubMed] [Google Scholar]
- 3. Kepplinger K. J., Förstner G., Kahr H., Leitner K., Pammer P., Groschner K., Soldatov N. M., Romanin C. (2000) J. Physiol. 529, 119–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Minobe E., Hao L. Y., Saud Z. A., Xu J. J., Kameyama A., Maki M., Jewell K. K., Parr T., Bardsley R. G., Kameyama M. (2006) Biochem. Biophys. Res. Commun. 348, 288–294 [DOI] [PubMed] [Google Scholar]
- 5. Saud Z. A., Minobe E., Wang W. Y., Han D. Y., Horiuchi M., Hao L. Y., Kameyama M. (2007) Biochem. Biophys. Res. Commun. 364, 372–377 [DOI] [PubMed] [Google Scholar]
- 6. Hoeflich K. P., Ikura M. (2002) Cell 108, 739–742 [DOI] [PubMed] [Google Scholar]
- 7. Pate P., Mochca-Morales J., Wu Y., Zhang J. Z., Rodney G. G., Serysheva I. I., Williams B. Y., Anderson M. E., Hamilton S. L. (2000) J. Biol. Chem. 275, 39786–39792 [DOI] [PubMed] [Google Scholar]
- 8. Zühlke R. D., Pitt G. S., Tsien R. W., Reuter H. (2000) J. Biol. Chem. 275, 21121–21129 [DOI] [PubMed] [Google Scholar]
- 9. Pitt G. S., Zühlke R. D., Hudmon A., Schulman H., Reuter H., Tsien R. W. (2001) J. Biol. Chem. 276, 30794–30802 [DOI] [PubMed] [Google Scholar]
- 10. Erickson M. G., Liang H., Mori M. X., Yue D. T. (2003) Neuron 39, 97–107 [DOI] [PubMed] [Google Scholar]
- 11. Kim J., Ghosh S., Nunziato D. A., Pitt G. S. (2004) Neuron 41, 745–754 [DOI] [PubMed] [Google Scholar]
- 12. Dzhura I., Wu Y., Colbran R. J., Balser J. R., Anderson M. E. (2000) Nat. Cell Biol. 2, 173–177 [DOI] [PubMed] [Google Scholar]
- 13. Han D. Y., Minobe E., Wang W. Y., Guo F., Xu J. J., Hao L. Y., Kameyama M. (2010) J. Pharmacol. Sci. 112, 310–319 [DOI] [PubMed] [Google Scholar]
- 14. Lee W. J., Ma H., Takano E., Yang H. Q., Hatanaka M., Maki M. (1992) J. Biol. Chem. 267, 8437–8442 [PubMed] [Google Scholar]
- 15. Ding S., Kuroki S., Kameyama A., Yoshimura A., Kameyama M. (1999) J. Biochem. 125, 750–759 [DOI] [PubMed] [Google Scholar]
- 16. Frangioni J. V., Neel B. G. (1993) Anal. Biochem. 210, 179–187 [DOI] [PubMed] [Google Scholar]
- 17. Peterson B. Z., DeMaria C. D., Adelman J. P., Yue D. T. (1999) Neuron 22, 549–558 [DOI] [PubMed] [Google Scholar]
- 18. Asmara H., Minobe E., Saud Z. A., Kameyama M. (2010) J. Pharmacol. Sci. 112, 397–404 [DOI] [PubMed] [Google Scholar]
- 19. Xu J. J., Hao L. Y., Kameyama A., Kameyama M. (2004) Am. J. Physiol. Cell Physiol. 287, C1717–C1724 [DOI] [PubMed] [Google Scholar]
- 20. Hao L. Y., Kameyama A., Kameyama M. (1999) J. Physiol. 514, 687–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guo F., Minobe E., Yazawa K., Asmara H., Bai X. Y., Han D. Y., Hao L. Y., Kameyama M. (2010) Biochem. Biophys. Res. Commun. 391, 1170–1176 [DOI] [PubMed] [Google Scholar]
- 22. O'Rourke B., Backx P. H., Marban E. (1992) Science 257, 245–258 [DOI] [PubMed] [Google Scholar]
- 23. Yazawa K., Kameyama A., Yasui K., Li J. M., Kameyama M. (1997) Pflügers Arch. 433, 557–562 [DOI] [PubMed] [Google Scholar]
- 24. Zhou H., Yu K., McCoy K. L., Lee A. (2005) J. Biol. Chem. 280, 29612–29619 [DOI] [PubMed] [Google Scholar]
- 25. Xiong L., Kleerekoper Q. K., He R., Putkey J. A., Hamilton S. L. (2005) J. Biol. Chem. 280, 7070–7079 [DOI] [PubMed] [Google Scholar]
- 26. Seydl K., Karlsson J. O., Dominik A., Gruber H., Romanin C. (1995) Pflügers Arch. 429, 503–510 [DOI] [PubMed] [Google Scholar]
- 27. Takano E., Ma H., Yang H. Q., Maki M., Hatanaka M. (1995) FEBS Lett. 362, 93–97 [DOI] [PubMed] [Google Scholar]
- 28. Fallon J. L., Baker M. R., Xiong L., Loy R. E., Yang G., Dirksen R. T., Hamilton S. L., Quiocho F. A. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 5135–5140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim E. Y., Rumpf C. H., Van Petegem F., Arant R. J., Findeisen F., Cooley E. S., Isacoff E. Y., Minor D. L., Jr. (2010) EMBO J. 29, 3924–3938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Evans T. I., Hell J. W., Shea M. A. (2011) Biophys. Chem. 159, 172–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zühlke R. D., Pitt G. S., Deisseroth K., Tsien R. W., Reuter H. (1999) Nature 399, 159–162 [DOI] [PubMed] [Google Scholar]
- 32. Qin N., Olcese R., Bransby M., Lin T., Birnbaumer L. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 2435–2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Romanin C., Gamsjaeger R., Kahr H., Schaufler D., Carlson O., Abernethy D. R., Soldatov N. M. (2000) FEBS Lett. 487, 301–306 [DOI] [PubMed] [Google Scholar]
- 34. Barrett C. F., Tsien R. W. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 2157–2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mouton J., Feltz A., Maulet Y. (2001) J. Biol. Chem. 276, 22359–22367 [DOI] [PubMed] [Google Scholar]
- 36. Ivanina T., Blumenstein Y., Shistik E., Barzilai R., Dascal N. (2000) J. Biol. Chem. 275, 39846–39854 [DOI] [PubMed] [Google Scholar]
- 37. Tang W., Halling D. B., Black D. J., Pate P., Zhang J. Z., Pedersen S., Altschuld R. A., Hamilton S. L. (2003) Biophys. J. 85, 1538–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mori M. X., Erickson M. G., Yue D. T. (2004) Science 304, 432–435 [DOI] [PubMed] [Google Scholar]
- 39. Höfer G. F., Hohenthanner K., Baumgartner W., Groschner K., Klugbauer N., Hofmann F., Romanin C. (1997) Biophys. J. 73, 1857–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Peterson B. Z., Lee J. S., Mulle J. G., Wang Y., de Leon M., Yue D. T. (2000) Biophys. J. 78, 1906–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Otsuka Y., Goll D. E. (1987) J. Biol. Chem. 262, 5839–5851 [PubMed] [Google Scholar]
- 42. Lane R. D., Mellgren R. L., Mericle M. T. (1985) J. Mol. Cell Cardiol. 17, 863–872 [DOI] [PubMed] [Google Scholar]
- 43. Takano E., Maki M., Mori H., Hatanaka M., Marti T., Titani K., Kannagi R., Ooi T., Murachi T. (1988) Biochemistry 27, 1964–1972 [DOI] [PubMed] [Google Scholar]
- 44. De Tullio R., Averna M., Salamino F., Pontremoli S., Melloni E. (2000) FEBS Lett. 475, 17–21 [DOI] [PubMed] [Google Scholar]
- 45. Enns D., Karmazyn M., Mair J., Lercher A., Kountchev J., Belcastro A. (2002) Mol. Cell Biochem. 241, 29–35 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.