

Background: NOSs possess a highly conserved tryptophan residue, proximal to the heme-thiolate bond.
Results: Replacement of this Trp by His or Phe in Bacillus subtilis NOS altered both thermodynamic and kinetic parameters and NO synthesis.
Conclusion: B. subtilis NOS control catalysis by tuning the electron density of its heme-thiolate bond.
Significance: This is the first study to investigate these relationships in a bacterial NOS.
Keywords: Enzyme Catalysis, Enzyme Kinetics, Nitric-oxide Synthase, Reductase, Spectroscopy, Bacillus subtilis, Bacterial NOS, Midpoint Potentials, NO Synthesis, Oxygen Activation
Abstract
Nitric-oxide synthases (NOS) are heme-thiolate enzymes that generate nitric oxide (NO) from l-arginine. Mammalian and bacterial NOSs contain a conserved tryptophan (Trp) that hydrogen bonds with the heme-thiolate ligand. We mutated Trp66 to His and Phe (W66H, W66F) in B. subtilis NOS to investigate how heme-thiolate electronic properties control enzyme catalysis. The mutations had opposite effects on heme midpoint potential (−302, −361, and −427 mV for W66H, wild-type (WT), and W66F, respectively). These changes were associated with rank order (W66H < WT < W66F) changes in the rates of oxygen activation and product formation in Arg hydroxylation and N-hydroxyarginine (NOHA) oxidation single turnover reactions, and in the O2 reactivity of the ferrous heme-NO product complex. However, enzyme ferrous heme-O2 autoxidation showed an opposite rank order. Tetrahydrofolate supported NO synthesis by WT and the mutant NOS. All three proteins showed similar extents of product formation (l-Arg → NOHA or NOHA → citrulline) in single turnover studies, but the W66F mutant showed a 2.5 times lower activity when the reactions were supported by flavoproteins and NADPH. We conclude that Trp66 controls several catalytic parameters by tuning the electron density of the heme-thiolate bond. A greater electron density (as in W66F) improves oxygen activation and reactivity toward substrate, but decreases heme-dioxy stability and lowers the driving force for heme reduction. In the WT enzyme the Trp66 residue balances these opposing effects for optimal catalysis.
Introduction
Nitric oxide (NO) is a small molecule essential to life in higher organisms. In mammals, NO synthesis is performed by three isoforms (inducible, endothelial, and neuronal) of homodimeric nitric-oxide synthases (NOS, EC 1.14.13.39). Each monomer is comprised of a nitric-oxide synthase oxygenase domain (NOSoxy),3 with structurally defined binding sites for its substrate l-arginine, tetrahydrobiopterin (H4B), and a Cys-coordinated Fe-protoporphyrin IX moiety (heme), and a C-terminal reductase domain (NOSred) with binding sites for NADPH, FAD, and FMN. NOSoxy and NOSred are connected by a calmodulin binding sequence (1, 2).
The existence of NOS is not unique to animals. A number of bacterial NOS or NOS-like proteins have been isolated and characterized during the past 10 years (3–34). With the exception of the complete NOS present in Sorangium cellulosum (25), all of the bacterial NOSs characterized to date consist of a truncated NOSoxy domain that lacks a portion of the N-terminal region and has no attached reductase domain (30). Thus, NO synthesis by bacterial NOSs relies on their interaction with nondedicated reductases (3, 4, 20, 22). An exclusive feature of bacterial NOSs is their ability to utilize tetrahydrofolate (H4T) as the cofactor to support NO synthesis (3, 4). This is presumably an evolutionary adaptation, as microorganisms that bear a NOS gene do not usually have the complete set of genes required for the synthesis of tetrahydrobiopterin (30).
The sequence of reactions that leads to NO synthesis by bacterial NOS has been proposed to be very similar to that of the mammalian counterpart. Briefly, resting NOS is first reduced by electrons coming from the reductase domain or partner. Ferrous heme then reacts with molecular oxygen to form a transient FeO2 species (Fig. 1). This FeO2 intermediate is further reduced by the cofactor H4X (H4B in mammalian NOS, and either H4B or H4T in bacterial NOS) to form a ferric-peroxo complex. Scission of the O-O follows to form the highly valent compound I species, which ultimately oxidizes l-Arg to form NOHA. Conversion of NOHA into citrulline and NO requires another round of oxygen activation, in which H4X is proposed to operate as the electron donor (Fig. 1).
FIGURE 1.

Model for catalysis by mammalian and bacterial NOS. Ferric heme is first reduced by the flavoprotein domain of a reductase protein (NOS reductase in mammalian NOS, or an unidentified flavoprotein in bacterial NOS) enabling O2 binding and formation of a Fe(III)-O-O− species. A second electron derived from H4X generates a ferric peroxo species and yields H4X+·. Note, H4X represents H4B in mammalian NOS and either H4B or H4T in the case of bacterial NOS.
Both animal NOS and bacterial NOS possess a conserved tryptophan residue that forms stacking interactions with the porphyrin ring and hydrogen bonds with the heme-thiolate bond (Fig. 2). Replacement of this proximal Trp residue by histidine in murine iNOS (W188H) increased the midpoint potential of the heme group by +88 mV and reduced the rate of NO synthesis compared with wild-type iNOS. In addition, the W188H mutation stabilized a heme intermediate that formed downstream of the FeO2 species, which reacted slowly with l-Arg to form NOHA (35). Surprisingly, the W188F mutant of iNOS had a defective heme binding, thus hampering any further characterization (36). Substitution of Trp409 by Phe in rat nNOS reduced the heme midpoint potential of the protein, led to a faster formation of the FeO2 species and to greater rates of Fe(II)-NO oxidation compared with the wild-type protein (37–39). Thus, the proximal Trp residue may play a role in controlling the reactivity of the enzyme by tuning the properties and reactivity of its heme.
FIGURE 2.

The active site of BsNOS. The spatial arrangement of l-Arg, heme, and its axial Cys, H4T, and Trp66 are shown. The structure of the active site was created from the crystallographic data available for native BsNOS (PDB 1M7V (6)) using PyMol software. In the crystal structure utilized herein, the proximal Trp is in position 56 due to a truncation of the first 10 amino acids of the protein.
We set out to investigate these relationships in Bacillus subtilis NOS. Replacement of the conserved Trp residue (Trp66) by His or Phe led to the expression of stable proteins, with characteristic spectroscopic features as described recently (34). We utilized these mutations to increase (His) or decrease (Phe) the H-bonding properties of the heme-thiolate bond of B. subtilis NOS and inquired on its effects on: heme midpoint potential, enzyme oligomeric state, substrate binding, FeO2 stability, kinetics of heme transitions during catalysis, kinetics and extent of product formation in both steps, reactivity of the FeNO complex, and NO synthesis. This is the first comprehensive study to: (i) provide a side-by-side comparison of the effect of replacing the proximal Trp with an electron donating (His) versus an electron withdrawing (Phe) residue in catalysis by a bacterial NOS. (ii) Demonstrate that H4T enables the formation of a Fe(III)-NO species during NOHA-driven single turnover reactions and (iii) show that H4T supports NO synthesis by B. subtilis in an in vitro reconstitution system.
EXPERIMENTAL PROCEDURES
Reagents
H4B and H4T were purchased from Schircks Laboratories (Jona, Switzerland). CO gas was obtained from Praxair, Inc. (Danbury, CT). N5-CH3-H4T (Eprova, Switzerland) was a generous gift of Dr. Donald Jacobsen (Department of Cell Biology, Cleveland Clinic). N-Hydroxy-l-Arg (NOHA) and 14C-labeled l-arginine (l-[14C]Arg) were purchased from MP Biomedicals (Solon, OH). EPPS was purchased from Fisher Scientific (Pittsburgh, PA). DTT was purchased from RPI Corp. (Mount Prospect, IL). All other reagents were purchased from Sigma.
Site-directed Mutagenesis
BsNOS mutants were prepared by site-directed mutagenesis on a pET15b vector containing the cDNA of wild-type BsNOS (3). Site-directed mutagenesis was performed using the QuikChange XL mutagenesis kit (Stratagene, La Jolla, CA). Mutations were confirmed by DNA sequencing. Mutated plasmids (pET15b-BsNOS-W66H and pET15b-BsNOS-W66F) were transformed into BL21(DE3) Escherichia coli cells using the TransformAid bacterial transformation kit (Fermentas, Hanover, MD).
Protein Expression and Purification
Wild-type and mutant BsNOS proteins containing a His6 tag attached to their N termini were overexpressed in E. coli strain BL21(DE3). BsNOS proteins were expressed and purified as described (34). Protein concentration was determined from the absorbance at 444 nm of the ferrous heme-CO complex, using an extinction coefficient of 76 mm−1 cm−1 (Δϵ444–500 nm) (25). All proteins were purified to homogeneity (≥95%) as assessed by SDS-PAGE (supplemental Fig. S1A). The oligomeric state of wild-type and mutant B. subtilis NOSs reconstituted with H4T and l-Arg was examined by size-exclusion chromatography. Protein samples (∼150 μm) in EPPS buffer (40 mm, pH 7.6, 150 mm NaCl) were incubated with 2 mm l-Arg, 400 μm H4T, and 1.2 mm DTT for 15 min. Samples were injected on a Superdex 200 resin pre-equilibrated with EPPS buffer (40 mm, pH 7.6, 150 mm NaCl) supplemented with 100 μm l-Arg, 40 μm H4T, and 120 μm DTT. Under these conditions, all proteins existed predominantly in the dimeric state (80–97%) (supplemental Fig. S1B).
Imidazole and Arginine Binding
Imidazole and l-Arg binding affinities were studied at 25 °C by perturbation difference spectroscopy according to methods described previously (27, 28). NOS samples (around 5 μm) in 40 mm EPPS buffer, pH 7.6, with 10% glycerol, 0.6 mm DTT, 0.2 mm H4T were titrated by stepwise addition of imidazole, to a final concentration of 10 mm. The Kd of imidazole (Kd(imid)) was calculated by fitting the data to a simple saturation binding equation. The Kd of l-Arg (Kd) was determined under the same conditions, in the presence of 10 mm imidazole. The data were fit to a simple saturation binding equation, and Kd was calculated according to the Equation 1.
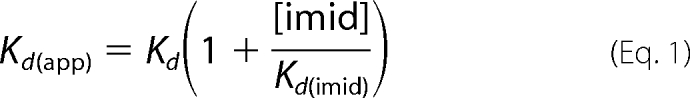 |
Single Turnover Reactions
l-Arg hydroxylation and NOHA oxidation experiments were carried out in a Hi-Tech SF61-DX2 stopped-flow instrument (Hi-Tech Scientific, Salisbury, UK) coupled to a diode array detector, as previously described (40). An anaerobic solution of 20 μm ferrous NOS, 2 mm l-Arg (or 1 mm NOHA), 0.2 mm H4T (or another pterin, where indicated), and 1 mm DTT in 40 mm EPPS, pH 7.6, containing 10% glycerol and 150 mm NaCl was mixed at 10 °C with a syringe containing oxygen-saturated buffer, 2 mm l-Arg, 0.2 mm H4T, and 1 mm DTT. Sequential spectral data were fitted to an A → B → C model using the Specfit/32 global analysis software, version 3.0 (Spectrum Software Associates, Marlborough, MA), which calculates the spectra of the different enzyme species and their concentration change versus time. The reported rates are the average of 5–10 measurements. The error associated to these measurements was 2–5%.
Kinetics of Product Formation by Rapid Quench
An anaerobic solution of 10 μm ferrous NOS, 15 μm l-[14C]Arg plus 5 μm unlabeled l-Arg, 0.2 mm H4T, and 0.6 mm DTT in 40 mm EPPS, pH 7.6, containing 10% glycerol and 150 mm NaCl was mixed at 10 °C with a syringe containing oxygen-saturated buffer and 15 μm l-[14C]Arg. The quenching solution was a solution of 0.5 m HCl, 20% 2-propanol, 1 mm l-Arg, and 1 mm NOHA. Quenched reaction samples were collected and stored at −80 °C. To determine the final dilution factors the protein samples included 200 μm l-glutamic acid as an internal standard. Infinite time reactions with l-Arg (supplemental Fig. S5) were carried out by incubating 10 μm NOS with 5 μm l-[14C]Arg and 95 μm unlabeled l-Arg in the presence of 200 μm H4T and 1 mm DTT. Ferrous protein was generated by addition of dithionite. The reactions were started by mixing with oxygen-saturated buffer and allowed to proceed at room temperature for 10 min. The reactions were quenched with a solution of 0.5 m HCl, 20% 2-propanol, 1 mm l-Arg, and 1 mm NOHA. Quenched samples were stored at −80 °C for further HPLC analysis. Infinite time reactions with NOHA were performed as described for the reactions of l-Arg, except the concentrations of reagents were: 150 μm NOS, 400 μm NOHA, 1 mm H4T, and 3 mm DTT. Quenched samples were stored at −80 °C and citrulline formation was determined using a published HPLC procedure (see below).
Determination of Reaction Products by HPLC
l-[14C]Arg and [14C]NOHA were extracted from the reaction mixtures (100 μl each) with 5 μl of 2-propanol 40 mm HCl, and were vortexed for 45 min at room temperature. These solutions were then centrifuged at 10,000 × g for 10 min to eliminate any precipitated materials. 50 μl was injected on a Nucleosil® C18-HPLC column (particle size 5 μm, 250 × 4.6 mm), and the amino acids were separated using an isocratic method with elution buffer (50 mm sodium acetate, pH 6.50). Under these conditions l-[14C]NOHA and l-[14C]Arg eluted at ∼13 and 18 min, respectively. The radioactivity of each sample was determined using a scintillation counter. Citrulline formation from NOHA was determined by derivatization of the amino acid products with naphthalene-2,3-dicarboxyaldehyde in the presence of cyanide followed by HPLC analysis, according to a published procedure (41).
Midpoint Potential Measurements
Spectroelectrochemical titrations were carried out in a glove-box (Belle Technology, Dorset, UK) under N2 atmosphere, as previously described (35, 42). Briefly, NOS proteins were made anaerobic by gel filtration in a Sephadex G-25 column (PD 10, GE Healthcare) equilibrated with anaerobic buffer (100 mm phosphate buffer, pH 7.0, 125 mm NaCl). Protein samples were diluted to a 3.5-ml final volume (final concentration ≈10 μm) and l-Arg (2 mm) and H4T (100 μm) were added. The following electron mediator dyes (0.5–1 μm) were used: phenosafranine (Em = −252 mV), benzyl viologen (Em = −358 mV), methyl viologen (−450 mV), and anthraquinone-2-sulfonate (Em = −225 mV). The titration was carried out at 15 °C by bolus additions of a sodium dithionite solution. Absorption spectra were recorded with a Cary 50 spectrophotometer equipped with a dip-probe detector, and the potentials were measured with an Accumet AB15 pH meter (Fisher Scientific) using a silver/silver chloride microelectrode saturated with 4 m KCl.
Ferrous Heme-NO Complex Oxidation (kox)
Wild-type, W66H, and W66F BsNOS proteins (∼5 μm) in 100 mm EPPS (pH 7.6, 150 mm NaCl, 10% glycerol) containing 2 mm l-Arg, 0.2 mm H4T, and 1 mm DTT were reduced with dithionite in an anaerobic cuvette. The Fe(II)-NO complex of W66F was unstable at neutral pH, hence these reactions were carried out under the conditions described above using CHES buffer (100 mm, pH 9.5, 150 mm NaCl, 10% glycerol). The Fe(II)-NO complexes were generated by adding successive aliquots of an anaerobic NO-saturated buffer. The samples were then transferred to an anaerobic stopped-flow instrument using a gas tight syringe, and the reactions were initiated by mixing the anaerobic Fe(II)-NO protein samples with air-saturated buffer at 10 °C. Spectra were taken during the course of the reactions and the data were fit to a single exponential model A → B using Specfit global analysis software.
NO Synthesis
NO synthesis by BsNOS and mutant proteins was assessed via a reconstitution assay as described previously (20). Briefly, 1 μm NOS, 2.5 μm flavodoxin reductase (FLDR), and 10 μm flavodoxin (YkuN) were incubated with 400 μm NOHA, 100 μm H4T or H4B, and 300 μm DTT in 40 mm EPPS, pH 7.6, containing 10% glycerol and 125 mm NaCl, at room temperature for 20 min. The reactions were started by addition of 1 mm NADPH. After 15 min the reactions were stopped by addition of 1 unit of lactate dehydrogenase and 10 mm sodium pyruvate. Total nitrite was determined using the Griess assay (43).
RESULTS
Spectrocoscopic Properties and Substrate Binding
We first examined the spectroscopic properties and the ability of the two mutant proteins to bind imidazole, the substrate l-Arg, and the stable reaction intermediate NOHA. UV-visible data for BsNOS, W66H, and W66F reconstituted with H4T are given under supplemental Table S1. First, the UV-visual spectral features of BsNOS reconstituted with H4T are almost identical to those observed for the protein reconstituted with H4B (11, 34). The UV-visible spectra of the Fe(III), Fe(II), and Fe(II)-CO complexes of BsNOS, W66H, and W66F reconstituted with H4T are given in Fig. 3. The spectra of the Fe(II), Fe(II)-CO, and Fe(III)-imidazole complexes of W66H and W66F are similar but not identical to that of wild-type BsNOS (supplemental Table S1). In the presence of l-Arg and H4T all three proteins exist in the typical Fe(III) high-spin configuration. Reduction with dithionite and exposure to CO resulted in the formation of the characteristic Fe(II)-CO complex with a Soret band appearing at 445–449 nm. The Fe(II)-CO species was unstable in the case of W66F (Fig. 3C), slowly converting to form an uncharacterized species with an absorption maximum at 420 nm. This could be a hexa-coordinated Fe(II)-CO complex, in which the axial Cys is protonated (44), however, the exact nature of this species in NOS is currently unknown. This was also observed for W66F reconstituted with H4B (34), as well as in the corresponding Trp → Phe mutants of Staphylococcus aureus NOS and eNOS (45), suggesting that the hexa-coordinated ferrous state in this mutant NOS is unstable compared with wild-type BsNOS. Binding dissociation constants, Kd, for imidazole, l-Arg, and NOHA in the presence of H4T were measured spectrophotometrically (supplemental Fig. S2). The affinity constants of BsNOS wild-type for imidazole and l-Arg in the presence of H4T were 160 ± 9 μm and 1.20 ± 0.06 μm, respectively. Kd values for imidazole and l-Arg binding to BsNOS in the presence of H4B have been reported previously as 384 and 4.8 μm (11), respectively. This suggests a slightly increased affinity of BsNOS for its substrate l-Arg and the ligand imidazole in the presence of H4T. A similar, yet more pronounced effect of H4T on substrate binding affinity has been reported for Drosophila melanogaster iNOS (28). Binding affinities of BsNOS and W66H for imidazole were very similar (160 ± 9 and 122 ± 7 μm, respectively), and higher (∼4-fold) than that observed in W66F (523 ± 29 μm). Both wild-type and mutant BsNOS proteins displayed competent binding for l-Arg, with Kd values following the order: KdWT < KdW66F < KdW66H (1.20 ± 0.06, 4.3 ± 0.2, and 8.5 ± 0.2 μm, respectively). In addition, all B. subtilis NOS proteins bound NOHA with Kd values of 23.5 ± 1.3, 138.2 ± 9.5, and 15.9 ± 0.7 μm for BsNOS, W66H, and W66F, respectively (supplemental Fig. S2). The lower affinity for both l-Arg and NOHA in W66H compared with wild-type BsNOS resembles the previously reported effects of the analogous mutation in iNOSoxy (W188H)(35) and P450 BM3 (F393H) (46).
FIGURE 3.

UV-visible spectral properties of wild-type BsNOS and mutant proteins W66H and W66F in the presence of H4T and l-Arg. The UV-visible spectral properties of the bacterial NOSs do not differ markedly from those of the mammalian NOS oxygenase. From left to right: BsNOS, W66H, and W66F. In W66F the Fe-CO species (449 nm) slowly decays to an uncharacterized species with absorption maximum at 420 nm (inset). A summary of the absorption maxima is given in Table S1.
Redox Potentiometry
A possible consequence of replacing the proximal Trp residue by His or Phe is an alteration of the heme midpoint potential. Redox titrations in the presence of l-Arg and H4T were performed for BsNOS, W66H, and W66F and the results are shown in Fig. 4. The calculated midpoint potentials were: −361, −302, and −427 mV for BsNOS, W66H, and W66F, respectively. Thus, substitution of Trp66 by His increased the midpoint potential by +59 mV with respect to the wild-type protein. A similar effect was observed for the corresponding mutations in iNOSoxy (W188H) (35) and cytochrome P450 BM3 (F393H) (46–48). In contrast, substitution of Trp66 with Phe resulted in a substantial decrease of the heme midpoint potential (−66 mV). Thus, changes in hydrogen bonding of the heme-thiolate in BsNOS have a direct effect on the redox properties of the heme.
FIGURE 4.

Determination of the redox potential of BsNOS wild-type, W66H, and W66F proteins. The fraction of oxidized protein at each redox potential value was fitted to the Nernst equation. The fitted values for the midpoint potentials are: BsNOS, −361 ± 5 mV; W66H, −302 ± 7 mV; and W66F, −427 ± 5 mV.
Stopped-flow Analysis of a Single Turnover l-Arg Hydroxylation Reaction in the Presence of H4T
We next investigated the heme transitions that occur in BsNOS, W66H, and W66F during catalysis in the presence of H4T and l-Arg. Anerobic samples of each protein in the presence of H4T and l-Arg were reduced with dithionite and mixed with air-saturated buffer containing H4T and l-Arg in a stopped-flow instrument. A minimum of 100 spectra were collected during the course of the reaction and subjected to global analysis. We found that l-Arg hydroxylation reactions of both wild-type and mutant proteins could be best fit to a two-exponential model A → B → C, with Fe(II), Fe(II)-O2, and Fe(III) as the only detectable species (supplemental Fig. S3). A summary of the observed rate constants is given in Table 1. Replacement of Trp66 to His resulted in a slower transition of the Fe(II)-O2 species to form Fe(III) during single turnover reactions. Unlike W188H iNOSoxy (35), conversion of the Fe(II)-O2 species in W66H BsNOS occurs without the buildup of a detectable downstream intermediate. In contrast, replacement of Trp66 by Phe resulted in a faster transition of the Fe(II)-O2 species to form Fe(III). These relationships held true for the reactions performed in the presence of N5-CH3-H4T as well (Table 1). Representative reaction profiles and the relative concentration of the different species during the course of the reactions performed in the presence of N5-CH3-H4T are given in Fig. 5. For all three proteins, formation and disappearance of the Fe(II)-O2 species was slower in the presence N5-CH3-H4T with respect to H4T (Table 1). An enhanced conversion rate of the Fe(II)-O2 species in the presence of the methylated cofactor at position N5 has been reported for the reactions of iNOS with N5-CH3-H4B (49). It was proposed that a faster electron transfer from N5-CH3-H4T to the heme-oxy species occurs via an increased H4B+· stability afforded by the presence of the N5-methyl group (49). Although this effect on Fe(II)O2 stability was not observed in B. subtilis NOSs, W66F displayed the fastest rates of Fe(II)O2 conversion with respect to BsNOS and W66H, regardless of the pterin used. Thus, replacement of Trp by Phe enhanced the processing of the heme-oxy intermediate in B. subtilis NOS, similar to the reported features of the corresponding mutant in rat nNOSoxy (W409F) (37–39). This implies that the properties of the Fe(II)O2 species in BsNOS are affected by the electronic structure of its heme, as observed in mammalian NOS. A comparison of single turnover reactions with l-Arg and H4B in BsNOS proteins versus the iNOSoxy and W188Hoxy mammalian counterparts showed that in general, the heme transition rates in the bacterial NOSs are slightly slower than those seen in the mammalian proteins (Table 1). The heme transitions observed for the reactions with H4B were comparable with the corresponding reactions performed with H4T for all three B. subtilis NOSs (Table 1). We next determined the formation and autoxidation rates of the Fe(II)O2 species in reactions where H4B and H4T were substituted with H2B and H2T, respectively. Formation of the Fe(II)O2 species was faster in the presence of the oxidized pterins, whereas its conversion to ferric was significantly slower than that observed in the reactions performed with reduced biopterin or folate (supplemental Tables S2 and S3). This suggests that the reduced pterins may play a role in l-Arg oxidation via electron transfer, as it occurs in mammalian NOSs. A graphic representation of Fe(II)O2 conversion rates for selected hemeproteins is provided in Fig. 6. A comparison with other heme proteins shows that the reactivity of the Fe(II)O2 species in B. subtilis NOS falls between the fast transition rates observed in mammalian NOSs and the extremely slow rates of Fe(II)O2 disappearance reported for most members of the cytochrome P450 family (supplemental Table S4). In B. subtilis NOSs the rates of conversion of the Fe(II)O2 species correlated well with the heme midpoint potentials. The W66H complex was the most long-lived in the presence of l-Arg and H4B, H4T, or N5-CH3-H4T, whereas the opposite was true for W66F (Table 1).
TABLE 1.
Observed rate constants in B. subtilis wild-type, W66H, and W66F NOS versus murine wild-type and W188H iNOSoxy

a ND, not determined. Only two heme transitions could be observed during single turnover reactions with NOHA and H4T: Fe(II)/FeO2 → FeNO → Fe(III).
FIGURE 5.

Single turnover reactions with l-Arg and N5-CH3-H4T of BsNOS wild-type (A–C) and mutant proteins W66H (D–F) and W66F (G–I). Three species were identified by global analysis using the SpecFit 3.0 software: Fe(II) (Soret peak ∼412 nm), Fe-O2 (Soret peak ∼427 nm), and Fe(III) (Soret peak ∼394 nm). Mutation of Trp66 to His results in a slower decay of the Fe(II)-O2 species to form Fe(III) during single turnover reactions. Unlike the mammalian counterpart W188H iNOSoxy, decay of the Fe(II)-O2 species in W66H occurs without the buildup of a detectable intermediate. Replacement of Trp66 by Phe results in a faster decay of the Fe(II)-O2 species to form Fe(III), as depicted by the time courses of the different species with respect to BsNOS wild-type.
FIGURE 6.

FeO2 autoxidation rates for various hemeproteins. FeO2 autoxidation rates are shown for mammalian and bacterial NOSs and some members of the cytochrome P450 family. In the case of NOSs, the observed rate constants were determined in the presence of oxidized pterin or folate or in their absence. The values and the corresponding references are listed under supplemental Table S4.
Stopped-flow Analysis of a Single Turnover NOHA Oxidation Reaction in the Presence of H4T
We next examined NOHA oxidation reactions under single turnover conditions. Earlier studies by our laboratory demonstrated that a Fe(III)-NO complex builds up in BsNOS during single turnover reactions with H4B and NOHA (3). Given the plasticity in pterin utilization proposed to be at play in bacterial NOSs (30), it was interesting to investigate whether H4T could support the formation of a Fe-NO species in B. subtilis NOS. Analysis of the reaction profiles of BsNOS and the W66 mutants shows that H4T also supports formation of a Fe(III)-NO complex (Fig. 7). Analysis of single turnover reactions in the presence of NOHA and H4T showed that formation of Fe(II)-O2 was followed by fast formation of a Fe(III)-NO complex, which reacted further to form Fe(III). Fit of the data to a three exponential sequential model (A → B → C → D) yielded the rates provided in Table 1. Identification of the Fe(III)-NO complex was accomplished through the detection of a heme intermediate that formed downstream of the Fe(II)-O2 species in NOHA-driven reactions. The spectral features of this intermediate were in good agreement with those of authentic heme Fe(III)-NO. Importantly, this species was not observed in reactions performed with H2T (supplemental Fig. S4).
FIGURE 7.

Single turnover reactions with NOHA and H4T of BsNOS wild-type (A–C) and mutant proteins W66H (D–F) and W66F (G–I). Four species were identified by global analysis using the SpecFit 3.0 software for a three-exponential, four species model: A → B → C → D (panel B): Fe(II) (Soret peak ∼412 nm), Fe-O2 complex (Soret peak ∼430 nm, shoulders at 564 and 588 nm), Fe(III)-NO (Soret peaks ∼416 and 437 nm), and Fe(III) resting enzyme (Soret peak ∼392 nm, high-spin). The reaction of Fe(II) with dioxygen to form FeO2 is very fast in W66F, thus the reaction could be best fit to a two-exponential model: A → B → C, where species A is likely a mixture of Fe(II) and FeO2.
Due to the elusive nature of the Fe(II)O2 species in W66F, the reaction was modeled to a double-exponential sequential model, A → B → C. Formation of the Fe(III)-NO complex was fast (apparent kobs ∼ 26 s−1, however, its rate of conversion to Fe(III) (0.30 s−1) did not differ markedly (∼1.5-fold) from that of BsNOS WT (0.19 s−1) or W66H (0.20 s−1) (Table 1).
Extent and Kinetics of l-Arg Hydroxylation
To determine whether substitution of Trp for His or Phe had an impact in the total yield and kinetics of l-Arg hydroxylation we measured the time course of NOHA formation in the presence of H4T by rapid-quench and HPLC, using l-[14C]Arg as the substrate (Fig. 8). The observed rates of [14C]NOHA formation were 4.7 ± 1.1, 2.9 ± 0.2, and 17 ± 2 s−1, for BsNOS, W66H, and W66F, respectively. These rates follow the trend W66F > BsNOS > W66H, which are consistent with the rates for the heme transitions observed by rapid UV-visible scan during single turnover reactions. NOHA formation from l-Arg, and citrulline formation from NOHA were also determined, for infinite incubation times (15 min) (supplemental Table S5). Production of NOHA from l-Arg was characterized by notably low yields in both BsNOS and W66 mutants. In contrast, production of citrulline from NOHA was a relatively efficient process; the extent of product formation was ∼0.6 citrulline per heme (supplemental Table S5).
FIGURE 8.

Kinetics of NOHA formation from l-Arg by rapid-quench experiments and HPLC analysis. Product formation was monitored by the formation of [14C]NOHA from l-[14C]Arg as described under “Experimental Procedures.” Mutant W66H catalyzed the conversion of l-Arg at a slower rate than wild-type BsNOS. In contrast, W66F displayed a faster conversion of the substrate to form NOHA compared with the native protein. These results are in agreement with the trends observed during single turnover reactions with l-Arg and H4T, and suggest that the mutations had a substantial impact on the kinetic properties of these proteins.
NO Synthesis
NO synthesis by BsNOS and mutant proteins was assessed using a three-component reconstitution assay (FLDR, YkuN, and NOS) as described previously (20). Our results indicate that both W66H and W66F are less proficient than wild-type BsNOS for NO synthesis (Fig. 9). In addition, H4B supported NO synthesis to a greater extent compared with H4T (Fig. 9). However, the relative NO synthesis efficiency of each Trp66 mutant compared with wild-type remained unchanged. This suggests that the diminished NO synthesis observed in the Trp66 background is likely caused by alterations in the heme-thiolate environment per se, rather than it being an isolated effect inherent to pterin preference or substrate affinity. The total yield of NO per heme indicates that under these experimental conditions B. subtilis NOS performs ∼20 turnovers, and suggest that in vivo, H4B might be more competent than H4T to drive NO synthesis.
FIGURE 9.

Nitric-oxide synthase activity in a reconstitution assay with the flavoproteins FLDR and YkuN. NO synthesis by wild-type and mutant proteins W66H and W66F was assessed by measuring total nitrite production in a reconstitution assay (FLDR/YkuN/NOS, 2.5:10:1.0), as described previously (20). The values correspond to reactions carried out in the presence of 400 μm NOHA and 100 μm H4B or H4T and 1 mm NAPDH. Values are expressed as mean ± S.D. of triplicate experiments for each condition. Both W66H and W66F displayed a lower yield of NO synthesis compared with wild-type BsNOS, either with H4B or H4T.
Oxidation Rate of the Ferrous-NO complex (kox)
During catalysis, the Fe(III)-NO forms as an immediate product complex and can be reduced by NOS reductase (or the corresponding reductase partner in bacterial NOSs) at rates comparable with that of Fe(III)-NO dissociation (50, 51). To return to the catalytic cycle, the ferrous-NO complex must react with O2. We went on to determine the observed rate constants for reaction of the Fe(II)-NO complex with O2 in BsNOS, W66H, and W66F at half-air saturation ([O2] ∼ 120 μm). The oxidation of anaerobic, pre-formed heme Fe(II)-NO complexes by O2 was investigated by stopped-flow spectroscopy. Spectral data are given in Fig. 10. In all cases, only two species were observed, namely Fe(II)-NO and Fe(III), and therefore the oxidation of Fe(II)-NO by O2 could be best fit to a single-step reaction (Fig. 10). We observed that at neutral pH the Fe(II)-NO complex in W66F (Soret maximum at 439 nm) converted quickly to form a species with a Soret maximum at 415 nm and a marked shoulder at 439 nm. This presumably corresponds to a Fe(II)-NO complex in which the axial Cys has been protonated. This phenomenon is thought to arise by weakening of the Fe-S via disruption of the H-bonding interaction brought about by replacement of the proximal Trp with Phe (52). A similar behavior was observed in the W409F mutant of rat nNOSoxy.4 To minimize this proton-mediated effect, the reactions of W66F were performed at pH 9.5 (note: previous studies in our laboratory indicated that pH has a negligible effect on the oxidation rates of Fe(II)-NO by O2).4 kox values were found to be 0.092, 0.063, and 1.17 s−1 for BsNOS, W66H, and W66F, respectively. Thus, the oxidation rates of Fe(II)-NO in B. subtilis NOS are slow in the range of those reported for nNOSoxy and W409F, respectively (supplemental Table S6).
FIGURE 10.

Kinetics of Fe(II)-NO oxidation by dioxygen. Reaction kinetics of Fe(II)-NO oxidation by O2 are given in panels A–C for BsNOS, W66H, and W66F, respectively. The reactions were started by mixing anaerobic Fe(II)-NO complex with air-saturated buffer in the presence of 2 mm l-Arg, 200 μm H4T, and 1 mm DTT in EPPS buffer (100 mm, pH 7.6) supplemented with 10% glycerol and 125 mm NaCl (BsNOS and W66H) or in CHES buffer (100 mm, pH 9.5) supplemented with 10% glycerol and 125 mm NaCl (W66F). The oxidation rate of Fe(II)-NO in W66F was more than an order of magnitude faster than the observed in BsNOS and W66H.
DISCUSSION
The effect of H-bonding on midpoint potentials and metalloprotein properties is not unprecedented. Chang and Traylor (54) were among the first groups to show that the basicity of the proximal nitrogen in myoglobin (His) influences the affinity of its heme for oxygen and carbon monoxide. A few years later, Jensen and co-workers (55) provided evidence that the NH-S hydrogen bond in ferredoxin and rubredoxin could be important to modulate the midpoint potentials of the Fe-S clusters of the enzyme. The elucidation of the first crystal structures of heme- and iron-sulfur proteins provided strong evidence that a relationship exists between changes in oxidation state of the iron atom and hydrogen bond geometry (56, 57). These early studies indicated that this proximal hydrogen bond could be the link between changes in midpoint potentials and the reactivity of the iron atom (56, 57). Later on, model studies conducted by Ueyama et al. (58) suggested that the NH-S hydrogen bond of ferredoxin model complexes contributed to the redox potential of the Fe-S center. More recent approaches utilizing site-directed mutagenesis confirmed that these relationships hold true for a number of enzyme systems, including peroxidases (59) and cytochrome P450 (46, 60–62).
A great advantage of studying these effects in NOS is that this hydrogen bond is provided by a Trp residue proximal to the heme-thiolate, hence mutagenesis of this Trp by residues with different H-bonding capacities are generally feasible (an exception is the Trp188 → Phe mutant of murine iNOS, which has a defective heme binding (36)). The expression of the stable, dimeric mutants W66H and W66F of B. subtilis NOS permitted a side-by-side characterization of the role of the H-bonding properties of the heme-thiolate in catalysis by a bacterial NOS. We found that replacement of Trp by His or Phe had profound effects on substrate binding affinity, the formation and disappearance of catalytically relevant heme intermediates, the heme midpoint potential, and the proficiency of the enzyme to drive NO synthesis.
Imidazole and Substrate Binding
Replacement of Trp by His or Phe did not disrupt Fe-Cys ligation in the mutant proteins, but had a measurable impact on the electronic properties of the heme and the binding affinity for the substrate l-Arg, and the stable intermediate, NOHA. These findings are in line with a recent spectroscopic study that showed that the proximal H-bond network modulates a number of electronic and structural properties in B. subtilis NOS (34).
Midpoint Potentials
The midpoint potential of wild-type BsNOS (−361 mV) is ∼100 mV lower than that of wild-type mammalian NOSs (−250 to −270 mV) (42). Replacement of the proximal Trp by His increased the heme midpoint potential by 57 mV, whereas a 66-mV decrease was achieved via replacement of Trp with the electron-withdrawing residue Phe. This has important implications for catalysis; lower midpoint potentials pose a thermodynamic challenge for bacterial NOSs, making their heme more difficult to reduce. Consequently, lower heme midpoint potentials limit the repertoire of reductases capable of furnishing electrons into the bacterial NOSs.
Stability of FeO2 during a Single Turnover l-Arg Hydroxylation Reaction in the Presence of H4T
Overall, it appears that the Fe(II)O2 species is destabilized in proteins with lower redox potentials, and vice versa. An elevated heme midpoint potential decreases the driving force for reduction of the Fe(II)-O2 complex, leading to a Fe(II)-O2 that is more stable than the alternate Fe(III)O2− (ferric superoxy) species. Therefore, oxygen activation is disfavored in W66H, due to stabilization of the Fe(II)-O2 complex, which in turn decreases the overall rate of catalysis. This general phenomenon has been referred as to a “thermodynamic trap” by Ost et al. (46). The observation that the Fe(II)O2 species has a prolonged half-life in W66H and a faster disappearance in W66F compared with wild-type BsNOS concurs with the notion that modulation of the heme midpoint potential is crucial for tuning NOS reactivity. Indeed, other heme-thiolate proteins featuring very negative midpoint potentials display an enhanced rate of oxygen activation and reactivity with their substrates (62, 63). Conversion of the FeO2 intermediate was accelerated in reactions driven by reduced pterins versus those performed in the presence of H2B or H2T, suggesting that the pterins may be employed as electron donors for oxygen activation, as it occurs in mammalian NOSs.
Formation of Fe(III)-NO during a Single Turnover NOHA Oxidation Reaction in the Presence of H4T
Our results show that tetrahydrofolate enables the formation of a Fe(III)-NO species downstream of the Fe(II)O2 intermediate during single turnover reactions with NOHA. The Fe(III)-NO species dissociated slowly to form high-spin Fe(III) (resting NOS). In contrast, NOHA single turnover reactions performed in the presence of the oxidized pterin did not support the formation of a Fe-NO complex. Thus, even though BsNOS does not have a well defined binding site for biopterin or folate compared with mammalian NOSs (6, 17), our results suggest the pterin could operate as a source of electrons in oxygen activation reactions. Buildup of the FeNO complex was faster in W66F compared with WT BsNOS and W66H, and so was its transition to form Fe(III) enzyme. These kinetic features are overall unfavorable for NO synthesis, because a rapid formation and a low rate of dissociation of the Fe(III)-NO complex may lead to a larger partitioning of the available NO into the futile NO-bound form (39, 50), thus enhancing the NO dioxygenase mode of the enzyme. A similar phenomenon has been observed in the corresponding mutant of nNOS (W409F) (38, 39) and in nNOS harboring Fe-mesoporphyrin IX (42), both of which possess lower heme midpoint potentials compared with their respective native counterparts.
Product Yield and NO Synthesis
An examination of NOHA formation from l-Arg and citrulline formation from NOHA indicated that the efficiency of product formation is very similar between wild-type BsNOS and the W66 variants (∼0.3 NOHA per heme and ∼0.6 citrulline per heme). This suggests that the mutations per se did not increase the extent of uncoupled O2 reduction compared with wild-type BsNOS. However, the relatively low yields of NOHA production from l-Arg compared with that observed in mammalian NOSs suggests that the first catalytic step of NO synthesis is somewhat less efficient in bacterial NOSs. Both H4B and H4T supported NO synthesis by B. subtilis NOSs in a reconstitution system comprised of FLDR and YkuN. Under our experimental conditions, H4B proved to be a better supporter of NO synthesis compared with H4T. The causes of this effect are presently unknown.
Oxidation of FeNO by Dioxygen
The slow dissociation rates of the Fe(III)-NO complex in B. subtilis NOS increase the tendency of the enzyme to engage into a nonproductive cycle where the Fe(III)-NO is reduced by the reductase partner before it can be released. To avoid this, the Fe(II)-NO complex must react with O2 (kox) to return to the catalytic cycle. We uncovered that oxidation of Fe(II)-NO by O2 is remarkably slow in B. subtilis NOSs compared with the native mammalian NOSs (Table 1). Two other NOSs display comparable kox values, namely D. melanogaster NOS (53) and Geobacillus stearothermophilus NOS (18). An immediate consequence of a slow kox is that a significant proportion of the enzyme may accumulate as Fe(II)-NO during steady-state NO synthesis. The higher kox observed in the W66F mutant is advantageous in that it diminishes buildup of the Fe(II)-NO species during catalysis, perhaps at the expense of a very negative heme midpoint potential.
A summary of the relevant thermodynamic and kinetic parameters for WT and W66 variants is given in Table 2. Despite the fact that the W66F mutant performed certain catalytic transitions (oxygen activation, Fe(III)-NO dissociation (koff), and Fe(II)-NO oxidation (kox)) faster than wild-type BsNOS and the W66H variant, its yield of NO synthesis was the lowest in the reconstitution system with FLDR and YkuN. Nonetheless, wild-type and both Trp66 NOS variants displayed equivalent yields of product formation under single turnover conditions. It is therefore plausible that a negative midpoint potential has dual, counterproductive effects; whereas it is beneficial for the NO synthesis reactions occurring at the heme, it becomes a thermodynamic obstacle by compromising heme reduction by the reductase partner. A similar relationship has been observed for Fe-mesoporphyrin IX-substituted nNOS whose heme midpoint potential is more negative than that of native nNOS (42). In bacterial NOSs, this thermodynamic barrier could be overcome if NOS proteins partnered with the ubiquitous ferredoxins, which are capable of reducing very low-midpoint potential redox centers.
TABLE 2.
Thermodynamic, kinetic, and catalytic parameters for WT and Trp66 variants of B. subtilis NOS
| Protein | Heme midpoint potentialsa | Autooxidation of FeO2 with H2Ta | Arg to NOHAa,b, kcat1 | NOHA to NOa,b, kcat2 | Rate of NOHA formationa,c | Rate of Fe(II)-NO oxidationa,b, kox | Product yield in single
turnover reactionsa,c
(product/heme) |
NO synthesis supported by FLDR/YkuNd (NO/heme, with H4T) | |
|---|---|---|---|---|---|---|---|---|---|
| l-Arg | NOHA | ||||||||
| mV | s−1 | ||||||||
| W66H | −302 | 0.12 | 2.7 | 2.0 | 2.9 | 0.063 | 0.31 | 0.58 | 7.5 |
| Protein | −361 | 0.42 | 15.7 | 4.3 | 4.3 | 0.092 | 0.27 | 0.60 | 9.9 |
| W66F | −427 | 2.8 | 37 | 26 | 17 | 1.17 | 0.23 | 0.59 | 3.9 |
a Determined at 10 °C.
b Results from stopped-flow spectroscopy.
c Results from chemical quench and HPLC.
d Determined at room temperature.
This scenario comprising a low heme midpoint potential (favorable for oxygen activation but thermodynamically uphill toward heme reduction), slow Fe(III)-NO dissociation, and slow Fe(II)-NO oxidation compromises the NO synthesis activity of B. subtilis NOS, which may partly explain earlier and present observations regarding the poor NO synthesis capacity of B. subtilis NOS compared with mammalian NOSs. Furthermore, because most bacterial NOSs lack a covalently attached reductase domain, it is conceivable that the steady-state NO synthesis reaction is largely uncoupled compared with mammalian NOSs.
On the grounds of our observations of how modifying the heme-thiolate environment of B. subtilis NOS affected its catalytic behavior, we propose that low midpoint potentials, slow Fe(III)-NO dissociation, and slow Fe(II)-NO oxidation are key factors underlying the poor NO synthesis output of certain bacterial NOSs. Whether this is an evolutionary trait developed to satisfy perhaps slimmer NO needs in microbial metabolism remains to be investigated. Despite an increasing body of evidence suggesting that bacterial NOSs are well fit for NO synthesis, other functions for these proteins in vivo are also plausible. The remarkable flexibility regarding cofactor usage and their obligated promiscuity in terms of reductase partners may be ultimately, an unusual advantage.
Supplementary Material
Acknowledgments
We thank Dr. Donald W. Jacobsen for providing N5-CH3-H4T and technical support with HPLC experiments. We also thank Dr. Zhihao Yu for assistance with size exclusion chromatography and the generation of Fig. 2, and members of the Stuehr laboratory for general technical support.
This work was supported, in whole or in part, by National Institutes of Health Grants CA53914, GM51491, and HL76491 (to D. J. S.) and American Heart Association Postdoctoral Fellowship Grant 11POST650034 (to L. H.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1–S6 and Figs. S1–S5.
J. Tejero, J. Santolini, and D. J. Stuehr, manuscript in preparation.
- NOSoxy
- nitric-oxide synthase oxygenase domain
- EPPS
- 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid
- H4B
- (6R)-5,6,7,8-tetrahydro-l-biopterin
- H4T
- tetrahydrofolate
- N5-CH3-H4T
- N5-methyltetrahydrofolate
- iNOS
- inducible nitric-oxide synthase
- nNOS
- neuronal nitric-oxide synthase
- FLDR
- flavodoxin reductase
- YkuN
- flavodoxin
- WT
- wild-type
- NOHA
- N-hydroxyarginine
- CHES
- 2-(cyclohexylamino)ethanesulfonic acid.
REFERENCES
- 1. Crane B. R., Arvai A. S., Gachhui R., Wu C., Ghosh D. K., Getzoff E. D., Stuehr D. J., Tainer J. A. (1997) Science 278, 425–431 [DOI] [PubMed] [Google Scholar]
- 2. Crane B. R., Arvai A. S., Ghosh D. K., Wu C., Getzoff E. D., Stuehr D. J., Tainer J. A. (1998) Science 279, 2121–2126 [DOI] [PubMed] [Google Scholar]
- 3. Adak S., Aulak K. S., Stuehr D. J. (2002) J. Biol. Chem. 277, 16167–16171 [DOI] [PubMed] [Google Scholar]
- 4. Adak S., Bilwes A. M., Panda K., Hosfield D., Aulak K. S., McDonald J. F., Tainer J. A., Getzoff E. D., Crane B. R., Stuehr D. J. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 107–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bird L. E., Ren J., Zhang J., Foxwell N., Hawkins A. R., Charles I. G., Stammers D. K. (2002) Structure 10, 1687–1696 [DOI] [PubMed] [Google Scholar]
- 6. Pant K., Bilwes A. M., Adak S., Stuehr D. J., Crane B. R. (2002) Biochemistry 41, 11071–11079 [DOI] [PubMed] [Google Scholar]
- 7. Buddha M. R., Keery K. M., Crane B. R. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 15881–15886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buddha M. R., Tao T., Parry R. J., Crane B. R. (2004) J. Biol. Chem. 279, 49567–49570 [DOI] [PubMed] [Google Scholar]
- 9. Chartier F. J., Couture M. (2004) Biophys. J. 87, 1939–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kers J. A., Wach M. J., Krasnoff S. B., Widom J., Cameron K. D., Bukhalid R. A., Gibson D. M., Crane B. R., Loria R. (2004) Nature 429, 79–82 [DOI] [PubMed] [Google Scholar]
- 11. Wang Z. Q., Wei C. C., Sharma M., Pant K., Crane B. R., Stuehr D. J. (2004) J. Biol. Chem. 279, 19018–19025 [DOI] [PubMed] [Google Scholar]
- 12. Buddha M. R., Crane B. R. (2005) J. Biol. Chem. 280, 31965–31973 [DOI] [PubMed] [Google Scholar]
- 13. Gusarov I., Nudler E. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 13855–13860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pant K., Crane B. R. (2005) J. Mol. Biol. 352, 932–940 [DOI] [PubMed] [Google Scholar]
- 15. Gautier C., Mikula I., Nioche P., Martasek P., Raman C. S., Slama-Schwok A. (2006) Nitric Oxide 15, 312–327 [DOI] [PubMed] [Google Scholar]
- 16. Pant K., Crane B. R. (2006) Biochemistry 45, 2537–2544 [DOI] [PubMed] [Google Scholar]
- 17. Santolini J., Roman M., Stuehr D. J., Mattioli T. A. (2006) Biochemistry 45, 1480–1489 [DOI] [PubMed] [Google Scholar]
- 18. Sudhamsu J., Crane B. R. (2006) J. Biol. Chem. 281, 9623–9632 [DOI] [PubMed] [Google Scholar]
- 19. Chartier F. J., Couture M. (2007) Biochem. J. 401, 235–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang Z. Q., Lawson R. J., Buddha M. R., Wei C. C., Crane B. R., Munro A. W., Stuehr D. J. (2007) J. Biol. Chem. 282, 2196–2202 [DOI] [PubMed] [Google Scholar]
- 21. Crane B. R. (2008) Biochem. Soc. Trans. 36, 1149–1154 [DOI] [PubMed] [Google Scholar]
- 22. Gusarov I., Starodubtseva M., Wang Z. Q., McQuade L., Lippard S. J., Stuehr D. J., Nudler E. (2008) J. Biol. Chem. 283, 13140–13147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Johnson E. G., Sparks J. P., Dzikovski B., Crane B. R., Gibson D. M., Loria R. (2008) Chem. Biol. 15, 43–50 [DOI] [PubMed] [Google Scholar]
- 24. Kabir M., Sudhamsu J., Crane B. R., Yeh S. R., Rousseau D. L. (2008) Biochemistry 47, 12389–12397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Agapie T., Suseno S., Woodward J. J., Stoll S., Britt R. D., Marletta M. A. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 16221–16226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Davydov R., Sudhamsu J., Lees N. S., Crane B. R., Hoffman B. M. (2009) J. Am. Chem. Soc. 131, 14493–14507 [DOI] [PubMed] [Google Scholar]
- 27. Gusarov I., Shatalin K., Starodubtseva M., Nudler E. (2009) Science 325, 1380–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reece S. Y., Woodward J. J., Marletta M. A. (2009) Biochemistry 48, 5483–5491 [DOI] [PubMed] [Google Scholar]
- 29. Sudhamsu J., Crane B. R. (2009) Trends Microbiol. 17, 212–218 [DOI] [PubMed] [Google Scholar]
- 30. Crane B. R., Sudhamsu J., Patel B. A. (2010) Annu. Rev. Biochem. 79, 445–470 [DOI] [PubMed] [Google Scholar]
- 31. Kinloch R. D., Sono M., Sudhamsu J., Crane B. R., Dawson J. H. (2010) J. Inorg. Biochem. 104, 357–364 [DOI] [PubMed] [Google Scholar]
- 32. Maréchal A., Mattioli T. A., Stuehr D. J., Santolini J. (2010) FEBS J. 277, 3963–3973 [DOI] [PubMed] [Google Scholar]
- 33. Montgomery H. J., Dupont A. L., Leivo H. E., Guillemette J. G. (2010) Biochem. Res. Int. 2010, 489892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brunel A., Wilson A., Henry L., Dorlet P., Santolini J. (2011) J. Biol. Chem. 286, 11997–12005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tejero J., Biswas A., Wang Z. Q., Page R. C., Haque M. M., Hemann C., Zweier J. L., Misra S., Stuehr D. J. (2008) J. Biol. Chem. 283, 33498–33507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wilson D. J., Rafferty S. P. (2001) Biochem. Biophys. Res. Commun. 287, 126–129 [DOI] [PubMed] [Google Scholar]
- 37. Adak S., Crooks C., Wang Q., Crane B. R., Tainer J. A., Getzoff E. D., Stuehr D. J. (1999) J. Biol. Chem. 274, 26907–26911 [DOI] [PubMed] [Google Scholar]
- 38. Adak S., Stuehr D. J. (2001) J. Inorg. Biochem. 83, 301–308 [DOI] [PubMed] [Google Scholar]
- 39. Adak S., Wang Q., Stuehr D. J. (2000) J. Biol. Chem. 275, 17434–17439 [DOI] [PubMed] [Google Scholar]
- 40. Wang Z. Q., Wei C. C., Stuehr D. J. (2002) J. Biol. Chem. 277, 12830–12837 [DOI] [PubMed] [Google Scholar]
- 41. de Montigny P., Stobaugh J. F., Givens R. S., Carlson R. G., Srinivasachar K., Sternson L. A., Higuchi T. (1987) Anal. Chem. 59, 1096–1101 [DOI] [PubMed] [Google Scholar]
- 42. Tejero J., Biswas A., Haque M. M., Wang Z. Q., Hemann C., Varnado C. L., Novince Z., Hille R., Goodwin D. C., Stuehr D. J. (2011) Biochem. J. 433, 163–174 [DOI] [PubMed] [Google Scholar]
- 43. Griess P. (1879) Ber. Deutsch. Chem. Ges. 12, 426–428 [Google Scholar]
- 44. Hanson L. K. (1979) Intl. J. Quantum. Chem. 16, 73–87 [Google Scholar]
- 45. Lang J., Driscoll D., Gélinas S., Rafferty S. P., Couture M. (2009) J. Inorg. Biochem. 103, 1102–1112 [DOI] [PubMed] [Google Scholar]
- 46. Ost T. W., Miles C. S., Munro A. W., Murdoch J., Reid G. A., Chapman S. K. (2001) Biochemistry 40, 13421–13429 [DOI] [PubMed] [Google Scholar]
- 47. Chartier F. J., Couture M. (2007) J. Biol. Chem. 282, 20877–20886 [DOI] [PubMed] [Google Scholar]
- 48. Ost T. W., Munro A. W., Mowat C. G., Taylor P. R., Pesseguiero A., Fulco A. J., Cho A. K., Cheesman M. A., Walkinshaw M. D., Chapman S. K. (2001) Biochemistry 40, 13430–13438 [DOI] [PubMed] [Google Scholar]
- 49. Wei C. C., Wang Z. Q., Arvai A. S., Hemann C., Hille R., Getzoff E. D., Stuehr D. J. (2003) Biochemistry 42, 1969–1977 [DOI] [PubMed] [Google Scholar]
- 50. Santolini J., Adak S., Curran C. M., Stuehr D. J. (2001) J. Biol. Chem. 276, 1233–1243 [DOI] [PubMed] [Google Scholar]
- 51. Santolini J., Meade A. L., Stuehr D. J. (2001) J. Biol. Chem. 276, 48887–48898 [DOI] [PubMed] [Google Scholar]
- 52. Couture M., Adak S., Stuehr D. J., Rousseau D. L. (2001) J. Biol. Chem. 276, 38280–38288 [DOI] [PubMed] [Google Scholar]
- 53. Ray S. S., Tejero J., Wang Z. Q., Dutta T., Bhattacharjee A., Regulski M., Tully T., Ghosh S., Stuehr D. J. (2007) Biochemistry 46, 11857–11864 [DOI] [PubMed] [Google Scholar]
- 54. Chang C. K., Traylor T. G. (1973) J. Am. Chem. Soc. 95, 8477–8479 [DOI] [PubMed] [Google Scholar]
- 55. Adman E., Watenpaugh K. D., Jensen L. H. (1975) Proc. Natl. Acad. Sci. U.S.A. 72, 4854–4858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Carter C. W., Jr. (1977) J. Biol. Chem. 252, 7802–7811 [PubMed] [Google Scholar]
- 57. Valentine J. S., Sheridan R. P., Allen L. C., Kahn P. C. (1979) Proc. Natl. Acad. Sci. U.S.A. 76, 1009–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ueyama N., Yamada Y., Okamura Ta., Kimura S., Nakamura A. (1996) Inorg. Chem. 35, 6473–6484 [DOI] [PubMed] [Google Scholar]
- 59. Choudhury K., Sundaramoorthy M., Hickman A., Yonetani T., Woehl E., Dunn M. F., Poulos T. L. (1994) J. Biol. Chem. 269, 20239–20249 [PubMed] [Google Scholar]
- 60. Clark J. P., Miles C. S., Mowat C. G., Walkinshaw M. D., Reid G. A., Daff S. N., Chapman S. K. (2006) J. Inorg. Biochem. 100, 1075–1090 [DOI] [PubMed] [Google Scholar]
- 61. Ost T. W., Clark J., Mowat C. G., Miles C. S., Walkinshaw M. D., Reid G. A., Chapman S. K., Daff S. (2003) J. Am. Chem. Soc. 125, 15010–15020 [DOI] [PubMed] [Google Scholar]
- 62. Yoshioka S., Takahashi S., Ishimori K., Morishima I. (2000) J. Inorg. Biochem. 81, 141–151 [DOI] [PubMed] [Google Scholar]
- 63. Matsumura H., Wakatabi M., Omi S., Ohtaki A., Nakamura N., Yohda M., Ohno H. (2008) Biochemistry 47, 4834–4842 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.