

Abstract
Maf1 is negative regulator of RNA polymerase III in yeast. We observed high levels of both primary transcript and end-matured, intron-containing pre-tRNAs in the maf1Δ strain. This pre-tRNA accumulation could be overcome by transcription inhibition, arguing against a direct role of Maf1 in tRNA maturation and suggesting saturation of processing machinery by the increased amounts of primary transcripts. Saturation of the tRNA exportin, Los1, is one reason why end-matured intron-containing pre-tRNAs accumulate in maf1Δ cells. However, it is likely possible that other components of the processing pathway are also limiting when tRNA transcription is increased. According to our model, Maf1-mediated transcription control and nuclear export by Los1 are two major stages of tRNA biosynthesis that are regulated by environmental conditions in a coordinated manner.
Keywords: RNA Polymerase III, RNA Transport, Transcription Repressor, Transfer RNA (tRNA), Yeast, Maf1, tRNA Nuclear Export
Introduction
The existence of three RNA polymerases (Pol)2 is documented for all investigated eukaryotes. Pol I synthesizes ribosomal RNA (rRNA), Pol II produces mainly mRNAs, and Pol III generates tRNAs, 5 S rRNA, and other small noncoding RNAs. Each major category of nuclear-encoded RNA has unique processing, nucleus/cytoplasmic dynamics, and decay pathways. Generally, the regulation of individual categories of RNA occurs by mechanisms that are not shared with the other categories, probably providing a selective advantage by separately controlling mRNA, rRNA, and tRNA synthesis in response to environmental changes. Recent studies have uncovered relationships between RNA transcription and RNA maturation. Co-transcriptional processing occurs during rRNA synthesis and most mRNAs (1–5), but it remains unknown whether there is co-regulation of Pol III transcription with any of the tRNA processing steps.
tRNA transcription is controlled by a general negative regulator, Maf1, conserved from yeast to man (6). Maf1 mediates several signaling pathways by repressing Pol III transcription in response to diverse stresses such as nutrient deprivation, rapamycin treatment, secretory defects, or DNA damage (7, 8). Maf1 is the only negative Pol III regulator known in yeast; it is regulated by both PKA and TOR signaling pathways (for review see Refs. 9 and 10). Under favorable growth conditions, Maf1 is phosphorylated by PKA kinase, CK2 kinase, TORC1-dependent Sch9 kinase, and TORC1 kinase itself (11–13). In conditions that slow growth, Maf1 is dephosphorylated by PP2A phosphatase. The dephosphorylated form of Maf1 interacts with the Pol III complex and is thereby active as a repressor (8). Both phosphorylation and dephosphorylation of Maf1 may occur inside the nucleus. Phosphorylated Maf1 is exported to cytoplasm by the Msn5 carrier, but nuclear export is not fundamental for appropriate regulation of Pol III (14).
Because Maf1 is a negative regulator of Pol III, its absence causes an increase in tRNA transcripts in yeast and mammals (6, 15). Elevated activity of mammalian Pol III is a recurring feature of mouse and human tumors (15). It has been shown that overexpression of Maf1 suppresses anchorage-independent growth of U87 glioblastoma cells (16). This suggests that Maf1 is a potential tumor suppressor. Because of overexpression of the Pol III product, tRNAiMet, is sufficient to drive tumorigenesis in mice (17), it is therefore of great interest to better understand the involvement of Maf1 in global regulation of tRNA biosynthesis. We employed yeast as a powerful tool to study this regulation.
Inactivation of the MAF1 gene in yeast leads also to increased translation fidelity (18). An explanation for how an excess of tRNA in maf1Δ affects translation remains unclear. The simplest hypothesis is that tRNAs transcribed in maf1Δ cells are incorrectly or incompletely processed, or they fail to be appropriately delivered to ribosomes.
tRNA processing involves multiple steps that occur in yeast at different subcellular locations (for review see Ref. 19). Initial transcripts are extended both at 5′ and 3′ termini and may contain introns. The 5′ leader is removed from initial transcripts by RNase P endonuclease located in the nucleolus. Processing of the 5′ leader generally precedes trimming of 3′ trailer, which is followed by CCA addition at 3′ termini. End-processed tRNAs are transported to the cytoplasm by the exportins Los1 and/or Msn5 (19–23). In yeast, 10 tRNA families are encoded by intron-containing genes. Introns are removed from end-processed transcripts by the splicing machinery consisting of the heterotetrameric tRNA splicing endonuclease located at the outer surface of mitochondria and the cytoplasmic pools of tRNA ligase and 2′ phosphotransferase (24–26). tRNAs are modified, and modifications are added throughout tRNA processing, both in the nucleus and in the cytoplasm. Thus, unlike pre-mRNA processing, which is largely, if not completely, co-transcriptional, pre-tRNA processing occurs in multiple subcellular locations.
To assess whether tRNA transcription is coordinated with any part of the post-transcriptional tRNA biosynthesis process, we explored the possibility that perturbation of Maf1-mediated control of transcription could affect tRNA processing. Here we show accumulation of both the initial transcripts and end-processed, intron-containing tRNA precursors in the absence of Maf1, reflecting the imbalance between the rate of tRNA synthesis and efficiency of its maturation. These observations indicate that Maf1-dependent Pol III regulation plays an important role in synchronization of transcription with post-transcriptional events. Moreover, we have shown that saturation of Los1-dependent nuclear export of intron-containing tRNA precursors is responsible for the accumulation of pre-tRNA in maf1Δ nuclei.
EXPERIMENTAL PROCEDURES
Strains, Media, and Plasmids
Yeast strains BY4742 (MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0) and isogenic BY4742 maf1Δ (BY4742 maf1::KanMX4) were purchased from Euroscarf. The MSN5 locus in BY4742 was replaced by the natMX4 deletion cassette amplified from pAG25 plasmid (Euroscarf) creating BY4742 msn5Δ::NATR. maf1Δ msn5Δ was obtained by genetic cross. W303 maf1Δ (W303 maf1::KanMX4) was created in W303a (MATα ade2-1 leu2-3 ura3-1 his3-11 trp1-1 can1-100; Euroscarf). sen2-42 mutant (MAT a sen2Δ::NATR leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15) (CEN6-URA3 sen2-42) was kindly provided by Dr. Yoshihisa. To express the Los1-Myc fusion protein, a cassette encoding 13 repeats of the myc epitope an ADH1 terminator, and a kanMX6 selectable marker was amplified by PCR with target gene-specific primers from pF6a-13myc-kanMx6 plasmid (27) and integrated immediately upstream of the stop codon of LOS1 genomic locus in BY4742 strain. The MAF1 gene was subsequently deleted in LOS1-Myc strain by a URA3 cassette (amplified from MB159–4D maf1Δ::URA3) (18). The sequences of the primers used are available upon request.
Yeast media contained 1% yeast extract, 2% peptone, and 2% glucose (YPD) or 2% glycerol (YPGly). The minimal medium (SC-ura) containing 0.67% yeast nitrogen base and 2% glucose or glycerol was supplemented with all necessary requirements without uracil (28). Solid media contained 2% agar. All of the reagents were from Difco. Temperature-sensitive cultures of sen2-42 were transferred to nonpermissive temperatures at 37 °C for 2 h before RNA extraction. Induction of yeast cultures was done by galactose addition to 2% concentration and further incubation for 2 h.
The plasmids used in this study were pFL44L (URA3, 2μ), YEp24 (URA3, 2μ), pLR233 (pFL44L-RPR1) (29), and YEpLOS1 (YEp24-LOS1) (30). The plasmids encoding inducible tRNA splicing endonuclease subunits and tRNA ligase were created in Escherichia coli strain XL1 blue using pRS412, pRS413, pRS414, pRS415, and pRS416 (American Type Culture Collection) as backbones. Plasmids contained a GAL1 promoter to allow inducible gene expression and GFP, both derived from pGP54a plasmid by restriction digest with XhoI and EagI, with the insertion of a stop codon after the GFP gene. Each digestion product was ligated into the pRS backbones. Sequences encoding splicing endonuclease subunit proteins and tRNA ligase were PCR-amplified using high fidelity Taq polymerase (Invitrogen). Each PCR product was ligated into the pGEM-T (Promega) to obtain high quantities of the product, digested with XmaI and EcoRI, and ligated into the appropriate vectors. Vectors encoding only the galactose-inducible GFP were used as controls. All of the plasmid sequences were confirmed by DNA sequencing. Strains were transformed with all five inducible plasmids expressing tRNA splicing endonuclease subunits and tRNA ligase or with five control plasmids. Selection of yeast carrying the plasmids was accomplished by auxotrophic markers on the plasmids.
Northern Analysis
Cells (30 ml of liquid culture; grown to 0.6 A600 nm) were harvested by centrifugation and resuspended in 50 mm sodium acetate, pH 5.3, 10 mm EDTA. Total RNA was isolated by heating and freezing the cells in the presence of SDS and phenol as described previously (31). For the experiment presented in Fig. 5, small RNAs were extracted as described previously (32). RNA (10 μg/sample) was resolved by electrophoresis and transferred from gel to Hybond N+ membrane (Amersham Biosciences) with 0.5 × TBE by electroblotting and cross-linked by UV radiation (1200 mJ/cm2) (33). The membrane was prehybridized in phosphate buffer (7% SDS, 0.5 m sodium phosphate, pH 7.2, 1 mm EDTA, pH 7.0, 1% BSA) and hybridized at 37 °C in the same solution with oligonucleotide probes labeled with [γ-32P]ATP and T4 polynucleotide kinase (New England Biolabs). Following probes were employed: tRNALeu(CAA) intron, 5′-TATTCCCACAGTTAACTGCGGTCA-3; mature tRNALeu(CAA), 5′-CGATACCTGAGCTTGAATCAGG-3′; tRNALeu(CAA), 5′-GACCGCTCGGCCAAACAAC-3 (recognizing precursors as well as the mature form); tRNAPhe(GAA), 5′-GCGCTCTCCCAACTGAG CT-3′ (recognizing precursors as well as the mature form); tRNAIle(UAU), 5′-GTGGGGATTGAACCCACGACGGTCGCGTTATAAGCACGAAGCTCTAACCACTGGCTACA-3′; tRNAiMet(CAU), 5′-GTTTCGATCCGAGGACATCAG 3′; tRNAHis(GUG), 5′-GCCATCTCCTAGAATCGAAC-3′; tRNAGly(GCC), 5′-AAGCCCGGAATCGAACCGG-3′; 5.8 S rRNA, 5′-GCGTTGTTCATCGATGC-3′; U3 snoRNA, 5′-GGATTGCGGACCAAGCTAA-3′; and RPR1, 5′-GGCTCCACTGTGTTCCACCGAATTTCCCAC-3′.
FIGURE 5.

Cytoplasmic splicing endonuclease is not limiting for processing of intron-containing pre-tRNA that accumulate in maf1Δ cells. Wild type W303 (wt), W303 maf1Δ (maf1Δ), or control W303 sen2-24 (sen2-24) strains were transformed with plasmids bearing cytoplasmic tRNA splicing endonuclease genes under control of galactose-inducible promoter. As a control, we used the same strains transformed with the empty vectors. Transformants were grown in minimal glucose medium supplemented as required. + indicates induction by galactose addition to final concentration 2% for 2 h; − indicates no induction; 23 and 37 °C are the temperature conditions. Northern hybridization was performed with probes specific to tRNAIle(UAU). Lane 1, maf1Δ with induced plasmids bearing cytoplasmic tRNA splicing endonuclease genes; lane 2, maf1Δ with induced empty vectors; lane 3, maf1Δ with uninduced plasmids bearing cytoplasmic tRNA splicing endonuclease genes; lane 4, maf1Δ with uninduced empty vectors; lane 5, wild type with induced plasmids bearing cytoplasmic tRNA splicing endonuclease genes; lane 6, wild type with induced empty vectors; lane 7, wild type with uninduced plasmids bearing cytoplasmic tRNA splicing endonuclease genes; lane 8, wild type with uninduced empty vectors; lane 9, sen2-42 with uninduced plasmids bearing cytoplasmic tRNA splicing endonuclease genes at 23 °C; lane 10, sen2-42 with uninduced plasmids bearing cytoplasmic tRNA splicing endonucleases genes at 37 °C; lane 11, sen2-42 with induced plasmids bearing cytoplasmic tRNA splicing endonuclease genes at 23 °C; lane 12, sen2-42 with induced plasmids bearing cytoplasmic tRNA splicing endonuclease genes at 37 °C. RNAs for lanes 9–12 were extracted and probed at the same times as those in lanes 1–8 but were run on a separate gel.
After hybridization, blots were washed 2 × 10 min with 1 × SSC, 1% SDS, and 3 × 10 min with 0.5 × SSC, 0.1% SDS at 37 °C and exposed to film or to a phosphorimaging screen (Fujifilm). RNA was quantified using FLA-7000 PhosphoImager (Fujifilm). Band intensities were quantified using Multi Gauge V3.0 Software.
Fluorescence in Situ Hybridization
The cells were grown at 30 °C to early log phase in YPD, shifted to glycerol medium (YPGly), and cultivated at 37 °C for the indicated period of time. The cells were prefixed, washed, and converted to spheroplasts as previously described (34). Spheroplasts were attached to the poly-l-lysine-coated slides. Prehybridization was conducted at 39 °C for 3 h in a buffer containing 10% dextran sulfate, 2× SSC, 1× Denhardt's solution, 0.1% BSA, 0.1 mg/ml E. coli tRNA, 1 mg/ml salmon sperm DNA, and 0.8 unit/μl RNasin. Hybridization was conducted at 43 °C overnight in the same buffer with the addition of Cy3-labeled probes in concentration of 0.1 pmol/μl. The sequence of probes specific for pre-tRNALeu(CAA) was 5′-TATTCCCACAGTTAACTGCGGTCA-3. The cells were washed three times with 2× SSC at 50 °C, once with 1× SSC at 50 °C, and two times with 1× SSC at room temperature. The cell nuclei were stained with 0.1 μg/ml DAPI. The samples were viewed using a Zeiss microscope, and the images were captured through a 100× objective.
Immunofluorescence
The cells were cultivated to early log phase in YPD at 30 °C. Immunofluorescence microscopy was performed according to a standard protocol (35). The primary anti-Myc antibody (Millipore) at a 1:500 dilution and the secondary anti-mouse Cy3 conjugated antibody (Millipore) at a 1:250 dilution were used for detection of Los1-Myc fusion protein. The cell nuclei were stained with 0.1 μg/ml DAPI. The samples were viewed using a Zeiss microscope, and images were captured through a 100× objective.
Quantitative β-Galactosidase Activity Assay
Cells transformed with p180 reporter plasmid (pGCN4-lacZ URA3 CEN) (36) and grown overnight in SC-ura were inoculated to YPD, incubated to early log phase, and split for two parts. One part of cells was harvested, and the second part was shifted to YPGly and cultivated for 2 h at 37 °C. Protein extracts were prepared from cells, and β-galactosidase activity was measured as described previously (37).
Western Analysis
The cells were broken, and the proteins were isolated as described previously (14). Protein extracts were separated on 10% or 4–15% gradient SDS-PAGE and transferred to PVDF membrane by electroblotting. The membrane was blocked for 30 min in 10 mm Tris, 150 mm NaCl, and 0.05% Tween 20 containing 5% fat-free dry milk and then incubated with the appropriate antibodies. The following antibodies were used: rabbit Gcn4-specific antibody (Santa Cruz Biotechnology) at a 1:1000 dilution, mouse anti-Act1 antibody (Millipore) at a 1:2000, mouse anti-Myc tag antibody (clone 9E10, Millipore) at a 1:5000 dilution; rabbit antibodies specific for phosphorylated Ser-51 in eIF2α (Invitrogen) at 1:2000, and mouse anti-Vma1 antibody (Molecular Probes) at a 1:5000. To detect Act1, Los1-Myc, and Vma1, the membranes were incubated with the secondary HRP-conjugated anti-mouse antibody (DAKO). Gcn4 and eIF2α-P were detected by the use of secondary HRP-conjugated anti-rabbit antibody (DAKO). The proteins were visualized by chemiluminescence using the ECL detection kit (Millipore).
RESULTS
Inactivation of Maf1 Results in Accumulation of tRNA Precursors
Despite its role as the sole negative regulator of Pol III transcription, Maf1 is not essential in yeast. The growth defect caused by maf1Δ is observed when cells are propagated on nonfermentable carbon sources at elevated temperatures (38). Initially, tRNA levels in maf1Δ cells incubated at nonpermissive conditions were monitored by EtBr staining of RNA extracted from wild type and mutant cells (6). The increased levels were verified by analyses employing microarrays that contained sequences of all of the different yeast tRNA families and other genes transcribed by the Pol III (33). Comparison of Pol III transcribed RNAs from maf1Δ and wild type cells grown in medium with a nonfermentable carbon source shifted to an elevated temperature by microarray analysis showed that the levels of RNAs were, on average, ∼4-fold higher in the mutant cells. Importantly, not all tRNAs were elevated in maf1Δ to the same extent because they varied from over 10-fold to less than 2-fold different in abundance. Surprisingly, most of the tRNAs encoded by intron-containing tRNA genes were more elevated than were the tRNAs encoded by intron-lacking genes. For example, intron-containing tRNAPhe(GAA) was increased over 11-fold, tRNATrp(CCA) was increased over 10-fold, tRNALeu(CAA) was increased nearly 9-fold, but tRNAIle(UAU) was increased only 2.5-fold (33).
Because both pre-tRNAs and mature tRNAs hybridize to the microarray and the absence of Maf1 seemed to preferentially affect tRNA products encoded by intron-containing genes, we wondered whether there might be a defect in splicing of these transcripts. To evaluate tRNA processing, we employed Northern hybridization using tRNA-specific oligonucleotide probes. The levels of U3 snoRNA or 5.8 S rRNA were comparable in maf1Δ and wild type cells and thus serve as internal controls. First, we monitored the processing of two representative intron-containing tRNAs, tRNAPhe(GAA) and tRNALeu(CAA) (Fig. 1, A and B). Both unprocessed initial transcripts (designated —▭–▭—) and end-matured intron-containing pre-tRNAs (designated ▭–▭) were increased in maf1Δ mutant relative to wild type cells. It is noteworthy that the relative amount of tRNA precursors varied depending on the carbon source utilized. Quantification of the Northern blots revealed that maf1Δ cells shifted to glycerol medium have over 10-fold elevated levels of tRNAPhe initial transcript over its level in wild type cells cultivated under standard conditions in glucose medium. End-processed intron-containing pre-tRNAPhe was also elevated, ∼2.5-fold, over wild type. Lower fold of increase was detected in maf1Δ for both tRNALeu precursors; however, the numbers varied depending on the probe specificity (Fig. 1, compare A and B). In contrast, the levels of spliced mature forms were only slightly affected by Maf1. It appears that, in the absence of Maf1, tRNA processing lags behind the elevated rate of Pol III transcription. Note that under restrictive conditions in glycerol medium, removal of pre-tRNA termini in maf1Δ lags behind more than in favorable conditions in glucose.
FIGURE 1.

maf1Δ cells accumulate tRNA precursors. maf1Δ and wild type (wt) control strain BY4742 were grown to mid-log phase in rich glucose medium (YPD), and one-half of each culture was shifted to rich medium containing glycerol (YPGly) and cultivated for 2 h at 37 °C. A–C, Northern analysis of intron-containing tRNAPhe(GAA) and tRNALeu(CAA) using probes that recognize both pre-tRNA and mature tRNA (A); intron-containing tRNALeu(CAA) using distinct probes, specific to precursors or mature tRNA (B); and intron-less tRNAiMet(CAU) and tRNAHis(GUG) (C). The sequences of tRNA-specific oligonucleotide probes are specified under “Experimental Procedures.” The positions of initial transcripts, end-processed intron-containing pre-tRNA, and mature tRNA are indicated. Pre-tRNAs and mature tRNAs were quantified. The bars represent levels for primary transcript retaining 5′ and 3′ termini, intron-containing precursor, and mature form normalized to loading control. The fold of change in maf1Δ cells cultivated under indicated conditions was calculated relative to expression in control strain (wt) at standard conditions (YPD). Standard deviation was calculated on the basis of three independent experiments.
tRNAs encoded by genes lacking introns are also processed at 5′ and 3′ termini in the nucleus Thus, we monitored by Northern hybridization the effect of Maf1 on end processing for representative intron-free tRNAs: tRNAiMet(CAU), tRNAHis(GUG), tRNAGly(GCC), and tRNAVal(AAC) Initial transcripts of tRNAHis (Fig. 1C), tRNAGly, and tRNAVal (not shown) were hardly detectable by Northern blotting (even upon prolonged exposure), indicating efficient processing of 5′ and 3′ extensions. However, pre-tRNAiMet precursors can be detected, and they are significantly increased in maf1Δ cells (Fig. 1C). The results show that maturation of both categories of tRNAs encoded by intron-containing genes and intron-less tRNAs are defective in the absence of Maf1. It is noteworthy that maf1Δ cells shifted to glycerol medium have ∼3-fold elevated levels of mature tRNAiMet and tRNAHis (Fig. 1C).
Accumulation of Pre-tRNA Is Correlated with Loss of Nuclear Maf1 Function
Maf1 shuttles between nucleus and cytoplasm. The function of Maf1 in cytoplasm is unknown, but one possibility is that cytoplasmic Maf1 could be directly involved in tRNA splicing. To investigate this possibility, we sought to examine pre-tRNA accumulation in msn5Δ cells, in which localization of Maf1 is limited to the nucleus because of a lack of Maf1-specific exportin (14).
As assessed by Northern analyses, retention of Maf1 in the nucleus in msn5Δ cells resulted in a low level of pre-tRNALeu, especially when cells were cultivated under restrictive conditions (Fig. 2). Because Msn5 is responsible for nuclear export of other proteins beside Maf1, theoretically their nuclear localization could indirectly cause increased tRNA processing in msn5Δ cells. We excluded this option by analyzing maf1Δ msn5Δ double mutant. The results showed that the double mutant accumulated elevated levels of both initial transcript and intron-containing end-matured precursor (Fig. 2). These results indicate that accumulation of pre-tRNA is correlated with a loss of nuclear Maf1 function.
FIGURE 2.

pre-tRNA levels depend on nuclear Maf1. A, wild type BY4742 (wt), maf1Δ, msn5Δ, and maf1Δ msn5Δ cells were grown to mid-log phase in rich glucose medium (YPD), and one-half of each culture was shifted to rich medium containing glycerol (YPGly) and cultivated for 2 h at 37 °C. Northern hybridization was performed with probes specific to pre-tRNALeu and mature tRNALeu(CAA). 5.8 S rRNA served as a loading control. B, quantification of primary transcripts (–▭–▭–) and end-matured, intron-containing pre-tRNALeu(CAA) (▭–▭) levels. The fold of change in each strain cultivated under indicated conditions was calculated relative to expression in control strain (wild type) at the same growth conditions. Standard deviation was calculated on the basis of three independent experiments.
Pre-tRNA Accumulation in maf1Δ Cells Is Due to Elevated Pol III Transcription
The increased pre-tRNA levels in maf1Δ cells raised the question as to how a lack of nuclear Maf1 affected tRNA maturation. In principle, Maf1 could function dually in the nucleus by regulating tRNA transcription by Pol III and by regulating pre-tRNA processing. Alternatively, the observed pre-tRNA accumulation could be a secondary effect caused by high levels of primary tRNA transcripts saturating the processing and/or the nuclear export machinery.
If the pre-tRNA processing defects in maf1Δ cells were caused by saturation of the processing or nuclear export machinery, they should be suppressed by a reduction of Pol III transcripts. To reduce de novo synthesis of tRNA, the cells were treated with thiolutin, which inhibits RNA polymerases, including Pol III (39). As expected, the thiolutin treatment resulted in a gradual disappearance of pre-tRNALeu in wild type cells (Fig. 3A) with no detectable changes of mature tRNALeu levels (supplemental Fig. S1). Both primary transcripts and intron-containing end-matured tRNALeu precursors, initially elevated in maf1Δ cells, were reduced upon thiolutin treatment. Finally, after 2 h of incubation in the presence of thiolutin, pre-tRNALeu transcripts disappeared in maf1Δ cells similarly as in the control wild type (Fig. 3B). We thus concluded that tRNA precursors accumulate in maf1Δ cells because their transcription is elevated and that maturation simply lags behind. Consequently, we focused our further study on the identification of the rate-limiting steps in tRNA maturation.
FIGURE 3.

Transcription inhibition prevents accumulation of pre-tRNA in maf1Δ cells. maf1Δ and wild type cells were grown to mid-log phase in rich glucose medium (YPD) and then were treated with 5 μg/ml thiolutin. The cells were harvested just before (time 0) and after thiolutin treatment at the indicated time points. A, Northern analysis was performed with probe recognizing pre-tRNALeu(CAA). The positions of primary transcript and end-matured, intron-containing precursor were indicated. U3 snoRNA served as a loading control. wt, wild type. B, quantification of pre-tRNALeu(CAA) levels in wild type (solid lines and closed squares) and maf1Δ (dashed lines and open squares). The curves show the ratio of primary transcripts (–▭–▭–) and end-matured, intron-containing pre-tRNALeu(CAA) (▭–▭) at each time point relative to its levels in the wild type strain immediately before thiolutin addition (time 0). Each value itself was first normalized to U3 snoRNA. Standard deviation was calculated on the basis of three independent experiments.
Saturation of RNase P RNA Is Not a Reason for Pre-tRNA Accumulation in the Absence of Maf1
First, we explored the possibility that accumulation of untrimmed initial transcripts in maf1Δ cells is due to saturation of RNase P, which removes the 5′ leader preceding removal of the 3′ trailer (40). RPR1, the gene encoding the RNA subunit of RNase P, is transcribed by Pol III (41). It is known that Maf1 does not have the same effects on all Pol III genes. In fact, RPR1 RNA levels are only slightly enhanced in maf1Δ cells compared with the substantially increased tRNA pool (33). Therefore, it is possible that the amount of RPR1 required for RNase P activity could be disproportionately low in maf1Δ cells, causing inefficient processing of 5′ termini. To increase levels of RPR1 RNA, we transformed wild type and maf1Δ cells with a multicopy plasmid carrying RPR1. The resulting transformants showed increased levels of RPR1 precursor and mature forms as observed previously by others (42) (Fig. 4A). RPR1 is a limiting factor for 5′ end removal in wild type cells because RPR1 overexpression resulted in a decrease of the initial 5′-extended tRNALeu transcript (Fig. 4B). However, RPR1 overexpression did not reduce the level of 5′-extended pre-tRNA transcripts in maf1Δ cells (Fig. 4B). Thus, the elevated level of 5′-extended pre-tRNA transcripts in maf1Δ cells is unlikely to be due to limiting quantities of RPR1. In contrast, a shortage of RPR1 RNA was previously reported as the reason for initial transcript accumulation in a mutant defective in Pol III initiation (43). Although the elevated level of 5′-extended pre-tRNA transcripts in maf1Δ cells is unlikely to be due to limiting quantities of RPR1, it still could result from limiting one or more of the nine RNase P protein subunits.
FIGURE 4.
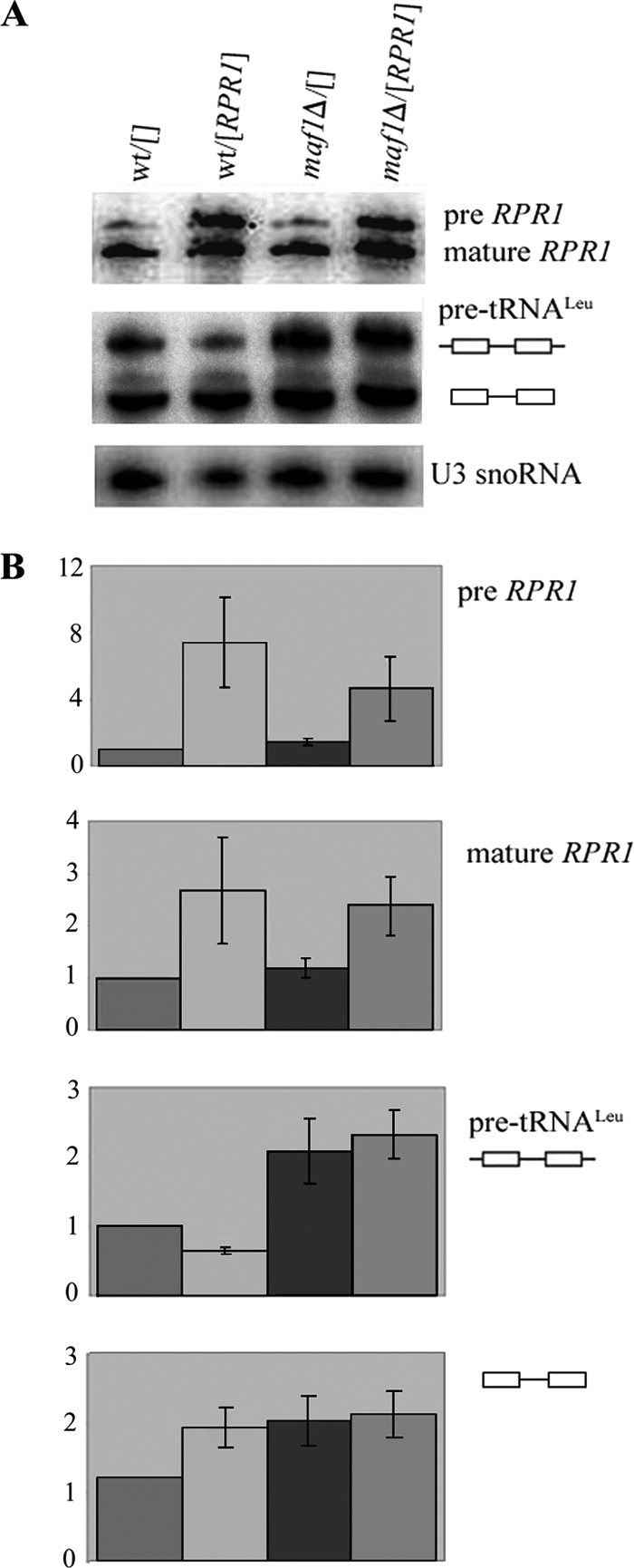
Overexpression of RPR1 gene has no effect on pre-tRNA accumulation in maf1Δ cells. Wild type BY4742 (wt) and maf1Δ cells transformed with control plasmid pFL44L (designated []) or overexpressing RPR1 ([RPR1]) were grown in SC-ura medium containing 2% glucose, shifted to SC-ura medium containing 2% glycerol, and cultivated for 2 h at 37 °C. A, Northern hybridization was performed with probes specific to RNA RPR1, pre-tRNALeu(CAA), or U3 snoRNA (used as a control). B, bars indicate the ratio of initial transcript and end-matured, intron-containing pre-tRNALeu(CAA) levels relative to their levels in wild type cells bearing control plasmid pFL44L (designated wt/[]). The levels of RPR1 precursor and its mature form were quantified. The bars represent the ratio of both RPR1 transcripts relative to their levels in wild type cells bearing control plasmid pFL44L (designated wt/[]). Each value itself was first normalized to U3 snoRNA. Standard deviation was calculated on the basis of three independent experiments.
Cytoplasmic Sen Complex Is Not the Limiting Factor for Processing of Intron-containing Pre-tRNA Accumulating in maf1Δ Cells
In Saccharomyces cerevisiae, unlike in vertebrates, the intron-containing tRNA precursors are exported from the nucleus, and pre-tRNA splicing occurs in the cytoplasm at mitochondrial membrane (25). Significantly, strains lacking Maf1 have growth defects in medium with a nonfermentable carbon source, indicating a defect in mitochondrial function. This raised the possibility that accumulation of intron-containing tRNA in maf1Δ may be due to deficiency of mitochondrially localized tRNA splicing endonuclease.
To address whether end-matured intron-containing pre-tRNAs might accumulate in maf1Δ cells because of the saturation of cytoplasmic splicing endonuclease, we expressed galactose-inducible splicing endonuclease in wild type and maf1Δ cells. We generated strains bearing each of the four genes specifying GFP-tagged splicing endonuclease subunits, SEN2, SEN15, SEN34, and SEN54, as well as the tRNA ligase TRL1 on plasmids under galactose regulation. As anticipated, the tRNA splicing components are appropriately located in wild type and maf1Δ cells, Sen subunits to mitochondria, and ligase to the cytoplasm (not shown). To ensure that splicing endonuclease proteins being expressed from the plasmids were functional, we transformed plasmids expressing inducible splicing endonuclease subunits into a temperature-sensitive splicing mutant, sen2-42, that is known to accumulate intron-containing tRNAs at the nonpermissive temperature (44). Under galacatose induction, the sen2-42 mutant expressing inducible Sen subunits and tRNA ligase no longer displayed an accumulation of end-matured, intron-containing pre-tRNAs at nonpermissive conditions, although this accumulation was observed when expression of splicing factors was not induced (Fig. 5, lane 10 compared with lane 12). Therefore, the overexpressed splicing endonuclease proteins are functional. We then assessed whether overexpression of these genes alleviates the accumulation of intron-containing pre-tRNAs in maf1Δ cells. When expression of splicing endonuclease proteins was induced by the addition of galactose to the medium, maf1Δ cells continued to accumulate intron-containing pre-tRNAs at levels comparable to those in cells that were transformed with empty vectors or with not induced splicing machinery (Fig. 5, compare lanes 1–4). Wild type cells transformed with the same vectors did not show any difference in pre-tRNA processing when compared with these transformed with empty vectors. The data indicate that limitation of the cytoplasmic splicing machinery does not account for accumulation of intron-containing pre-tRNAs in maf1Δ cells.
Saturation of Nuclear Export Machinery in maf1Δ
Because the yeast tRNA splicing factors are located in the cytoplasm, accumulation of intron-containing end-matured precursors in maf1Δ cells may be caused by saturation of nuclear export machinery. Inefficient export of pre-tRNAs to the cytoplasm would cause intron-containing tRNAs to be physically separated from splicing machinery (45). Employing fluorescence in situ hybridization analyses and probe specific to the intron of pre-tRNALeu, we showed elevated levels of pre-tRNA in nuclei of maf1Δ cells as compared with wild type cells (Fig. 6). We assumed that initial transcript together with the end-matured intron-containing tRNA transcript account for this effect because these pre-tRNA forms are indistinguishable by fluorescence in situ hybridization. We thus explored whether the elevated level of Los1 exportin in maf1Δ saturates the tRNA nuclear export machinery.
FIGURE 6.

maf1Δ cells accumulate pre-tRNALeu(CAA) in the nucleus. maf1Δ and control wild type cells were grown in glucose medium (YPD) at 30 °C to early log phase. Part of the culture was shifted to glycerol medium (YPGly) for 2 h at 37 °C. The cultures were harvested and subjected to fluorescence in situ hybridization. The Cy3-labeled probe specific to sequence of pre-tRNALeu(CAA) intron and DAPI staining were applied.
We overproduced Los1 by transformation of yeast with a LOS1-containing multicopy plasmid and examined the level of pre-tRNA accumulation by Northern blotting (Fig. 7). As anticipated (46), control los1Δ cells accumulated end-matured intron-containing, pre-tRNA (Fig. 7A). Quantification of the Northern blot revealed 3-fold elevated levels of intron-containing end-matured pre-tRNALeu in los1Δ cells over that of the wild type when corrected for the U3 snoRNA levels (Fig. 7B). Plasmid-encoded Los1 fully compensated the elevated levels of pre-tRNA in los1Δ and did not affect cell growth. Importantly, LOS1 overexpression partially suppressed pre-tRNA accumulation in maf1Δ cells grown in minimal glucose medium (Fig. 7A). Both initial transcript and end-matured intron-containing pre-tRNALeu were reduced in maf1Δ glucose cells by a LOS1-containing multicopy plasmid ∼2- and 3-fold, respectively (Fig. 7B). We thus conclude that the Los1 level is limiting for processing of intron-containing pre-tRNAs in maf1Δ cells under standard growth conditions. It is noteworthy that an increased amount of Los1 affects also the levels of initial transcript by an unknown mechanism.
FIGURE 7.

Los1 is limiting for intron-containing tRNA export and processing in maf1Δ cells. Wild type (wt) BY4742, los1Δ, and maf1Δ cells, transformed with control plasmid Yep24 (designated []) or overexpressing LOS1 ([LOS1]) were grown in SC-ura glucose medium. Part of the culture was shifted to minimal glycerol medium (Sc-ura + glycerol) for 2 h at 37 °C. A, Northern hybridization was performed with probes specific to pre-tRNALeu(CAA) or U3 snoRNA (used as a control). B, quantification of the pre-tRNALeu(CAA) levels. The bars represent levels for primary transcript retaining 5′ and 3′ termini and intron-containing precursor of tRNALeu(CAA) normalized to U3 snoRNA transcript. The fold of change in each strain was calculated relative to expression in wild type strain bearing control plasmid YEp24 (designated as wt/[]). The standard deviation was calculated on the basis of three independent experiments.
Under restrictive conditions in glycerol medium, LOS1-containing multicopy plasmid had, however, no detectable effect on pre-tRNALeu accumulation in maf1Δ cells (Fig. 7). We explored the possibility that growth in glycerol medium may influence expression or localization of Los1. It has been previously reported that Los1 is mislocalized to cytoplasm upon DNA damage, causing increased nuclear accumulation of intron-containing tRNA (47). We therefore examined localization of C-terminally Myc-tagged Los1 in control and maf1Δ cells grown under standard (YPD) or restrictive (YPGly, 37 °C) conditions (Fig. 8). Los1-Myc was detected primarily in the nucleus of cells cultivated on glucose, and no obvious difference in its location was observed between wild type and maf1Δ cells (Fig. 8A). By contrast, a large part of Los1-Myc became redistributed to the cytoplasm in wild type and maf1Δ cell populations grown in glycerol medium (Fig. 8B). Note that neither growth conditions nor the absence of Maf1 affect Los1 expression (supplemental Fig. S2).
FIGURE 8.

Localization of Los1 exportin is affected by growth conditions but not by Maf1. Strains BY4742 LOS1-Myc (wt) and BY4742 maf1Δ LOS1-Myc (maf1Δ) were grown to early logarithmic phase in YPD. Part of the culture was shifted to glycerol medium (YPGly) for 2 h at 37 °C. Localization of Los1 was examined by indirect immunofluorescence microscopy using anti-Myc antibodies. Nuclear DNA was stained with DAPI.
Thus, it is most likely that under normal conditions, an insufficient amount rather than inappropriate location of Los1 accounts for pre-tRNA accumulation in maf1Δ. Growth in medium with a nonfermentable carbon source additionally limits tRNA export by mislocalization of Los1 to cytoplasm, and therefore its overproduction does not prevent pre-tRNA accumulation in maf1Δ.
Induction of Gcn4 Expression in maf1Δ Cells
Both aberrant tRNA processing and limited tRNA export have marked effects upon the translation of the transcription factor, Gcn4 (47, 48). Conditions that activate Gcn4 reduce the rate of general protein synthesis while simultaneously activating expression of proteins required under conditions of starvation and stress (reviewed in Ref. 49). Hence, we investigated whether accumulation of pre-tRNA in maf1Δ similarly results in elevated Gcn4 translation. We utilized a widely employed CEN-based reporter plasmid carrying a GCN4 promoter and GCN4 UTR translation regulatory sequences driving the expression of a lacZ gene that provided a quantitative measure for Gcn4 by assessing β-galactosidase activity (36). The GCN4-lacZ reporter plasmid was introduced into wild type and maf1Δ cells, and resulting transformants were grown under standard conditions with a shift to glycerol medium and incubation at elevated temperatures. There was greater than 2-fold increased expression of GCN4-lacZ in maf1Δ cells under restrictive conditions compared with wild type cells (Fig. 9A). The induction of Gcn4 expression in maf1Δ was confirmed by Western blot analysis. Total proteins isolated from wild type and maf1Δ cells were separated on SDS-polyacrylamide gel, followed by incubation with anti-Gcn4 antibodies. Elevated levels of the Gcn4 protein were detected in maf1Δ cells, after a shift to restrictive conditions (YPGly 37 °C) (Fig. 9B). Although the effect of Maf1 inactivation on Gcn4 expression is relatively weak, increased Gcn4 translation correlates with GCN4-lacZ induction in maf1Δ cells and is linked to pre-tRNA nuclear accumulation observed in the same growth conditions. Moreover, accumulation of tRNA precursors in maf1Δ nuclei activate Gcn4 in a manner independent on phosphorylation of eIF2α (supplemental Fig. S3). These observations are consistent with results of Qiu et al. (48) and support the existence of a system communicating immature tRNA in the nucleus to the translation machinery.
FIGURE 9.
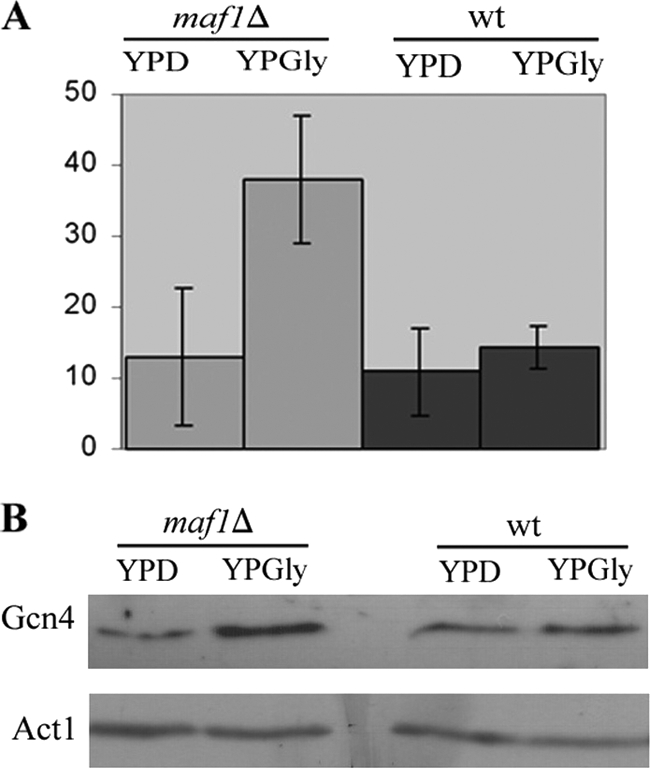
Gcn4 induction in maf1Δ cells. A GCN4-lacZ (CEN, URA) reporter plasmid was introduced into wild type and maf1Δ mutant cells. Transformants were grown in YPD grown to early log phase in rich glucose medium (YPD) at 30 °C, then transferred to glycerol medium (YPGly), and incubated at 37 °C for 2 h. A, β-galactosidase activity was measured in yeast extracts and correlated to protein concentration (arbitrary units). B, total proteins were isolated as described under “Experimental Procedures.” Equal amounts of proteins were resolved on 10% SDS-polyacrylamide gel and subjected to immunoblot analysis using specific rabbit anti-Gcn4 or mouse anti-Act1 antibodies (used as a loading control).
DISCUSSION
tRNAs are essential components of the translation machinery, and therefore their biosynthesis is tightly coupled to cell growth and metabolism. A mechanism for this coordination is provided by Maf1, a general negative regulator of RNA polymerase III transcription. Maf1 is essential for down-regulation of tRNA transcription during a transition from fermentative to glycerol-based medium. Previously we showed that tRNAs encoded by intron-containing genes are the most sensitive to Maf1-mediated regulation (33). Those results led us to examine the potential effect of Maf1-mediated regulation on tRNA processing. Because the 5′ and 3′ ends and splicing junctions in the intron-containing tRNAs are not conserved, and the introns themselves vary in sequence and length, we probed two intron-containing tRNAs to verify the ubiquity of the Maf1 effects. Both primary transcripts and end-processed intron-containing tRNA precursors were abnormally abundant in the absence of Maf1. It is noteworthy that maf1Δ cells have also elevated primary transcript of intron-less tRNAiMet, indicating an effect on the end-processing of this particular pre-tRNA. Initial transcripts of other tRNAs encoded by genes lacking introns are hardly detectable, but the mature forms are more abundant in maf1Δ cells than wild type cells. These data are consistent with our previously published results, which showed increased levels of mature tRNAHis in maf1Δ cells grown in medium with a nonfermentable carbon source (6). Because 80% of the tRNAs in yeast are encoded by intron-less genes, they account for the global increase of tRNA in maf1Δ observed earlier on ethidium bromide-stained gels (6). Additionally, accumulation of unprocessed intron-containing transcript accounts for relatively high increase of intron-containing tRNAs (33).
In response to changing growth conditions, Maf1 shuttles between nucleus and cytoplasm (14). The cytoplasmic location provides a mechanism to prevent Maf1 from interaction with the transcription machinery under favorable growth circumstances. One tempting hypothesis, excluded here, was that Maf1 might be involved in tRNA splicing in the cytoplasm independent from its known nuclear role in negatively regulating Pol III transcription. Instead we have shown that Maf1-mediated repression of Pol III transcription in the nucleus indirectly influences pre-tRNA maturation.
Further, we sought to identify the step in tRNA maturation that is saturated by the high levels of primary transcripts produced when Maf1 is missing. Our data argue against saturation of RPR1, the RNA component of RNase P machinery in maf1Δ nuclei, found before as the limiting for RNase P-mediated processing (42, 43, 47, 48). In addition, we excluded saturation of cytoplasmic splicing machinery in the absence of Maf1 and detected an increased amount of intron-containing tRNA in maf1Δ nuclei (Fig. 6). We found that maf1Δ cells are defective in nuclear export of intron-containing tRNAs.
We then addressed the issue of why mainly processing of tRNAs encoded by intron-containing tRNA genes is altered in maf1Δ cells. First, maf1Δ may preferentially transcribe intron-containing tRNA genes. Second, end maturation could be more efficient in tRNAs encoded by intron-less genes. Finally, there are more pathways for nuclear export of intron-less tRNA than for intron-containing tRNA. So far, intron-containing tRNAs can be exported to the cytoplasm by only exportin, Los1. In contrast tRNAs lacking introns can be exported by two exportins, Los1 and Msn5 (50). Thus, two different categories of tRNAs may be differently regulated at the nuclear export step.
It is quite possible that in addition to Los1-limited tRNA nuclear export in maf1Δ cells, other processes may also be limited for efficient pre-tRNA processing, especially under unfavorable conditions. Systematic regulation of tRNA processing by environmental growth conditions has not been experimentally addressed so far. Possibly growth of cells in glycerol medium at 37 °C negatively affects early steps of tRNA processing. Nevertheless, among the various steps of tRNA maturation, Los1-mediated tRNA trafficking seems to be a main target for selective control. It was recently shown that inactivation of Xpo-t, the Los1 homologue in mammals, resulted in elevated autophagy and reduced activity of mTORC1 pathway. The authors suggested that the subcellular localization of tRNA plays a role in the signaling a nutritional stress in mammalian cells (51). In yeast, regulation of tRNA export contributes to the proper execution of G1 checkpoint (47). This response is mediated via differential cellular localization of Los1. Normally Los1 shuttles between the nucleus and the cytoplasm, whereas after DNA damage Los1 is mainly found in the cytoplasm. Although the mechanism is not clear, localization of Los1 is controlled by the cell cycle checkpoint proteins Rad53 and Mec1. Los1 intracellular distribution is also dependent on nutrient supply and Snf1-mediated signaling (52). As a consequence of the Los1 mislocalization, intron-containing pre-tRNAs accumulate in the nucleus (47).
According to our model (Fig. 10), transcription and nuclear export are two major controls of tRNA biosynthesis. Both Maf1-mediated Pol III regulation and Los1-dependent nuclear export are regulated by environment circumstances in coordinated manner. Under favorable growth conditions in glucose medium, Maf1 is phosphorylated, thus preventing Pol III repression, and Los1 is localized in nuclear membrane, providing active tRNA export. Following a shift to repressive conditions, tRNA transcription is inhibited because of Maf1 dephosphorylation, and concurrently tRNA export is lowered by redistribution of Los1 to the cytoplasm. By mutual regulation of transcription and export, differences in growth conditions have a substantial effect on the relative levels of pre-tRNA and mature tRNA species. In contrast, transcription and export are uncoupled in strain lacking Maf1 resulting in abnormal high levels of pre-tRNA. It will be interesting to assess the joint regulation of Maf1 and Los1 by cellular signaling.
FIGURE 10.

Maf1-mediated transcription control and nuclear export by Los1 are regulated by environmental conditions in coordinated manner. A, under favorable growth conditions in glucose medium Maf1 is phosphorylated, thus preventing Pol III repression, and Los1 is localized in nuclear membrane providing active export of intron-containing tRNA precursors (wt panel). tRNA synthesis is increased in the absence of Maf1, but export is saturated (maf1Δ panel). B, under repressive conditions on glycerol medium, both tRNA synthesis and tRNA export are decreased, respectively, by Maf1-Pol III interaction and Los1 redistribution to the cytoplasm (wt panel). This coupling is lost in the absence of Maf1, resulting in nuclear accumulation of intron containing pre-tRNAs (maf1Δ panel).
Supplementary Material
Acknowledgments
We thank Dr. T. Yoshihisa for the sen2-42 constructs and Dr. O. Lefebvre for RPR1 encoding plasmid.
This work was supported, in whole or in part, by National Institutes of Health Grant 27930 (to A. K. H.). This work was also supported by Ministry of Science and Higher Education (Poland) Grant MNSIW N301 298837.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- Pol
- polymerase
- rRNA
- ribosomal RNA
- snoRNA
- small nucleolar RNA.
REFERENCES
- 1. Osheim Y. N., French S. L., Keck K. M., Champion E. A., Spasov K., Dragon F., Baserga S. J., Beyer A. L. (2004) Mol. Cell 16, 943–954 [DOI] [PubMed] [Google Scholar]
- 2. Buratowski S. (2009) Mol. Cell 36, 541–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kos M., Tollervey D. (2010) Mol. Cell 37, 809–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alexander R. D., Innocente S. A., Barrass J. D., Beggs J. D. (2010) Mol. Cell 40, 582–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carrillo Oesterreich F., Preibisch S., Neugebauer K. M. (2010) Mol. Cell 40, 571–581 [DOI] [PubMed] [Google Scholar]
- 6. Pluta K., Lefebvre O., Martin N. C., Smagowicz W. J., Stanford D. R., Ellis S. R., Hopper A. K., Sentenac A., Boguta M. (2001) Mol. Cell. Biol. 21, 5031–5040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Upadhya R., Lee J., Willis I. M. (2002) Mol. Cell 10, 1489–1494 [DOI] [PubMed] [Google Scholar]
- 8. Oficjalska-Pham D., Harismendy O., Smagowicz W. J., Gonzalez de Peredo A., Boguta M., Sentenac A., Lefebvre O. (2006) Mol. Cell 22, 623–632 [DOI] [PubMed] [Google Scholar]
- 9. Moir R. D., Lee J., Haeusler R. A., Desai N., Engelke D. R., Willis I. M. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 15044–15049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boguta M. (2009) Cell Cycle 8, 4023–4024 [DOI] [PubMed] [Google Scholar]
- 11. Huber A., Bodenmiller B., Uotila A., Stahl M., Wanka S., Gerrits B., Aebersold R., Loewith R. (2009) Genes Dev. 23, 1929–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Graczyk D., Debski J., Muszyńska G., Bretner M., Lefebvre O., Boguta M. (2011) Proc. Natl. Acad. Sci. U.S.A. 108, 4926–4931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wei Y., Tsang C. K., Zheng X. F. (2009) EMBO J. 28, 2220–2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Towpik J., Graczyk D., Gajda A., Lefebvre O., Boguta M. (2008) J. Biol. Chem. 283, 17168–17174 [DOI] [PubMed] [Google Scholar]
- 15. Johnson S. S., Zhang C., Fromm J., Willis I. M., Johnson D. L. (2007) Mol. Cell 26, 367–379 [DOI] [PubMed] [Google Scholar]
- 16. White R. J. (2005) Nat. Rev. Mol. Cell Biol. 6, 69–78 [DOI] [PubMed] [Google Scholar]
- 17. Marshall L., Kenneth N. S., White R. J. (2008) Cell 133, 78–89 [DOI] [PubMed] [Google Scholar]
- 18. Kwapisz M., Smagowicz W. J., Oficjalska D., Hatin I., Rousset J. P., Zoładek T., Boguta M. (2002) Curr. Genet. 42, 147–152 [DOI] [PubMed] [Google Scholar]
- 19. Phizicky E. M., Hopper A. K. (2010) Genes Dev. 24, 1832–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hellmuth K., Lau D. M., Bischoff F. R., Künzler M., Hurt E., Simos G. (1998) Mol. Cell. Biol. 18, 6374–6386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Arts G. J., Kuersten S., Romby P., Ehresmann B., Mattaj I. W. (1998) EMBO J. 17, 7430–7441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lipowsky G., Bischoff F. R., Izaurralde E., Kutay U., Schäfer S., Gross H. J., Beier H., Görlich D. (1999) RNA 5, 539–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cook A. G., Fukuhara N., Jinek M., Conti E. (2009) Nature 461, 60–65 [DOI] [PubMed] [Google Scholar]
- 24. Huh W. K., Falvo J. V., Gerke L. C., Carroll A. S., Howson R. W., Weissman J. S., O'Shea E. K. (2003) Nature 425, 686–691 [DOI] [PubMed] [Google Scholar]
- 25. Yoshihisa T., Yunoki-Esaki K., Ohshima C., Tanaka N., Endo T. (2003) Mol. Biol. Cell 14, 3266–3279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shaheen H. H., Hopper A. K. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 11290–11295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Longtine M. S., McKenzie A., 3rd, Demarini D. J., Shah N. G., Wach A., Brachat A., Philippsen P., Pringle J. R. (1998) Yeast 14, 953–961 [DOI] [PubMed] [Google Scholar]
- 28. Sherman F. (2002) Methods Enzymol. 350, 3–41 [DOI] [PubMed] [Google Scholar]
- 29. Lefebvre O., Rüth J., Sentenac A. (1994) J. Biol. Chem. 269, 23374–23381 [PubMed] [Google Scholar]
- 30. Hurt D. J., Wang S. S., Lin Y. H., Hopper A. K. (1987) Mol. Cell. Biol. 7, 1208–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schmitt M. E., Brown T. A., Trumpower B. L. (1990) Nucleic Acids Res. 18, 3091–3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hurto R. L., Tong A. H., Boone C., Hopper A. K. (2007) Genetics 176, 841–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ciela M., Towpik J., Graczyk D., Oficjalska-Pham D., Harismendy O., Suleau A., Balicki K., Conesa C., Lefebvre O., Boguta M. (2007) Mol. Cell. Biol. 27, 7693–7702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sarkar S., Hopper A. K. (1998) Mol. Biol. Cell 9, 3041–3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pringle J. R., Adams A. E., Drubin D. G., Haarer B. K. (1991) Methods Enzymol. 194, 565–602 [DOI] [PubMed] [Google Scholar]
- 36. Conesa C., Ruotolo R., Soularue P., Simms T. A., Donze D., Sentenac A., Dieci G. (2005) Mol. Cell. Biol. 25, 8631–8642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gajda A., Towpik J., Steuerwald U., Müller C. W., Lefebvre O., Boguta M. (2010) J. Biol. Chem. 285, 35719–35727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Boguta M., Czerska K., Zoładek T. (1997) Gene 185, 291–296 [DOI] [PubMed] [Google Scholar]
- 39. Jimenez A., Tipper D. J., Davies J. (1973) Antimicrob. Agents Chemother. 3, 729–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kufel J., Tollervey D. (2003) RNA 9, 202–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Harismendy O., Gendrel C. G., Soularue P., Gidrol X., Sentenac A., Werner M., Lefebvre O. (2003) EMBO J. 22, 4738–4747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ishiguro A., Kassavetis G. A., Geiduschek E. P. (2002) Mol. Cell. Biol. 22, 3264–3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Deprez E., Arrebola R., Conesa C., Sentenac A. (1999) Mol. Cell. Biol. 19, 8042–8051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yoshihisa T., Ohshima C., Yunoki-Esaki K., Endo T. (2007) Genes Cells 12, 285–297 [DOI] [PubMed] [Google Scholar]
- 45. Hopper A. K. (2006) Crit. Rev. Biochem. Mol. Biol. 41, 3–19 [DOI] [PubMed] [Google Scholar]
- 46. Shen W. C., Selvakumar D., Stanford D. R., Hopper A. K. (1993) J. Biol. Chem. 268, 19436–19444 [PubMed] [Google Scholar]
- 47. Ghavidel A., Kislinger T., Pogoutse O., Sopko R., Jurisica I., Emili A. (2007) Cell 131, 915–926 [DOI] [PubMed] [Google Scholar]
- 48. Qiu H., Hu C., Anderson J., Björk G. R., Sarkar S., Hopper A. K., Hinnebusch A. G. (2000) Mol. Cell. Biol. 20, 2505–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hinnebusch A. G., Natarajan K. (2002) Eukaryot. Cell 1, 22–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Murthi A., Shaheen H. H., Huang H. Y., Preston M. A., Lai T. P., Phizicky E. M., Hopper A. K. (2010) Mol. Biol. Cell. 21, 639–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Huynh L. N., Thangavel M., Chen T., Cottrell R., Mitchell J. M., Praetorius-Ibba M. (2010) Cell Cycle 9, 3112–3118 [DOI] [PubMed] [Google Scholar]
- 52. Quan X., Yu J., Bussey H., Stochaj U. (2007) Biochim. Biophys. Acta 1773, 1052–1061 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}