

Background: Chemerin (chem163S) is proteolytically cleaved to generate active isoforms.
Results: Chem157S has the highest activity in stimulating calcium transients and chemotaxis, chem158K and chem163S have much lower activity, and chem155A is not an agonist. Chem157S stimulates signaling in glioblastoma cells.
Conclusion: Chem163S proteolysis to chem157S is needed for its activity.
Significance: Chemerin may play a role in glioblastoma.
Keywords: Calcium Intracellular Release, Chemokines, Chemotaxis, G Protein-coupled Receptor (GPCR), Proteolytic Enzymes, Glioblastoma
Abstract
Chemerin is a chemoattractant involved in innate and adaptive immunity as well as an adipokine implicated in adipocyte differentiation. Chemerin circulates as an inactive precursor in blood whose bioactivity is closely regulated through proteolytic processing at its C terminus. We developed methodology for production of different recombinant chemerin isoforms (chem163S, chem157S, and chem155A) which allowed us to obtain large quantities of these proteins with purity of >95%. Chem158K was generated from chem163S by plasmin cleavage. Characterization by mass spectrometry and Edman degradation demonstrated that both the N and C termini were correct for each isoform. Ca2+ mobilization assays showed that the EC50 values for chem163S and chem158K were 54.2 ± 19.9 nm and 65.2 ± 13.2 nm, respectively, whereas chem157S had a ∼50-fold higher potency with an EC50 of 1.2 ± 0.7 nm. Chem155A had no agonist activity and weak antagonist activity, causing a 50% reduction of chem157S activity at a molar ratio of 100:1. Similar results were obtained in a chemotaxis assay. Because chem158K is the dominant form in cerebrospinal fluid from patients with glioblastoma (GBM), we examined the significance of chemerin in GBM biology. In silico analysis showed chemerin mRNA was significantly increased in tissue from grade III and IV gliomas. Furthermore, U-87 MG cells, a human GBM line, express the chemerin receptors, chemokine-like receptor 1 and chemokine receptor-like 2, and chem157S triggered Ca2+ flux. This study emphasized the necessity of appropriate C-terminal proteolytic processing to generate the likely physiologic form of active chemerin, chem157S, and suggested a possible role in malignant GBM.
Introduction
Chemerin, also called tazarotene-induced gene 2 or retinoic acid receptor responder 2, was first identified as an up-regulated gene in human skin raft culture by the antipsoriatic retinoid, tazarotene (1). Using human hemofiltrate and ascitic fluids, chemerin was identified as a ligand for the orphan G protein-linked chemokine-like receptor 1 (CMKLR1),3 also known as chemR23 (2, 3). A number of cell types involved in innate and adaptive immunity, such as myeloid dendritic cells, plasmacytoid dendritic cells, natural killer cells, and macrophages, express CMKLR1. Indeed chemerin functions as a chemoattractant that promotes the recruitment of these cells to lymphoid organs and sites of tissue injury (4, 5). Recent studies have demonstrated that chemerin also serves as a ligand for at least two additional receptors, chemokine receptor-like 2 (CCRL2) and G protein-coupled receptor 1 (GPR1) (6, 7). CCRL2 serves to concentrate chemerin on the cell surface and facilitate its presentation to CMKLR1 (7) whereas the function of GPR1 remains undefined. In addition to immune functions, chemerin has been shown to be an adipokine that regulates adipocyte development and metabolic function such as glucose metabolism (8–11).
Chemerin is synthesized as a 163-amino acid precursor that contains a hydrophobic signal peptide sequence, a cystatin fold-containing domain, and a labile C terminus. After removal of the signal sequence, the secreted form is a 143-amino acid proform (prochemerin, chem163S) with low biological activity. Chem163S undergoes further proteolytic processing at its C terminus into a potent agonist by plasmin, carboxypeptidases, or serine proteases of the coagulation, fibrinolytic, and inflammatory cascades (12–14). Different cleavage sites have been found in chemerin depending on the location from which it is isolated as well as the methods used to isolate it, e.g. chemerin isoforms terminating at Ser157, Ala155, and Phe154 are found in ascitic fluids, serum, and hemofiltrate, respectively (15).
The most active form, chem157S, can be generated in vitro either by direct elastase cleavage of prochemerin or by sequential cleavages with plasmin and carboxypeptidase N or carboxypeptidase B2 (also termed thrombin-activatable fibrinolysis inhibitor) (13). Enzymatic proteolysis also inactivates bioactive chemerin. Neutrophil-derived PR3, mast cell chymase, and angiotensin-converting enzyme can all convert active chemerin into inactive derivatives (16). Furthermore, mouse chem156S, terminating in the site homologous to human chem157S, exerts antiinflammatory activities on macrophage activation that is dependent on an unknown cysteine protease both in vitro (17) and in a LPS-induced acute lung injury model (18). This strongly suggests that proteolytic processing is a key regulatory mechanism for generating local chemerin bioactivity.
Properties of chemerin isoforms have been largely determined by synthetic peptides derived from the labile C-terminal region (17–20). Part of the reason for this is that recombinant prochemerin tends to get partially proteolyzed during the expression and/or the purification process. The resulting mixture of prochemerin and partially cleaved and activated chemerin isoforms makes it difficult to fully characterize and standardize the various chemerin isoforms in a biochemically rigorous manner. In addition, the use of synthetic C-terminal peptides may not accurately recapitulate the functional properties of the intact proteins. For example, chemerin149–157, YFPGQFAFS, exerts >2 orders of magnitude higher potency than chemerin150–157, FPGQFAFS, even though both peptides end at the same C-terminal amino acid residue (19). There have also been conflicting reports regarding the antiinflammatory activity of a chemerin peptide (Cys15, Ala141-Ala155; the mouse homolog being Ala140-Ala154), which is homologous to the C terminus of human chem155A (18, 19). In this paper, we described a new method for the expression and purification of recombinant prochemerin (chem163S), chem158K, chem157S, and chem155A in large quantities that allow us to characterize their biological activities in detail. In the accompanying paper (31), we have identified chem158K as a dominant form in cerebrospinal fluid from patients with GBM. We therefore also examined the possible role of chemerin signaling in GBM biology.
EXPERIMENTAL PROCEDURES
Reagents
Protease-free BSA (A3059) and heparin-agarose were purchased from Sigma. Recombinant human chemerin 21–157 and human chemerin DuoSet ELISA kit were products of R&D Systems (Minneapolis, MN). Human cathepsin G and H-d-Phe-Pro-Arg-chloromethyl ketone were obtained from Calbiochem. Peptides listed in Table 1 were synthesized by Elim Biopharmaceuticals (Hayward, CA). Human plasmin was from Hematologic Technologies (Essex Junction, VT).
TABLE 1.
Properties of recombinant chemerin isoforms and chemerin-derived peptides
The C-terminal sequences of all chemerin isoforms and the equivalent peptides tested in this study were displayed, as well as the EC50 values on human CMKLR1/L1.2 cells determined by Ca2+ mobilization and chemotaxis assay. Data are shown as mean ± S.D. and calculated from at least four independent experiments.
| C-terminal Sequence | Amino acids | Molecular mass | pI | Ca2+ flux | Chemotaxis | |
|---|---|---|---|---|---|---|
| Da | nm | nm | ||||
| Chem163S | –HSFYFPGQFAFSKALPRS | 143 | 16,529.9 | 8.89 | 54.2 ± 19.9 | 68.4 ± 46.2 |
| Chem158K | –HSFYFPGQFAFSK | 138 | 16,005.3 | 8.81 | 65.2 ± 13.2 | 45.3 ± 25.0 |
| Chem157S | –HSFYFPGQFAFS | 137 | 15,877.1 | 8.60 | 1.17 ± 0.74 | 3.15 ± 1.57 |
| Chem156F | –HSFYFPGQFAF | 136 | 15,790.0 | 8.60 | NDa | ND |
| Chem155A | –HSFYFPGQFA | 135 | 15,642.8 | 8.60 | >1,000 | 381 ± 253 |
| 15-mer | YFPGQFAFSKALPRS | 15 | 1,715.9 | 9.99 | >1,000 | >1,000 |
| 10-mer | YFPGQFAFSK | 10 | 1,191.3 | 8.59 | >1,000 | 849 ± 740 |
| 9-mer | YFPGQFAFS | 9 | 1,063.1 | 5.52 | 5.92 ± 3.26 | 6.38 ± 3.03 |
| 11-mer | HSFYFPGQFAF | 11 | 1,347.5 | 6.74 | 343 ± 107 | 288 ± 200 |
| FA | HSFYFPGQFA | 10 | 1,200.3 | 6.74 | >1,000 | >1,000 |
a ND, not determined.
Cell Culture
CHO-S cells were purchased from Invitrogen and cultured in CD-CHO medium supplemented with 8 mm l-glutamine, 1% HT supplement (Invitrogen), 50 units/ml penicillin, and 50 mg/ml streptomycin under a humidified atmosphere of 8% CO2 at 37 °C. Human cell lines DBTRG-05MG, Hs 683, T98G, U-87 MG, and Jurkat (T lymphocyte) were purchased from the American Type Culture Collection (ATCC, Manassas VA), and the 1321N1 cell line was from the European Collection of Cell Cultures (ECACC, Wiltshire, UK). 1321N1, Hs 683, and U-87 MG cells were maintained in DMEM with 10% heat-inactivated FBS, 50 units/ml penicillin, and 50 μg/ml streptomycin. T98G cells were cultured in Earle's minimal essential medium supplemented with additional 1% nonessential amino acids, 1 mm sodium pyruvate. DBTRG-05MG cells were grown in RPMI 1640 medium with an additional 0.1 mm sodium hypoxanthine, 16 mm thymidine, 1 mm sodium pyruvate, and Jurkat cells in RPMI 1640 medium with an additional 1% nonessential amino acids and 55 mm 2-mercaptoethanol. The murine pre-B lymphoma cell line L1.2 and the derivative L1.2 cells transfected with pcDNA3.1 encoding CMKLR1 were provided by Dr. Brian Zabel and Dr. Eugene C. Butcher (Stanford University School of Medicine and Veterans Affairs Palo Alto Health Care System) and cultured in RPMI 1640 medium with 10% FBS and 1 mg/ml Geneticin.
Generation of CHO-S Stable Cell Clones Expressing Human Chemerins
A cDNA clone encoding human chemerin (NM_002889) was obtained from Open Biosystems (Huntsville, AL). cDNA fragments encoding various chemerin isoforms (chem163S, chem158K, chem157S, chem156F, and chem155A) were amplified by PCR using Platinum Pfx DNA polymerase (Invitrogen) with termination codons inserted into the primers (listed in supplemental Table 1) used to amplify the chemerin cDNA to create the different forms, before cloning into a ubiquitous chromatin opening element (UCOE®) vector, pCET-1019AS-puro, provided by Millipore. The resultant expression plasmids containing a UCOE® element upstream of a guinea pig CMV that drives expression of the chemerin cDNA were verified by sequencing. The chemerin constructs were transfected into CHO-S cells by DMRIE-C reagent (Invitrogen) according to the manufacturer's protocol. After 48 h, the cells were seeded at a density of 5 × 104 cells/ml in 100 μl/well in 96-well plates. Clones were selected with 10 μg/ml puromycin. After 12 days, expression of human chemerins was assessed by a human chemerin ELISA kit. High expressing clones were chosen on the basis of their specific productivity rate (micrograms of chemerin/cell per day).
Protein Production and Purification
CHO-S cells stably expressing the chemerin isoforms were seeded into 800 ml of CD-CHO medium at 2 × 105 cells/ml in a Spinner flask. After 4 days of culture at 37 °C, the conditioned medium was collected by centrifugation, filtered through a 0.22-mm filter, and applied to a HiTrap SP HP column (GE Healthcare) equilibrated with PBS (pH 7.4), washed with PBS containing 150 mm NaCl, and chemerin proteins were eluted with a 150–500 mm NaCl linear gradient in PBS using an AKTA fast protein liquid chromatography system (GE Healthcare) in 5-ml fractions. Fractions containing chemerin were identified by SDS-PAGE analysis and pooled. The purified proteins were analyzed by silver-stained SDS-PAGE, and purity was quantitated by scanning the gels. The molecular mass of the chemerin forms was confirmed by MALDI-TOF mass spectrometry with internal standards of 2,466, 3,660, and 5,734 Da included in each analysis (PAN Facility, Stanford University). For storage, 0.1% protease-free BSA was added, and aliquots were stored at −20 °C.
Chemotaxis Assay
hCMKLR1/L1.2 cells were treated with 5 mm sodium butyrate for 24 h before seeding 2 × 105 cells in serum-free RPMI 1640 medium in the Transwell insert of a Transwell 24-well plate with 5.0-μm pore size (Corning). Different concentrations of chemerin forms were added to 600 μl of the same medium in the bottom well. After 4 h of incubation at 37 °C, the cells that passed through the membrane were labeled with CKK-8 (Dojindo, Kumamoto, Japan) and measured at a wavelength of 450 nm in a SPECTRAMAX plus plate reader (Molecular Devices, Sunnyvale, CA). The data were expressed as a percentage of the total input cells based on a standard curve generated from a known number of the labeled cells. The whole assay was repeated on at least four independent occasions for each protein or peptide using three independent preparations of protein.
Calcium Mobilization Assay
hCMKLR1/L1.2 cells were labeled with Quest Fluo-8 AM dye (AAT Bioquest, Sunnyvale, CA) in HHBS buffer (Hanks' buffered saline plus 20 mm Hepes (pH 7.0)) containing 2.5 mm probenecid and 0.04% Pluronic F-127 at 37 °C for 30 min in the wells of a 96-well plate (12.5 × 104/well). The dye-loaded cells were stimulated with chemerin forms at room temperature, and the fluorescence intensity was followed at excitation/emission 495/535 nm using a FLUOROSKAN ASCENT FL fluorescent plate reader (Thermo Electron Corp., Waltham, MA). The whole assay was repeated on at least four independent occasions for each protein or peptide using three independent preparations of protein.
In Silico Analysis of Chemerin, CMKLR1, and CCRL2 Expression in Glioma
The European Bioinformatics Institute (EBI) ArrayExpress was used to identify experiments analyzing gene expression in human GBM in which the expression of chemerin, CMKLR1, or CCRL2 was significantly altered. In experiment E-GEOD-4290, the difference in chemerin expression between GBM and the controls was highly significant with p = 4.35 × 10−5. One other experiment was identified in which GBM was compared with other tumors (E-GEOD-4412) in which the difference in chemerin expression was also significant (p = 0.008). Data on expression levels of chemerin, CMKLR1, and CCRL2 were extracted from the data files for E-GEOD-4290 and analyzed by disease type.
RNA Extraction and Quantitative RT-PCR
Total RNA was isolated by direct lysis of adherent cells or pelleted Jurkat cells grown in each well of 6-well plates for 2 days using the Qiagen RNeasy Mini kit (Valencia, CA). cDNA was synthesized from 1 μg of total RNA using iScript cDNA Synthesis kit (Bio-Rad) according to the manufacturer's protocol. Gene expression levels were quantified with SYBR Green-based real-time PCR using the 7900 HT Fast Real-time PCR Systems (Applied Biosystems). Real-time PCR was performed using iQ SYBR Green Supermix (Bio-Rad) with predesigned and validated QuantiTect Primer assays (Qiagen). The PCR conditions were 50 °C for 2 min, 95 °C for 10 min, and 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The values were normalized to the expression of GAPDH. The data were expressed as mean ± S.D. of relative gene expression compared with the values obtained from the Hs 683 cell line using the ΔΔCt method.
Analysis of CMKLR1 and CCRL2 Expression by Flow Cytometry
U-87 MG cells collected by trypsinization were fixed with 4% paraformaldehyde. After extensive washing with PBS, the cells were stained with anti-human CMKLR1 (eBioscience, San Diego, CA) and anti-human CCRL2 antibody (R&D Systems) and analyzed using FACS Calibur (BD Biosciences).
Statistical Analysis
Values for 50% effective concentration (EC50) were determined with Prism V4 (GraphPad Software, San Diego, CA) using nonlinear regression applied to a sigmoidal dose-response model. The maximum value used for the equation for the four-parameter fit was the maximal value obtained for chem157S in the chemotaxis assay. In comparisons between two values, Student's t test was used, and values of p < 0.05 were considered significant.
RESULTS
Expression and Purification of Recombinant Human Chem163S, Chem158K, Chem157S, and Chem155A
Inclusion of a UCOE® in a mammalian expression plasmid allows higher productivity clones to be isolated and grown stably (21, 22). We utilized this system to produce different forms of chemerin. Human chemerin isoforms listed in Table 1 were cloned into pCET-1019AS-puro that has a UCOE® element upstream of a guinea pig CMV promoter before transfection into CHO-S cells. After 2 days of incubation, the stable cell clones were selected by limiting dilution with 10 μg/ml puromycin, and 90 clones for each form were screened. We identified the clones producing >0.5 μg/ml chemerin in the conditioned media which were then rescreened for their specific productivity rate. One clone was identified for each chemerin form that was used for subsequent production and purification. The productivity of the clones that were used to produce chem163S, 157S, and 155A was 6.45, 6.81, and 5.87 pg/cell per day, respectively.
Recombinant chemerins were purified by a single-step cation exchange chromatography and analyzed by UV absorption, SDS-PAGE followed by silver staining and mass spectrometry (Fig. 1). Chem163S, chem157S, and chem155A were successfully obtained at >95% purity with no single impurity contributing > 2% based on quantitating scans of silver stained gels. To determine the N termini of the recombinant proteins, Edman sequencing was performed, and every protein had a uniform N terminus, ELTEAQRRGL, in the expected yield, confirming that the signal peptide had been cleaved at the amino acid identified previously (2). Analysis of the proteins by MALDI-TOF-MS showed that the observed mass for chem163S was 16,521, for chem157S was 15,867, and for chem155A, 15,636. These values were almost identical to the estimate by the ProtParam tool of 16,529, 15,877, and 15,642 for chem163S, chem157S, and chem155A respectively (Table 1). On the other hand, chem158K and chem156F did not possess uniform C-terminal sequences, suggesting that they are susceptible to proteolytic cleavages in the CHO-S conditioned medium (data not shown).
FIGURE 1.

Purification and characterization of human chemerin isoforms. Following centrifugation and filtration, conditioned cell culture medium was applied to an anion exchange column equilibrated with PBS (pH 7.4) and then chemerin proteins (left, chem163S; center, chem157S; right, chem155A) were eluted by gradients of increasing ionic strength. A, absorbance at 280 nm (mAU, blue line), NaCl gradient from 150 to 500 mm (green), and conductivity (brown). B, silver stained SDS-PAGE of fractions from the anion exchange column of a purification of chem163S (left), chem157S (center), and chem155A (right). S, postculture medium supernatant; F, flow-through; W, wash. Fraction numbers are shown. C, MALDI-TOF-MS spectrum of chem163S (left), chem157S (center), and chem155A (right). Internal standards (a, 2,466 Da; b, 3,660 Da; c, 5734 Da) are shown. The molecular mass of the principal peak is indicated.
To investigate whether chem158K and 156F could be generated from chem163S by proteolytic cleavage, chem163S was digested with plasmin or cathepsin G and analyzed by SDS-PAGE and MALDI-TOF-MS. Incubation with 10 milliunits of plasmin (55:1 substrate:enzyme) for 10 min successfully converted chem163S into chem158K with an observed mass of 16,006 compared with a predicted molecular mass of 16,005 (Fig. 2, A and B). However, further incubation resulted in the production of a nonspecific smaller fragment. When chem163S was treated with cathepsin G even at a lower substrate:enzyme ratio of 156:1, two bands corresponding to chem156F (observed molecular mass of 15,789 versus predicted molecular mass of 15,790) and 148F (observed molecular mass of 14,844 versus predicted molecular mass of 14,832) appeared in parallel (Fig. 2, C and D). We were unable to identify conditions that allowed production of pure chem156F. To obtain pure chem158K, following treatment with 10 milliunits of plasmin at 37 °C for 10 min, the reaction was terminated by 100 μm H-d-Phe-Pro-Arg-chloromethyl ketone. The mixture was then applied to a heparin-agarose column, and the bound protein eluted with 0.8 m NaCl (Fig. 3A). Thus, chem163S, chem158K, chem157S, and chem155A were successfully purified (Fig. 3B).
FIGURE 2.

Cleavage of chem163S by plasmin and cathepsin G. A, 10 μm chem163S (total 1 nmol) was digested with 10 and 30 milliunits (180 or 546 nm) of plasmin for the indicated time at 37 °C and analyzed by silver-stained SDS-PAGE. Arrows show the plasmin bands. B, mass spectrometric analysis of chem163S cleaved with 10 milliunits of plasmin for 10 min is shown. C, 10 μm chem163S (total 1 nmol) was digested with 0.3 and 1 milliunits (64 or 213 nm) of cathepsin G for the indicated times at 37 °C and analyzed by silver stained SDS-PAGE. Arrows show the cathepsin G bands. D, mass spectrometric analysis of the products resulting from cleavage of chem163S with 0.3 milliunit (64 nm) of cathepsin G for 30 min was performed.
FIGURE 3.

Analysis of purified chemerin forms by SDS-PAGE. A, generation of chem158K by plasmin cleavage followed by heparin affinity chromatography. The reaction mixture was adsorbed onto to a heparin-agarose column following elution with 0.8 m NaCl. Silver-stained SDS-PAGE of the starting material, chem163S (163S), followed plasmin treatment (163S + plasmin), flow-through, and wash, and material eluted with 0.8 m NaCl (0.8 m NaCl). B, different purified chemerin isoforms were analyzed by silver-stained SDS-PAGE.
Functional Characterization of Chem163S, Chem158K, Chem157S, and Chem155A
We next evaluated the functional properties of purified chemerins using hCMKLR1/L1.2 cells and compared them with the equivalent C-terminal peptides. These assays were run in the absence of serum to prevent interconversion of chemerin isoforms. Chemotaxis assays showed that the EC50 values for chem163S and chem158K were 68.4 ± 46.2 nm and 45.3 ± 25.0 nm, respectively, whereas chem157S had about 20-fold higher potency with an EC50 of 3.15 ± 1.57 nm (Fig. 4A). In contrast, chem155A that has been reported earlier to be a potent antiinflammatory molecule was poorly active. Comparison with the corresponding peptides showed that the full size proteins were significantly more active than the peptides, with the EC50 of the 15-mer (corresponding to chem163S) being >1 μm, 10-mer (chem158K) 849 ± 740 nm, and 9-mer (chem157S) being 6.38 ± 3.03 nm. Of note is the low but detectable activity of 11-mer, equivalent to chem156F. When the activity of the chemerin protein was compared with its equivalent C-terminal peptide, in all cases the protein was more active, although the precise ratio varied. This suggests that the N-terminal portion of the intact protein plays a role in engaging the receptor, and this is particularly apparent in the less biologically active chemerin isoforms (Fig. 4B and Table 1). The FA peptide, equivalent to the C terminus of chem155A, displayed no detectable activity in the chemotaxis assay (EC50 >1 μm).
FIGURE 4.

Biological activities of the purified chemerin isoforms and their equivalent C-terminal peptides. A, the indicated concentrations of chem163S (■), 158K (♦), 157S (▴), and 155A (●) were assayed for their chemotactic activity on hCMKLR1/L1.2 cells using the Transwell chemotaxis assay. B, chemotactic activity was assessed in the same assay for the chemerin-derived peptides corresponding to chem163S (15-mer, □), chem158K (10-mer, ♢), chem157S (9-mer, △), chem156F (11-mer, ▿), and chem155A (FA, ○). C and D, calcium flux in hCMKLR1/L1.2 cells in response to the above proteins and peptides, respectively, in which the different proteins and peptides are represented by the same symbols. The maximal fluorescence intensities triggered by addition of each protein or peptide were plotted and used to determine the EC50 value. One experiment that is representative of at least three independent experiments is shown.
To exclude the possibility of overestimating the actual potency due to additional processing of the proteins into more potent form(s) during the chemotaxis assay (4 h at 37 °C), the proteins were tested for their ability to trigger transient release of intracellular calcium (Fig. 4C). The EC50 values for chem163S and chem158K were 54.2 ± 19.9 nm and 65.2 ± 13.2 nm, respectively. In contrast, chem157S was 50-fold higher more potent (EC50 of 1.17 ± 0.74 nm). In the calcium mobilization assay, chem155A had no detectable agonist activity when tested at concentrations up to 3 μm. Comparison with the activity of the equivalent peptides from the C terminus again showed that the proteins were significantly more active. The EC50 in this calcium transient assay for the 15-mer and 10-mer was >1 μm, 9-mer was 5.92 ± 3.26, 11-mer 343 ± 107, and again there was no detectable activity with FA peptide (EC50 >1 μm). Thus, similar results were obtained in both assays, demonstrating that proteolytic processing to generate chem157S is necessary for maximal activation.
Given that no agonistic effect of chem155A or FA peptide was detected in the calcium mobilization assay, we investigated whether chem155A and FA could display their antiinflammatory role through antagonism toward the same receptor. Fluo-8-loaded cells were pretreated with or without FA, scrambled FA, or chem155A for 10 min, and their antagonistic effects on chem157S or 9mer-elicited calcium influx were examined (Fig. 5A). Calcium release by 100 nm 9-mer was reduced by 34% in the presence of 100-fold molar excess of FA, whereas no inhibition was observed with the scrambled FA peptide, showing specificity of the FA peptide. Interestingly, chem155A showed significantly more competitive potency against 9-mer (by ∼3-fold), suggesting that the N terminus of chemerin contributes to binding to the receptor, consistent with the observation that the active chemerin isoforms are more potent than the corresponding peptides. To confirm the antagonistic effect further, chem157S was utilized as a stimulant in the same experiment (Fig. 5B). A 100-fold higher molar ratio of chem155A to chem157S, but not FA, inhibited chem157S-triggered calcium influx by 50%. Simultaneous stimulation with chem157S and chem155A showed similar results, suggesting that the reduced activity observed for the agonist was not due to desensitization of the receptor or reduction of intracellular calcium pool (data not shown). These data indicated that chem155A has minimal antagonist activity.
FIGURE 5.
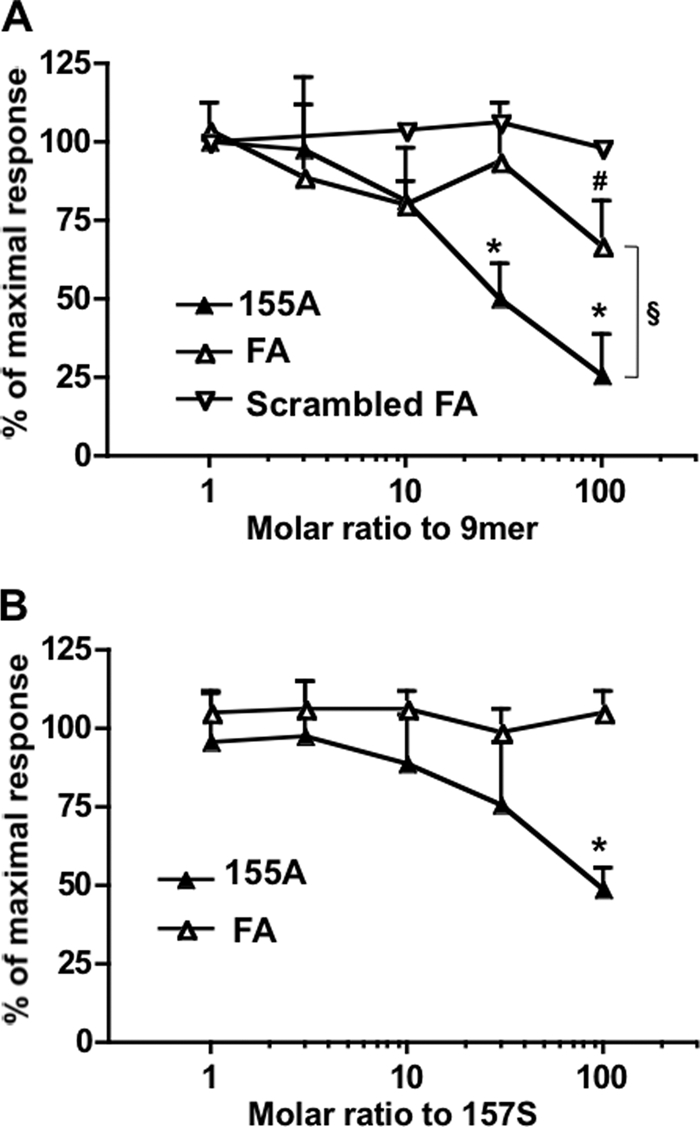
Chem155A and its equivalent C-terminal peptide are weak antagonists of chem157S by calcium flux assay. A, Fluo-8-labeled cells were pretreated with the indicated molar ratio of chem155A protein (▴), FA peptide (△), and scrambled FA peptide, AFQGPFYFSH (▿), for 10 min and then stimulated with 100 nm 9-mer (C-terminal peptide of chem157S). The data are shown as the fluorescence intensity relative to that in the absence of antagonist (n ≥ 3). B, Fluo-8-labeled cells were pretreated with the indicated molar ratio of chem155A protein (▴) and FA peptide (△) for 10 min and then stimulated with 30 nm chem157S. The curves represent the mean ± S.D. of at least three independent experiments. *, p < 0.01; #, p < 0.05 compared with no antagonist by paired Student's t test; §, p < 0.05 when chem155A is compared with FA peptide by paired Student's t test.
Chem157S Elicited Calcium Transients in Human GBM U87-MG Cells
In the accompanying paper (31), we showed that chem158K was markedly elevated in cerebrospinal fluid samples from patients with malignant GBM compared with its level in normal plasma using specific ELISAs for chem163S, 158K, and 157S. This observation led us to investigate whether chemerin plays a role in GBM biology. To compare expression levels of chemerin and its receptors, CMKLR1 and CCRL2 in the different WHO grades of human glioma, an in silico analysis on expression levels of chemerin, CMKLR1, and CCRL2 was performed by retrieving published microarray gene expression data sets containing samples from epilepsy (n = 23), grade II glioma (n = 45), grade III glioma (n = 31), and grade IV glioma (n = 81). Chemerin mRNA was significantly up-regulated in grade III and IV glioma (grade IV is equivalent to malignant GBM) compared with epilepsy and grade II glioma (Fig. 6). In contrast, the CMKLR1 and CCRL2 mRNA levels were unchanged.
FIGURE 6.

In silico analysis of mRNA expression of chemerin, CMKLR1, and CCRL2 in GBM tissues. The raw data for signal intensity (relative fluorescent units, RFU) from the microarray experiment, E-GEOD-4290 available in ArrayExpress at EBI was plotted for chemerin (top), CMKLR1 (middle), and CCRL2 (bottom) compared with their disease classification.
To see whether chemerin could function as a signaling molecule, we first tested expression of chemerin and its receptors in five different GBM cell lines and Jurkat cells (Fig. 7A) by quantitative PCR. Chemerin expression was detected in DBTRG-05MG cells at a relatively high level whereas its receptors, CMKLR1 and CCRL2, were expressed at the highest levels in U-87 MG cells among the cell lines tested. Expression of CMKLR1 and CCRL2 at the protein level was confirmed by flow cytometric analysis (Fig. 7B). These results led us to utilize U-87 MG as a cell model.
FIGURE 7.

Human U-87 MG cells expresses CMKLR1 and CCRL2, and chem157S causes calcium signaling. A, mRNA levels of chemerin and its receptors, CMKLR1, CCRL2, and GPR1 in human glioma cell lines 1321N1, DBTRG-05MG, Hs 683, T98G, U-87 MG, and Jurkat cells were quantified with SYBR Green-based real-time quantitative PCR. The data are expressed as mean ± S.D. of relative gene expression to the Hs 683 cell line. B, U-87 MG cells were analyzed for the expression of human CMKLR1 and CCRL2 by flow cytometry. Isotype-labeled control is shown in purple, and anti-CMKLR1 or anti-CCRL2 shown in green. C, Fluo-8-loaded U-87 MG cells were stimulated with the indicated concentrations of chem157S (100 nm, ■; 30 nm, □; 10 nm, ▴; 3 nm, △; 1 nm, ●; and control, ○) and the fluorescent intensity monitored. The inset shows the maximum fluorescence intensity observed plotted as a function of the chem157S concentration. The data shown are representative of at least three independent experiments. The EC50 was calculated based on the maximal fluorescence intensities observed at each concentration after stimulation.
Addition of chem157S to U-87 MG cells stimulated a transient increase of intracellular calcium in a dose-dependent manner (Fig. 7C). The EC50 calculated for chem157S-induced calcium transients in U-87 MG was 4.45 ± 3.8 nm, which was very close to that found in CMKLR1/L1.2 cells (Table 1). This result demonstrated that active chemerin triggered an intracellular signaling pathway through its functional receptors in U-87 MG cells.
DISCUSSION
In the present study, we developed a single-step purification for large quantities of pure chemerin proteins from mammalian cells using the UCOE® technology. Characterization of the purified proteins using human CMKLR1-transfected L1.2 cells demonstrated that (i) chem157S is the most potent and likely the physiologically active form of chemerin; (ii) chem155A has no agonist activity and weak antagonist activity; (iii) the full-length proteins are more active than the equivalent peptides showing that the N-terminal portion of the molecule plays a role in ligand-receptor binding although the ratio of activity between the protein and peptide depends on the chemerin isoform and the assay. Based on our observation that the level of chem158K was markedly increased in GBM cerebrospinal fluid, we furthermore demonstrated that physiological concentrations of chem157S stimulated intracellular calcium increase through its functional receptors endogenously expressed in human U-87 MG cells. These observations provide additional molecular links among coagulation, inflammation, and cancer.
Previous studies on the function of the different isoforms of chemerin have mainly employed peptides or recombinant proteins that have been refolded from Escherichia coli (12, 23, 24), or yeast (3), or from the suspension culture of insect cells using insect-derived signal peptide (12, 24). The small peptides do not necessarily have the same properties such as solubility and pI as the full-length protein whereas use of the refolded protein is problematic because there is no agreed standard for chemerin activity. In addition, partial proteolysis of recombinant full-length prochemerin can occur during its production and purification preventing rigorous biochemical characterization. To overcome these issues, we produced several of the reported isoforms of chemerin by mammalian expression. This allowed us to fully characterize those proteins in terms of their purity and sequence uniformity and provide a standard definition of mammalian chemerin activity. Additionally, as shown in the accompanying report (31), these purified chemerin isoforms have allowed us to develop ELISAs specific for each isoform.
Consistent with previous reports, in vitro processing of prochemerin showed that plasmin (12) and cathepsin G (5) cleaved chem163S at positions Lys158 and Phe156, respectively. In our study, however, the higher concentrations and longer incubations of both resulted in cleavage at Phe148. To our knowledge, this is a new C-terminal cleavage site for these two proteases in chemerin although a chemerin isoform corresponding to chem148F has not yet been reported in human samples.
When these chemerin isoforms were tested for their agonist activity in Ca2+ mobilization and chemotaxis assays, it was found that the only form with significant activity was chem157S. The EC50 values obtained from these assays were similar to the value of 1.51 nm obtained for binding of chem157S to its receptor, CMKLR1 (19). Both chem163S and 158K possessed only modest activity in agreement with previous reports (13, 19). Similar results were obtained from both assays in terms of the EC50 for chem163S, chem158K, and chem157S. However, no agonist activity was detectable with chem155A in the calcium mobilization assay; but chem155A at a high concentration (300 nm) showed modest activity in a chemotaxis assay. We cannot exclude the possibility that chem155A was being processed into a shorter but more bioactive form during the chemotaxis assay such as chem154F, which was isolated from human hemofiltrate and has been suggested to be active (3).
Because a mouse peptide ending at the equivalent of human chem155A has been shown to be anti-inflammatory both in vitro and in a mouse model of leukocyte infiltration (17, 25), we tested whether chem155A is an antagonist of the most active chemerin form, chem157S. We showed that chem155A is a weak antagonist of chem157S in the calcium mobilization assay. The molar ratio required was very high. Similarly, FA, the peptide equivalent to chem155A, was a weak antagonist of 9-mer, the peptide equivalent to chem157S. FA peptide was, however, unable to antagonize full-length chem157S protein. Full-length chem155A was a better antagonist of 9-mer than FA, confirming the hypothesis that the N-terminal domain of chemerin plays a role in the interactions with its receptors.
An in silico analysis showed that expression of chemerin mRNA was significantly increased in grade III and IV brain gliomas compared with grade II or brain samples from patients with epilepsy, consistent with the observation in mesothelioma (26). Moreover, a positive correlation of expression of chemerin mRNA with tumor size of adrenocortical carcinoma has been reported (27). Similar to the human glioma cell line DBTRG-05MG (28), we also detected expression of chemerin receptors including CMKLR1, CCRL2, and GPR1 in human GBM U-87 MG cells at the mRNA level, and flow cytometry showed that U-87 MG cells expressed chemerin receptors. Treatment with chem157S led to activation of calcium-triggered downstream signaling, demonstrating the functionality of the chemerin receptors. Future studies will determine whether chemerin signaling contributes to GBM cancer biology.
Other chemokines require proteolytic processing to generate the forms with full activity. Human CCL15 and CCL23 along with murine CCL6 and CCL9 form a subfamily of β-chemokines that possess an N-terminal extension and an extra disulfide bond (29). For their full activity the N terminus needs to be cleaved by proteases released by inflammatory cells such as neutrophil elastase or cathepsin G or mast cell chymase. Thus, infiltrating inflammatory cells will modify the local chemokine environment by proteolysis of chemokines (29, 30). Proteolytic cleavage of G protein-coupled receptor ligands could be a more general mechanism for controlling local inflammation.
Prochemerin has been reported to be cleaved by a variety of serine proteases (12); however, the biochemical efficiencies of these cleavages are unclear. By use of a large quantity of recombinant chem163S reported in this study, we can address in detail the proteolytic processing by the different proteases and the activity of the resultant C-terminal cleaved isoforms. These data, coupled with the generation of specific ELISAs for the cleaved chemerin isoforms as demonstrated in the accompanying paper (31), will allow us to define the activation and inactivation pathways of prochemerin in vivo.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants HL057530 and AI085268.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1.
- CMKLR1
- chemokine-like receptor 1
- CCRL2
- chemokine receptor-like 2
- FA peptide
- HSFYFPGQFA
- GBM
- glioblastoma
- GPR1
- G protein-coupled receptor 1
- UBOE
- ubiquitous chromatin opening element.
REFERENCES
- 1. Nagpal S., Patel S., Jacobe H., DiSepio D., Ghosn C., Malhotra M., Teng M., Duvic M., Chandraratna R. A. (1997) J. Invest. Dermatol. 109, 91–95 [DOI] [PubMed] [Google Scholar]
- 2. Wittamer V., Franssen J. D., Vulcano M., Mirjolet J. F., Le Poul E., Migeotte I., Brézillon S., Tyldesley R., Blanpain C., Detheux M., Mantovani A., Sozzani S., Vassart G., Parmentier M., Communi D. (2003) J. Exp. Med. 198, 977–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Meder W., Wendland M., Busmann A., Kutzleb C., Spodsberg N., John H., Richter R., Schleuder D., Meyer M., Forssmann W. G. (2003) FEBS Lett. 555, 495–499 [DOI] [PubMed] [Google Scholar]
- 4. Vermi W., Riboldi E., Wittamer V., Gentili F., Luini W., Marrelli S., Vecchi A., Franssen J. D., Communi D., Massardi L., Sironi M., Mantovani A., Parmentier M., Facchetti F., Sozzani S. (2005) J. Exp. Med. 201, 509–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wittamer V., Bondue B., Guillabert A., Vassart G., Parmentier M., Communi D. (2005) J. Immunol. 175, 487–493 [DOI] [PubMed] [Google Scholar]
- 6. Barnea G., Strapps W., Herrada G., Berman Y., Ong J., Kloss B., Axel R., Lee K. J. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 64–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zabel B. A., Nakae S., Zúñiga L., Kim J. Y., Ohyama T., Alt C., Pan J., Suto H., Soler D., Allen S. J., Handel T. M., Song C. H., Galli S. J., Butcher E. C. (2008) J. Exp. Med. 205, 2207–2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bozaoglu K., Bolton K., McMillan J., Zimmet P., Jowett J., Collier G., Walder K., Segal D. (2007) Endocrinology 148, 4687–4694 [DOI] [PubMed] [Google Scholar]
- 9. Goralski K. B., McCarthy T. C., Hanniman E. A., Zabel B. A., Butcher E. C., Parlee S. D., Muruganandan S., Sinal C. J. (2007) J. Biol. Chem. 282, 28175–28188 [DOI] [PubMed] [Google Scholar]
- 10. Roh S. G., Song S. H., Choi K. C., Katoh K., Wittamer V., Parmentier M., Sasaki S. (2007) Biochem. Biophys. Res. Commun. 362, 1013–1018 [DOI] [PubMed] [Google Scholar]
- 11. Takahashi M., Takahashi Y., Takahashi K., Zolotaryov F. N., Hong K. S., Kitazawa R., Iida K., Okimura Y., Kaji H., Kitazawa S., Kasuga M., Chihara K. (2008) FEBS Lett. 582, 573–578 [DOI] [PubMed] [Google Scholar]
- 12. Zabel B. A., Allen S. J., Kulig P., Allen J. A., Cichy J., Handel T. M., Butcher E. C. (2005) J. Biol. Chem. 280, 34661–34666 [DOI] [PubMed] [Google Scholar]
- 13. Du X. Y., Zabel B. A., Myles T., Allen S. J., Handel T. M., Lee P. P., Butcher E. C., Leung L. L. (2009) J. Biol. Chem. 284, 751–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ernst M. C., Sinal C. J. (2010) Trends Endocrinol. Metab. 21, 660–667 [DOI] [PubMed] [Google Scholar]
- 15. Zabel B. A., Zuniga L., Ohyama T., Allen S. J., Cichy J., Handel T. M., Butcher E. C. (2006) Exp. Hematol. 34, 1021–1032 [DOI] [PubMed] [Google Scholar]
- 16. Guillabert A., Wittamer V., Bondue B., Godot V., Imbault V., Parmentier M., Communi D. (2008) J. Leukocyte Biol. 84, 1530–1538 [DOI] [PubMed] [Google Scholar]
- 17. Cash J. L., Hart R., Russ A., Dixon J. P., Colledge W. H., Doran J., Hendrick A. G., Carlton M. B., Greaves D. R. (2008) J. Exp. Med. 205, 767–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Luangsay S., Wittamer V., Bondue B., De Henau O., Rouger L., Brait M., Franssen J. D., de Nadai P., Huaux F., Parmentier M. (2009) J. Immunol. 183, 6489–6499 [DOI] [PubMed] [Google Scholar]
- 19. Wittamer V., Grégoire F., Robberecht P., Vassart G., Communi D., Parmentier M. (2004) J. Biol. Chem. 279, 9956–9962 [DOI] [PubMed] [Google Scholar]
- 20. Shimamura K., Matsuda M., Miyamoto Y., Yoshimoto R., Seo T., Tokita S. (2009) Peptides 30, 1529–1538 [DOI] [PubMed] [Google Scholar]
- 21. Benton T., Chen T., McEntee M., Fox B., King D., Crombie R., Thomas T. C., Bebbington C. (2002) Cytotechnology 38, 43–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Williams S., Mustoe T., Mulcahy T., Griffiths M., Simpson D., Antoniou M., Irvine A., Mountain A., Crombie R. (2005) BMC Biotechnol. 5, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xiang D., Zhang J., Chen Y., Guo Y., Schalow A., Zhang Z., Hu X., Yu H., Zhao M., Zhu S., Lu H., Wu M., Yu Y., Moldenhauer A., Han W. (2010) Protein Expr. Purif. 69, 153–158 [DOI] [PubMed] [Google Scholar]
- 24. Zabel B. A., Silverio A. M., Butcher E. C. (2005) J. Immunol. 174, 244–251 [DOI] [PubMed] [Google Scholar]
- 25. Cash J. L., Christian A. R., Greaves D. R. (2010) J. Immunol. 184, 5315–5324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mohr S., Bottin M. C., Lannes B., Neuville A., Bellocq J. P., Keith G., Rihn B. H. (2004) Biochimie 86, 13–19 [DOI] [PubMed] [Google Scholar]
- 27. Fernandez-Ranvier G. G., Weng J., Yeh R. F., Khanafshar E., Suh I., Barker C., Duh Q. Y., Clark O. H., Kebebew E. (2008) Arch. Surg. 143, 841–846 [DOI] [PubMed] [Google Scholar]
- 28. Arita M., Bianchini F., Aliberti J., Sher A., Chiang N., Hong S., Yang R., Petasis N. A., Serhan C. N. (2005) J. Exp. Med. 201, 713–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Berahovich R. D., Miao Z., Wang Y., Premack B., Howard M. C., Schall T. J. (2005) J. Immunol. 174, 7341–7351 [DOI] [PubMed] [Google Scholar]
- 30. Miao Z., Premack B. A., Wei Z., Wang Y., Gerard C., Showell H., Howard M., Schall T. J., Berahovich R. (2007) J. Immunol. 178, 7395–7404 [DOI] [PubMed] [Google Scholar]
- 31. Zhao L., Yamaguchi Y., Sharif S., Du X.-Y., Lee D. M., Recht L. D., Robinson W. H., Song J. J., Morser J., Leung L. L. K. (2011) J. Biol. Chem. 286, 39520–39527 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.