

Abstract
Normal human colonic epithelial cells (HCECs) are not immortalized by telomerase alone but also require CDK4. Some human cell types growth-arrest due to stress- or aberrant signaling-induced senescence (stasis). Stasis represents the consequences of growth conditions culture that are inadequate to maintain long-term proliferation. Overexpressed CDK4 titers out p16 and allows cells to ignore the growth arrest signals produced by stasis. To identify factors contributing to the inadequate culture environment, we used a 62,000-member shRNA library to knock down factors cooperating with human telomerase reverse transcriptase (hTERT) in the immortalization of HCECs. Knockdown of Klotho gamma (KLG; also known as KLPH and LCTL) allowed hTERT to immortalize HCECs. KLG is one isoform of the Klotho family of factors that coordinate interaction between different FGF ligands and the FGF receptor. We also found that knockdown of KLG induced another member of the Klotho family, Klotho beta (KLB). Induction of KLB was maintained and could activate ERK1/2 in immortalized cells. Supplementation of the culture medium with the KLB ligand FGF19 had a similar effect on hTERT-expressing HCECs as knockdown of KLG regarding both immortalization and down-regulation of the tumor suppressor Klotho alpha. Together, these data suggest that KLB is an important regulator in the immortalization of HCECs by facilitating FGF19 growth factor signaling.
Keywords: Cellular Senescence, Fibroblast Growth Factor (FGF), Fibroblast Growth Factor Receptor (FGFR), Growth Factors, Signaling, Immortalization, Stasis, Telomerase
Introduction
Human somatic cells have a limited replicative life span in vitro, known as the Hayflick limit (1). Our laboratory has proposed a two-stage model of cellular senescence. Normal human fibroblasts undergo a limited number of divisions in culture and eventually enter a non-dividing state designated replicative senescence or mortality stage 1 because telomeres have shortened enough to induce a DNA damage signal. In the absence of cell cycle check points (e.g. p53 or p16/Rb), cells can continue to divide until extreme telomere shortening initiates a second block to cell division, mortality stage 2 (2). During mortality stage 2 (crisis), end fusions and chromosome breakage cycles result in cell death. The expression of telomerase at levels sufficient to maintain or elongate telomeres can prevent the telomere shortening responsible for both mortality stages 1 and 2 and results in the immortalization of some cell types such as retinal pigment epithelial cells and foreskin fibroblasts (3, 4). However, other cell types growth-arrest due to inadequate culture conditions (lack of growth factors, micronutrients, or cell-cell interactions), termed stasis (stress- or aberrant signaling-induced senescence) (5). This telomere length-independent growth arrest can be bypassed either by adjusting the culture conditions to the needs of the particular cell type or by abrogating the stress-induced check point that triggers growth arrest. WI-38 fetal lung fibroblasts cannot be immortalized by ectopic human telomerase reverse transcriptase (hTERT)2 under standard conditions but are immortalized in low oxygen with media supplements (4). Keratinocytes and mammary epithelial cells up-regulate p16 and growth-arrest when cultivated on plastic dishes. This can be prevented by the use of feeder layers, and on feeder layers, they can be immortalized with hTERT alone (6). For bronchial and colonic epithelial cells, culture conditions allowing immortalization with hTERT have not yet been found. Exogenous CDK4 can bind and prevent p16 inhibition, and in many cases, CDK4, together with hTERT, is sufficient to immortalize cells (7, 8).
Human colonic epithelial cells (HCECs) show premature growth arrest due to p16 proteins under normal culture conditions (8). This HCEC strain can be immortalized by coexpression of hTERT and CDK4. To identify factors or pathways causing stasis in HCECs, we performed an shRNA library screen for factors that allow immortalization in cells expressing hTERT without CDK4. We found that knockdown of Klotho gamma (KLG) was able to cooperate with hTERT to immortalize HCEC. KLG is a member of the Klotho family of receptor coactivators that include Klotho beta (KLB) and the tumor suppressor Klotho alpha (KLA). The ligand for KLG (FGF19) was also able to cooperate with hTERT in HCEC immortalization, even in the absence of KLG knockdown. We suggest a model in which KLG knockdown or exogenous FGF19 produces up-regulation of KLB and sufficient growth stimulatory activity to down-regulate KLA and prevent the tumor suppressor function of KLA from inducing stasis. FGF19 is thus an important growth factor for the culture of HCECs.
EXPERIMENTAL PROCEDURES
Cell Culture and RNA Preparation
HCEC strains 1 and 2 (HCEC1 and HCEC2) were derived and maintained as described previously (8). HCEC1-hTERT and HCEC2-hTERT cells (expressing cDNAs for the catalytic subunit of telomerase) were cultured in medium consisting of medium-X (4:1 DMEM/Medium 199, HyClone), 2% Cosmic calf serum (HyClone), insulin, and gentamycin. HCEC1-hTERT cells were cultured at 37 °C in low oxygen (2–5%). Cells were subcultured when 80% confluent. Total RNA was isolated from 1 × 106 cells using an RNeasy mini kit (Qiagen) according to the manufacturer's procedures. The RNA yield from each replicate was calculated based on the absorbance at 260 nm.
Production of Virus-packaged shRNA Library
The Open Biosystems pGIPZ viral shRNA library was produced as described following the manufacturer's protocol with the following modifications. 293FT cells were propagated in medium-X with 10% Cosmic calf serum. 15 μg of the pGIPZ lentiviral shRNA library plasmids were cotransfected with 7.5 μg of the pAX2 packaging plasmid and 7.5 μg of the pMD2G envelope plasmid using calcium phosphate. Virus-containing supernatant was collected at multiple time points after transfection and used to infect target cells after filtration through a 0.45-μm filter. As the pGIPZ backbone contains GFP, infection efficiency was determined by measuring the percentage of GFP-positive cells by flow cytometry.
Identification of shRNA Clones
The 62,000-member shRNA library contains zero to five shRNAs for different genes. We divided the 62,000-member library into pools of 20,000 shRNAs and infected three sets of duplicate plates of HCEC2-hTERT cells (seven to eight population doublings (PDs)) prior to when they would enter stasis. Cells were infected at a low multiplicity of infection designed to introduce one to two shRNAs/cell. After day 5, these dishes were plated at low density (10,000 cells/15-cm plate) and cultured until control HCEC2-hTERT cells reached stasis.
Any knockdown of a gene contributing to stasis in HCEC2-hTERT cells will have a growth advantage (see Fig. 1B). About 20 colonies/plate were isolated (total of 135 colonies). These were expanded individually until they reached confluency in a T25 flask, and then genomic DNA was isolated for the identification of shRNAs. Individual shRNA cassettes were recovered from the genomic DNA using forward primer TGCCTGAGTTTGTTTGAATGAGGCTT and reverse primer CATAGTTAAGAATACCAGTCAATCTT. The PCR products were then gel-purified using the Qiagen QIAquick PCR purification kit following the manufacturer's protocol. The amplified PCR fragments were subcloned into the TA cloning vector (pGEMT) and sequenced. The recovered shRNA was identified by sequence comparison with the pGIPZ shRNA library database.
FIGURE 1.

Knockdown of KLG cooperates with telomerase to immortalize HCECs. A, HCEC2 cells require CDK4 to overcome stasis in culture and need both hTERT and CDK4 to proliferate indefinitely. B, schematic outline of shRNA screen. Near-stasis HCEC2 cells expressing only telomerase were infected with the shRNA library. After 40 days, the shRNAs in growing HCEC2-hTERT cells were recovered by PCR amplification and identified by sequence analysis. C, shKLG1 recovered in the screen was validated as sufficient to overcome stasis in HCEC2 cells. D, knockdown of KLG immortalizes a second HCEC line. Off-target effects were eliminated by showing that two additional shRNAs to KLG also immortalized the HCEC1-hTERT cell line.
Effects of FGFs on Proliferation of HCEC1-hTERT Cells
To determine the effect of FGFs on HCEC1-hTERT cells, four different FGF ligands (FGF19, FGF21, FGF23, and basic FGF; R&D Systems) were applied to HCEC1-hTERT cells. 5 ng/ml each FGF ligand was added to 10-cm plates, and the medium was changed with fresh FGF ligand every 3 days during the cell culture period.
Western Blotting
Cell lysates were separated by 10% SDS-PAGE. Proteins were transferred to PVDF membranes and incubated with primary antibodies as indicated. Primary antibodies were detected using a secondary HRP-conjugated antibody.
Detection of mRNA Levels
First-strand cDNA of HCEC mRNA was synthesized using 1 μg of total RNA in a 20-μl reverse transcriptase reaction mixture as recommended by the manufacture (Roche Applied Science). A region of the mRNA was amplified using oligo(dT) primers and hexamer primers. The amplified cDNA fragment was generated in 20 μl, and one-twentieth was used to generate the CT value. All real-time PCRs were performed in a 20-μl mixture containing 1 μl of cDNA, 2× reaction buffer (Roche Applied Science), 4 mm MgCl2, 1 μm primers (Table 1), 0.1 μl of Universal probe (Table 1), and 0.2 mm dNTP mixture (Roche Applied Science). The CT value from each template was calculated using Roche 480 software based on the first maximum of the second derivative of the amplification curve of the template. Relative expression levels were normalized with respect to GAPDH.
TABLE 1.
List of primers and Roche Universal probes used in this study
| Gene | Forward primer | Reverse primer | Universal probe |
|---|---|---|---|
| KLG | 5′-ACGGCAGATGTAGCCTGTG-3′ | 5′-GCAGTTCCCTCAGCAGAATG-3′ | 79 |
| Fbx36 | 5′-GCCGGAGACTCTTTGAAAC-3′ | 5′-TCTTCCACCATCTAAAGATTACCTG-3′ | 70 |
| LYN | 5′-GGAGGTGCATCTTCCTCATTT-3′ | 5′-GCTTCATAATCATATAAGGCCACA-3′ | 18 |
| KLB | 5′-ACGGCGACATGGACATTTAC-3′ | 5′-CATCCTCCAGAGCCTGGTC-3′ | 59 |
| c-fos | 5′-GGGGCAAGGTGGAACAGT-3′ | 5′-TCTCCGCTTGGAGTGTATCA-3′ | 46 |
| KLA | 5′-TCCAATGGAATCGATGACG-3′ | 5′-CCATCCAGTATGTGGGCTTT-3′ | 73 |
| GAPDH | 5′-AGCCACATCGCTCAGACAC-3′ | 5′-GCCCAATACGACCAAATCC-3′ | 60 |
| FGF19 | 5′-CGTGCGGTACCTCTGCAT-3′ | 5′-TCGGTACACATTGTAGCCATCT-3′ | 63 |
RESULTS
shRNA Library Screening of HCEC2-hTERT Cells
HCECs were derived from biopsies from two different patients undergoing colonoscopy (8). HCECs stopped dividing after 20–30 PDs in culture and showed minimal additional PDs after expressing telomerase (hTERT). This indicates the presence of inadequate culture conditions producing stasis. Stasis can often be overcome by expressing CDK4, and the cultured life span of the cells was extended by 10–20 PDs by the overexpression of CDK4 alone. HCEC2 cells were immortalized only after expressing CDK4 to block stasis and hTERT to overcome replicative senescence (Fig. 1A). To identify factors that might be contributing to stasis, we infected HCEC2-hTERT cells with a 62,000-member shRNA library. We used three pools of 20,000 shRNAs to infect HCEC2-hTERT cells (seven to eight PDs) prior to stasis. Knockdown of any gene contributing to stasis would confer a growth advantage and potentially permit the cells to immortalize (Fig. 1B). A total of 21 different shRNAs were recovered from 135 colonies. Sequence data indicated that two to three shRNAs were present in most individual cellular clones. Of the 21 gene targets, 12 failed to have any effect when individually tested, six extended the life span of HCEC2-hTERT cells by ∼10 PDs but failed to immortalize the cells, and three were sufficient to cooperate with hTERT and produce continuous cell growth. One of these was against an oncogene homologue (LYN), one was against a protein involved in ubiquitination of multiple products (FBX36), and one was against KLG. The results of studies on KLG (Fig. 1C) are described in detail here. This shRNA against KLG produced a 50% reduction in KLG mRNA compared with uninfected cells.
To rule out off-target effects, several different shRNAs against these genes were tested. As shown in Fig. 1D, two different KLG shRNAs (shKLG2 and shKLG3) also overcame stasis and cooperated with telomerase to immortalize HCEC1-hTERT cells, in contrast to the empty vector (control). shKLG2 and shKLG3 produced a 70 and 50% reduction in KLG mRNA compared with uninfected cells, respectively. Knockdown of KLG did not affect stasis in human skeletal muscle cells (data not shown).
Knockdown of KLG Increases KLB Expression and Activates ERK1/2 in Immortalized Cells
The shRNAs used in these experiments also express GFP as a marker of shRNA expression. Interestingly, the GFP intensity of HCEC1-hTERT-shKLG cells gradually decreased even though the cells continued to grow exponentially (0.9 PD/day) (Fig. 1D) at the later PDs. We examined KLG mRNA levels during the immortalization of HCEC1-hTERT cells. In early HCEC1-hTERT-shKLG2 and HCEC1-hTERT-shKLG3 cells (PD54), the expression levels of KLG were reduced by 50 and 70%, respectively. However, expression levels returned to those in uninfected control cells at PD100 (Table 2, Part A). This suggested that knockdown of KLG is required initially to overcome stasis but that maintaining the knockdown is inhibitory for growth and that ultimately some other compensatory change selects for a return to normal levels of KLG expression. This suggested that additional factor(s) are necessary for the long-term maintenance of proliferation of HCEC1-hTERT cells.
TABLE 2.
Changes in expression by quantitative PCR normalized to pre-stasis HCEC1-hTERT cells (±S.D.)
| Part A. KLG after shKLG | |
| PD54 | 1.0 ± 0.02 |
| shKLG2 PD50 | 0.5 ± 0.05 |
| shKLG3 PD50 | 0.3 ± 0.15 |
| shKLG2 PD100 | 1.1 ± 0.12 |
| shKLG3 PD100 | 1.1 ± 0.32 |
| Part B. KLB after shKLG | |
| PD58 | 1.0 ± 0.13 |
| shEmptya PD58 | 1.1 ± 0.36 |
| shKLG3 PD117 | 8.9 ± 0.31 |
| Part C. KLB in FGF19-immortalized cells | |
| PD53 | 1.0 ± 0.04 |
| FGF19 PD109 | 29.1 ± 1.51 |
| Part D. c-fosexpression | |
| PD56 | 1.00 ± 0.08 |
| shEmpty PD59 | 0.80 ± 0.02 |
| FGF19-immortalized PD103.3 | 0.05 ± 0.00 |
| shKLG3 PD117.7 | 2.67 ± 0.08 |
| Part E. c-fosafter acute FGF19 stimulation | |
| PD50.3 | 1.00 ± 0.10 |
| PD50.3, 40-min FGF19 | 0.89 ± 0.04 |
| FGF19 PD103.3 | 0.21 ± 0.03 |
| FGF19 PD103.3, 40-min FGF19 | 2.92 ± 0.48 |
| Part F. KLB after acute FGF19 | |
| PD53 | 1.0 ± 0.14 |
| PD53, 40-min FGF19 stimulation | 0.9 ± 0.09 |
| shEmpty PD58 | 1.3 ± 0.16 |
| FGF19 PD103.3 | 7.3 ± 1.29 |
| FGF19 PD103.3, 40-min FGF19 stimulation | 5.1 ± 0.65 |
| shKLG3 PD117 | 7.7 ± 1.37 |
| Part G. KLG | |
| PD56.1 | 1.0 ± 0.17 |
| shEmpty PD58.3 | 0.9 ± 0.39 |
| shKLG PD129.1 | 1.0 ± 0.05 |
| FGF19 PD103.1 | 1.0 ± 0.07 |
a shEmpty, empty shRNA.
KLG is a single-pass transmembrane protein that binds to FGF receptors (FGFRs) and increases their affinity for FGF19 (9). KLB, another member of the Klotho gene family, has the same function as KLG (10). We next investigated whether there had been any changes in KLB or its downstream targets. RT-PCR showed that KLB mRNA levels increased by 9-fold in the immortalized HCEC1-hTERT cells (Table 2, Part B). ERK1/2 is a direct downstream target of KLB. Lysates from HCEC1-hTERT cells were harvested before stasis and after immortalization by three different KLG shRNAs. Fig. 2 shows that after KLG levels had returned to normal levels (PD100), cells infected with all three KLG shRNAs produced an up-regulation of the phosphorylated (activated) forms of ERK1/2, whereas the levels of total ERK1/2 remained constant.
FIGURE 2.
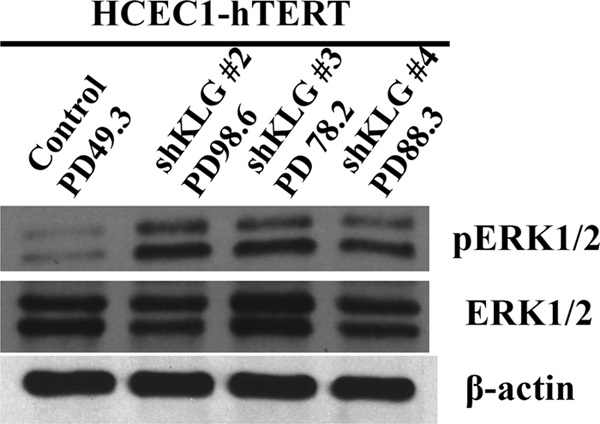
Changes in signaling after HCEC-hTERT immortalization. Phospho-ERK1/2 levels were increased at a time when KLG levels had returned to normal. ERK1/2 is a downstream target of KLB.
FGF19 Signaling Is Sufficient to Activate KLB, Which Is Required for Immortalization of HCEC1-hTERT Cells
KLG and KLB are co-receptors for FGF19 (9, 10). We thus tested excess amounts of four different FGF ligands (FGF19, FGF21, FGF23, and basic FGF) on HCEC1-hTERT cells. Only FGF19 cooperated with hTERT to immortalize cells and produced significant colony growth formation (Fig. 3). Cells immortalized with FGF19 exhibited a 29-fold increase in KLB levels (Table 2, Part C) but only 8-fold in other experiments, supporting the evidence that KLB signal transduction is involved in avoiding stasis. Knockdown of either KLG or KLB in FGF19-immortalized HCEC1-hTERT cells slowed their growth, confirming the contribution of both factors to proliferation in these cells.
FIGURE 3.

FGF19 cooperates with telomerase to immortalize HCEC-hTERT cells. A, cells were cultured in 5 ng/ml each FGF ligand. Only FGF19 overcame stasis. bFGF, basis FGF. B, HCEC1-hTERT cells were plated at 1000 cells/10-cm dish in the presence of 5 ng/ml each FGF ligand. Only FGF19 prevented stasis and permitted vigorous clonal growth.
We examined whether endogenous FGF19 expression is maintained during the immortalization of HCEC1-hTERT cells by shKLG or exogenous FGF19. Endogenous FGF19 expression levels were maintained prior to stasis but were undetectable in immortalized cells (Fig. 4A). FGF19 expression was thus altered even in the shKLG cells that were not exposed to feedback inhibition by exposure to exogenous FGF19.
FIGURE 4.

Changes in expression after HCEC-hTERT immortalization. A, FGF19 expression levels were suppressed after immortalization with either exogenous FGF19 or shKLG. shEmpty, empty shRNA. B, KLA levels were suppressed after immortalization with either shKLG or exogenous FGF19.
KLB Target Genes Can Be Activated during Immortalization of HCEC1-hTERT Cells
FOS is a well characterized KLB target gene (11) and an important gene for continuous cell proliferation in cancer cells (12). We examined mRNA expression levels of c-fos in both immortalized cell lines (HCEC1-hTERT-shKLG and HCEC1-hTERT-FGF19) by real-time PCR. Relative expression levels of c-fos were increased by ∼2.7-fold in HCEC1-hTERT-shKLG cells compared with prestasis control cells (Table 2, Part D). Surprisingly, the relative expression level of c-fos was decreased by 20-fold in cells immortalized with FGF19. This was unexpected, and we hypothesized that it might be related to the specific time point of this cell harvest (2 days after feeding with FGF19). c-fos is an early-response gene, so we examined early time points (before and 40 min after FGF19 stimulation). In this experiment, not only was the basal level of c-fos only 5-fold lower than in prestasis cells, but it increased by 14-fold following FGF19 stimulation (Table 2, Part E). This confirmed that FGF19 stimulation activates the KLB target gene c-fos. We also found that acute FGF19 stimulation did not affect KLB expression (Table 2, Part F).
Klotho Family Members Can Be Coordinately Activated during Immortalization of HCEC1-hTERT Cells
Klotho family members form tertiary complexes with multiple FGFRs and endocrine FGFs (FGF19, FGF21, and FGF23) to activate downstream signaling events (13). We asked whether immortalization of HCEC1-hTERT cells requires all of the Klotho family members. As described above, mRNA levels of KLB were increased following both methods of immortalization (HCEC1-hTERT-shKLG and HCEC1-hTERT-FGF19) (Table 2, Parts B and C). In contrast, although KLA mRNA was present in prestasis cells, it was not detectable in either immortalized cell line (Fig. 4B). The same result was confirmed by quantitative PCR (data not shown). This suggests that down-regulation of KLA may be contributing to cell proliferation after cells overcome stasis. We also examined KLG expression in the cells immortalized by FGF19 (Table 2, Part G). KLG levels remained unchanged. Table 2 (Part G) also confirms the previous result that KLG levels in the shKLG cells had returned to normal levels after immortalization.
DISCUSSION
In this study, we have demonstrated that knockdown of KLG overcame stasis and allowed hTERT to immortalize two different colonic epithelial cell strains, HCEC1-hTERT and HCEC2-hTERT. KLG is one isoform of the Klotho family of factors that interact with the FGFR. Knockdown of KLG led to the disappearance of KLA and increased the expression of KLB. We found evidence that the increase in KLB expression activated the ERK1/2 proteins in the immortalized cells. Together, these data suggest that the dynamic regulation of KLA and KLB is involved in the immortalization of HCEC1-hTERT cells by accelerating growth factor signaling.
KLB Signaling Is Involved in Senescence Signaling
It is well known that up-regulation of p21 is associated with cell cycle arrest at the G1/S boundary (14, 15). Several studies have indicated that decreased Klotho (KLB) expression can produce p53/p21-induced stasis. RNAi with KLB in MRC-5 human primary fibroblasts results in premature senescence with a concomitant up-regulation of p21 (16). Human vascular endothelial cells exposed to recombinant soluble Klotho are resistant to the senescence induced by hydrogen peroxide or etoposide (17). Neither p21 nor p53 levels were increased in immortalized HCEC1-hTERT-shKLG or HCEC1-hTERT-FGF19 cells (data not shown), consistent with the overexpression of KLB that we observed. There may be differences in Klotho signaling during stasis in different cell types because we found that knockdown of KLG did not overcome stasis in human skeletal muscle cells expressing hTERT.
Effect of FGF19 in HCEC1-hTERT Cells
It is not clear how shKLG immortalizes HCEC1-hTERT cells in the absence of endogenous FGF19 expression. Exogenous FGF19 was able to cooperate with hTERT to immortalize HCECs. It is possible that the serum we used contained sufficient FGF19, which could then interact with the increased KLB protein to activate the downstream signaling.
The Klotho family forms a binary complex with multiple FGFRs in the presence of FGF ligands. KLB can facilitate the interaction between FGF19 or FGF21 and FGFRs in the membrane to activate the downstream signaling (10, 18). KLA can facilitate the interaction between a different FGF ligand (FGF23) and FGFRs. It remains undetermined why FGF19 but not FGF21 was able to immortalize HCEC1-hTERT cells. FGF19 and FGF21 signal through KLB-FGFR4 and KLB-FGFR1, respectively (18, 19). Quantitative PCR analysis indicated that HCEC1-hTERT cells expressed ∼100 times more FGFR1 mRNA than FGFR4 mRNA, so failure to express FGFR1 transcripts cannot explain this result. Given that neither shKLG- nor FGF19-treated HCEC1-hTERT cells expressed KLA (Fig. 4), it is obvious why FGF23 had no effect, but it is not clear why KLA expression disappeared. FGF2 (basic FGF) has been found to be required for the immortalization of some cell types by TERT (20–23). However, basic FGF could not immortalize the HCEC1-hTERT cells, suggesting that canonical FGF signaling is not involved in this immortalization pathway. We anticipate that many specialized cell types will require different constellations of specific growth factors for their long-term proliferation given the diversity of growth factors expressed during development.
Klotho Family and Cancer
Recent studies have revealed a link between FGF19 and colon cancer. FGF19 is expressed in colon tumors and in a subset of human colon cancer cell lines and is required for the growth of colon cancer xenografts (24). FGF19 expression is induced after treatment with chemotherapeutic agents in a pregnane X receptor-dependent manner and is required for regrowth of colon cancer cells (25). The mRNA for KLA was expressed (low CT value in quantitative PCR) prior to stasis, and KLA expression then later disappeared after immortalization (Fig. 4). This is consistent with evidence that KLA can function as a tumor suppressor that is activated during stasis. Loss of KLA mRNA has been observed in several cervical cancer cell lines, and the promoter region of KLA showed CpG hypermethylation in 41% (9/22) of invasive carcinoma cases (26). KLA has also been reported to function as a secreted Wnt antagonist and as a tumor suppressor (27). Silencing of KLA mRNA may occur during the late phase of cervical tumorigenesis, and the consequent functional loss of KLA as a secreted Wnt antagonist may contribute to aberrant activation of the canonical Wnt pathway in cervical carcinoma.
KLB Signaling and Effector Gene(s)
Immortalized HCEC1-hTERT cells exhibited a significant increase in the phosphorylation of ERK1/2 compared with control (empty shRNA) cells. Cells immortalized with either shKLG or FGF19 also up-regulated c-fos expression (Fig. 2 and Table 2, Part E). Other cell cycle regulators could also be important. It is unknown whether c-fos gene expression levels are different in colon cancer cells and these normal colonic cells.
Model for Klotho and FGF19 Interactions in Immortalizing HCEC-hTERT Cells
Normal early passage HCECs express KLG, KLB, and KLA. KLG levels would be low, whereas KLA levels would be insufficient to activate the growth-arresting tumor suppressor pathway (Fig. 5A). Growth in the absence of excess FGF19 (inadequate culture conditions; partial occupancy by FGF19 is indicated by shadowing and dashed underlining of the FGF19 label) would produce an up-regulation of the tumor suppressor activity of KLA, resulting in stasis (Fig. 5B). Knockdown of KLG would induce compensatory up-regulation of KLB, a redundant co-receptor for FGF19. Because the ability of KLB to support FGF19 signaling is more potent than that of KLG (9, 10), up-regulation of KLB would enhance the effect of endogenous or exogenous FGF19. Cells that have regained KLG expression would have a growth advantage over those that have not (Fig. 5C). Adding FGF19 would increase KLG and KLB signaling and remove the stimulus for KLA expression (Fig. 5D). The mechanism for the up-regulation of KLB following exposure to exogenous FGF19 is unknown.
FIGURE 5.

Suggested model of immortalization by KLG knockdown or FGF19 treatment in HCEC-hTERT cells. A, before cells reach stasis, KLG and KLB both contribute proliferative signals. KLA signaling is weak in the absence of cellular stress. B, stasis is the result of inadequate FGF19-induced signaling and increased activity of the tumor suppressor KLA. C, knockdown of KLG induces a compensatory up-regulation of KLB, which is a more potent transducer of FGF19 than KLG. The disappearance of KLA also removes negative growth signals. D, excess exogenous FGF19 increases KLB levels and KLG and KLB signaling. This is accompanied by the down-regulation of KLA and its tumor suppressor effects.
It is important to have culture conditions that support the long-term proliferation of normal specialized cell types so that their study is not confounded by stress responses that could alter experimental results. This study has defined FGF19 as a growth factor sufficient to overcome stasis and permit telomerase to immortalize colonic epithelial cells. These cells now provide a common cellular reagent that can be used by many different laboratories to study HCEC biology without the difficulties and lack of reproducibility encountered using primary cultures. This should greatly facilitate advances in understanding normal HCEC behavior and how it is altered during cancer progression and in other diseases.
Note Added in Proof
LYN was a second shRNA hit in our screen. LYN down-regulates the stem cell growth factor (SCF) receptor. 10 ng/ml SCF also immortalized HCEC1-hTERT cells (in the absence of FGF19).
This work was supported, in whole or in part, by National Institutes of Health Grants AG01228 (to W. E. W.) and AG019712 and DK091392 (to M. K.).
- hTERT
- human telomerase reverse transcriptase
- HCEC
- human colonic epithelial cell
- KLG
- Klotho gamma
- KLB
- Klotho beta
- KLA
- Klotho alpha
- PD
- population doubling
- shKLG
- KLG shRNA
- FGFR
- FGF receptor.
REFERENCES
- 1. Hayflick L. (1979) J. Invest. Dermatol. 73, 8–14 [DOI] [PubMed] [Google Scholar]
- 2. Shay J. W., Wright W. E. (2005) Carcinogenesis 26, 867–874 [DOI] [PubMed] [Google Scholar]
- 3. Bodnar A. G., Ouellette M., Frolkis M., Holt S. E., Chiu C. P., Morin G. B., Harley C. B., Shay J. W., Lichtsteiner S., Wright W. E. (1998) Science 279, 349–352 [DOI] [PubMed] [Google Scholar]
- 4. Forsyth N. R., Evans A. P., Shay J. W., Wright W. E. (2003) Aging Cell 2, 235–243 [DOI] [PubMed] [Google Scholar]
- 5. Drayton S., Peters G. (2002) Curr. Opin. Genet. Dev. 12, 98–104 [DOI] [PubMed] [Google Scholar]
- 6. Ramirez R. D., Morales C. P., Herbert B. S., Rohde J. M., Passons C., Shay J. W., Wright W. E. (2001) Genes Dev. 15, 398–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ramirez R. D., Sheridan S., Girard L., Sato M., Kim Y., Pollack J., Peyton M., Zou Y., Kurie J. M., Dimaio J. M., Milchgrub S., Smith A. L., Souza R. F., Gilbey L., Zhang X., Gandia K., Vaughan M. B., Wright W. E., Gazdar A. F., Shay J. W., Minna J. D. (2004) Cancer Res. 64, 9027–9034 [DOI] [PubMed] [Google Scholar]
- 8. Roig A. I., Eskiocak U., Hight S. K., Kim S. B., Delgado O., Souza R. F., Spechler S. J., Wright W. E., Shay J. W. (2010) Gastroenterology 138, 1012–1021e1–5 [DOI] [PubMed] [Google Scholar]
- 9. Fon Tacer K., Bookout A. L., Ding X., Kurosu H., John G. B., Wang L., Goetz R., Mohammadi M., Kuro-o M., Mangelsdorf D. J., Kliewer S. A. (2010) Mol. Endocrinol. 24, 2050–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kurosu H., Choi M., Ogawa Y., Dickson A. S., Goetz R., Eliseenkova A. V., Mohammadi M., Rosenblatt K. P., Kliewer S. A., Kuro-o M. (2007) J. Biol. Chem. 282, 26687–26695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lin B. C., Wang M., Blackmore C., Desnoyers L. R. (2007) J. Biol. Chem. 282, 27277–27284 [DOI] [PubMed] [Google Scholar]
- 12. Ashida R., Tominaga K., Sasaki E., Watanabe T., Fujiwara Y., Oshitani N., Higuchi K., Mitsuyama S., Iwao H., Arakawa T. (2005) Inflammopharmacology 13, 113–125 [DOI] [PubMed] [Google Scholar]
- 13. Goetz R., Nakada Y., Hu M. C., Kurosu H., Wang L., Nakatani T., Shi M., Eliseenkova A. V., Razzaque M. S., Moe O. W., Kuro-o M., Mohammadi M. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 407–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sherr C. J., Roberts J. M. (1995) Genes Dev. 9, 1149–1163 [DOI] [PubMed] [Google Scholar]
- 15. Zhu H., Zhang L., Wu S., Teraishi F., Davis J. J., Jacob D., Fang B. (2004) Oncogene 23, 4984–4992 [DOI] [PubMed] [Google Scholar]
- 16. de Oliveira R. M. (2006) FEBS Lett. 580, 5753–5758 [DOI] [PubMed] [Google Scholar]
- 17. Ikushima M., Rakugi H., Ishikawa K., Maekawa Y., Yamamoto K., Ohta J., Chihara Y., Kida I., Ogihara T. (2006) Biochem. Biophys. Res. Commun. 339, 827–832 [DOI] [PubMed] [Google Scholar]
- 18. Wu X., Li Y. (2009) Aging 1, 1023–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kharitonenkov A., Dunbar J. D., Bina H. A., Bright S., Moyers J. S., Zhang C., Ding L., Micanovic R., Mehrbod S. F., Knierman M. D., Hale J. E., Coskun T., Shanafelt A. B. (2008) J. Cell. Physiol. 215, 1–7 [DOI] [PubMed] [Google Scholar]
- 20. Nisato R. E., Harrison J. A., Buser R., Orci L., Rinsch C., Montesano R., Dupraz P., Pepper M. S. (2004) Am. J. Pathol. 165, 11–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Techangamsuwan S., Imbschweiler I., Kreutzer R., Kreutzer M., Baumgärtner W., Wewetzer K. (2008) Brain Res. 1240, 31–38 [DOI] [PubMed] [Google Scholar]
- 22. Techangamsuwan S., Kreutzer R., Kreutzer M., Imbschweiler I., Rohn K., Wewetzer K., Baumgärtner W. (2009) J. Neurosci. Methods 176, 112–120 [DOI] [PubMed] [Google Scholar]
- 23. Kurz D. J., Hong Y., Trivier E., Huang H. L., Decary S., Zang G. H., Lüscher T. F., Erusalimsky J. D. (2003) Arterioscler. Thromb. Vasc. Biol. 23, 748–754 [DOI] [PubMed] [Google Scholar]
- 24. Desnoyers L. R., Pai R., Ferrando R. E., Hötzel K., Le T., Ross J., Carano R., D'Souza A., Qing J., Mohtashemi I., Ashkenazi A., French D. M. (2008) Oncogene 27, 85–97 [DOI] [PubMed] [Google Scholar]
- 25. Wang H., Venkatesh M., Li H., Goetz R., Mukherjee S., Biswas A., Zhu L., Kaubisch A., Wang L., Pullman J., Whitney K., Kuro-o M., Mohammadi M., Mank S. (2011) J. Clin. Invest., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee J., Jeong D. J., Kim J., Lee S., Park J. H., Chang B., Jung S. I., Yi L., Han Y., Yang Y., Kim K. I., Lim J. S., Yang I., Jeon S., Bae D. H., Kim C. J., Lee M. S. (2010) Mol. Cancer 9, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu H., Fergusson M. M., Castilho R. M., Liu J., Cao L., Chen J., Malide D., Rovira, Schimel D., Kuo C. J., Gutkind J. S., Hwang P. M., Finkel T. (2007) Science 317, 803–806 [DOI] [PubMed] [Google Scholar]