

Background: Intracellular trafficking of MT1-MMP is essential for its role in tumor cell invasion.
Results: Mutation of Thr567 in MT1-MMP altered its internalization and recycling and associated biochemical signaling.
Conclusion: Phosphorylation of MT1-MMP at Thr567 regulates its intracellular trafficking, which is coupled to integrin trafficking and ERK phosphorylation.
Significance: Phosphorylation of MT1-MMP may be a regulatory point for control of tumor cell invasion.
Keywords: ERK, Invasion, Matrix Metalloproteinase (MMP), Phosphorylation, Snare
Abstract
In multicellular organisms, uncontrolled movement of cells can contribute to pathological conditions, such as multiple sclerosis and cancer. In highly aggressive tumors, the expression of matrix metalloproteinases (MMPs) is linked to the capacity of tumor cells to invade surrounding tissue and current research indicates that the membrane-anchored membrane type 1-matrix metalloproteinase (MT1-MMP) has a central role in this process. Endocytosis and trafficking of MT1-MMP are essential for its proper function, and here we examine the phosphorylation, internalization, and recycling of this enzyme, and the associated biochemical signaling in HeLa and HT-1080 fibrosarcoma cells. Activation of protein kinase C with phorbol 12-myristate 13-acetate resulted in phosphorylation of endogenous MT1-MMP at Thr567 in vivo. Mutation of Thr567 to alanine (to mimic non-phosphorylated MT1-MMP) reduced internalization of MT1-MMP, whereas mutation of Thr567 to glutamic acid (to mimic phosphorylation) resulted in decreased levels of MT1-MMP on the cell surface. The endosomal trafficking and recycling of MT1-MMP was found to be dependent upon Rab7 and VAMP7, and blocking the function of these proteins reduced cell migration and invasion. Intracellular trafficking of MT1-MMP was observed to be coupled to the trafficking of integrin α5 and phosphorylation of ERK that coincided with this was dependent on phosphorylation of MT1-MMP. Together, these results reveal important roles for MT1-MMP phosphorylation and trafficking in both cell signaling and cell invasion.
Introduction
The ability of cells to invade into and migrate through their surrounding environment is a crucial component in the development and homeostasis of all multicellular organisms and can be a primary contributor to the progression of disease (1). The extracellular matrix (ECM)3 acts as both a supportive scaffold and a physical barrier, maintaining tissue architecture (2), and degradation of ECM proteins disrupts this structure, enabling cell invasion through the matrix. Matrix metalloproteinases (MMPs) constitute a family of zinc-dependent proteolytic enzymes that function to degrade the ECM (3, 4). MMP family members can be subdivided into membrane type MMPs and soluble MMPs. Membrane type MMPs contain either a transmembrane domain or glycosylphosphatidylinositol anchors, which tether them to the plasma membrane (5), whereas soluble MMPs are secreted by cells into the extracellular space, where they can diffuse into the ECM.
The expression of MMPs has been intimately linked with highly aggressive cancers, such as ovarian, melanoma, breast, and colorectal (6–9), and recent research indicates that the membrane-anchored membrane type 1 matrix metalloproteinase (MT1-MMP) has a central role in the aggressive behavior of some tumor types (2, 8, 10, 11). MT1-MMP can directly degrade the ECM by proteolytic cleavage of collagen types I, II, and III, laminins 1 and 5, fibronectin, vitronectin, fibrin, and aggrecan and can indirectly degrade the ECM through activating pro-MMP-2 (12). Activation of MT1-MMP is accomplished intracellularly by the Golgi-associated enzyme furin, which recognizes the unique amino acid sequence (RRKR) located between the prodomain and catalytic domain (7, 12). Once activated, MT1-MMP is trafficked to the plasma membrane through a Rab8-dependent pathway (13). At the plasma membrane, MT1-MMP-mediated proteolytic degradation of ECM proteins facilitates cell invasion. The proteolytic activity of MT1-MMP can be attenuated through internalization, which occurs through both clathrin-mediated and caveolin-mediated pathways (14, 15). It has also been demonstrated that internalized MT1-MMP can be recycled back to the plasma membrane (5). Importantly, deletion of the cytoplasmic tail of MT1-MMP inhibits internalization, and this can perturb cell invasion, demonstrating the importance of recycling for proper enzyme function (16).
Trafficking of cellular material between the plasma membrane and intracellular compartments is dependent upon SNARE (soluble N-ethylmaleimide-sensitive factor-activating protein receptor) proteins, which facilitate the fusion of vesicles with target membranes. Several recent studies support a role for SNAREs in MT1-MMP localization; SNAP23, syntaxin 13, VAMP3 (17), TI-VAMP/VAMP7 (18), and syntaxin4 (19) have been shown to be involved in trafficking of MT1-MMP and cell invasion. These functions of SNAREs complement their established roles in cell-ECM adhesion and migration (20–22).
Having characterized the requirement for SNAREs in trafficking of MT1-MMP and efficient tumor cell invasion, in the present study we examine the internalization of MT1-MMP and its trafficking from endosomal compartments back to the plasma membrane. We report that MT1-MMP is phosphorylated at Thr567 in a protein kinase C-dependent manner and that this phosphorylation is required for MT1-MMP internalization. Once internalized, MT1-MMP is recycled through the late endosome in a VAMP7-dependent manner. Inhibiting either the internalization or the recycling of MT1-MMP significantly reduced the ability of cells to invade through a Matrigel matrix. Interestingly, mutation of Thr567 to alanine resulted in increased cell surface expression of MT1-MMP but impaired cellular invasive and migratory abilities. This suggests that the internalization and recycling of MT1-MMP functions to promote invasion and migration through mechanisms beyond ECM degradation. Consistent with this, we found evidence that trafficking/recycling of MT1-MMP is coupled to trafficking of α5 integrin and stimulates phosphorylation of ERK.
EXPERIMENTAL PROCEDURES
Reagents and cDNA Constructs
All chemicals were purchased from Sigma or Fisher unless otherwise indicated. Antibodies to the following proteins were obtained from the indicated suppliers: MT1-MMP, GFP, and Rab7 (Abcam; ab3644, ab290, and ab50533); phospho-Ser/Thr, Rab5, Rab11, and α5 integrin (BD Biosciences; 610281, 610656, and 610634); α5β1 (Chemicon; ab1950); and ERK and phospho-ERK (Cell Signaling; 9107 and 4377). Secondary antibodies and rhodamine-labeled phalloidin were purchased from Invitrogen.
The pEGFP Rab5WT, Rab7WT, Rab8WT, Rab11WT, and pECFP Rab7DN were kind gifts from Dr. John Brumell (Hospital For Sick Children, Toronto). GFP MT1-MMP-WT, T567A, and T567E were created through PCR amplification of MT1-MMP (OpenBiosystems) and cloned into pEGFP-N1. mCherry MT1-MMP was generated by inserting the XhoI/AgeI fragment from MT1-MMP-GFP into mCherry-N1. VAMP7FL and VAMP7C were created through PCR amplification of VAMP7 (OpenBiosystems) and cloned into pEGFP-N1. The following oligonucleotides were used as primers: MT1-MMP-FOR (5′-TATAAAGAATTCGGTGGTCTCGGACCATGTC-3′), MT1-MMP-WT-REV (5′-TTTATAATCCCGCGGGACCTTGTCCAGCAGGGAAC-3′), MT1-MMP-T567A-REV (5′-ATAAAAAAGCTTTTATTATTTATCATCATCATCTTTATAATCCCGCGGGACCTTGTCCAGCAGGGAACGCTGGCAGTAGAGCAGTCGCCTGGGGGCCCC-3′), MT1-MMP-T567E-REV (5′-ATAAAAAAGCTTTTATTATTTATCATCATCATCTTTATAATCCCGCGGGACCTTGTCCAGCAGGGAACGCTGGCAGTAGAGCAGTCGCCTGGGCTCCCC-3′), VAMP7-FOR (5′-ATAAAAAGCTTAAGCCATGGCGATTCTTTTT-3′), VAMP7FL-REV (5′-ATAAAAGGATCCCCGCGGTTTCTTCACACAGCTTGG-3′), and VAMP7C-REV (5′-ATAAAAGGATCCCCGCGGCATGGCTCGAGCAAGATTTC-3′).
Cell Culture and Transfection
HeLa cells and HT-1080 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Sigma) supplemented with 10% fetal bovine serum (Sigma) under 5% CO2 at 37 °C. Cells were transfected with Polyfect (Qiagen) transfection reagent as described by the manufacturer's protocol. All transfected constructs were expressed for 24–36 h.
Immunofluorescence Microscopy
Cells were grown on glass coverslips coated with fibronectin-coated (20 μg/ml), serum-starved, and treated with PMA where indicated and subsequently fixed and permeabilized with 2% (w/v) paraformaldehyde, 0.2% Triton X-100 in PBS. Samples were then blocked with 5% (w/v) bovine serum albumin (BSA) powder/PBS before staining with primary and secondary antibody, followed by washing and mounting. Samples were imaged using a 63× (numerical aperture 1.4) lens on a Leica DM-IRE2 inverted microscope with a Leica TCS SP2 system (Leica, Heidelberg, Germany). Images were captured, and three-dimensional reconstructions were performed using the Leica Confocal Software package.
Immunoprecipitation
Cyanogen bromide-activated Sepharose beads (Sigma) were coated with antibody as per the manufacturer's instructions. Cells were lysed in 1% Nonidet P-40, 2 mm EDTA, 10% glycerol, 137 mm NaCl, 20 mm Tris-HCl, pH 8.0, 10 mm NaF, 10 mm Na4P2O7, 0.2 mm Na3VO4, and protease inhibitor mixture (Sigma). Lysate was incubated with antibody-bound beads overnight at 4 °C and washed three times with lysis buffer followed by four washes with 0.1% Tween/PBS. Bound proteins were eluted using 2.5× SDS running buffer, heated to 100 °C. Proteins were separated using SDS-PAGE and analyzed by Western blotting.
MT1-MMP Trafficking
Cells were grown on fibronectin-coated glass coverslips and serum-starved for 3 h in DMEM. 500 nm PMA was added to cells to induce trafficking of MT1-MMP to the cell surface. Cells were washed with ice-cold PBS and incubated with 1% BSA/PBS for 20 min at 4 °C on ice to prevent internalization of MT1-MMP. Anti-MT1-MMP antibodies were added to the cells at 8 μg/ml for 1 h and/or anti-α5 antibodies at 5 μg/ml. Cells were rinsed with 0.2 m glycine/HCl, pH 5.0, followed by successive rinses with PBS to remove any unbound antibody and either fixed and permeabilized with 2% (w/v) paraformaldehyde, 0.2% Triton X-100 in PBS, or slowly warmed in serum-free medium to 37 °C to monitor internalization. After fixation, cells were incubated with AlexaFluor647 and/or AlexaFluor594 secondary antibody in 1% BSA/PBS for 1 h. MT1-MMP surface expression was examined using the 63× (numerical aperture 1.4) lens of a Leica inverted microscope with constant gain and pin hole parameters. Cell surface expression was quantified for 15–25 cells per experiment using ImageJ software. For removal of TIMP-2 (tissue inhibitor of matrix metalloproteinase-2) from the cell surface, cells grown on coverslips were washed with 150 mm NaCl, and 50 mm glycine/HCl, pH 3.0 (5 min on ice). Cells were then rinsed with PBS and incubated with TIMP-2 antibody for 1 h at 4 °C prior to fixation with 4% PFA. After fixation, cells were incubated with rhodamine-phalloidin and/or AlexaFluor488 secondary antibody in 1% BSA/PBS for 1 h. MT1-MMP surface expression was examined using the 63× (numerical aperture 1.4) lens of a Leica inverted microscope with constant gain and pin hole parameters.
Cell Invasion Assay
Cell culture inserts, in 24-well dishes (Costar), were prepared with and without (control) Matrigel. The bottom chamber was coated with 20 μg/ml fibronectin, and the upper chamber was coated with 0.125 mg/ml growth factor-reduced Matrigel (BD Biosciences). HT-1080 or HeLa cells were transfected for 12 h, at which point they were lifted and seeded onto Matrigel-coated and control (without Matrigel) upper surface in 0.1% BSA/serum-free medium (80,000 cells/well). The cells that invaded toward chemoattractant (10% FBS, 0.1% BSA) in the lower chamber and penetrated the Matrigel were fixed with 4% paraformaldehyde, stained with DAPI, and counted. Cells that did not invade were removed with a cotton swab prior to fixation of sample. Ten fields of cells per membrane were counted. The data are presented as the number of cells that invaded through the Matrigel divided by the number of cells that migrated through the control insert (setting mock-treated, GFP-transfected cells at 100%.)
Cell Migration Assays
Boyden transwell migration chambers (Costar) were coated with 20 μg/ml fibronectin on the bottom chamber. Transfected cells were counted, and 20,000 cells/well in serum-free medium were added to the top well. Cells were serum-starved for 3 h, and then the lower wells were filled with DMEM, 10% FBS, and cells were allowed to migrate overnight. The top and bottom of the membrane were fixed in 4% PFA, stained with DAPI, and mounted on coverslips. 10 fields of cells on the membrane were counted per experiment, using fluorescence microscopy. The data are presented as the number of transfected cells that migrated to the bottom membrane divided by the number of cells that remain on the top membrane.
Sucrose Gradient Centrifugation
A method was established to isolate endosomal compartments using sucrose gradient centrifugation modified from previous work (23). Briefly, cells were kept on ice, washed three times in ice-cold PBS, scrapped, and pelleted at 200 × g for 5 min. The pellet was resuspended in a homogenization buffer (250 mm sucrose, 1 mm EDTA, 3 mm imidazole) and pelleted at 1,300 × g for 10 min. The pellet was resuspended in the homogenization buffer supplemented with 0.03 μm cycloheximide and passed through a 22-gauge needle 5–10 times and pelleted at 2,000 × g for 10 min. The postnuclear supernatant was then loaded on top of a sucrose gradient (10–40%) and centrifuged using a SW41 rotor at 100,000 × g for 18 h. All centrifugation was done at 4 °C. 500-μl fractions were extracted from the gradient, and 100 μl was added to 5× SDS running buffer, heated to 100 °C. Proteins were separated using SDS-PAGE and analyzed by Western blotting.
RESULTS
PMA Stimulates MT1-MMP Phosphorylation
Phorbol 12-myristate 13-acetate (PMA) induces the activation of conventional (α, β, and γ) and novel (η, θ, ϵ, and δ) isoforms of protein kinase C (PKC) and is one of the most potent and widely used tumor promoters (24). When stimulated by PMA, isoforms of PKC regulate the machinery that controls the intracellular trafficking and transport of proteins; for example, PKCϵ stimulates membrane trafficking through binding of β'COP, a coatomer protein, and regulates the function of cytoskeletal components (25). We and others have shown that PMA stimulates the trafficking of MT1-MMP to the plasma membrane (17, 26). Trafficking of MT1-MMP occurs rapidly because cell surface levels of MT1-MMP transiently increase within 20 min of PMA treatment (17, 26). Recently, it was shown using an in vitro kinase assay that MT1-MMP is phosphorylated in a PKC-dependent manner (27). Here, we tested whether MT1-MMP is phosphorylated in vivo upon PMA treatment and examined the effects of this phosphorylation on MT1-MMP trafficking, cell migration, and cell invasion.
HT-1080 cells were treated with PMA and extracted, and MT1-MMP immunoprecipitates were probed for phospho-Ser/Thr (Fig. 1A). Phosphorylated, endogenous MT1-MMP was detected 10 min after PMA treatment, and this increased significantly within 20 min of treatment (Fig. 1A). Extracts from HT-1080 cells (with or without PMA treatment) were also immunoprecipitated with anti-phospho-Ser/Thr and probed for MT1-MMP. In these experiments, an increase in MT1-MMP phosphorylation was also observed after treatment of cells with PMA (Fig. 1B). To determine the site of phosphorylation, we mutated the only threonine residue, Thr567, in the cytoplasmic tail of MT1-MMP to alanine (T567A-MT1-MMP), a site previously shown to be phosphorylated in vitro (27). HT-1080 cells transfected with GFP-tagged wild-type MT1-MMP (GFP-WT-MT1-MMP) or T567A mutant MT1-MMP (GFP-T567A-MT1-MMP) were treated with or without PMA and extracted, and GFP-tagged MT1-MMP was immunoprecipitated with antibodies to GFP. The immunoprecipitates were subsequently probed by Western blot for phospho-Ser/Thr. Fig. 1C shows that the mutation of Thr567 to alanine abolishes the PMA-dependent phosphorylation of MT1-MMP.
FIGURE 1.

PMA induces phosphorylation of MT1-MMP on Thr567. Serum-starved HT-1080 cells were treated with vehicle alone (DMSO) or 500 nm PMA for 0, 10, and 20 min. Cells were then lysed and immunoprecipitated (IP) using antibody against either MT1-MMP (A and D) or phospho-Ser/Thr (B). Immunoprecipitated proteins were separated by SDS-PAGE and analyzed by Western blot. Membranes were probed for MT1-MMP (A and D, upper blots; B), stripped, and reprobed for phospho-Ser/Thr (A and D, lower blots). C, HT-1080 cells were transfected with either wild-type MT1-MMP-GFP (WT) or T567A-MT1-MMP-GFP (TA) and then serum-starved and treated with vehicle alone (DMSO) or 500 nm PMA for 0 or 20 min. Cells were lysed and immunoprecipitated with anti-GFP antibody, and immunoprecipitates were then separated by SDS-PAGE and analyzed by Western blot. Membranes were probed for MT1-MMP (upper blot), stripped, and reprobed for phospho-Ser/Thr (lower blot). D, replicate of A, using cells in which cell surface TIMP-2 was washed away prior to PMA treatment. In A–D, lysate lanes were loaded with 10% of the quantity of protein used for immunoprecipitations. E, immunostaining of cell surface TIMP2 in control cells (top panels) and cells after acid wash (bottom panels). Cell surface TIMP2 is shown in the left panels (Alexa488-conjugated secondary antibody), and rhodamine-phalloidin-labeled F-actin is shown in the right panels.
TIMP-2, a member of the TIMP family of MMP inhibitors, can directly bind and modulate MT1-MMP activity on the cell surface (28, 29). TIMP-2 is expressed in HT-1080 cells, and we examined the possibility that the interaction of MT1-MMP with TIMP-2 could influence the phosphorylation of MT1-MMP. Therefore, we removed cell surface-associated TIMP-2 by acid wash, as described previously (30), prior to inducing MT1-MMP phosphorylation. Removal of TIMP-2 from the cell surface (Fig. 1E) had no effect on the PMA-induced phosphorylation of MT1-MMP (Fig. 1D).
Phosphorylation of Thr567 Is Required for Trafficking of MT1-MMP
To assess the role of Thr567 phosphorylation in MT1-MMP trafficking, we introduced a threonine to glutamic acid substitution at amino acid position 567 of MT1-MMP (T567E-MT1-MMP). This substitution produces a form of MT1-MMP that mimics permanently phosphorylated MT1-MMP and can be compared with the non-phosphorylatable T567A-MT1-MMP. These constructs were transfected into HeLa cells, which express very low levels of MT1-MMP, so that distribution and trafficking of the mutant proteins could be analyzed in the near absence of endogenous protein. Cell surface levels of GFP-tagged WT-, T567A-, and T567E-MT1-MMP were determined using antibody labeling at 4 °C prior to fixation. Cell surface levels of T567A-MT1-MMP were higher than those of WT-MT1-MMP (Fig. 2A). Interestingly, expression of T567E-MT1-MMP resulted in decreased cell surface levels. Equal levels of expression of the transfected GFP-MT1-MMP constructs were observed via Western blot (data not shown). Quantification of cell surface MT1-MMP revealed a 59 ± 12% increase in cell surface MT1-MMP in T567A-MT1-MMP-transfected samples and a 38.5 ± 6% decrease in cell surface expression in T567E-MT1-MMP-transfected samples relative to WT-MT1-MMP-transfected controls (Fig. 2B).
FIGURE 2.

Cell surface expression of Thr567-MT1-MMP mutants. HeLa cells were transfected with wild-type MT1-MMP-GFP (WT), T567A-MT1-MMP-GFP (TA), or T567E-MT1-MMP-GFP (TE). Cells were grown overnight on fibronectin-coated coverslips, incubated with MT1-MMP antibody at 4 °C, fixed, permeabilized, and stained with AlexaFluor647-conjugated secondary antibody and rhodamine-phalloidin. A, cell surface MT1-MMP, total MT1-MMP-GFP, and the actin cytoskeleton were imaged using confocal microscopy. z series stacks are shown. Saturation of signal in the GFP panels was to allow visualization of MT1-MMP-GFP in the peripheral area of cells. Scale bar, 10 μm. B, fluorescence of cell surface MT1-MMP on transfected cells was analyzed in images represented by those in the far left column of A and quantified using ImageJ. Images were acquired using consistent acquisition parameters to allow comparison between samples. Means ± S.E. from three independent experiments in which 10–20 cells/sample were measured are shown. *, value significantly different from wild-type cells; p < 0.05.
To test whether the observed changes in cell surface MT1-MMP levels were the result of altered internalization of MT1-MMP, transfected cells were treated with PMA, cell surface-labeled with MT1-MMP antibodies at 4 °C, and then warmed to 37 °C to allow internalization. Under these conditions, internalized WT-MT1-MMP and T567E-MT1-MMP could be detected within 15 min, whereas T567A-MT1-MMP remained abundant on the cell surface and could not be detected in obvious endosomal structures at this time point (Fig. 3, A and B). Internalization was assessed by monitoring the presence of MT1-MMP, which had been labeled on the cell surface, within small vesicular structures that were located more centrally within the cell, away from the F-actin network near the edge of the cells (Fig. 3). These results are consistent with the notion that T567A-MT1-MMP is more abundant on the cell surface (Fig. 2) because it is not internalized as efficiently as WT-MT1-MMP (note the lack of MT1-MMP-containing vesicles in the sample transfected with T567A-MT1-MMP compared with the sample transfected with WT-MT1-MMP in Fig. 3). T567E-MT1-MMP appeared to be internalized efficiently (Fig. 3).
FIGURE 3.

Mutation of Thr567 alters internalization of MT1-MMP. HeLa cells were transfected with wild-type MT1-MMP-GFP (WT), T567A-MT1-MMP (TA), or T567E-MT1-MMP-GFP (TE). Cells were grown overnight on fibronectin-coated coverslips, serum-starved for 3 h, and treated with PMA for 20 min. Cells were then incubated with MT1-MMP antibody at 4 °C and slowly warmed to 37 °C for a total of 15 min to allow internalization of MT1-MMP. After fixation, samples were fixed, permeabilized, stained using AlexaFluor-647-conjugated secondary antibody and rhodamine-phalloidin, and then imaged using confocal microscopy. A, images (z series stacks) of cell surface MT1-MMP, total MT1-MMP-GFP, and F-actin and overlay for each sample. Saturation of signal in the GFP panels was to allow visualization of MT1-MMP-GFP in peripheral areas of cells. Scale bar, 10 μm. B, zoom of selected regions in each overlay panel in A showing internalized MT1-MMP that had been labeled on the cell surface (blue) co-localized with MT1-MMP-GFP (green) in small vesicular punctae (light blue; refer to arrows), away from the peripheral membrane (red). The arrows indicate compartments (light blue), in WT and T567E samples only, containing MT1-MMP-GFP that was labeled on the cell surface, internalized, and no longer closely associated with F-actin structures at the edge of the cell.
Phosphorylation Mutants of MT1-MMP Affect Cell Migration and Invasion
To test the role of phosphorylation of MT1-MMP in cell migration and invasion, HeLa cells were transfected with WT-, T567A-, or T567E-MT1-MMP and analyzed using transwell migration assays and Matrigel invasion assays. HeLa cells were used again here because of their low endogenous level of MT1-MMP expression, resulting in the fact that their capacity to invade in vitro is dependent upon transgenic expression of MT1-MMP (31). Expression of T567E-MT1-MMP increased both cell migration (by 24%) and cell invasion (by 63%) compared with WT-MT1-MMP (Fig. 4, A and B). Cells expressing T567A-MT1-MMP exhibited a decrease in both migration (36% reduction) and invasion (42% reduction) compared with WT-MT1-MMP (Fig. 4A and B). The observed changes to cell migration were consistent whether migration assays were conducted for 12 h (Fig. 6B) or 6 h (Fig. 6C). It is interesting that expression of T567A-MT1-MMP reduced migration and invasion, given that expression of this construct caused an increase in cell surface expression of the MT1-MMP (Fig. 2). These observations suggest that MT1-MMP internalization and trafficking are essential for this enzyme to promote cell migration and invasion.
FIGURE 4.
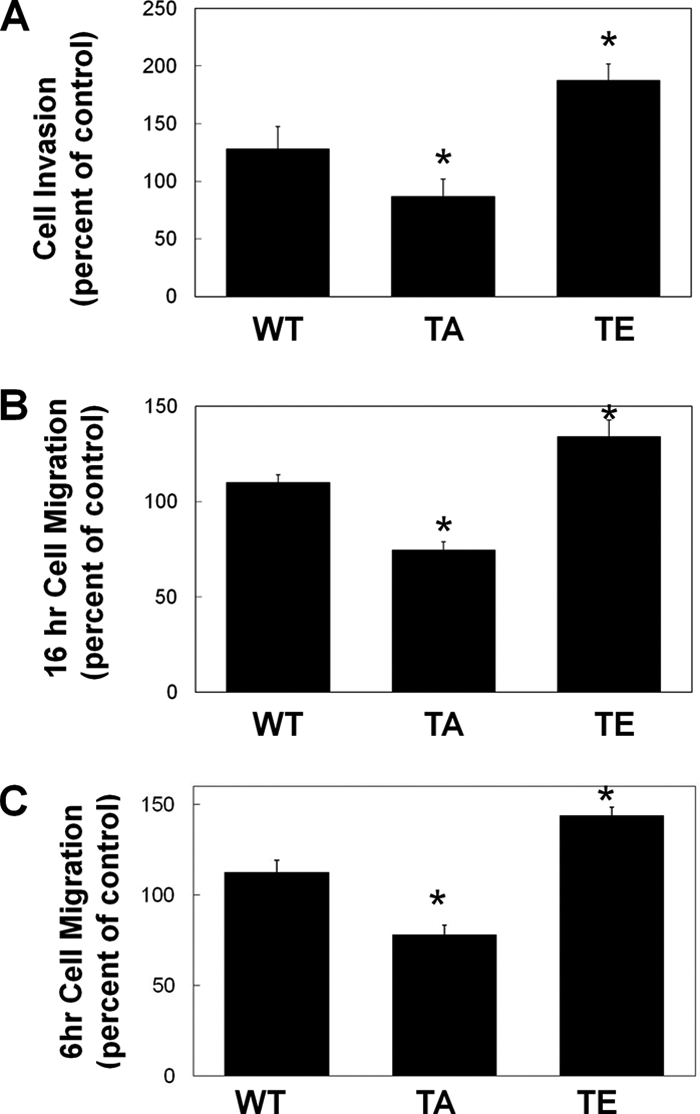
Expression of Thr567-MT1-MMP alters cell migration and invasion. HeLa cells were transfected with wild-type MT1-MMP-GFP (WT), T567A-MT1-MMP (TA), or T567E-MT1-MMP-GFP (TE). 16 h after transfection, cells were collected, and transwell invasion assays (A) or migration assays (B and C) were performed. A, cells invaded through Matrigel toward 10% FBS for 24 h and were then fixed and counted. B and C, cells migrated toward 10% FBS for 16 h (B) or 6 h (C), and transfected cells that migrated to the underside of the membrane were counted. In A–C, means ± S.E. (error bars) from three independent experiments are shown. *, a value significantly different from control cells (p < 0.05).
FIGURE 6.

MT1-MMP is trafficked to the late endosome and recycled in a VAMP7-dependent manner. HT-1080 cells were transfected with GFP-tagged full-length VAMP7 (GFP-Vamp7FL), VAMP7 cytoplasmic domain (GFP-Vamp7C), wild-type Rab7 (GFP-Rab7), or CFP-tagged dominant negative Rab7 (CFP-Rab7DN). Transfected cells were grown overnight on fibronectin-coated coverslips and serum-starved for 3 h prior to treatment with PMA for 20 min. Cells were then incubated with MT1-MMP antibody at 4 °C followed by incubation in serum-free medium at 37 °C for 2 h to allow internalization of MT1-MMP and subsequently fixed, permeabilized, and stained with Alexa-647-conjugated secondary antibody. A–D, GFP-VAMP7FL-transfected cells, co-stained with anti-Rab7 and Alexa-594-conjugated secondary antibody. The arrow in D indicates the compartment where VAMP7, MT1-MMP, and Rab7 co-localize (white). E–H, GFP-Rab7-transfected cells, co-stained with anti-LAMP-2 and Alexa-594-conjugated secondary antibody. The arrow in H indicates the compartment where MT1-MMP and LAMP-2 co-localize (magenta). I–T, 2 h post-MT1-MMP internalization, 500 nm PMA was added again for 20 min to induce recycling of MT1-MMP to the plasma membrane. I–K, cells transfected with GFP-VAMP7FL. L–N, cells transfected with GFP-VAMP7C. O–Q, cells transfected with GFP-Rab7. R–T, cells transfected with CFP-Rab7DN. Images are z series stacks and are representative of three independent experiments. Arrows in K and Q indicate MT1-MMP recycled to the plasma membrane; arrows in N and T indicate MT1-MMP still retained in the endosomal compartment. Panels with the prime symbol are single confocal slices from series, and panels with the double prime symbol are magnifications of the regions selected in the single slice panel. Saturation of signal in the GFP panels was done to allow visualization of GFP-tagged protein in peripheral areas of cells. Scale bar, 10 μm. U, quantification of MT1-MMP localization at the plasma membrane was performed on z series stacks using ImageJ. Plasma membrane MT1-MMP was measured, in non-saturated images, at the cell boundary (dotted outline in J, M, P, and S) and is represented as a percentage of total MT1-MMP. Measurements of MT1-MMP intensity were made in a minimum of 10 cells/experimental condition; means ± S.E. (error bars) from three independent experiments are shown.
MT1-MMP Is Recycled through the Late Endosome in a VAMP7-dependent Manner
To elucidate the pathway through which MT1-MMP is internalized and recycled in response to PMA-stimulated phosphorylation, we used antibodies to label cell surface MT1-MMP and subsequently follow its intracellular trafficking. This approach has been used effectively by others to monitor recycling of MT1-MMP (5). HT-1080 cells were used for these experiments to exploit their high level of endogenous MT1-MMP expression. MT1-MMP was observed on the surface of cells, but internal MT1-MMP was not detected where GFP-tagged Rab5 served as a marker of the early endosome (Fig. 5, A–C). After PMA stimulation under serum-free conditions, MT1-MMP was internalized into a Rab5 compartment within 20 min (Fig. 5, D–F), and 40 min later, some MT1-MMP remained in the Rab5 compartment (Fig. 5, G, H, and J), whereas some was localized in a Rab7 compartment (Fig. 5, G, I, J, and K–M). By 2 h, a substantial portion of MT1-MMP was localized in the Rab7 compartment (Fig. 5, N–P). Consistent with the localization of MT1-MMP in the late endosome, the distributions of MT1-MMP and Rab7 were found to partly overlap with the endosomal SNARE VAMP7 (Fig. 6, A–D). This pathway of MT1-MMP trafficking was consistently observed, including in experiments in which cell surface TIMP-2 had been washed away as described in the legend to Fig. 1 (data not shown).
FIGURE 5.

PMA induces trafficking of MT1-MMP through Rab5 and Rab7 compartments. HT-1080 cells were transfected with either GFP-Rab5 or GFP-Rab7. Transfected cells were grown overnight on fibronectin-coated coverslips and serum-starved for 3 h prior to treatment with PMA for 20 min. Cells were then incubated with MT1-MMP antibody at 4 °C, followed by incubation in serum-free medium at 37 °C for the indicated time periods to allow internalization of MT1-MMP. Cells were fixed, permeabilized, and stained using AlexaFluor647-conjugated secondary antibody. MT1-MMP localization following internalization was analyzed using confocal microscopy. A–F, cells transfected with GFP-Rab5 and fixed after treatment with PMA for 20 min to observe cell surface MT1-MMP (A–C) or after treatment with PMA for 20 min followed by an additional 20 min in serum-free medium to observe internalization of cell surface MT1-MMP (D–F). G–J, cells transfected with GFP-Rab5, fixed, and stained using anti-Rab7 and Alexa-594-conjugated secondary antibody. The cells were treated with PMA for 20 min followed by an additional 40 min in serum-free medium to monitor internalization of MT1-MMP. K–P, cells transfected with GFP-Rab7, treated with PMA for 20 min, and then fixed after MT1-MMP internalization for either 40 min (K–M) or 2 h (N–P). All images are z series stacks, and results are representative of three independent experiments. Panels with the prime symbol are single confocal slices from series, and panels with the double prime symbol are magnifications of regions selected in the single slice panel. Scale bar, 10 μm. The arrows in F, J, M, and P indicate areas of co-localization of Rab protein and MT1-MMP.
Internalized MT1-MMP was not trafficked exclusively into a Rab7 compartment, and some could be detected in a compartment containing the lysosomal marker LAMP-2 but not Rab7 (Fig. 6, E–H). This localization might correspond to a population of internalized MT1-MMP that is targeted for degradation. The MT1-MMP that is delivered to the Rab7/VAMP7 compartment does not appear to be destined for degradation as some of the MT1-MMP co-localizing with Rab7 is not localized with (Fig. 6, E–H). Also, we detected MT1-MMP within this compartment up to 4 h postinternalization (data not shown). Furthermore, if trafficking was restimulated with PMA, we observed that MT1-MMP recycled back to the plasma membrane, as indicated by decreased signal in the late endosome and increased signal at the plasma membrane; compare Fig. 6D (after internalization) with Fig. 6K (after restimulation with PMA). Expression of dominant negative mutants of VAMP7 (VAMP7C) and Rab7 (Rab7DN) inhibited this trafficking back to the plasma membrane (Fig. 6, compare N with K and compare T with Q). Recycling of MT1-MMP was quantified in these experiments by measuring the amount of MT1-MMP at the plasma membrane after PMA treatment to restimulate MT1-MMP trafficking out of Rab7 endosomes. Image stacks like those in Fig. 6 were analyzed using ImageJ to determine levels of MT1-MMP at the plasma membrane (dotted outline in Fig. 6, J, M, P, and S) as a percentage of total MT1-MMP (Fig. 6U). MT1-MMP was never observed to appear at the plasma membrane uniformly but rather was delivered to restricted regions of the membrane, possibly at sites where membrane protrusions are forming. Overall, the results are consistent with a model of MT1-MMP trafficking in which both VAMP7 and Rab7 are involved in a recycling pathway.
We also examined the endosomal markers Rab8 (Fig. 7, A–C) and Rab11 (Fig. 7, D–F) and found no significant overlap of these two markers with internalized MT1-MMP. In all experiments with antibody-labeled MT1-MMP, the distribution of the enzyme closely resembled that of unlabeled endogenous MT1-MMP. As reported by others (15, 32), there is a large pool of intracellular MT1-MMP, shown in Fig. 7, G–I, co-localizing with the Golgi SNARE VAMP4. Following PMA treatment, a portion of this MT1-MMP is transported to the cell periphery, where it no longer co-localizes with VAMP4 (Fig. 7, J–L). To ensure that antibody labeling of MT1-MMP was not altering its normal trafficking pattern, we assessed total MT1-MMP trafficking under identical conditions. Confirming our observations made with antibody-labeled MT1-MMP, staining of total endogenous MT1-MMP revealed that, during PMA-induced recycling, MT1-MMP co-localizes first with Rab5 (Fig. 7, M–O) and subsequently with Rab7 (Fig. 7, P–R).
FIGURE 7.

MT1-MMP does not localize to a Rab8 or Rab11 compartment. HT-1080 cells were transfected with GFP-Rab8 (A–C), GFP-Rab11 (D–F), or GFP-VAMP4 (G–L) or stained for endogenous Rab5 (M–O) or Rab7 (P–R). Cells were grown overnight on fibronectin-coated coverslips, serum-starved for 3 h, and subsequently stimulated with PMA for 20 min. A–F, samples were incubated with MT1-MMP antibody at 4 °C followed by incubation in serum-free medium at 37 °C for 2 h to allow internalization of MT1-MMP. Cells were then fixed, permeabilized, and stained using AlexaFuor647-conjugated secondary antibody. Negligible overlap is observed between MT1-MMP and Rab8 or Rab11. G–L, after treatment with DMSO alone (G–I) or PMA (J–L), cells were then fixed, permeabilized, and stained for total MT1-MMP. MT1-MMP partly co-localizes with VAMP4 in perinuclear compartments. After stimulation with PMA, a portion of MT1-MMP moves out of central compartments to the cell periphery, where it does not colocalize with VAMP4 (L). M–O, non-transfected samples were treated as in A–F and subsequently stained with antibodies to MT1-MMP and either Rab5 or Rab7. Co-localization of MT1-MMP was observed with Rab5 (M–O) and with Rab7. All images are representative of three independent experiments. Panels with the prime symbol are single confocal slices from z series stacks, and panels with the double prime symbol are magnification of regions selected in the single slice panel. Saturation of signal in some panels was done to allow visualization of protein in peripheral areas of cells. Scale bar, 10 μm.
To verify the subcellular compartmentalization of MT1-MMP following PMA stimulation, we used sucrose gradient centrifugation to isolate subcellular organelle fractions. Fig. 8A shows that, 30 min post-PMA treatment, MT1-MMP was abundant in fractions 18–20, which are enriched in Rab5. 2 h post-PMA treatment, MT1-MMP is abundant in fractions 14–17, which are enriched in Rab7 (Fig. 8B). These observations are consistent with the distributions of MT1-MMP documented using confocal microscopy above.
FIGURE 8.

MT1-MMP and integrin α5 are internalized into a Rab5 compartment and trafficked to a Rab7 compartment. HT-1080 cells were serum-starved, treated with 500 nm PMA for 20 min, and then incubated in serum-free medium to allow internalization for the indicated time periods. Cells were homogenized, loaded onto the top of a discontinuous sucrose gradient (10–40%), and centrifuged overnight at 100,000 × g using a swinging bucket rotor. Twenty fractions were collected from the gradient, and the proteins in each fraction were separated by SDS-PAGE and analyzed by Western blot. Membranes were probed for MT1-MMP, α5 integrin, and Rab5, Rab7, and/or Rab11. Internalization was assessed at 30 min (A) and 2 h (B) after PMA stimulation.
Dominant Negative VAMP7 and Rab7 Impair Cell Migration and Invasion
To determine if perturbation of MT1-MMP trafficking through the late endosome would have an impact on cell migration and invasion, similar to that caused by expression of the T567A-MT1-MMP mutant, we transfected HT-1080 cells with wild-type or dominant negative constructs for VAMP7 and Rab7 and assessed migration and invasion in vitro. Inhibition of late endosomal trafficking, through expression of either VAMP7C or Rab7DN, reduced migration and invasion in transfected cells (Fig. 9). This is consistent with results observed after transfection of T567A-MT1-MMP and suggests that trafficking of MT1-MMP is essential for normal function of this enzyme during cell migration and invasion.
FIGURE 9.
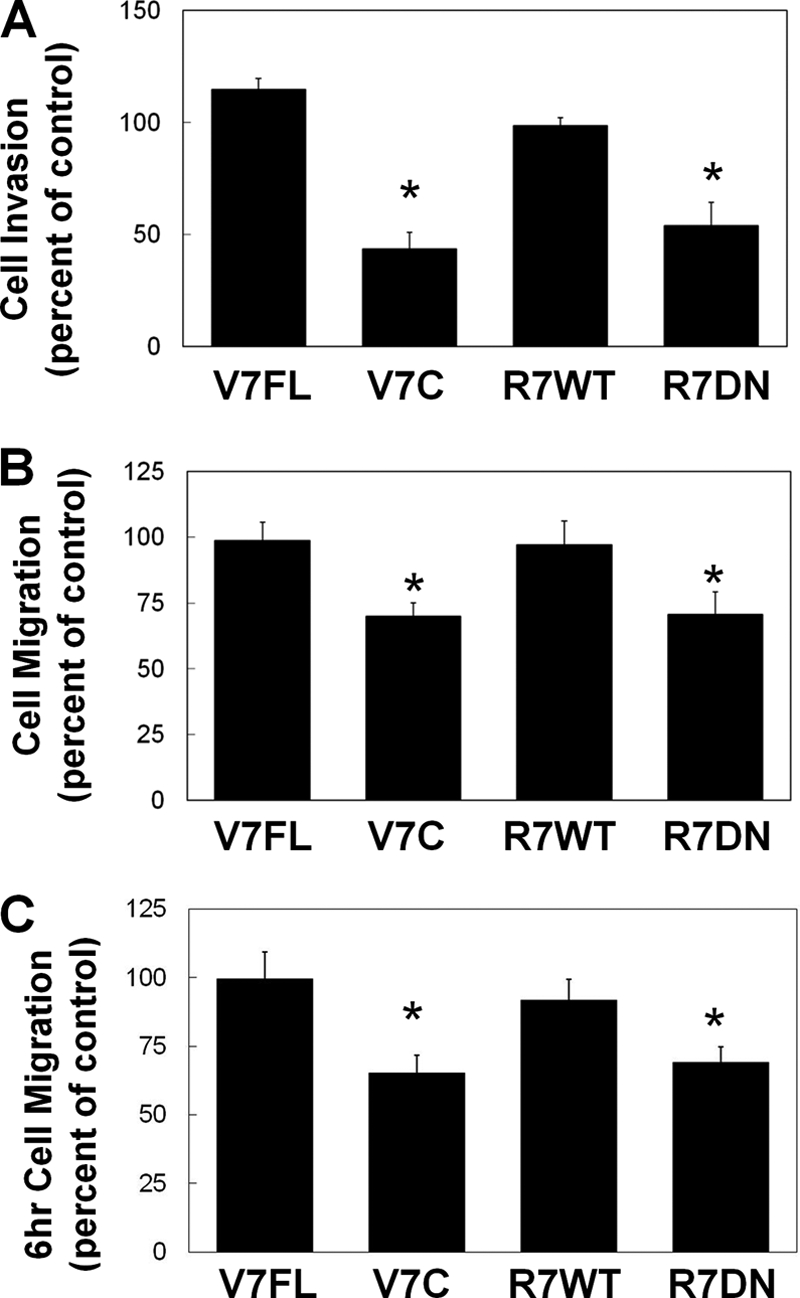
HT-1080 cell invasion and migration are impaired by VAMP7C and Rab7DN. HT-1080 cells were transfected with GFP-tagged constructs of wild-type VAMP7 (VAMP7FL), cytoplasmic domain of VAMP 7 (VAMP7C), wild-type Rab7 (Rab7WT), or CFP-tagged dominant negative Rab7 (Rab7D). 14 h after transfection, cells were collected, and transwell invasion assays (A) or migration assays (B and C) were performed. A, cells invaded through Matrigel toward 10% FBS for 24 h and were then fixed and counted. B and C, cells migrated toward 10% FBS for 16 h (B) or 6 h (C), and transfected cells that migrated to the underside of the membrane were counted. A–C, mean ± S.E. (error bars) from three independent experiments. *, value significantly different from control cells (p < 0.05).
PMA Induces Association and Co-trafficking of MT1-MMP with Integrin α5 and Phosphorylation of ERK
The impaired migration and invasion of cells expressing T567A-MT1-MMP, VAMP7C, or Rab7DN led us to investigate if PMA treatment was possibly enabling an association between MT1-MMP and α5 integrin. MT1-MMP has been shown to process a number of integrin subunits, including α3, α5, and αv (33, 34), and α5β1 has been implicated in cell migration and enhanced cell invasion (35, 36). We examined immunoprecipitates of MT1-MMP for the presence of α5 integrin by Western blot. 20 min after PMA treatment, we detected an increase in the amount of α5 integrin pulled down with MT1-MMP (Fig. 10). This observation suggested the possibility that MT1-MMP and α5 integrin associate and are trafficked together, and to further investigate this possibility, we examined the localization of these proteins in HT-1080 cells. Cell surface MT1-MMP as well as intracellular MT1-MMP co-localized with α5 integrin (Fig. 11, A–D). When internalization and trafficking of cell surface MT1-MMP and α5 integrin were monitored following PMA treatment, it was observed that MT1-MMP and α5 co-localize at the cell surface (Fig. 11, F–H) and internalize together within 20 min (Fig. 11, J–L), and after 2 h, they are observed together in a compartment consistent with a late endosome (Fig. 11, M–P). This was confirmed using sucrose gradient centrifugation to assess the intracellular localization of MT1-MMP and α5. 30 min post-PMA treatment, MT1-MMP and α5 integrin were found to be abundant in fractions 17–20, enriched in Rab5, and 2 h post-PMA treatment, MT1-MMP and α5 integrin were found in fractions 14–17, enriched in Rab7 (Fig. 8).
FIGURE 10.
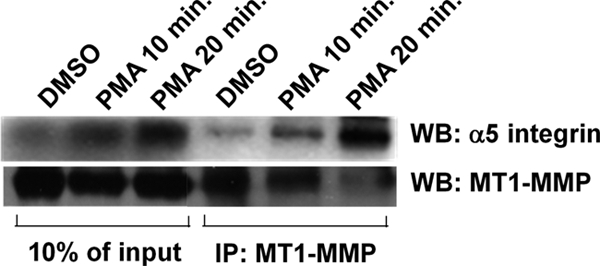
MT1-MMP associates with α5 integrin in a PMA-dependent manner. Serum-starved HT-1080 cells were treated with vehicle alone (DMSO) or 500 nm PMA for 0, 10, and 20 min. Cells were then lysed and immunoprecipitated (IP) using antibody against MT1-MMP. Immunoprecipitated proteins were separated by SDS-PAGE and analyzed by Western blot (WB). Membranes were probed for MT1-MMP and α5 integrin. Lysate lanes contain 10% of the quantity of protein used for immunoprecipitations.
FIGURE 11.

MT1-MMP co-localizes and traffics with α5 integrin. HT-1080 cells transfected with GFP-α5 integrin were serum-starved for 3 h, treated with PMA for 20 min, and then incubated with primary α5 integrin and/or MT1-MMP antibody at 4 °C. Cells were fixed, permeabilized, and stained with AlexaFluor594- and/or AlexaFluor647-conjugated secondary antibody. A–D, GFP-α5 integrin, cell surface MT1-MMP, and total MT1-MMP-mCherry after PMA stimulation. E–H, total GFP-α5 integrin, cell surface MT1-MMP, and cell surface α5 integrin after PMA stimulation. I–L, total GFP-α5 integrin, internalized MT1-MMP, and internalized α5 integrin after incubation at 37 °C for 20 min in serum-free medium. M–P, GFP-Rab7, internalized MT1-MMP, and internalized integrin α5 after incubation at 37 °C for 2 h in serum-free medium. Co-trafficking and compartment localization were assessed using confocal microscopy. The arrows indicate areas of co-localization. All images are z series stacks, and results are representative of three independent experiments. Panels with the prime symbol are single confocal slices from series, and panels with the double prime symbol are magnifications of regions selected in the single slice panel. Saturation of signal in some panels was done to allow visualization of protein in peripheral areas of cells. Scale bar, 10 μm.
It has previously been demonstrated that ERK phosphorylation is linked with MT1-MMP-dependent cell migration (37), and increased ERK signaling enhances tumorigenesis (38). We therefore analyzed changes to ERK phosphorylation during PMA-stimulated MT1-MMP phosphorylation and internalization. In this system, ERK phosphorylation was increased 20 min after PMA treatment (Fig. 12, A and B), coinciding with MT1-MMP phosphorylation (Fig. 1) and internalization (Fig. 3). To examine the dependence of ERK phosphorylation on MT1-MMP phosphorylation, HeLa cells were transfected with GFP-tagged WT-, T567A-, or T567E-MT1-MMP, and ERK phosphorylation was measured. ERK phosphorylation was dramatically increased in cells transfected with T567E-MT1-MMP, compared with untransfected control cells or cells transfected with WT- or T567A-MT1-MMP (Fig. 12, C and D). These results are consistent with the notion that phosphorylation-dependent internalization and recycling of MT1-MMP is required for efficient cell migration and invasion, possibly because this trafficking is needed for an association with α5 integrin that contributes to activation of ERK.
FIGURE 12.
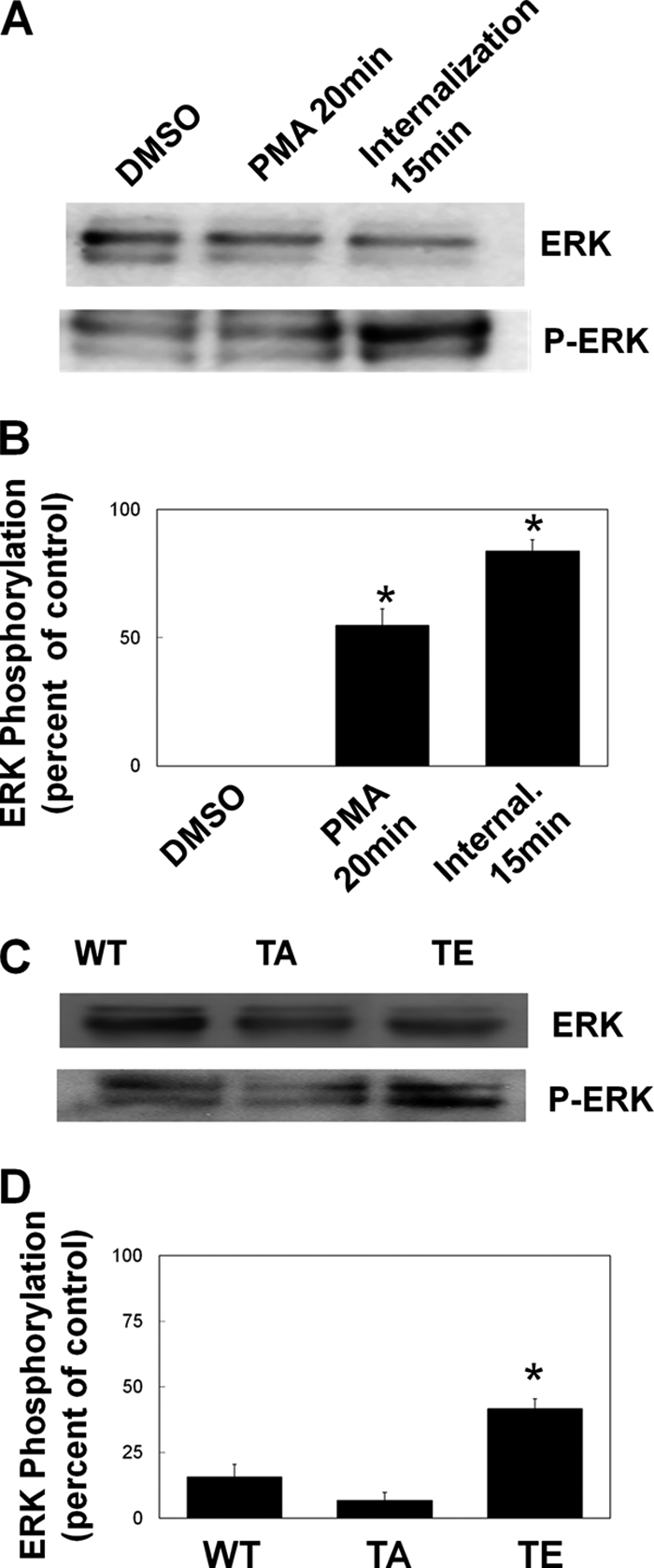
PMA stimulation or expression of T567E-MT1-MMP increases ERK phosphorylation. A and B, HT-1080 cells grown on fibronectin were serum-starved for 3 h prior to treatment with vehicle alone (DMSO), treatment with 500 nm PMA for 20 min (PMA 20 min), or treatment with PMA for 20 min followed by a 15-min incubation in serum-free medium to allow internalization of MT1-MMP (Internal. 15 min). Cells were then lysed, and extracted proteins were separated by SDS-PAGE prior to analysis by Western blot for ERK (A, upper blot) and phospho-ERK (A, lower blot). B, Western blots from at least three independent experiments were analyzed by ImageJ to quantify the increase in ERK phosphorylation. Means ± S.E. (error bars) from three independent experiments are shown. *, value significantly different from control cells (p < 0.05). C and D, HeLa cells transfected with GFP-tagged MT1-MMP or mutant MT1-MMP (WT, T567A, or T567E) were grown for 24 h on fibronectin, serum-starved for 3 h, and lysed, and extracted proteins were separated by SDS-PAGE and analyzed by Western blot. C, membranes were probed for ERK (upper blot) and phospho-ERK (lower blot). D, Western blots were analyzed by ImageJ to quantify the increase in ERK phosphorylation. Means ± S.E. from three independent experiments are shown. *, value significantly different from control cells (p < 0.05).
DISCUSSION
Studies on the endocytosis and trafficking of MT1-MMP reveal the importance of these processes to both cellular invasion and migration (11, 16, 27, 39, 40). It has been established that MT1-MMP is phosphorylated at Tyr573 in a Src-dependent manner and that this event regulates the endocytosis of MT1-MMP in cells stimulated with epidermal growth factor (EGF) (27, 32). MT1-MMP has also been shown to be phosphorylated at Thr567 using an in vitro kinase assay (27), and here we observed the phosphorylation of MT1-MMP at Thr567 in a PMA-dependent manner in vivo. Mutation of Thr567 to alanine (T567A), creating a non-phosphorylatable form of MT1-MMP, significantly impaired invasion and migration in cells expressing this mutant, although cell surface levels of this MT1-MMP were increased. Interestingly, T567E-MT1-MMP (which mimics phosphorylated MT1-MMP) was less abundant on the cell surface and appeared to be endocytosed faster than WT-MT1-MMP. These results implicate phosphorylation of Thr567, in addition to that of Tyr573, in regulating internalization and trafficking of MT1-MMP. The observed phosphorylation of MT1-MMP at Thr567 in a PKC-dependent manner is consistent with the presence of a consensus sequence (TXR) for PKC as predicted from sequence analysis using the Web tool NetPhos (41).
The internalization of MT1-MMP following PMA stimulation led to delivery of the enzyme to a Rab5-containing compartment, followed by transition into a late endosomal compartment, as characterized by the presence of both Rab7 and VAMP7. Moss et al. (27) reported a lack of colocalization between MT1-MMP and Rab7 in the context of EGF-stimulated Tyr573 phosphorylation, whereas Remacle et al. (15) showed MT1-MMP localization in the late endosome after PMA treatment, in this case characterized by Lamp-1. Collectively, these findings suggest the possibility of alternative trafficking routes that might be influenced by specific phosphorylation events on MT1-MMP. We monitored trafficking up to 4 h post-MT1-MMP internalization and found that after 1 h, the majority of MT1-MMP is localized to a late endosomal compartment and can be stimulated to recycle back to the plasma membrane. This suggests that trafficking to the late endosome is not a means of down-regulating MT1-MMP expression, but rather it is a recycling pathway, allowing for redelivery of MT1-MMP to the plasma membrane. The delivery of MT1-MMP to the plasma membrane was dependent upon Rab7 and VAMP7, and inhibiting this step using dominant negative forms of Rab7 and VAMP7 reduced both invasion and migration, similar to the results seen in the T567A mutant. To our knowledge, this is the first published report of a role for Rab7 in a recycling pathway in mammalian cells. Although we cannot formally exclude the possibility that inhibition of Rab7 or VAMP7 might have more general effects on intracellular trafficking, the results of these experiments do support a model wherein Rab7 and VAMP7 are involved in trafficking of MT1-MMP during cell migration and invasion. Consistent with previous studies showing that VAMP7 is required for MT1-MMP localization to invadopodia (18), our findings support the notion that VAMP7 is a key regulator of MT1-MMP trafficking because we observed dramatic effects on invasion following its inhibition.
We have found that intracellular trafficking and recycling of MT1-MMP to the plasma membrane is crucial for this enzyme to promote not only cell invasion but migration as well. It has been established that MT1-MMP colocalizes with integrin and has a role in processing α3, α5, and αv integrin subunits (34, 42, 43). Here, we observed that MT1-MMP co-localizes with α5 and co-traffics with α5β1 to the late endosome, and we then considered the possibility that the observed decrease in migration and invasion of cells transfected with T567A or dominant negative Rab7 and VAMP7 was due, in part, to changes in integrin-MT1-MMP association. We assessed the co-localization of MT1-MMP and α5 as well as the trafficking of α5β1 and found an association between MT1-MMP and α5 that increased upon PMA stimulation and coincided with the phosphorylation of Thr567. It is thus possible that phosphorylation of Thr567 results in an increased association between MT1-MMP and α5 integrin. Interestingly, we also observed an increase in ERK phosphorylation correlating with MT1-MMP phosphorylation and internalization as well as in T567E mutants; thus, it is reasonable to speculate that MT1-MMP and α5 integrin may interact to promote ERK activation. There is an established link between ERK and MT1-MMP; for example, it has been demonstrated that MT1-MMP-dependent cell migration requires ERK activation (33). The ERK pathway has also been shown to increase transcription of MT1-MMP (44). Furthermore, it has been shown that binding of TIMP-2 by MT1-MMP can activate ERK and up-regulate cell migration (45). This activation of ERK was found to be dependent on the cytoplasmic tail but not the catalytic activity of MT1-MMP and required the 573YCQR576 sequence. Tyr573 of MT1-MMP is phosphorylated in an Src-dependent manner, and it is thus possible that this phosphorylation event could affect ERK activation. It is further plausible that MT1-MMP phosphorylation at Thr567 may function in a similar manner through binding of integrin.
MT1-MMP is one of the best characterized MMPs and has been extensively studied since its discovery in 1994 (46). Overexpression of MT1-MMP has been shown in vitro and in vivo to promote invasion, migration, and metastasis of tumor cells (47). Nonetheless, the regulation of MT1-MMP activity, as well as other MMPs, in cancer is complex and not fully understood (48). Much recent research has gone into the development of pharmacological inhibitors to target MMP synthesis, secretion, and activity (49), and a more detailed understanding of the molecular mechanisms underlying MMP trafficking will benefit this effort. The findings here show, for the first time, the in vivo phosphorylation of MT1-MMP at Thr567, which is accompanied by a decrease in MT1-MMP cell surface expression, possibly due to an increase in endocytosis and trafficking. We also highlight the importance of MT1-MMP trafficking through a late endosome as part of a recycling pathway rather than a degradative pathway. Moreover, MT1-MMP association with integrin α5 and subsequent internalization may be a means to up-regulate the ERK pathway and increase cell migration and invasion. Collectively, this work points to the importance of MT1-MMP phosphorylation in regulating MT1-MMP activity at the cell surface and the activation of intracellular signaling.
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Ontario Ministry of Research and Innovation.
- ECM
- extracellular matrix
- MMP
- metalloproteinase
- MT1-MMP
- membrane type 1 MMP
- VAMP
- vesicle-associated membrane protein
- PMA
- phorbol 12-myristate 13-acetate.
REFERENCES
- 1. Ridley A. J., Schwartz M. A., Burridge K., Firtel R. A., Ginsberg M. H., Borisy G., Parsons J. T., Horwitz A. R. (2003) Science 302, 1704–1709 [DOI] [PubMed] [Google Scholar]
- 2. Sabeh F., Ota I., Holmbeck K., Birkedal-Hansen H., Soloway P., Balbin M., Lopez-Otin C., Shapiro S., Inada M., Krane S., Allen E., Chung D., Weiss S. J. (2004) J. Cell Biol. 167, 769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Weaver A. M. (2006) Clin. Exp. Metastasis 23, 97–105 [DOI] [PubMed] [Google Scholar]
- 4. Linder S. (2007) Trends Cell Biol. 17, 107–117 [DOI] [PubMed] [Google Scholar]
- 5. Wang X., Ma D., Keski-Oja J., Pei D. (2004) J. Biol. Chem. 279, 9331–9336 [DOI] [PubMed] [Google Scholar]
- 6. Seftor R. E., Seftor E. A., Koshikawa N., Meltzer P. S., Gardner L. M., Bilban M., Stetler-Stevenson W. G., Quaranta V., Hendrix M. J. (2001) Cancer Res. 61, 6322–6327 [PubMed] [Google Scholar]
- 7. Sato H., Takino T., Miyamori H. (2005) Cancer Sci. 96, 212–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sodek K. L., Ringuette M. J., Brown T. J. (2007) Br. J. Cancer 97, 358–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zucker S., Hymowitz M., Conner C., Zarrabi H. M., Hurewitz A. N., Matrisian L., Boyd D., Nicolson G., Montana S. (1999) Ann. N.Y. Acad. Sci. 878, 212–227 [DOI] [PubMed] [Google Scholar]
- 10. Sabeh F., Shimizu-Hirota R., Weiss S. J. (2009) J. Cell Biol. 185, 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nyalendo C., Sartelet H., Gingras D., Béliveau R. (2010) Anticancer Res. 30, 1887–1895 [PubMed] [Google Scholar]
- 12. Itoh Y. (2006) IUBMB Life 58, 589–596 [DOI] [PubMed] [Google Scholar]
- 13. Bravo-Cordero J. J., Marrero-Diaz R., Megías D., Genís L., García-Grande A., García M. A., Arroyo A. G., Montoya M. C. (2007) EMBO J. 26, 1499–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gálvez B. G., Matías-Román S., Yáñez-Mó M., Vicente-Manzanares M., Sánchez-Madrid F., Arroyo A. G. (2004) Mol. Biol. Cell 15, 678–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Remacle A., Murphy G., Roghi C. (2003) J. Cell Sci. 116, 3905–3916 [DOI] [PubMed] [Google Scholar]
- 16. Uekita T., Itoh Y., Yana I., Ohno H., Seiki M. (2001) J. Cell Biol. 155, 1345–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kean M. J., Williams K. C., Skalski M., Myers D., Burtnik A., Foster D., Coppolino M. G. (2009) J. Cell Sci. 122, 4089–4098 [DOI] [PubMed] [Google Scholar]
- 18. Steffen A., Le Dez G., Poincloux R., Recchi C., Nassoy P., Rottner K., Galli T., Chavrier P. (2008) Curr. Biol. 18, 926–931 [DOI] [PubMed] [Google Scholar]
- 19. Miyata T., Ohnishi H., Suzuki J., Yoshikumi Y., Ohno H., Mashima H., Yasuda H., Ishijima T., Osawa H., Satoh K., Sunada K., Kita H., Yamamoto H., Sugano K. (2004) Biochem. Biophys. Res. Commun. 323, 118–124 [DOI] [PubMed] [Google Scholar]
- 20. Proux-Gillardeaux V., Gavard J., Irinopoulou T., Mège R. M., Galli T. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 6362–6367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Skalski M., Coppolino M. G. (2005) Biochem. Biophys. Res. Commun. 335, 1199–1210 [DOI] [PubMed] [Google Scholar]
- 22. Tayeb M. A., Skalski M., Cha M. C., Kean M. J., Scaife M., Coppolino M. G. (2005) Exp. Cell Res. 305, 63–73 [DOI] [PubMed] [Google Scholar]
- 23. de Araùjo M. E., Huber L. A., Stasyk T. (2008) Methods Mol. Biol. 424, 317–331 [DOI] [PubMed] [Google Scholar]
- 24. Griner E. M., Kazanietz M. G. (2007) Nat. Rev. Cancer 7, 281–294 [DOI] [PubMed] [Google Scholar]
- 25. Newton P. M., Messing R. O. (2010) Biochem. J. 427, 189–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zucker S., Hymowitz M., Conner C. E., DiYanni E. A., Cao J. (2002) Lab. Invest. 82, 1673–1684 [DOI] [PubMed] [Google Scholar]
- 27. Moss N. M., Wu Y. I., Liu Y., Munshi H. G., Stack M. S. (2009) J. Biol. Chem. 284, 19791–19799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sounni N. E., Janssen M., Foidart J. M., Noel A. (2003) Matrix Biol. 22, 55–61 [DOI] [PubMed] [Google Scholar]
- 29. Strongin A. Y. (2010) Biochim. Biophys. Acta 1803, 133–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cho J. A., Osenkowski P., Zhao H., Kim S., Toth M., Cole K., Aboukameel A., Saliganan A., Schuger L., Bonfil R. D., Fridman R. (2008) J. Biol. Chem. 283, 17391–17405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhai Y., Hotary K. B., Nan B., Bosch F. X., Muñoz N., Weiss S. J., Cho K. R. (2005) Cancer Res. 65, 6543–6550 [DOI] [PubMed] [Google Scholar]
- 32. Nyalendo C., Michaud M., Beaulieu E., Roghi C., Murphy G., Gingras D., Béliveau R. (2007) J. Biol. Chem. 282, 15690–15699 [DOI] [PubMed] [Google Scholar]
- 33. Takino T., Miyamori H., Watanabe Y., Yoshioka K., Seiki M., Sato H. (2004) Cancer Res. 64, 1044–1049 [DOI] [PubMed] [Google Scholar]
- 34. Gálvez B. G., Matías-Román S., Yáñez-Mó M., Sánchez-Madrid F., Arroyo A. G. (2002) J. Cell Biol. 159, 509–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McKenzie J. A., Liu T., Goodson A. G., Grossman D. (2010) Cancer Res. 70, 7927–7937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mierke C. T., Frey B., Fellner M., Herrmann M., Fabry B. (2011) J. Cell Sci. 124, 369–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Takino T., Watanabe Y., Matsui M., Miyamori H., Kudo T., Seiki M., Sato H. (2006) Exp. Cell Res. 312, 1381–1389 [DOI] [PubMed] [Google Scholar]
- 38. Shin S., Dimitri C. A., Yoon S. O., Dowdle W., Blenis J. (2010) Mol. Cell 38, 114–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Langlois S., Nyalendo C., Di Tomasso G., Labrecque L., Roghi C., Murphy G., Gingras D., Béliveau R. (2007) Mol. Cancer Res. 5, 569–583 [DOI] [PubMed] [Google Scholar]
- 40. Moss N. M., Barbolina M. V., Liu Y., Sun L., Munshi H. G., Stack M. S. (2009) Cancer Res. 69, 7121–7129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Blom N., Sicheritz-Pontén T., Gupta R., Gammeltoft S., Brunak S. (2004) Proteomics 4, 1633–1649 [DOI] [PubMed] [Google Scholar]
- 42. Baciu P. C., Suleiman E. A., Deryugina E. I., Strongin A. Y. (2003) Exp. Cell Res. 291, 167–175 [DOI] [PubMed] [Google Scholar]
- 43. Wolf K., Mazo I., Leung H., Engelke K., von Andrian U. H., Deryugina E. I., Strongin A. Y., Bröcker E. B., Friedl P. (2003) J. Cell Biol. 160, 267–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tanimura S., Asato K., Fujishiro S. H., Kohno M. (2003) Biochem. Biophys. Res. Commun. 304, 801–806 [DOI] [PubMed] [Google Scholar]
- 45. D'Alessio S., Ferrari G., Cinnante K., Scheerer W., Galloway A. C., Roses D. F., Rozanov D. V., Remacle A. G., Oh E. S., Shiryaev S. A., Strongin A. Y., Pintucci G., Mignatti P. (2008) J. Biol. Chem. 283, 87–99 [DOI] [PubMed] [Google Scholar]
- 46. Sato H., Takino T., Okada Y., Cao J., Shinagawa A., Yamamoto E., Seiki M. (1994) Nature 370, 61–65 [DOI] [PubMed] [Google Scholar]
- 47. Seiki M., Yana I. (2003) Cancer Sci. 94, 569–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hadler-Olsen E., Fadnes B., Sylte I., Uhlin-Hansen L., Winberg J. O. (2011) FEBS J. 278, 28–45 [DOI] [PubMed] [Google Scholar]
- 49. Gialeli C., Theocharis A. D., Karamanos N. K. (2011) FEBS J. 278, 16–27 [DOI] [PubMed] [Google Scholar]