

Background: Tau inhibits kinesin on GDP-microtubules in vitro, but the physiological significance in neurons is unclear.
Results: On GTP-microtubules, Tau loses its inhibitory effect, and kinesin becomes less processive.
Conclusion: The nucleotide-binding state of the microtubule influences the behavior of both kinesin and Tau.
Significance: Tau has different functions, both inhibitory and non-inhibitory, in regulating axonal transport.
Keywords: Kinesin, Microtubules, Neurons, Nucleotide, Tau
Abstract
The ability of Tau to act as a potent inhibitor of kinesin's processive run length in vitro suggests that it may actively participate in the regulation of axonal transport in vivo. However, it remains unclear how kinesin-based transport could then proceed effectively in neurons, where Tau is expressed at high levels. One potential explanation is that Tau, a conformationally dynamic protein, has multiple modes of interaction with the microtubule, not all of which inhibit kinesin's processive run length. Previous studies support the hypothesis that Tau has at least two modes of interaction with microtubules, but the mechanisms by which Tau adopts these different conformations and their functional consequences have not been investigated previously. In the present study, we have used single molecule imaging techniques to demonstrate that Tau inhibits kinesin's processive run length in an isoform-dependent manner on GDP-microtubules stabilized with either paclitaxel or glycerol/DMSO but not guanosine-5′-((α,β)-methyleno)triphosphate (GMPCPP)-stabilized microtubules. Furthermore, the order of Tau addition to microtubules before or after polymerization has no effect on the ability of Tau to modulate kinesin motility regardless of the stabilizing agent used. Finally, the processive run length of kinesin is reduced on GMPCPP-microtubules relative to GDP-microtubules, and kinesin's velocity is enhanced in the presence of 4-repeat long Tau but not the 3-repeat short isoform. These results shed new light on the potential role of Tau in the regulation of axonal transport, which is more complex than previously recognized.
Introduction
Neurons are electrically excitable cells responsible for receiving and transmitting information throughout the nervous system. These cells have a distinct polarity and a unique architecture, consisting of a complicated system of input processes known as dendrites and a single extremely long output process known as the axon, which can be up to a meter in length in extreme cases. Such extraordinarily long distances pose a unique set of problems for neurons, because most of the proteins, organelles, and other cellular materials required for axonal function are produced in the neuronal cell body (1). Because these distances are far too great for diffusion to move cargo efficiently along the length of the axon, neurons take advantage of the microtubule-based molecular motors kinesin and dynein to facilitate anterograde and retrograde axonal transport, respectively. Kinesin is particularly well suited for this function, because it is a highly processive motor capable of transporting cargo produced in the cell body over long distances down the axon without dissociating from the underlying microtubule track. However, this presents a new challenge as to how the processive behavior of kinesin can be modulated to ensure that cargo is delivered to the appropriate locations within the axon, either at intermediate points along its length (e.g. nodes of Ranvier in myelinated nerve cells) or at presynaptic terminals, which may require navigating through numerous axonal branch points. Given the number of heterogeneous intracellular cargo and destinations within the axon, there are likely to be several different mechanisms for regulating kinesin's motile function in vivo, including posttranslational modifications of specific kinesin subunits (2) and the underlying microtubule track (e.g. polyglutamylation, tyrosination/detyrosination, and acetylation) (3–5). Recent work has indicated that the nucleotide-binding state (i.e. GTP versus GDP) of tubulin subunits in the microtubule may be important in modulating interactions with kinesin as well (6). A fourth possible level of regulation involves microtubule-associated proteins, which may directly or indirectly influence the function of kinesin. For example, Tau is a neuron-specific microtubule-associated protein that has previously been shown to inhibit kinesin mediated transport in an isoform-specific manner, both in vitro (7, 8) and in vivo (9, 10). However, mechanisms underlying the inhibition of kinesin-based axonal transport by Tau remain unknown (11).
In humans, there are six known Tau isoforms, which are found primarily in the axonal compartment of neurons (12). Isoforms of Tau differ by possessing either three or four microtubule binding motifs in the C-terminal microtubule binding domain and by the presence or absence of one or two acidic inserts in their N-terminal projection domain (13). Tau has been shown to inhibit kinesin motility in vitro, with the 3-repeat short (3RS)2 isoform (possessing three microtubule binding repeats and no acidic inserts) having a greater effect than the 4-repeat long (4RL) isoform (possessing four microtubule binding repeats and two acidic inserts) (7, 8). Given these results, it is unclear how axonal transport can efficiently operate in neurons, because the 3- and 4-repeat isoforms of Tau are highly expressed at approximately equal levels in mature axons (14, 15). Reports from more physiologically relevant model systems have been conflicting, with the overexpression of Tau disrupting mitochondrial transport in cortical neurons (10) but the addition of supraphysiological levels of Tau having no effect on fast axonal transport in extruded axoplasm (11). One possible explanation that reconciles these disparate observations is that the manner in which Tau itself interacts with the microtubule lattice may be regulated, because there is both structural (16) and biochemical (17) evidence that Tau may have more than one binding site on the microtubule. In addition to the external binding site occupied by Tau when bound to preformed, paclitaxel-stabilized microtubules, there appears to be an interior (luminal side) binding site for Tau when copolymerized with free tubulin and stabilized with the slowly hydrolyzable GTP analog GMPCPP (16). Consistent with these results, Tau copolymerized with tubulin in the presence of GTP has also been observed to exist in two populations, one being more stably bound than the other, whereas preformed microtubules stabilized with paclitaxel only possess the more dynamic population of Tau (17).
The existence of multiple populations of Tau opens up the intriguing possibility that, depending on its mode of interaction with the microtubule, Tau can adopt different conformations that result in different functions within the neuron. However, because most previous in vitro studies of the effect of Tau on kinesin motility have been done using paclitaxel-stabilized microtubules (7, 8), the role of alternative Tau-tubulin complexes involving copolymerization or different nucleotide-binding states of the microtubule (i.e. GDP versus GTP) has not been investigated. Thus, in the current work, we have directly examined the processive run length and velocity of kinesin-quantum dot complexes on microtubules in different combinations of nucleotide-binding state (GDP or GMPCPP), paclitaxel-stabilization, isoforms of Tau (3RS and 4RL), and order of Tau addition (during or after tubulin polymerization). We show for the first time that Tau is not only a negative regulator of kinesin motility, as previously reported (7, 8), but can adopt a non-inhibitory conformation on GMPCPP-microtubules and even enhance kinesin velocity in an isoform specific manner. These results have important implications for the process of axonal transport in nerve cells, which have recently been shown to be rich in GTP-tubulin (6), and suggest a mechanism by which changes to the microtubule lattice can dictate the function of microtubule binding partners, including kinesin and Tau, in complex and interesting ways.
EXPERIMENTAL PROCEDURES
Reagents
Paclitaxel, anti-β III tubulin monoclonal antibodies, PIPES, ATP, glucose oxidase, catalase, and glucose were purchased from Sigma-Aldrich. Streptavidin-coated 655 quantum dots (Qdots) and Alexa Fluor 488-C5 maleimide were purchased from Invitrogen. GMPCPP was purchased from Jena Bioscience (Jena, Germany). All other reagents were of the highest quality available.
Protein Expression and Purification
3RS- and 4RL-Tau isoforms were expressed in BL21-CodonPlus(DE3)-RP Escherichia coli cells (Stratagene, La Jolla, CA) using the isopropyl 1-thio-β-d-galactopyranoside-inducible pET vector system (Novagen, Madison, WI). Cells were lysed, and Tau was purified as described previously (16). Briefly, extracted proteins were boiled, clarified by centrifugation, passed through a 0.22-μm filter, and isolated by consecutive Q Sepharose® and SP Sepherose® Fast Flow columns (Sigma). Purified Tau was dialyzed in BRB80 buffer (80 mm PIPES, pH 6.9, at room temperature, 1 mm EGTA, 2 mm MgSO4), and purity was assessed by SDS-PAGE. Protein concentration was determined with the bicinchoninic acid protein assay (Pierce) using desalted, lyophilized 3RS- or 4RL-Tau as standards. Proteins were snap-frozen in liquid nitrogen and stored at −80 °C. Bovine brain was obtained from Vermont Livestock, Slaughter, and Processing (Ferrisburgh, VT), and tubulin was purified by two cycles of temperature-regulated polymerization and depolymerization in high molarity PIPES buffer (1 m PIPES, pH 6.9, at room temperature, 10 mm MgCl, and 20 mm EGTA) as described previously (18). Sf9 cells were co-infected with recombinant baculovirus containing a constitutively active truncated rat KIF5C kinesin heavy chain, ending in amino acid Ala888 with a C-terminal biotin tag (for attachment to streptavadin Qdots) followed by a FLAG epitope and YFP-tagged kinesin light chain 2 (kind gifts from Dr. Kathy Trybus, University of Vermont). Expressed dimeric kinesin constructs were purified as described previously (19). Briefly, cells were grown in suspension for 72 h, lysed, and clarified, and kinesin was isolated on a FLAG affinity resin column (Sigma), followed by elution with FLAG peptide (Sigma). Purified kinesin was dialyzed against 10 mm HEPES, pH 7.3, at 4 °C, 200 mm NaCl, 1 mm DTT, 10 μm MgATP, 50% glycerol, and 1 μg/ml leupeptin for storage at −20 °C.
Fluorescent Labeling of Tau
Tau was thawed on ice and incubated with a 10-fold molar excess of DTT for 2 h at room temperature. DTT was removed by passing Tau through a 2-ml 7,000 molecular weight cut-off ZebaTM spin desalting column (Pierce). After desalting, Tau was incubated in a 10-fold molar excess of Alexa Fluor 488-C5 maleimide for an additional 2 h at room temperature. Excess fluorophore was removed using a second desalting column. Labeling efficiency of Alexa Fluor 488-Tau was determined by comparing the concentration of fluorophore to protein. Tau concentration was determined as described above, and dye concentration was determined using an extinction coefficient of 71,000 cm−1 m−1 at 488 nm in a NanoDrop® ND-1000 spectrophotometer (Thermo Scientific, Rockford, IL) or by comparing the absorbance at 488 nm of the labeled protein with a standard curve created from various concentrations of free fluorophore. Both methods determined the labeling efficiency to be 75–80%. Alexa Fluor 488-labeled Tau was snap-frozen in liquid nitrogen and stored at −80 °C.
Microtubule Preparation
For all experiments, tubulin was thawed on ice and centrifuged at 350,000 × g for 20 min at 4 °C prior to use. For labeled microtubules, rhodamine-labeled tubulin (Cytoskeleton Inc., Denver, CO) was mixed with unlabeled tubulin at a 1:10 labeled/unlabeled tubulin ratio before polymerization. For paclitaxel-stabilized microtubules, tubulin was incubated in the presence of 1 mm GTP for 20 min at 37 °C, followed by the addition of an equal volume of motility assay buffer (MAB) (10 mm PIPES, pH 7.4, at room temperature, 50 mm potassium acetate, 4 mm magnesium acetate, 1 mm EGTA, 10 mm DTT, 1 mg/ml BSA, and an oxygen-scavenging system composed of 0.1 mg/ml glucose oxidase, 0.15 mg/ml catalase, and 3.0 mg/ml glucose) at a final concentration of 20 μm paclitaxel. The paclitaxel-stabilized microtubules were incubated for an additional 20 min at 37 °C before centrifugation at 16,000 × g at room temperature for 30 min. Microtubule pellets were resuspended in MAB buffer supplemented with 20 μm paclitaxel and stored at room temperature. For GMPCPP-microtubules, tubulin was incubated at 37 °C for 15 min, at which time GMPCPP was added to 1 mm. Microtubules were incubated for an additional 30 min at 37 °C and diluted 50% with 37 °C MAB buffer and 1 mm GMPCPP, followed by a final 30 min of 37 °C incubation. Microtubules were centrifuged at 16,000 × g at room temperature and resuspended in 37 °C MAB supplemented with 1 mm GMPCPP. Alternatively, lyophilized tubulin was resuspended in 1 mm GMPCPP buffer, and 1 μm GMPCPP-tubulin was incubated at 37 °C for 20 min. Additional 1 μm aliquots of GMPCPP-tubulin were added every 20 min until the final microtubule concentration was ∼5 μm. Both methods of polymerization produced equivalent results in the TIRF assays. In the absence of stabilizing agents, microtubules were prepared at the microscope. Tubulin, supplemented with 1 mm GTP, was mixed with 10% (v/v) each of glycerol and DMSO prior to a 30-min incubation at 37 °C. For all experiments using Tau, Tau was added after polymerization and before centrifugation except for copolymer experiments, where Tau was added prior to polymerization.
Protein Attachment to Qdots
Streptavidin-coated Qdots, emitting at 655 nm, were attached to kinesin by incubating at a 16:1 Qdot/kinesin ratio for ∼20 min at room temperature and then stored on ice, similar to previously described methods (20–22). At this molar ratio, 95% of moving quantum dots should only have a single kinesin motor bound (23).
Single Molecule TIRF Assay
TIRF assays were performed at room temperature on a Nikon TE2000-U microscope with a PlanApo lens (100×; numerical aperture, 1.49). Qdots, Alexa Fluor 488-labeled Tau, and rhodamine tubulin were excited with a 488-nm argon laser and emission filters (wavelength/band pass) of 630/60, 515/30, and 560/55 nm, respectively. Single color images were collected using a Turbo 620G high resolution 12-bit digital camera (Stanford Photonics, Palo Alto, CA) and a Video Scope VS4-1845 intensifier (Video Scope International, Dulles, VA). Images were processed using Piper ControlTM software (Stanford Photonics). 1000 images for Qdots and 100 images for microtubules were captured at 10 frames/s. Flow chambers were prepared by adhering ARTUS shims (ARTUS, Englewood, NJ) to siliconized glass coverslips using Norland optical adhesive (Norland Products Inc., Cranbury, NJ) followed by UV irradiation for 10 min. Samples were prepared by incubating the flow chamber with monoclonal anti-β III (neuronal) antibodies, diluted to 1–3% in BRB80 buffer, for 5 min. The chambers were washed and blocked with MAB (containing 1 mg/ml BSA) for an additional 5 min before the addition of microtubules for 15 min. For microtubules without a stabilizing agent, slides and buffers were kept at 37 °C until viewed. Kinesin-Qdot conjugates were diluted to 1 nm in MAB with 2 mm ATP and flowed through the observation cell just prior to data collection.
Microtubule-activated ATPase Assay
Detection of inorganic phosphate resulting from the hydrolysis of ATP by kinesin in the presence of various concentrations of microtubules was used to determine the microtubule-activated ATPase activity at room temperature of preparations in the absence and presence of 3RS- or 4RL-Tau at a 1:5 Tau/tubulin ratio. 50 nm kinesin was incubated with paclitaxel, ATP, and 2–35 μm paclitaxel-stabilized microtubules in ATPase buffer (20 mm MOPS, pH 7.2, at room temperature, 50 mm potassium acetate, 5 mm magnesium acetate, 0.1 mm EGTA, 0.1 mm EDTA, 1 mm DTT) for 5 min. Every minute, 5 μl of the reaction was placed in 200 μl of a solution composed of 1.6% ammonium molybdate (in 6 n HCl), 0.67% polyvinyl alcohol, and 0.023% malachite green. After 30 s, the reaction was quenched using 50 μl of 34% sodium citrate. The color was allowed to develop for 1 h, at which time the samples were read in a 96-well BioTek® plate reader (BioTek, Winooski, VT) at 595 nm. Rates of ATP hydrolysis were determined by the increase in phosphate concentration over time and plotted as a function of microtubule concentration. Microtubule-activated ATPase data were fit to Michaelis-Menten kinetics using GraphPad Prism software, version 5.0 (GraphPad Software, La Jolla, CA) to determine values of Vmax and Km ± S.E. of the fit under each condition studied.
Data Analysis
The processive run length and velocity of kinesin were measured using the manual object tracking plugin, MTrackJ, in ImageJ software, version 1.44 (National Institutes of Health, Bethesda, MD). Processive run lengths greater than 0.5 μm were recorded, and velocities were calculated as run length/time of the run. The 100 microtubule images were z-projected by average intensity, and kinesin tracks were overlaid on the averaged microtubule field. Only runs with clear beginnings and endings on visible microtubule tracks were counted and plotted in 0.5-μm bins. In addition, runs with significant pauses (>5 frames), runs over 10 μm, and runs that were perceived to encounter road blocks were discarded. All data sets reported are composites of data collected over multiple days from at least four different preparations of kinesin and each isoform of Tau. Processive run length distribution histograms were fit to a single exponential decay constant, which defines the kinesin characteristic run length and are reported with the S.E. of the fit (20). Velocity measurements are reported as the mean and S.D., assuming a Gaussian distribution of values. Statistical significance at p < 0.05 was determined using the Mann-Whitney test, a nonparametric bin width-independent statistical hypothesis test, for processive run length comparisons, and an unpaired two-tailed Student's t test was used to compare velocity measurements. All nonlinear regression fitting and statistical analysis were performed using GraphPad Prism software, version 5.0 (GraphPad Software).
Microtubule Pelleting Assay
20 μm microtubules were prepared with labeled Tau under various experimental conditions as described above and centrifuged for 30 min at 16,000 × g and 37 °C. The supernatant was removed, and the pellet was resuspended in MAB (without BSA and oxygen scavengers) of equal volume to the supernatant. The Tau in each fraction was assessed by NanoDrop absorption spectroscopy as described above. Labeled Tau was examined at 1 and 2 μm by SDS-PAGE. Gels were viewed by Coomassie staining and fluorescence using a Pharos FXTM Plus molecular imager (Bio-Rad) with laser excitation at 488 nm (supplemental Fig. S1, A and B, lanes 4–7).
RESULTS
In the Presence of Paclitaxel, Tau Modulates Kinesin-mediated Transport in an Isoform-specific Manner
Single molecule imaging was used to track the processive run length and velocity of Qdot-conjugated kinesin molecules on paclitaxel-stabilized microtubule tracks with and without 3RS- and 4RL-Tau. We observed processive run lengths of 1.31 ± 0.10 μm (Fig. 1A) in the absence of Tau, 0.62 ± 0.03 μm (Fig. 1C) in the presence of 3RS-Tau at a 1:5 Tau/tubulin ratio, and 0.84 ± 0.04 μm (Fig. 1E) in the presence of 4RL-Tau at a 1:5 Tau/tubulin ratio. Tau inhibited kinesin run lengths in an isoform-specific manner, with the 3RS-Tau isoform being more inhibitory than the 4RL-Tau isoform. This is precisely the inhibitory pattern demonstrated by other groups (7, 8).
FIGURE 1.

Characteristic run length and velocity data of kinesin on paclitaxel-stabilized microtubules in the absence and presence of 3RS-Tau or 4RL-Tau. Qdot-kinesin complexes were tracked on rhodamine-labeled paclitaxel-stabilized microtubules. Processive run length (plotted in 0.5-μm bins) and velocity (plotted in 0.1 μm/s bins) values were determined in the absence of Tau (A and B), presence of Alexa Fluor 488-labeled 3RS-Tau (1:5 Tau/tubulin ratio) (C and D), or presence of Alexa Fluor 488-labeled 4RL-Tau (1:5 Tau/tubulin ratio) (E and F). The resulting processive run length histograms were fit by a single exponential decay function describing the characteristic run length ± S.E. of the fit, whereas the Gaussian frequency distributions of the velocity data were used to calculate the mean velocity ± S.D.
We also observed a small but statistically significant (p < 0.0001) increase in kinesin's velocity in the presence of 4RL-Tau (0.69 ± 0.25 μm/s (Fig. 1F)) relative to that observed in the absence of Tau (0.46 ± 0.22 μm/s (Fig. 1B)) or in the presence of 3RS-Tau (0.50 ± 0.21 μm/s (Fig. 1D)), which has not been described previously (7, 8). In order to verify this result, we assayed microtubule-activated ATPase activity in the presence and absence of 4RL- and 3RS-Tau (Fig. 2). As expected, we observed an increase in the values of both Vmax (78.2 ± 4.7 s−1) and Km (11.7 ± 1.6 μm) on microtubules in the presence of 4RL-Tau compared with those without Tau (Vmax = 65.5 ± 3.0 s−1 and Km = 5.9 ± 0.7 μm). In the presence of 3RS-Tau, we obtained a similar Vmax value (67.3 ± 5.0 s−1) as in the absence of Tau, whereas the Km value (13.8 ± 2.2 μm) decreased in a similar manner as seen in the presence of 4RL-Tau. These results correlate well with our motility data, where we observed an approximately 20% increase in velocity in the presence of 4RL-Tau but not in the absence of Tau or in the presence of 3RS-Tau.
FIGURE 2.
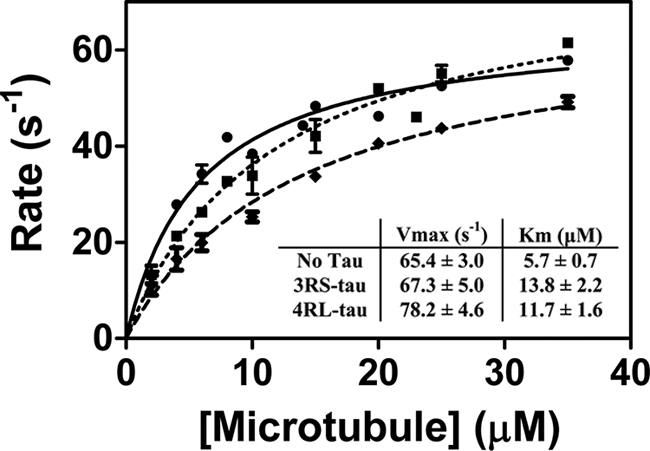
Microtubule-activated ATPase activity of kinesin in the presence and absence of 3RS- or 4RL-Tau. The rate of ATP hydrolysis by kinesin was measured and plotted as a function of microtubule concentration. Michaelis-Menten kinetics were used to determine the values of Vmax and Km for the microtubule-activated ATPase activity of kinesin in the absence (●) and presence of 3RS-Tau (♦) or 4RL-Tau (■).
Kinesin Run Lengths Are Reduced, and Tau Loses Its Ability to Inhibit Kinesin on GMPCPP-stabilized Microtubules
Using cryo-EM, Kar et al. (16) demonstrated that microtubules copolymerized with Tau in the presence of the GTP analog GMPCPP possess Tau density on the interior of the microtubule in addition to the normal binding site on the microtubule exterior. We wanted to determine if preparing Tau-microtubule complexes in this manner had an impact on the ability of Tau to modulate kinesin motility. In the absence of Tau, kinesin had a characteristic run length of 1.08 ± 0.05 μm (Fig. 3A and supplemental Movie S1) and a velocity of 0.50 ± 0.18 μm/s (Fig. 3B) on GMPCPP-stabilized microtubules, representing a significant (p = 0.035) decrease in processive run length relative to that observed on paclitaxel-stabilized microtubules. Furthermore, the inhibitory effect of Tau decreased greatly on GMPCPP microtubule-Tau copolymers. In the presence of 3RS-Tau, at a 1:5 Tau/tubulin ratio, we observed no change in processive run length (1.15 ± 0.17 μm (supplemental Fig. S2A)) or velocity (0.52 ± 0.20 μm/s (supplemental Fig. S2B)) relative to the no Tau control. In the presence of 4RL-Tau, we also saw no change in kinesin's processive run length (0.96 ± 0.10 μm (supplemental Fig. S2C)) on GMPCPP microtubule-Tau copolymers; however, velocity (0.65 ± 0.22 μm/s (supplemental Fig. S2D)) was again higher than observed in the no Tau and 3RS-Tau experiments (p < 0.0001).
FIGURE 3.

Characteristic run length and velocity data of kinesin on GMPCPP-stabilized microtubules in the absence and presence of 3RS-Tau or 4RL-Tau. Qdot-kinesin complexes were tracked on rhodamine-labeled GMPCPP-stabilized microtubules. Processive run length (plotted in 0.5-μm bins) and velocity (plotted in 0.1-μm/s bins) values were determined in the absence of Tau (A and B), in the presence of Alexa Fluor 488-labeled 3RS-Tau (1:5 Tau/tubulin ratio) (C and D), or in the presence of Alexa Fluor 488-labeled 4RL-Tau (1:5 Tau/tubulin ratio) (E and F). The resulting processive run length histograms were fit by a single exponential decay function describing the characteristic run length ± S.E. of the fit, whereas the Gaussian frequency distributions of the velocity data were used to calculate the mean velocity ± S.D.
We next directly compared the motility results from the GMPCPP microtubule-Tau copolymers with those from Tau added to preformed GMPCPP-microtubules to see if the order of Tau addition determines its ability to modulate kinesin motility. The processive run length and velocity of kinesin in the presence of 3RS-Tau added to preformed GMPCPP-microtubules at a 1:5 Tau/tubulin ratio were 1.09 ± 0.04 μm (Fig. 3C and supplemental Movie S2) and 0.49 ± 0.22 μm/s (Fig. 3D), respectively, comparable with the values observed on GMPCPP microtubules alone or copolymerized with Tau. Likewise, the processive run length and velocity of kinesin in the presence of 4RL-Tau added to preformed GMPCPP-microtubules at a 1:5 Tau/tubulin ratio were 1.08 ± 0.07 μm (Fig. 3E and supplemental Movie S3) and 0.64 ± 0.24 μm/s (Fig. 3F), respectively, again comparable with the values observed on GMPCPP microtubules alone or copolymerized with Tau. Thus, in contrast to the results obtained with the addition of Tau to preformed paclitaxel-stabilized microtubules, the addition of 3RS- or 4RL-Tau to preformed GMPCPP-microtubules at the identical 1:5 Tau/tubulin ratio does not inhibit kinesin motility relative to GMPCPP-microtubules in the absence of Tau. Our results also demonstrate that there is no quantifiable difference in kinesin's processive run length or velocity based on the order of Tau addition (during or after polymerization) to GMPCPP-microtubules.
Tau Does Not Inhibit Kinesin Run Lengths on Microtubules Stabilized with both Paclitaxel and GMPCPP
Our findings indicate at least two possible mechanisms for the observed pattern of Tau inhibition of kinesin motility on microtubules under different stabilizing conditions. It could be that Tau is inhibitory only in the presence of paclitaxel, and the inhibition is an artifact of the drug. There is previous evidence that paclitaxel may alter Tau interaction with microtubules (24). An alternative explanation is that the nucleotide-binding state of the tubulin subunits alters Tau interaction with the microtubule lattice, and Tau is less inhibitory on GTP (or GMPCPP)-microtubules than on GDP-microtubules. To discriminate between these two possibilities, we examined the effect of Tau added to preformed microtubules stabilized with both paclitaxel and GMPCPP on kinesin processive run length and velocity. The addition of 3RS-Tau at a 1:5 Tau/tubulin ratio to paclitaxel-stabilized, GMPCPP-microtubules had no effect on processive run length (1.06 ± 0.09 μm (supplemental Fig. S3C)) or velocity (0.54 ± 0.25 μm/s (supplemental Fig. S3D)) relative to the no Tau control, which had a characteristic run length of 0.97 ± 0.04 μm (supplemental Fig. S3A) and a mean velocity of 0.56 ± 0.29 μm/s (supplemental Fig. S3B). In the presence of 4RL-Tau on paclitaxel-stabilized GMPCPP-microtubules, the characteristic run length of kinesin (1.09 ± 0.07 μm (supplemental Fig. S3E)) was also indistinguishable from the no Tau control, but there was again a small but significant (p < 0.001) increase in mean velocity (0.69 ± 0.20 μm/s (supplemental Fig. S3F)). These results suggest that the nucleotide-binding state of the microtubule, and not the presence of paclitaxel, is responsible for determining the ability of Tau to modulate kinesin motility.
Tau Inhibits Kinesin Run Lengths in an Isoform-specific and Concentration-dependent Manner in the Absence of GMPCPP and Paclitaxel
To further investigate whether paclitaxel or the nucleotide-binding state was responsible for the ability of Tau to inhibit kinesin, we measured the processive run length and velocity of kinesin on microtubules in the absence of GMPCPP and paclitaxel. In order to maintain microtubule stability, this procedure required slightly different conditions, including the presence of 10% glycerol and 10% DMSO in the motility buffer. In the absence of GMPCPP, paclitaxel, or Tau, kinesin's characteristic run length on microtubules was 1.32 ± 0.07 μm (supplemental Fig. S4A and Movie S4), with a mean velocity of 0.28 ± 0.09 μm/s (supplemental Fig. S4B). The processive run length of kinesin under these conditions was not significantly different than in the presence of paclitaxel (p = 0.825); however, it was significantly longer than that observed on microtubules stabilized with GMPCPP (p = 0.039). Both the 3RS- and 4RL-Tau isoforms inhibited kinesin motility under these conditions in a manner analogous to the paclitaxel case, where 3RS-Tau had a greater effect than 4RL-Tau. In addition, both isoforms inhibit kinesin in a concentration-dependent manner. In the presence of 3RS-Tau at a 1:8 Tau/tubulin ratio, the characteristic run length of kinesin was 0.49 ± 0.03 μm (supplemental Fig. S4C) with a mean velocity of 0.34 ± 0.17 μm/s. At a lower Tau/tubulin ratio of 1:15, 3RS-Tau was less inhibitory, and we observed an increase in run length to 0.75 ± 0.06 μm (supplemental Fig. S4D) with a mean velocity of 0.37 ± 0.10 μm/s (supplemental Fig. S4E). In the presence of 4RL-Tau, we saw a similar trend, albeit with less inhibition than with 3RS-Tau. At a 1:8 4RL-Tau/tubulin ratio, we observed a characteristic run length of 0.73 ± 0.03 μm (supplemental Fig. S4F) with a mean velocity of 0.35 ± 0.15 μm/s, whereas at a 1:15 4RL-Tau/tubulin ratio, we observed a run length of 1.00 ± 0.05 μm (supplemental Fig. S4G) and a mean velocity of 0.26 ± 0.08 μm/s (supplemental Fig. S4H). Given that microtubules in the absence of GMPCPP, whether stabilized by paclitaxel or not, are most likely in a GDP state (25, 26), these results further support the conclusion that Tau is capable of inhibiting kinesin motility on GDP- but not GMPCPP-microtubules.
We also wanted to determine if the order of Tau addition before or after tubulin polymerization had any effect on kinesin motility in the absence of GMPCPP and paclitaxel. 3RS-Tau copolymerized with tubulin at a 1:15 Tau/tubulin ratio resulted in a characteristic run length of 0.77 ± 0.08 μm (supplemental Fig. S5A) and a mean velocity of 0.24 ± 0.07 μm/s (supplemental Fig. S5B), whereas 4RL-Tau, copolymerized with tubulin at the same 1:15 Tau/tubulin ratio, resulted in a characteristic run length of 0.96 ± 0.07 μm (supplemental Fig. S5C) and a mean velocity of 0.32 ± 0.12 μm/s (supplemental Fig. S5D). Thus, in both cases, 3RS-Tau and 4RL-Tau produced no significant difference in the processive run length or velocity of kinesin regardless of the order of Tau addition to the microtubules, before or after polymerization, in the absence of GMPCPP or paclitaxel.
Although the addition of 10% glycerol and 10% DMSO as stabilizing agents in the absence of paclitaxel had no effect on the processive run length of kinesin, we did observe a perceptible reduction in velocity relative to paclitaxel-stabilized microtubules. This appeared to be directly related to the addition of glycerol to the motility buffer, which we assume is due to an increase in viscosity and not an intrinsic property of microtubules in the absence of GMPCPP or paclitaxel. Velocity values in the presence of GMPCPP and 10% glycerol and 10% DMSO were reduced to 0.31 ± 0.08 μm/s (supplemental Fig. S6B) with no apparent change in the characteristic run length (1.02 ± 0.11 μm; supplemental Fig. S6A) relative to GMPCPP-microtubules in the absence of glycerol. We also observed that the velocity of kinesin in the presence of 4RL-Tau did not appear to be significantly higher than that in the absence of Tau or in the presence of 3RS-Tau under these conditions. Again this is likely to be a function of the specific conditions used for the experiment. It was also noted that Tau appeared to be slightly more inhibitory when glycerol and DMSO were used as the stabilizing agent rather than paclitaxel. To determine if this was also a product of our conditions, we measured kinesin's motility on paclitaxel-stabilized microtubules in the presence of 10% glycerol, 10% DMSO, and a 1:8 3RS-Tau/tubulin ratio. This produced results similar to those obtained at 10% glycerol and 10% DMSO with no paclitaxel, with a characteristic run length of 0.44 ± 0.01 μm (supplemental Fig. S6C) and a velocity of 0.37 ± 0.13 μm/s (supplemental Fig. S6D). To ensure that our conditions were not forcing Tau to become inhibitory on microtubules in the absence of paclitaxel, we measured kinesin motility on GMPCPP-microtubules in the presence of 3RS-Tau at a 1:8 Tau/tubulin ratio and 10% glycerol plus 10% DMSO. These experiments produced characteristic run length values (1.02 ± 0.08 μm (supplemental Fig. S6E)) similar to those observed on GMPCPP-microtubules in the absence of glycerol and DMSO but again with a reduction in velocity (0.39 ± 0.12 μm/s (supplemental Fig. S6F)). The results of all of the motility conditions tested are summarized in Table 1.
TABLE 1.
Summary of kinesin characteristic run length and velocity data
Characteristic run length values and S.E. values of the fits were obtained from single exponential fits to the binned histogram data. Velocity is reported as the mean and S.D. n is the number of processive runs analyzed in a given data set.
| Microtubule preparation | Run length | Velocity | n |
|---|---|---|---|
| μm | μm/s | ||
| Paclitaxel | 1.31 ± 0.10 | 0.46 ± 0.22 | 250 |
| + 3RS-Tau | 0.62 ± 0.03 | 0.50 ± 0.21 | 284 |
| + 4RL-Tau | 0.84 ± 0.04 | 0.69 ± 0.25 | 262 |
| GMPCPP | 1.08 ± 0.05 | 0.50 ± 0.18 | 688 |
| + 3RS-Tau | 1.09 ± 0.04 | 0.49 ± 0.22 | 411 |
| 3RS-Tau copolymers | 1.15 ± 0.17 | 0.52 ± 0.20 | 324 |
| + 4RL-Tau | 1.08 ± 0.07 | 0.64 ± 0.24 | 202 |
| 4RL-Tau copolymers | 0.96 ± 0.10 | 0.65 ± 0.22 | 200 |
| GMPCPP + paclitaxel | 0.97 ± 0.04 | 0.56 ± 0.29 | 288 |
| + 3RS-Tau | 1.06 ± 0.09 | 0.54 ± 0.25 | 213 |
| + 4RL-Tau | 1.09 ± 0.07 | 0.69 ± 0.20 | 199 |
| Glycerol/DMSO | 1.32 ± 0.07 | 0.28 ± 0.09 | 357 |
| + 3RS-Tau (1:8 Tau/tubulin ratio) | 0.49 ± 0.03 | 0.34 ± 0.17 | 214 |
| + 3RS-Tau (1:15 Tau/tubulin ratio) | 0.75 ± 0.06 | 0.37 ± 0.10 | 333 |
| 3RS copolymers | 0.77 ± 0.08 | 0.24 ± 0.07 | 243 |
| + 4RL-Tau (1:8 tau/tubulin ratio) | 0.73 ± 0.03 | 0.35 ± 0.15 | 241 |
| + 4RL-Tau (1:15 tau/tubulin ratio) | 1.00 ± 0.05 | 0.26 ± 0.08 | 282 |
| 4RL copolymers | 0.96 ± 0.07 | 0.32 ± 0.12 | 196 |
Tau Has a Similar Affinity for Microtubules under All of the Experimental Conditions Tested
Finally, we wanted to determine if the difference in the ability of Tau to inhibit kinesin under our various experimental conditions was due to a difference in affinity of Tau for the various microtubule preparations used. To examine this possibility, we polymerized tubulin with either glycerol/DMSO, GMPCPP, GMPCPP with paclitaxel, or paclitaxel alone and added Alexa Fluor 488-labeled 3RS- or 4RL-Tau at a 1:5 Tau/tubulin ratio. Samples were centrifuged, and the pellet and supernatant fractions were analyzed for Tau content. In all cases, the majority of Tau appeared in the pellet fraction, and there was no significant difference in Tau binding affinity for microtubules between the preparations (Fig. 4). We also qualitatively examined each fraction by SDS-PAGE analysis. Upon excitation of the gel, it was apparent that the supernatant fractions contained only small quantities of labeled Tau, whereas the bulk of the protein was contained in the pellet fractions (supplemental Fig. S1A). After staining the gel with Coomassie, it was clear that the majority of tubulin was located in the pellet fractions (supplemental Fig. S1B).
FIGURE 4.
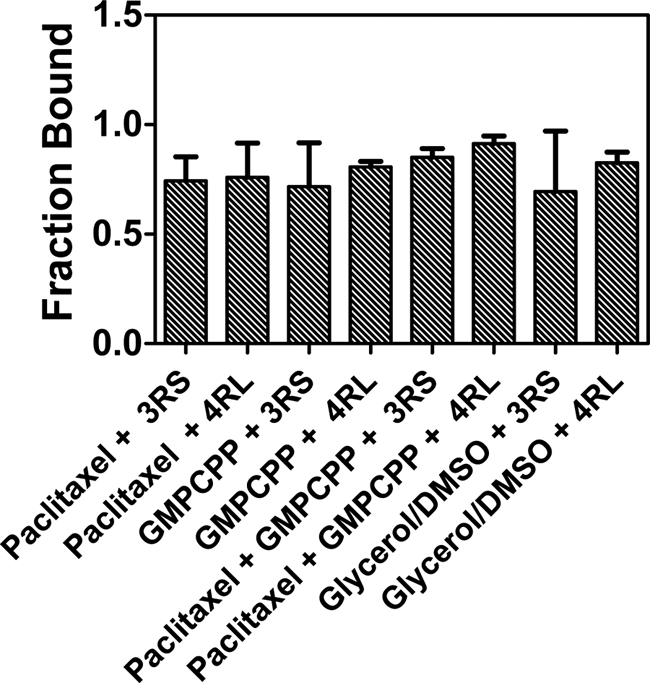
Fraction of Tau bound to microtubules under various stabilizing conditions. Microtubules were formed and stabilized with either paclitaxel (Taxol), GMPCPP, paclitaxel (Taxol) + GMPCPP, or glycerol/DMSO. Alexa Fluor 488-labeled 3RS- or 4RL-Tau was added at a 1:5 Tau/tubulin ratio, the microtubule-Tau complexes were centrifuged, and the resulting pellet and supernatant fractions were assessed for labeled Tau content. The fraction of Tau in the pellet is plotted for each stabilizing condition. Error bars, S.D.
DISCUSSION
Our results are consistent with previous reports of the ability of Tau to inhibit kinesin-mediated transport on paclitaxel-stabilized microtubules in an isoform-specific manner, with the 3RS-Tau isoform being more inhibitory than the 4RL-Tau isoform (7, 8). In striking contrast, Tau loses all ability to reduce kinesin's processive run length on GMPCPP-stabilized microtubules over the physiologically relevant range of Tau concentrations used in these experiments. At supraphysiological concentrations of Tau (1:1 Tau/tubulin ratio), we do see abolishment of kinesin motility on GMPCPP-microtubules (data not shown); however, it has been shown that Tau forms aggregates on the microtubule surface at such high concentrations, which may result in a completely different form of inhibition (27, 28). Because we did observe inhibition of kinesin by Tau on microtubules formed in the presence of glycerol/DMSO as an alternative stabilizing agent to paclitaxel, we doubt that paclitaxel is somehow inducing the inhibitory state of Tau. To completely preclude this possibility, we monitored kinesin motility on microtubules stabilized by both GMPCPP and paclitaxel. Again, Tau has no apparent effect on kinesin motility under these conditions. GMPCPP-microtubules closely resemble microtubules in the GTP nucleotide state, whereas microtubules stabilized with paclitaxel or glycerol/DMSO are presumably more representative of the GDP nucleotide state (29–31). We therefore conclude that Tau and/or kinesin can adopt a different mode of interaction with microtubules in the GTP state as compared with the GDP state, which abolishes the ability of Tau to inhibit kinesin motility.
In addition to Tau losing its ability to inhibit kinesin-mediated transport on GMPCPP-microtubules, we observe that kinesin's characteristic run length was reduced by ∼20% on GMPCPP-microtubules as compared with paclitaxel- or glycerol/DMSO-stabilized microtubules. This indicates that kinesin, like Tau, also interacts with GMPCPP-microtubules in a different manner than with paclitaxel- or glycerol/DMSO-stabilized microtubules. Previous work has demonstrated that small changes in the microtubule lattice can affect kinesin-mediated transport, where post-translational modifications, such as acetylation, detyrosination, and polyglutamylation, influence kinesin binding and motility on microtubules (5, 32, 33). Kinesin has also been shown to have an increased velocity in gliding assays on GMPCPP-microtubules as compared with GDP-microtubules (31) and to bind GMPCPP-microtubules 3.7-fold tighter than paclitaxel-stabilized GDP-microtubules. This discrimination toward the GTP state is facilitated by kinesin loop 11, which mediates the strong binding of kinesin in the ATP or apo states of its enzymatic cycle (6). Interestingly, this is similar to what has been observed with kinesin binding to subtilisin-treated microtubules in which the C-terminal tail of tubulin has been enzymatically removed (34). Not only does subtilisin treatment promote kinesin binding to microtubules, but it also leads to a reduction in kinesin's processive run length (35) much like we observe on GMPCPP-microtubules (35). Although we are not proposing direct structural links between microtubules in the GTP state and those missing their C-terminal tails, there may be a correlation between the stronger binding of kinesin on GMPCPP-stabilized microtubules and our observation that kinesin's characteristic run length is reduced under these conditions, just as in the case of subtilisin-treated microtubules.
Interestingly, in addition to the observed effects of the underlying microtubule lattice on kinesin motility, we see a small but significant increase in the velocity of kinesin in the presence of 4RL-Tau relative to that observed in the absence of Tau or the presence of 3RS-Tau, which has not been reported previously (7, 36). We cannot account for this discrepancy between our group and others; however, we consistently see this increase in velocity across all of our experimental conditions. We also observed a 20% increase in Vmax of the microtubule-activated ATPase activity of kinesin in the presence of 4RL-Tau compared with the 3RS-Tau and no Tau cases, which corresponds well with the 20% increase in velocity we saw in the motility assays in the presence of 4RL-Tau. Furthermore, it was recently shown in in vitro gliding assays of microtubules on a surface of kinesin that sliding velocity was reduced 17% in the presence of 4RS-Tau as compared with 3RS-Tau (24). The major difference between the 4RS-Tau used in the previous work and the 4RL-Tau used by our group in the current study is the inclusion of two 29-amino acid acidic inserts in the N-terminal projection domain of 4RL-Tau. Because we see an increase in velocity in our study, it is possible that these acidic inserts are influencing kinesin's interaction with the microtubule and are responsible for the isoform-specific effect on kinesin velocity and possibly reduction in processive run length. The N-terminal tails of Tau are acidic in general, and with the inclusion of the acidic inserts, the tails become highly acidic (13). Previous work has demonstrated that the acidic C-terminal tails of tubulin enhance kinesin processivity and velocity (35, 37). Because we see an increase in velocity with the 4RL-Tau isoform, it is tempting to speculate that when Tau is not in an inhibitory state, it may actually enhance kinesin motility via its acidic tail domain analogous to the C-terminal tails of tubulin.
Our results present a novel role for the microtubule lattice in modulating the interaction between kinesin and Tau and have important implications for the regulation of axonal transport in different developmental and pathological states of the neuron. The traditional view is that microtubules, with the exception of their GTP caps, exist primarily in the GDP form within the cell. Thus, considering the results of the present work and those reported previously (7, 8), one would expect Tau to bind microtubules mainly in its inhibitory conformation in vivo and, at the high levels at which it is expressed in neurons, significantly disrupt axonal transport. However, it has recently been demonstrated that there are significant populations of both GTP and GDP-tubulin in the axon of developing and mature neurons, and the microtubule lattice contains numerous segments predominantly composed of GTP-tubulin (6). In addition, kinesin localizes to these regions of GTP-tubulin, which may facilitate the localization of kinesin-1 to axons as opposed to dendrites (6). If GTP-tubulin is indeed the axonal localization cue for kinesin, it would be detrimental to the cell if Tau were inhibitory in regions of high GTP-tubulin content that promote kinesin binding, because kinesin could lose its ability to target cargo to their correct locations. This problem is potentially even more acute during the early stages of neuronal development when 3-repeat Tau, a more potent inhibitor of kinesin motility than 4-repeat Tau, is the predominant isoform expressed (38). During this time, one would expect kinesin-mediated transport to be essential for delivering materials to the growing axon and ancillary branches (39). The existence of significant GTP-tubulin populations along the length of the axon, which are present in mature neurons and appear to be enriched at early stages of development (6) and to which we observe that Tau binds in a non-inhibitory conformation, would explain how axonal transport mediated by kinesin could proceed unimpeded in the presence of high levels of Tau expression in the neuron. Thus, regulation of kinesin-mediated transport in the axon by Tau is likely to be a complex process dependent on the structural state of the microtubule (i.e. GTP versus GDP). Our results demonstrate that Tau is not simply an inhibitor of kinesin motility but that it can function in a non-inhibitory manner or even enhance axonal transport, depending on the specific isoform involved and its interaction with the underlying microtubule lattice.
Supplementary Material
Acknowledgments
We thank Dr. Steven King for the 3RS- and 4RL-Tau cDNA constructs and the laboratory of Dr. Kathy Trybus for the kinesin cDNA and ongoing support throughout this project. We also thank Gabrielle Anderson, Dr. Justin Decarreau, Dr. Nicole E. LaPointe, Dr. Gerardo A. Morfini, Dr. Jason Stumpff, and Dr. Andrew Thompson for help and guidance during the preparation of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health, NINDS, Grant R21 NS066249 (to C. L. B.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S6 and Movies S1–S4.
- 3RS
- 3-repeat short
- 4RL
- 4-repeat long
- GMPCPP
- guanosine-5′-((α,β)-methyleno)triphosphate
- TIRF
- total internal reflection fluorescence
- MAB
- motility assay buffer
- Qdot
- quantum dot.
REFERENCES
- 1. Hirokawa N., Noda Y. (2008) Physiol. Rev. 88, 1089–1118 [DOI] [PubMed] [Google Scholar]
- 2. Morfini G., Pigino G., Brady S. T. (2007) Methods Mol. Biol. 392, 51–69 [DOI] [PubMed] [Google Scholar]
- 3. Dunn S., Morrison E. E., Liverpool T. B., Molina-París C., Cross R. A., Alonso M. C., Peckham M. (2008) J. Cell Sci. 121, 1085–1095 [DOI] [PubMed] [Google Scholar]
- 4. Konishi Y., Setou M. (2009) Nat. Neurosci. 12, 559–567 [DOI] [PubMed] [Google Scholar]
- 5. Reed N. A., Cai D., Blasius T. L., Jih G. T., Meyhofer E., Gaertig J., Verhey K. J. (2006) Curr. Biol. 16, 2166–2172 [DOI] [PubMed] [Google Scholar]
- 6. Nakata T., Niwa S., Okada Y., Perez F., Hirokawa N. (2011) J. Cell Biol. 194, 245–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dixit R., Ross J. L., Goldman Y. E., Holzbaur E. L. (2008) Science 319, 1086–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vershinin M., Carter B. C., Razafsky D. S., King S. J., Gross S. P. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 87–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stamer K., Vogel R., Thies E., Mandelkow E., Mandelkow E. M. (2002) J. Cell Biol. 156, 1051–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stoothoff W., Jones P. B., Spires-Jones T. L., Joyner D., Chhabra E., Bercury K., Fan Z., Xie H., Bacskai B., Edd J., Irimia D., Hyman B. T. (2009) J. Neurochem. 111, 417–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morfini G., Pigino G., Mizuno N., Kikkawa M., Brady S. T. (2007) J. Neurosci. Res. 85, 2620–2630 [DOI] [PubMed] [Google Scholar]
- 12. Andreadis A., Brown W. M., Kosik K. S. (1992) Biochemistry 31, 10626–10633 [DOI] [PubMed] [Google Scholar]
- 13. Goode B. L., Chau M., Denis P. E., Feinstein S. C. (2000) J. Biol. Chem. 275, 38182–38189 [DOI] [PubMed] [Google Scholar]
- 14. Aronov S., Aranda G., Behar L., Ginzburg I. (2001) J. Neurosci 21, 6577–6587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Trojanowski J. Q., Schuck T., Schmidt M. L., Lee V. M. (1989) J. Histochem. Cytochem. 37, 209–215 [DOI] [PubMed] [Google Scholar]
- 16. Kar S., Fan J., Smith M. J., Goedert M., Amos L. A. (2003) EMBO J. 22, 70–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Makrides V., Massie M. R., Feinstein S. C., Lew J. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 6746–6751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Castoldi M., Popov A. V. (2003) Protein Expr. Purif. 32, 83–88 [DOI] [PubMed] [Google Scholar]
- 19. Lu H., Ali M. Y., Bookwalter C. S., Warshaw D. M., Trybus K. M. (2009) Traffic 10, 1429–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ali M. Y., Lu H., Bookwalter C. S., Warshaw D. M., Trybus K. M. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 4691–4696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Higuchi H., Muto E., Inoue Y., Yanagida T. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 4395–4400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Svoboda K., Block S. M. (1994) Cell 77, 773–784 [DOI] [PubMed] [Google Scholar]
- 23. Hodges A. R., Krementsova E. B., Trybus K. M. (2007) J. Biol. Chem. 282, 27192–27197 [DOI] [PubMed] [Google Scholar]
- 24. Peck A., Sargin M. E., LaPointe N. E., Rose K., Manjunath B. S., Feinstein S. C., Wilson L. (2011) Cytoskeleton 68, 44–55 [DOI] [PubMed] [Google Scholar]
- 25. Tran P. T., Joshi P., Salmon E. D. (1997) J. Struct. Biol. 118, 107–118 [DOI] [PubMed] [Google Scholar]
- 26. Xiao H., Verdier-Pinard P., Fernandez-Fuentes N., Burd B., Angeletti R., Fiser A., Horwitz S. B., Orr G. A. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 10166–10173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ackmann M., Wiech H., Mandelkow E. (2000) J. Biol. Chem. 275, 30335–30343 [DOI] [PubMed] [Google Scholar]
- 28. Santarella R. A., Skiniotis G., Goldie K. N., Tittmann P., Gross H., Mandelkow E. M., Mandelkow E., Hoenger A. (2004) J. Mol. Biol. 339, 539–553 [DOI] [PubMed] [Google Scholar]
- 29. Meurer-Grob P., Kasparian J., Wade R. H. (2001) Biochemistry 40, 8000–8008 [DOI] [PubMed] [Google Scholar]
- 30. Munson K. M., Mulugeta P. G., Donhauser Z. J. (2007) J. Phys. Chem. B 111, 5053–5057 [DOI] [PubMed] [Google Scholar]
- 31. Vale R. D., Coppin C. M., Malik F., Kull F. J., Milligan R. A. (1994) J. Biol. Chem. 269, 23769–23775 [PubMed] [Google Scholar]
- 32. Bulinski J. C. (2007) Curr. Biol. 17, R18–R20 [DOI] [PubMed] [Google Scholar]
- 33. Hammond J. W., Huang C. F., Kaech S., Jacobson C., Banker G., Verhey K. J. (2010) Mol. Biol. Cell 21, 572–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Skiniotis G., Cochran J. C., Müller J., Mandelkow E., Gilbert S. P., Hoenger A. (2004) EMBO J. 23, 989–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Z., Sheetz M. P. (2000) Biophys. J. 78, 1955–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Seitz A., Kojima H., Oiwa K., Mandelkow E. M., Song Y. H., Mandelkow E. (2002) EMBO J. 21, 4896–4905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lakämper S., Meyhöfer E. (2005) Biophys. J. 89, 3223–3234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Takuma H., Arawaka S., Mori H. (2003) Brain Res. Dev. Brain Res. 142, 121–127 [DOI] [PubMed] [Google Scholar]
- 39. Yu W., Baas P. W. (1994) J. Neurosci. 14, 2818–2829 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.