

Background: The biological role of miR-31 in vascular smooth muscle cell (VSMC) growth is currently unknown.
Results: rno-miR-31 up-regulated by the extracellular regulated kinase (ERK) could increase VSMC proliferation via LATS2 (large tumor suppressor homolog 2).
Conclusion: ERK/miR-31/LATS2 may represent a novel signaling pathway in VSMC growth.
Significance: The study may provide a new molecular mechanism in vascular disease.
Keywords: Cellular Regulation, Gene Regulation, MicroRNA, Signal Transduction, Smooth Muscle, Proliferation, Vascular Biology
Abstract
Aberrant growth of vascular smooth muscle cells (VSMCs) is a major cellular event in the pathogenesis of many proliferative vascular diseases. Recently, microRNA-31 (miR-31) has been found to play a critical role in cancer cell proliferation. However, the biological role of miR-31 in VSMC growth and the mechanisms involved are currently unknown. In the present study, the expression of rat mature miR-31 (rno-miR-31) was determined in cultured VSMCs and in rat carotid arteries. We identified that rno-miR-31 is an abundant miRNA in VSMCs, and its expression was significantly increased in proliferative VSMCs and in vascular walls with neointimal growth. The up-regulation of rno-miR-31 in proliferative VSMCs was inhibited by the inhibitor of mitogen-activated protein kinase/extracellular regulated kinase (MAPK/ERK). By both gain-of-function and loss-of-function approaches, we demonstrated that rno-miR-31 had a proproliferative effect on VSMCs. We further identified that LATS2 (large tumor suppressor homolog 2) is a downstream target gene product of rno-miR-31 that is involved in rno-miR-31-mediated effect on VSMC proliferation. The LATS2 as a target gene protein of rno-miR-31 is verified in vivo in balloon-injured rat carotid arteries. The results suggest that MAPK/ERK/miR-31/LATS2 may represent a novel signaling pathway in VSMC growth. miR-31 is able to enhance VSMC proliferation via its downstream target gene product, LATS2.
Introduction
MicroRNAs (miRNAs)2 have emerged as a novel class of endogenous, small, noncoding RNAs that regulate >30% of genes in a cell (1, 2). Functionally, an individual miRNA is important as a transcription factor because it is able to regulate the expression of its multiple target genes (3). It is therefore not surprising that miRNAs are involved in the regulation of approximately all major cellular functions such as cell proliferation, migration, and apoptosis (4).
Aberrant growth of vascular smooth muscle cells (VSMCs) is a major cellular event in the pathogenesis of many proliferative vascular diseases such as atherosclerosis and restenosis after angioplasty. Recent studies from us and other groups have identified a group of miRNAs that could control the growth of VSMCs (5–15). In this respect, miR-21, miR-221/222, miR-146a, and miR-1 are found to have a proproliferative effect on VSMCs (5–10). In contrast, an anti-proliferative effect of miR-143/145, and miR-26a was identified in VSMCs (11–15). The exciting results from these studies suggest that miRNAs could be novel therapeutic targets and devices in controlling the aberrant growth of VSMCs under disease conditions. However, the roles of many other aberrantly expressed miRNAs in VSMC growth as well as the mechanisms involved are still unknown.
Encoded by a single genomic locus, miR-31 is a broadly conserved miRNA expressed in a variety of tissues and cell types (16). miR-31 expression is up-regulated in many human cancers such as colorectal, hepatocellular, lung breast, tongue, and head-and-neck squamous tumors (17–21). In addition, a recent study showed that miR-31 is a pleiotropically acting inhibitor of breast cancer metastasis by targeting multiple genes (22). In our previous study, our microarray data revealed that rat mature miR-31 (rno-miR-31) is an abundant miRNA in VSMCs and arteries (5). However, the biological role of rno-miR-31 in VSMC biology and vascular disease as well as the mechanisms involved are currently unknown.
MAPK/ERK is a critical pathway responsible for extracellular growth stimuli-mediated gene regulation and cell proliferation (23). In a recent study, MAPK/ERK was found to be able to regulate miRNA expression such as Let-7 by phosphorylation of miRNA-generating complex (24). However, the role of MAPK/ERK in the expression of miR-31 in VSMCs is still unclear.
Encoding a putative serine/threonine kinase, Lats2 (large tumor suppressor homolog 2) has been identified as a novel tumor suppressor gene whose mutation accelerates cellular proliferation and tumorigenic development (25–28). Indeed, LATS2 is reported to inhibit tumor cell growth by causing G1/S and/or G2/M cycle arrest (29, 30). At the post-transcriptional level, LATS2 expression could be regulated by miR-373 in esophageal cancer (31) and by miR-372/373 in human testicular germ cell tumors (32). The biological functions of Lats2 and its post-transcriptional regulation in cardiovascular cells and cardiovascular diseases is still unexplored.
In the current study, the role of rno-miR-31 in VSMC growth and its mechanism involved are investigated. We have found that expression of rno-miR-31 is increased via MAPK/ERK in proliferative VSMCs. rno-miR-31 enhances VSMC growth via its target gene protein, LATS2.
EXPERIMENTAL PROCEDURES
Cell Culture
VSMCs were obtained from the aortic media of male Sprague-Dawley rats (5 weeks old) using an enzymatic dissociation method as described (5, 6). VSMCs and HEK293 were cultured with DMEM containing 10% FBS, 100 unit/ml penicillin, and 100 μg/ml streptomycin. VSMCs between passages 3 and 8 were applied for the experiments.
VSMC Proliferation Assay in Vitro
VSMC proliferation was determined by cell counting, cell proliferation kit MTT (Roche Applied Science) and BrdU incorporation assay (5–7). In addition, in some experiments, the expression of proliferating cell nuclear antigen (PCNA), a well known cell proliferation marker (33, 34), was also determined to further determine the cell proliferation of VSMCs. The cells were detached by trypsinization and resuspended in PBS and then counted under a microscope. The MTT assay was performed by using a cell proliferation kit (Roche Applied Science) according to the manufacturer's protocol. For BrdU incorporation assay, 10 mm BrdU was added to the culture medium for incorporation into the DNA of replicating cells. After 1 h of incubation, cells were fixed, and anti-BrdU antibody (in situ cell proliferation kit, Roche Applied Science) was added to each well for 45 min. Finally, the proliferative cells were detected under a fluorescence microscope.
Generation of Ad-miR-31 and Ad-GFP
The adenoviruses expressing rat miR-31 (Ad-miR-31) and control viruses expressing GFP (Ad-GFP) were generated using the ViraPowerTM adenoviral gateway expression system (Invitrogen) according to the manufacturer's instructions. Briefly, a fragment containing the rat precursor miR-31 was amplified with its primers (rno-miR-31 FP and rno-miR-31 RP) from rat genomic DNA and inserted into pENTR-3C vectors (Invitrogen) at EcoRI and XhoI sites. The construct, pENTR-miR-31 was sequenced to confirm the correct DNA sequences. Via Cre recombinase, the fragment was excised from the pENTR-miR-31 donor vector and inserted into the pAd/CMV/V5-DEST Gateway receptor vector, which was termed pAd-miR-31. To produce recombinant adenoviruses with Lipofectamine 2000 according to the manufacturer's protocols (Invitrogen), the plasmid pAd-miR-31 was digested by Pac and transfected into low passage HEK293 cells. Adenovirus expressing GFP was generated as described (7). The resulting adenoviruses (Ad-miR-31 and Ad-GFP) were further amplified by infection of HEK293A cells and purified by cesium chloride gradient ultracentrifugation. The titers of Ad-miR-31 and Ad-GFP were determined by using a Adeno-XTM rapid titer kit (Clontech).
Luciferase Assay
The reporter plasmid, a firefly luciferase reporter construct psiCHECK-2 (Promega) inserted with a fragment of the 3′-UTR of rat Lats2 mRNA containing the putative rno-miR-31 binding sequence, was transfected into HEK293 cells via Lipofectamine 2000 reagent (Invitrogen) with vehicle, an empty plasmid (pDNR-CMV, 0.2 μg/ml), a plasmid expressing rno-miR-31 (pmiR-31, 0.2 μg/ml), or a control plasmid expressing an unrelated miRNA, rno-miR-221 (pmiR-221, 0.2 μg/ml), following the transfection procedures provided by Invitrogen. In the truncated control construct, ∼400 bp containing the rno-miR-31 binding site was removed from the wild type 3′-UTR fragment (∼950 bp) of Lats2 mRNA. The sequences for luciferase assay are listed in Table 1. Forty-eight hours after transfection, cell extract was isolated to measure the luciferase expression on a scintillation counter by using a Dual-Luciferase reporter system (Promega).
TABLE 1.
Primers and oligonucleotides used for RT-PCR, vector construct, or miRNA knockdown
FP, forward primer; RP, reverse primer.
| Primer name | Sequence (5′-3′) |
|---|---|
| rno-GAPDH FP | aagctcactggcatggcctt |
| rno-GAPDH RP | cggcatgtcagatccacaac |
| rno-PCNA FP | agggctgaagataatgctgatacc |
| rno-PCNA RP | ctcctgttctgggattccaagttg |
| rno-Lats2 FP | ccaacagcaagcacccagag |
| rno-Lats2 RP | cgaccgcacctcctaactc |
| rno-miR-31 inhibitor | mCmAmGmCmUmAmUmGmCmCmAmGmCmAmUmCmUmUmGmCmCmU |
| pri-rno-mir-31 FP | attgccatctttcctgtctga |
| pri-rno-mir-31 RP | gaacattcctggtttgtcctag |
| U6 FP | ctcgcttcggcagcaca |
| U6 RP | aacgcttcacgaatttgcgt |
| rno-miR-31 FP | ttgaattctggacactaagggagactgttgc |
| rno-miR-31 RP | ttctcgagacctagtcgcagaacccctaca |
| rno-Lats2 3′ UTR FP | ttctcgagttgaggaaaccccaaaccagatg |
| rno-Lats2 3′ UTR RP | ttgcggccgctgtgggaaccctgtatgtgacga |
| rno-Lats2 truncated 3′-UTR RP | ttgcggccgctctgctcatgcagtactctctctc |
Oligonucleotide Transfection and Adenovirus Infection in Cultured VSMCs
Oligonucleotide transfection was performed as described previously (5–7). Briefly, cells were transfected using a transfection reagent (HiPerFect HTS Reagent, Qiagen) at 24 h after seeding into the wells. Transfection complexes were prepared according to the manufacturer's instructions. For rno-miR-31 knockdown, miR-31 inhibitor, 2′OMe-miR-31 (the antisense oligonucleotide for miR-31 that is modified at each nucleotide by an O-methyl moiety at the 2′-ribose position) (Integrated DNA Technologies) was added to the complexes at final oligonucleotide concentration of 100 nmol/liter. Vehicle and oligonucleotide control (Integrated DNA Technologies) were used as controls. Lats2 gene knockdown was performed using siRNA Lats2 (50 nm). The transfection medium was replaced 4 h post-transfection by the regular culture medium. Vehicle and scramble controls (Ambion, Inc.) were applied. For rno-miR-31 over-expression, Ad-miR-31 was added to the culture medium at 50 multiplicity of infection (MOI) or indicated MOI. Ad-GFP was used as an adenovirus control. Forty-eight and 72 h later, the cells were harvested for RNA or protein isolation, respectively.
RNA Isolation and qRT-PCR
RNA levels were determined by qRT-PCR. Briefly, RNAs from VSMCs and rat carotid arteries were isolated with TRIzol (Invitrogen). qRT-PCR for miRNAs was performed on cDNA generated from 100 ng of total RNA using TaqMan MicroRNA Reverse Transcription and TaqMan microRNA assays (Applied Biosystems). qRT-PCR for Lats2 and PCNA was performed on cDNA generated from 200 ng of total RNA using the protocol of a qRT-PCR mRNA detection kit (Roche Applied Science). Amplification and detection of specific products were performed with a Roche Lightcycler 480 Detection System. As an internal control, U6 was used for miRNA template normalization, and GADPH was used for other template normalizations. The sequences of the primers are shown in Table 1. Fluorescent signals were normalized to an internal reference, and the threshold cycle (Ct) was set within the exponential phase of the PCR. The relative gene expression was calculated by comparing cycle times for each target PCR. The target PCR Ct values were normalized by subtracting the U6 or GADPH Ct value, which provided the ΔCt value. The relative expression level between treatments was calculated using the following equation: relative gene expression = 2−(ΔCtsample−ΔCtcontrol).
Western Blotting
Proteins were isolated from cultured VSMCs, and carotid arteries and protein levels were determined by Western blot analysis. Briefly, proteins were prepared with radioimmune precipitation assay lysis buffer and quantified with BCA protein assay (Pierce). Equal amounts of protein were subjected to SDS-PAGE. Standard Western blot analysis was conducted using LATS2 (1:1000 dilution) (Abcam, Inc.), ERK (1:1000 dilution), pERK (1:1000 dilution) (Cell Signaling), PCNA monoclonal antibody (1:2000 dilution) (Invitrogen), and GADPH antibody (1:5000 dilution) (Cell Signaling) was used as a control.
Rat Carotid Artery Balloon Injury Model
Carotid artery balloon injury was induced in male Sprague-Dawley rats (230 to 300 g) as described in our previous studies (5–7). Briefly, rats were anesthetized with ketamine (80 mg/kg)/xylazine (5 mg/kg). Under a dissecting microscope, the right common carotid artery was exposed through a midline cervical incision. A 2F Fogarty catheter (Baxter Edwards) was introduced via an arteriotomy in the external carotid artery, and then the catheter was advanced to the proximal edge of the omohyoid muscle. To produce carotid artery injury, we inflated the balloon with saline and withdrew it three times from just under the proximal edge of the omohyoid muscle to the carotid bifurcation. After injury, the external carotid artery was permanently ligated with a 6-0 silk suture, and blood flow in the common carotid artery was restored. All protocols were approved by the Institutional Animal Care and Use Committee at the University of Medicine and Dentistry of New Jersey and were consistent with the Guide for the Care and Use of Laboratory Animals (NIH publication 85-23, revised 1985).
Local Oligonucleotide Delivery into Injured Vascular Walls
To deliver the rno-miR-31 inhibitor or control oligonucleotide into the injured vascular tissue and to avoid any potential systemic side effects, we applied an established local oligonucleotide delivery model via F-127 pluronic gel as described in our recent reports with a little modification (5, 6). Briefly, immediately after balloon injury of the right common carotid artery, transfection solutions (50 μl 0.2% transfection reagent in DMEM) was mixed with rno-miR-31 inhibitor or control oligonucleotides (10 μg) and infused into the ligated segment of the common carotid artery. Then, 90 μg of these oligonucleotides, preloaded into 200 μl 30% F-127 pluronic gel (Sigma) and 1% transfection reagent at 4 °C, was applied locally to the adventitia around injured artery segments.
Immunofluorescence of PCNA and Smooth Muscle α-Actin
Proliferating cells were evaluated in vessel sections by using the immunofluorescence of PCNA. The freeze (5 μm) sections were incubated with anti-PCNA antibodies (Santa Cruz Biotechnology) followed by fluorescein-conjugated secondary antibodies (1:200 dilution, Vector Laboratories). For the staining of VSMC marker, smooth muscle α-actin, the sections were incubated with anti-smooth muscle α-actin (1:400 dilution, Dako) followed by fluorescein conjugated secondary antibodies (1:200 dilution, Vector Laboratories). Cell nuclear was stained by DAPI (blue). The fluorescence was observed by Nikon Eclipse 80i immunofluorescence microscope.
Statistical Analysis
All of the experiments were repeated at least three times. All data are presented as means ± S.E. For relative gene expression, the mean value of vehicle control group is defined as 1 or 100%. Two-tailed unpaired Student's t tests and analysis of variance were used for statistical evaluation of the data. SPSS statistical analysis program (version 17.0) was used for data analysis. A p value < 0.05 was considered to be significant.
RESULTS
rno-miR-31 Is an Abundant miRNA in VSMCs and Its Expression Is Significantly Increased in Proliferative VSMCs
In our previous study, we identified that rno-miR-221 and rno-miR-222 are two abundant miRNAs in VSMCs and vascular walls (6). Our unpublished microarray data demonstrated that the expression of rno-miR-31 in VSMCs and rat carotid arteries was nearly 2-fold higher than that of rno-miR-221 and rno-miR-222.3 Thus, rno-miR-31 is an abundant miRNA in VSMCs and vessels. To investigate the potential link between VSMC growth and rno-miR-31, the expression of rno-miR-31 as well as its primary transcript, pri-rno-mir-31 was determined in nonproliferative VSMCs and in proliferative VSMCs induced by PDGF (20 ng/ml). As shown in Fig. 1, A and B, the expression of rno-miR-31 and pri-rno-mir-31 in PDGF-treated VSMCs was significantly higher than that in VSMCs without the treatment of PDGF, which was time-dependent. The dose-dependent response of rno-miR-31 expression to PDGF was also measured in cultured VSMCs at 24 h after treatment with different concentration of PDGF as shown in Fig. 1C. Moreover, the expression of rno-miR-31 could also be enhanced in VSMCs treated with FBS (Fig. 1C). These results demonstrate that the expression of the abundant rno-miR-31 and pri-rno-mir-31 is up-regulated in proliferative VSMCs. It should be noted that there was a delay (12 h) from the expression of pri-rno-miR-31 transcript (Fig. 1B) to rno-miR-31 (Fig. 1A). The delay time may be related to the processing time of rno-miR-31 by Drosha and Dicer in VSMCs.
FIGURE 1.
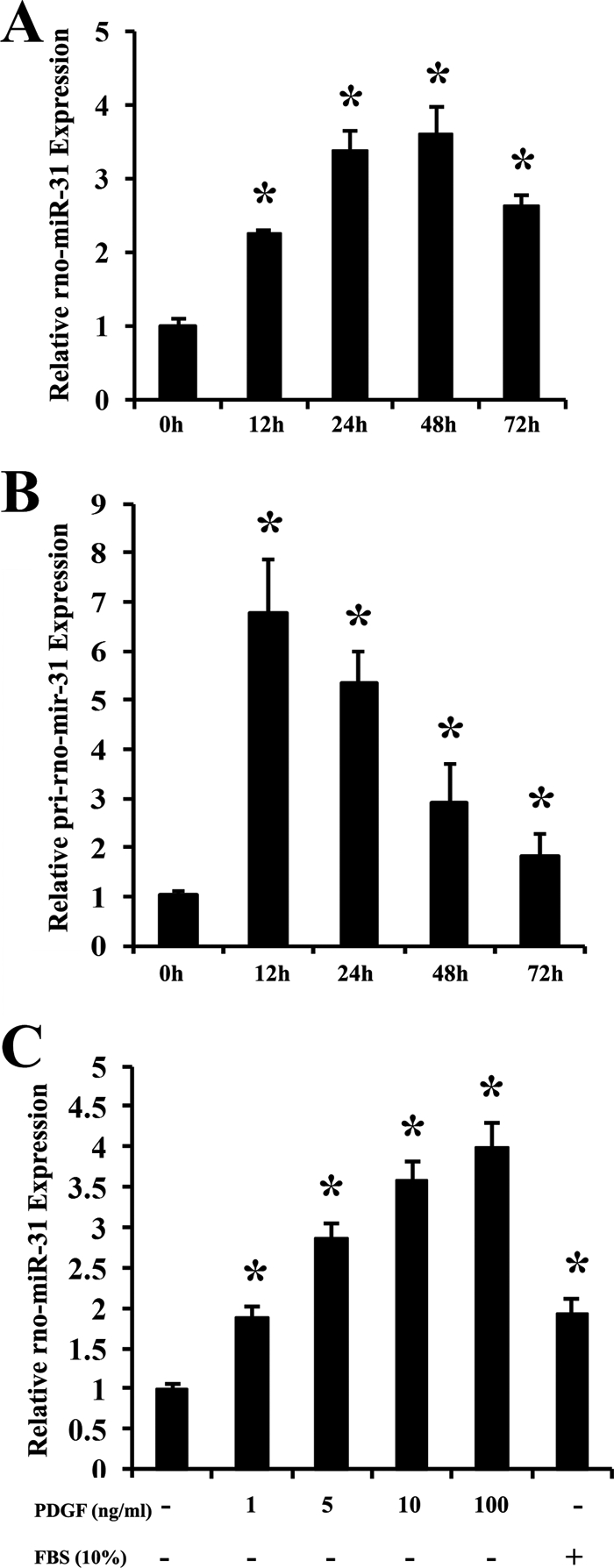
The expression of rno-miR-31 and pri-rno-mir-31 in cultured VSMCs. VSMCs were seeded into six-well plates for 24 h at 40∼60% confluent with 10% FBS. The medium was replaced with 0.1% FBS for 24 h, and the cells were then treated with PDGF (20 ng/ml) or 10% FBS for the indicated times. A, rno-miR-31 expression in VSMCs treated with PDGF (n = 6). *, p < 0.05 compared with 0 h group. B, pri-rno-mir-31 expression in VSMCs treated with PDGF (n = 6). *, p < 0.05 compared with 0 h group. C, rno-miR-31 expression in cultured VSMCs treated with different concentrations of PDGF or 10% FBS for 24 h (n = 6). *, p < 0.05 compared with that in VSMCs without PDGF and 10% FBS.
rno-miR-31 Is Mainly Expressed in VSMCs of Vascular Walls and Its Expression Is Up-regulated in Rat Carotid Arteries with Neointimal Lesion Growth
To determine the cellular distribution of rno-miR-31, we isolated VSMCs and endothelial cells, the two major cell types in normal vessels, from rat arteries. We found that rno-miR-31 was expressed preferentially in VSMCs, whereas its expression level in endothelial cells was low (Fig. 2A). In rat carotid arteries with neointimal lesion growth induced by balloon injury, the expression of rno-miR-31 was significantly increased (Fig. 2B). To further determine whether the up-regulated rno-miR-31 was due to its primary transcript increase, RNA was isolated from uninjured and balloon-injured arteries for qRT-PCR of pri-rno-mir-31. As displayed in Fig. 2C, pri-rno-mir-31 was significantly increased in injured vessels, compared with that in uninjured vessels.
FIGURE 2.

miR-31 expression in fresh isolated rat aortic VSMCs, endothelial cells, and in rat carotid arteries with or without balloon injury. A, relative expression of rno-miR-31 in rat VSMCs and endothelial cells (EC) determined by qRT-PCR (n = 6). *, p < 0.05 compared with that in VSMCs. B and C, the rat right carotid arteries were isolated at 3, 7, 14, and 28 days (d) after angioplasty for rno-miR-31 (B) or pri-rno-mir-31 (C) measurement by qRT-PCR. The uninjured right carotid arteries were used as the uninjured control group (uninjury) (n = 6). *, p < 0.05 compared with that in uninjured control.
MAPK/ERK Is Related to Up-regulation of rno-miR-31 in Proliferative VSMCs
To determine the role of MAPK in the up-regulation of rno-miR-31 in VSMCs, the specific inhibitors of the four major types of MAPK were applied in PDGF-stimulated VSMCs. PD98059, a selective cell-permeable inhibitor of MAPK/ERK, is widely used to inhibit ERK activation and has been shown to inhibit VSMC proliferation in response to PDGF (35, 36). The efficacy of PD98059 was confirmed by its ability to abolish PDGF-induced phosphorylation of ERK1/2 (Fig. 3A). Pretreatment of VSMCs with PD98059 (50 μm) significantly inhibited the PDGF-mediated up-regulation of rno-miR-31 and pri-rno-miR-31 (Fig. 3B). In contrast, inhibition of p38 with SB203580 (20 μm), JNK with SP600125 (20 μm), or PI3K with LY294002 (50 μm) had no significant effect on the up-regulation of rno-miR-31 and pri-rno-mir-31 (data not shown).
FIGURE 3.

The effect of MAPK/ERK inhibitor PD98059 on the expression of rno-miR-31 and pri-rno-mir-31. The cultured VSMCs were treated with vehicle or PD98059 (MAPK/ERK inhibitor) for 1 h and then stimulated with vehicle or PDGF (20 ng/ml) for 30 min in A and 24 h in B. A, phosphorylated ERK (pERK) and total ERK determined by Western blot. B, the expression of rno-miR-31 and pri-rno-mir-31 in VSMCs determined by qRT-PCR (n = 5). *, p < 0.05 compared with rno-miR-31 in PDGF group. #, p < 0.05 compared with pri-mir-31 in PDGF group.
Modulating rno-miR-31 Expression in Cultured VSMCs and in Injured Arteries
To overexpress the expression of rno-miR-31 in VSMCs, Ad-miR-31 was used. As shown in Fig. 4A, at 24 h after infection, rno-miR-31 expression was increased significantly in Ad-miR-31-treated VSMCs compared with that in VSMCs treated with either vehicle or Ad-GFP. For rno-miR-31 knockdown, VSMCs were cultured in medium containing rno-miR-31 inhibitor at final oligonucleotide concentrations of 0, 10, 30, and 100 nmol/liter. rno-miR-31 inhibitor decreased the rno-miR-31 expression in a dose-dependent manner (Fig. 4B). In addition, the expression of rno-miR-31 in PDGF-treated VSMCs was successfully inhibited by rno-miR-31 inhibitor (100 nmol/liter) (Fig. 4C). Moreover, the expression of rno-miR-31 in balloon-injured arteries was down-regulated by local delivery of rno-miR-31 inhibitor in the injured rat vessels (Fig. 4D) (5, 6).
FIGURE 4.

Modulation of rno-miR-31 expression in cultured VSMCs and in injured arteries. A, the expression of rno-miR-31 in VSMCs treated with vehicle, Ad-GFP and Ad-miR-31, at indicated MOI (n = 5). *, p < 0.05 compared with Ad-GFP group. B, knockdown of rno-miR-31 expression by rno-miR-31 inhibitor in cultured VSMCs (n = 5). *, p < 0.05 compared with 0 nm group. C, knockdown of rno-miR-31 expression by its inhibitor (100 nmol/liter) in VSMCs treated with PDGF (20 ng/ml) (n = 5). *, p < 0.05 compared with vehicle plus PDGF group. #, p < 0.05 compared with vehicle without PDGF group. D, inhibition of rno-miR-31 by its inhibitor in balloon-injured vascular walls (n = 6). *, p < 0.05 compared with the oligonucleotide control group.
rno-miR-31 Is a Critical Gene in VSMC Proliferation
Serum-induced VSMC proliferation was inhibited significantly by the rno-miR-31 inhibitor as determined by MTT assay, cell counting, and BrdU incorporation assay (Fig. 5, A, C, and E). In contrast, serum-induced VSMC proliferation was increased by overexpression of rno-miR-31 via Ad-miR-31 (Fig. 5, B and D). Representative BrdU-stained cell photomicrographs, their corresponding total cell photomicrographs stained by DAPI, and merged photomicrographs are shown in Fig. 5F. The effect of rno-miR-31 on the proliferation of VSMCs is not limited to serum-induced growth. In fact, PDGF-induced VSMC proliferation is also inhibited by its inhibitor (Fig. 5G) and is increased by Ad-miR-31 (Fig. 5H). The effect of rno-miR-31 on the proliferation of cultured VSMCs is further confirmed by the expression of a well known cell proliferation marker, PCNA. As shown in Fig. 5, I and J, the expression of PCNA is decreased in rno-miR-31 inhibitor-treated cells but is increased by the overexpression of rno-miR-31.
FIGURE 5.

The effect of rno-miR-31 on VSMC proliferation in cultured VSMCs in vitro and in balloon-injured rat carotid arteries in vivo. rno-miR-31 inhibitor (100 nmol/liter) decreased cell proliferation determined by MTT assay (A), cell number determined by cell accounting (C), and BrdU incorporation (E) at 48 h after culture in DMEM containing 5% FBS (n = 8). *, p < 0.05 compared with oligonucleotide (oligo) control group. Ad-miR-31 (30 MOI) increased cell proliferation determined by MTT assay (B) and cell number (D) at 48 h after culture with DMEM containing 5% FBS (n = 8). *, p < 0.05 compared with Ad-GFP group. Shown are representative BrdU-stained cell photomicrographs, their corresponding total cell photomicrographs stained by DAPI, and merged photomicrographs (F). G, rno-miR-31 inhibitor decreased cell proliferation stimulated by PDGF (10 ng/ml) determined by MTT assay (n = 6). *, p < 0.05 compared with oligonucleotide control group. H, Ad-miR-31 (30 MOI) increased cell proliferation stimulated by PDGF (10 ng/ml) determined by MTT assay (n = 6). *, p < 0.05 compared with Ad-GFP group. I, rno-miR-31 inhibitor decreased the expression of PCNA in cultured VSMCs. J, Ad-miR-31 increased the expression of PCNA in cultured VSMCs. K, rno-miR-31 inhibitor decreased the expression of PCNA in balloon-injured arteries. L, PCNA-positive VSMCs in balloon-injured arteries treated with rno-miR-31 inhibitor or control oligonucleotide. Note that green shading indicates the VSMC marker smooth muscle (SM) α-actin; red shading indicates PCNA; and blue shading indicates the cell nuclei stained by DAPI.
The expression of rno-miR-31 is increased in balloon-injured rat carotid arteries (Fig. 2B). To test the role of rno-miR-31 in VSMC proliferation in vivo, the up-regulated rno-miR-31 was inhibited in the balloon-injured arteries (Fig. 4D). As shown in Fig. 5K, the expression of PCNA in balloon-injured arteries is decreased by rno-miR-31 inhibition. Interestingly, less PCNA-positive VSMCs in rno-miR-31 inhibitor-treated vessels at 9 days after balloon injury were found compared with those in vessels treated with control oligonucleotides. Representative PCNA-positive cells (red), smooth muscle α-actin-positive cells (green), their corresponding total cell photomicrographs stained by DAPI (blue), and merged photomicrographs are shown in Fig. 5L.
Lats2 Is Direct Target Gene of rno-miR-31
The online software TargetScan (version 5.1) predicts that Lats2 has a miR-31 binding site in its 3′-UTR (37, 38). The “seed” sequence in miR-31 (5′-ggcaaga-3′) matches the nucleotides 603–609 from the first nucleotide of the 3′-UTR of the Lats2 mRNA (NM_001107267) retrieved from the National Center of Biotechnology Information (39), which are shown in Fig. 6A. To identify that rno-miR-31 is able to directly bind to Lats2 and inhibit its expression, a firefly luciferase reporter construct containing a fragment of the 3′-UTR of rat Lats2 mRNA with the putative rno-miR-31 binding sequence was transfected into HEK293 cells with either vehicle, an empty plasmid (pDNR-CMV), a plasmid expressing rno-miR-31 (pmiR-31), or a control plasmid expressing an unrelated miRNA, rno-miR-221 (pmiR-221). As shown in Fig. 6B, pmiR-31, but not pmiR-221 or pDNR-CMV, increased miR-31 expression in HEK293 cells. Consequently, pmiR-31, but not pDNR-CMV or pmiR-221, inhibited luciferase activity (Fig. 6C). In the truncated control group, the inhibitory effect of pmiR-31 disappeared (Fig. 6C). The results suggested that rno-miR-31 was able to bind to Lats2 directly and inhibit its expression.
FIGURE 6.

Lats2 is a direct target gene of rno-miR-31. A, Lats2 is one of the miR-31 target genes predicted by TargetScan (version 5.1). The seed sequences of miR-31 and the Lats2 binding sites were boxed. B, pmiR-31, but not pmiR-221 and pDNR-CMV, increased miR-31 expression in HEK293 cells (n = 6). *, p < 0.05 compared with pDNR-CMV group. C, a luciferase reporter construct, containing the putative miR-31 binding sequence from 3′-UTR of the rat Lats2 gene, was transfected into HEK293 with vehicle, pDNR-CMV, pmiR-31, or a control plasmid expressing an unrelated miRNA, pmiR-221. The construct without the miR-31 binding sequence was used as the truncated control. pmiR-31, but not pmiR-221 or pDNR-CMV, inhibited luciferase activity. In the truncated control group, the inhibitory effect of pmiR-31 disappeared (n = 6). *, p < 0.05 compared with pDNR-CMV group. D, rno-miR-31 inhibitor increased LATS2 protein level in VSMCs. E, Ad-miR-31 decreased LATS2 protein level in VSMCs. F, Lats2 mRNA levels in VSMCs treated with vehicle, Ad-GFP, or Ad-miR-31 (n = 6). *, p < 0.05 compared with Lats2 in Ad-GFP group. G, rno-miR-31 inhibitor increased LATS2 expression in the injured vascular walls.
To confirm that Lats2 is a target gene of rno-miR-31 in cultured VSMCs, both gain-of-function and loss-of-function approaches were applied. As expected, LATS2 was up-regulated by miR-31 inhibitor (Fig. 6D) and down-regulated by Ad-miR-31 (Fig. 6E) at the protein level. It is well known that miRNAs could regulate their target gene expression via inhibiting translation and/or promoting degradation of target genes (1, 2). To determine whether rno-miR-31 could cause the degradation of its target Lats2, we detected Lats2 mRNA levels in cultured VSMCs transfected with vehicle, Ad-GFP, or Ad-miR-31 respectively. Interestingly, overexpression of rno-miR-31 by Ad-miR-31 decreased the Lats2 mRNA level, whereas the vehicle, Ad-GFP, had no effect on the expression of Lats2 mRNA (Fig. 6F).
To further verify that LATS2 is a target gene protein of rno-miR-31 in vivo, the expression of rno-miR-31 in balloon-injured rat carotid arteries was inhibited by the rno-miR-31 inhibitor. As shown in Fig. 4D, rno-miR-31 is down-regulated by its inhibitor. Interestingly, in rno-miR-31 inhibitor-treated vessels, the expression of LATS2 is up-regulated (Fig. 6G). The results demonstrated that LATS2 is indeed a target gene protein of rno-miR-31 in vivo.
LATS2 Is Involved in VSMC Proliferation
Because LATS2 is a target gene product of rno-miR-31 and its biological functions are still unknown in VSMCs, we then determined the effect of LATS2 on the proliferation of VSMCs. As expected, the expression of Lats2 is down-regulated at both mRNA and protein levels in proliferative VSMCs stimulated by PDGF (Fig. 7, A and B) because the rno-miR-31 is up-regulated in these cells (Fig. 1). To determine the role of LATS2 in VSMC growth, Lats2 was knocked down by its siRNA (Lats2 siRNA). Knockdown of the gene is confirmed at mRNA and protein levels (Fig. 7, C and D). Interestingly, down-regulation of LATS2 increased the expression of PCNA in these cells (Fig. 7, C and D), suggesting that LATS2 might have an anti-proliferative effect on VSMCs. The inhibitory effect of LATS2 on the growth of VSMCs is further confirmed by the cell proliferation assay determined via MTT method (Fig. 7E).
FIGURE 7.

LATS2 in the proliferation of VSMCs. A, the mRNA level of Lats2 was down-regulated in proliferative VSMCs stimulated with PDGF (20 ng/ml) (n = 6). *, p < 0.05 compared with Lats2 of 0 h group. B, the protein level of LATS2 was decreased in proliferative VSMCs stimulated with PDGF (20 ng/ml). C, knockdown of Lats2 increased PCNA expression at mRNA level (n = 6). *, p < 0.05 compared with scramble group. #, p < 0.05 compared with scramble group. D, knockdown of LATS2 increased PCNA expression at protein level. E, knockdown of LATS2 increased the proliferation of VSMCs as determined by MTT assay (n = 6). *, p < 0.05 compared with scramble group.
DISCUSSION
miR-31 is encoded by a gene on the chromosome 9 in humans and on the chromosome 5 in rats. The seed sequence of miR-31 is highly conserved as shown in Fig. 6A. Recent studies have revealed that miR-31 may play important roles in many aspects of biology such as cancer development (17–21, 40–42), angiogenesis (43, 44), hair cycle control (45), and inflammation (46). Until now, the biological roles of miR-31 in VSMC biology are still unknown.
VSMC proliferation-mediated expansion of the neointima is unifying facet of a variety of proliferative vascular diseases. Recent studies have revealed that some miRNAs such as miR-221/222 may have strong biological functions in cell proliferation, including VSMC proliferation (5–15). Thus, identifying the miRNAs that may play a role in VSMC functions is a novel research field in vascular biology. In this study, we found that rno-miR-31 is an abundant miRNA in VSMCs, and its expression is significantly increased in proliferative VSMCs and in vascular walls with neointimal growth.
It is well established that MAPK/ERK is a critical signaling molecule in communicating the extracellular growth stimuli such as growth factors into nuclear events and cellular functions such as gene expression regulation and cell proliferation (23). In this study, the role of MAPK/ERK in the expression of rno-miR-31 in VSMCs is investigated. The results demonstrated that the up-regulation of rno-miR-31 in proliferative VSMCs is partially inhibited by the MAPK/ERK pathway. In contrast, no significant effect on the expression of rno-miR-31 was found by other kinase inhibitors. Thus, MAPK/ERK is a critical upstream signaling molecule that is related to the up-regulation of miR-31 in proliferative VSMCs.
Another aim of this study was to determine the potential role of rno-miR-31 in VSMC proliferation. We found that VSMC proliferation is inhibited significantly by knockdown of rno-miR-31. In contrast, the cell proliferation was increased by overexpression of rno-miR-31. The results suggested that rno-miR-31 is a novel gene involved in the growth control of VSMCs. The cellular effect of rno-miR-31 on VSMCs is consistent with that of lung cancer (40) and head-and-neck squamous cell carcinoma cell lines (21). However, Creihton and co-workers (47) reported that miR-31 expression could inhibit proliferation in ovarian cancer cells with a dysfunctional p53 signaling pathway. In cancer, miR-31 also has a strong effect on cell migration (16, 40, 46). However, no significant effect of miR-31 on VSMC migration is identified in our study (data not shown). Thus, the effects of miR-31 on cell proliferation and migration may be cell type-specific.
miRNAs achieve their cellular functions via their target genes. The negative correlation between rno-miR-31 expression and LATS2 expression in PDGF-stimulated VSMCs indicated that Lats2 mRNA could be a potential target gene product of rno-miR-31 in VSMCs. By both gain-of-function and loss-of-function approaches, we confirmed that Lats2 was indeed a target gene mRNA of miR-31 in VSMCs. The discovery is further verified in vivo in rat carotid arteries after angioplasty as the expression of LATS2 protein could be regulated by the rno-miR-31 inhibitor in the vascular walls. In addition, the biological role of LATS2 in VSMCs is identified for the first time, as having an inhibitory effect on VSMC proliferation. Other target genes of miR-31 currently identified in other cells include Lats2 (40), PP2A regulatory subunit B α isoform (Ppp2ra) (40), factor-inhibiting hypoxia-inducible factor (20), Satb2 (41), TIAM1 (42), Krt16 (45), Krt17 (45), Dlx3 (45), and Fgf10 (45).
PCNA is a cell cycle related protein and a widely used marker of cell proliferation (33, 34, 48, 49). Previous studies have revealed that PCNA is related to VSMC proliferation and atherosclerotic neointimal growth (33, 34). Moreover, the expression of PCNA can be regulated by the MAPK/ERK pathway (48, 49). However, the detailed molecular mechanism involved in ERK-mediated regulation of PCNA is still unclear. In this study, we identified that rno-miR-31 is a downstream gene of ERK. Moreover, rno-miR-31 has a strong effect on the expression of PCNA in VSMCs via its target LATS2 as determined by both gain-of-function and loss-of-function approaches.
Recent studies have revealed that multiple miRNAs are related to VSMC proliferation (5–15, 50). For example, miR-221/222 and miR-21 are proproliferative miRNAs in VSMCs (5–7). In contrast, miR-143 and miR-145 have an anti-proliferative effect on VSMCs (11–15). The interactions of these miRNAs may be complex; based on our unpublished data, they may have a synergistic, additive, or antagonistic action.3 For example, miR-31 and miR-221 have an additive action in VSMC proliferation (data not shown).
In summary, the present study reveals that rno-miR-31 is an abundant miRNA in VSMCs. Its expression is significantly increased in proliferative VSMCs and in vascular walls with neointimal growth via activation of MAPK/ERK. rno-miR-31 is able to enhance VSMC proliferation via its downstream target gene product, LATS2. As demonstrated in the diagram (Fig. 8), MAPK/ERK/miR-31/LATS2/PCNA may represent a novel signaling pathway in VSMC growth.
FIGURE 8.
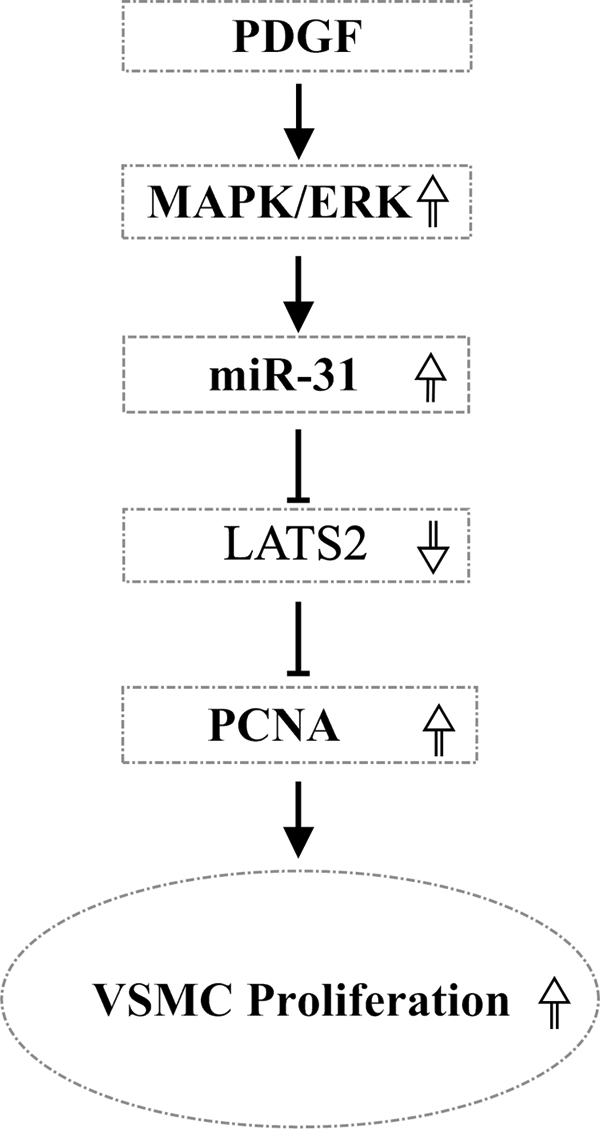
A model diagram of the role of miR-31 in VSMC growth and its mechanism. MAPK/ERK is activated in VSMCs stimulated by PDGF. The increased activation of MAPK/ERK results in the increased expression of miR-31. The increased miR-31 down-regulates the expression of its target gene protein, LATS2. The decreased LATS2 increases the PCNA and results in the increased proliferation of VSMCs.
This work was supported, in whole or in part, by National Institutes of Health Grants HL080133 and HL095707 (to C. Z.). This work was also supported by American Heart Association Grant 09GRNT2250567 (to C. Z.).
X. Liu, Y. Cheng, X. Chen, J. Yang, L. Xu, and C. Zhang, unpublished data.
- miRNA
- microRNA
- rno-miR-31
- rat mature miR-31
- VSMC
- vascular smooth muscle cell
- PCNA
- proliferating cell nuclear antigen
- MOI
- multiplicity of infection
- qRT-PCR
- quantitative RT-PCR
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
REFERENCES
- 1. Ambros V. (2004) Nature 431, 350–355 [DOI] [PubMed] [Google Scholar]
- 2. Lewis B. P., Burge C. B., Bartel D. P. (2005) Cell 120, 15–20 [DOI] [PubMed] [Google Scholar]
- 3. Chen K., Rajewsky N. (2007) Nat. Rev. Genet. 8, 93–103 [DOI] [PubMed] [Google Scholar]
- 4. Zhang C. (2008) Physiol. Genomics 33, 139–147 [DOI] [PubMed] [Google Scholar]
- 5. Ji R., Cheng Y., Yue J., Yang J., Liu X., Chen H., Dean D. B., Zhang C. (2007) Circ. Res. 100, 1579–1588 [DOI] [PubMed] [Google Scholar]
- 6. Liu X., Cheng Y., Zhang S., Lin Y., Yang J., Zhang C. (2009) Circ. Res. 104, 476–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Davis B. N., Hilyard A. C., Nguyen P. H., Lagna G., Hata A. (2009) J. Biol. Chem. 284, 3728–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sun S. G., Zheng B., Han M., Fang X. M., Li H. X., Miao S. B., Su M., Han Y., Shi H. J., Wen J. K. (2011) EMBO Rep. 12, 56–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen J., Yin H., Jiang Y., Radhakrishnan S. K., Huang Z. P., Li J., Shi Z., Kilsdonk E. P., Gui Y., Wang D. Z., Zheng X. L. (2011) Arterioscler. Thromb. Vasc. Biol. 31, 368–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leeper N. J., Raiesdana A., Kojima Y., Chun H. J., Azuma J., Maegdefessel L., Kundu R. K, Quertermous T., Tsao P. S., Spin J. M. (2011) J. Cell Physiol. 226, 1035–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cheng Y., Liu X., Yang J., Lin Y., Xu D. Z., Lu Q., Deitch E. A., Huo Y., Delphin E. S., Zhang C. (2009) Circ. Res. 105, 158–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cordes K. R., Sheehy N. T., White M. P., Berry E. C., Morton S. U., Muth A. N., Lee T. H., Miano J. M., Ivey K. N., Srivastava D. (2009) Nature 460, 705–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Elia L., Quintavalle M., Zhang J., Contu R., Cossu L., Latronico M. V., Peterson K. L., Indolfi C., Catalucci D., Chen J., Courtneidge S. A., Condorelli G. (2009) Cell Death Differ. 16, 1590–15098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boettger T., Beetz N., Kostin S., Schneider J., Krüger M., Hein L., Braun T. (2009) J. Clin. Invest. 119, 2634–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xin M., Small E. M., Sutherland L. B., Qi X., McAnally J., Plato C. F., Richardson J. A., Bassel-Duby R., Olson E. N. (2009) Genes Dev. 23, 2166–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Valastyan S., Weinberg R. A. (2010) Cell Cycle 9, 2124–2129 [DOI] [PubMed] [Google Scholar]
- 17. Bandrés E., Cubedo E., Agirre X., Malumbres R., Zárate R., Ramirez N., Abajo A., Navarro A., Moreno I., Monzó M., García-Foncillas J. (2006) Mol. Cancer 5, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Motoyama K., Inoue H., Takatsuno Y., Tanaka F., Mimori K., Uetake H., Sugihara K., Mori M. (2009) Int. J. Oncol. 34, 1069–1075 [DOI] [PubMed] [Google Scholar]
- 19. Slaby O., Svoboda M., Fabian P., Smerdova T., Knoflickova D., Bednarikova M., Nenutil R., Vyzula R. (2007) Oncology 72, 397–402 [DOI] [PubMed] [Google Scholar]
- 20. Liu X. Q., Chen Z. G., Yu J. S., Xia J., Zhou X. F. (2009) Comp. Funct. Genomics 2009, 837514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu C. J., Tsai M. M., Hung P. S., Kao S. Y., Liu T. Y., Wu K. J., Chiou S. H., Lin S. C., Chang K. W. (2010) Cancer Res. 70, 1635–1644 [DOI] [PubMed] [Google Scholar]
- 22. Valastyan S., Reinhardt F., Benaich N., Calogrias D., Szász A. M., Wang Z. C., Brock J. E., Richardson A. L., Weinberg R. A. (2009) Cell 137, 1032–1046 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23. Bornfeldt K. E., Krebs E. G. (1999) Cell Signal. 11, 465–477 [DOI] [PubMed] [Google Scholar]
- 24. Paroo Z., Ye X., Chen S., Liu Q. (2009) Cell 139, 112–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Justice R. W., Zilian O., Woods D. F., Noll M., Bryant P. J. (1995) Genes Dev. 9, 534–546 [DOI] [PubMed] [Google Scholar]
- 26. Hori T., Takaori-Kondo A., Kamikubo Y., Uchiyama T. (2000) Oncogene 19, 3101–3109 [DOI] [PubMed] [Google Scholar]
- 27. Yabuta N., Fujii T., Copeland N. G., Gilbert D. J., Jenkins N. A., Nishiguchi H., Endo Y., Toji S., Tanaka H., Nishimune Y., Nojima H. (2000) Genomics 63, 263–270 [DOI] [PubMed] [Google Scholar]
- 28. Xu T., Wang W., Zhang S., Stewart R. A., Yu W. (1995) Development 121, 1053–1063 [DOI] [PubMed] [Google Scholar]
- 29. Kamikubo Y., Takaori-Kondo A., Uchiyama T., Hori T. (2003) J. Biol. Chem. 278, 17609–17614 [DOI] [PubMed] [Google Scholar]
- 30. Li Y., Pei J., Xia H., Ke H., Wang H., Tao W. (2003) Oncogene 22, 4398–4405 [DOI] [PubMed] [Google Scholar]
- 31. Lee K. H., Goan Y. G., Hsiao M., Lee C. H., Jian S. H., Lin J. T., Chen Y. L., Lu P. J. (2009) Exp. Cell Res. 315, 2529–2538 [DOI] [PubMed] [Google Scholar]
- 32. Voorhoeve P. M., le Sage C., Schrier M., Gillis A. J., Stoop H., Nagel R., Liu Y. P., van Duijse J., Drost J., Griekspoor A., Zlotorynski E., Yabuta N., De Vita G., Nojima H., Looijenga L. H., Agami R. (2006) Cell 124, 1169–1181 [DOI] [PubMed] [Google Scholar]
- 33. Ahn J. D., Morishita R., Kaneda Y., Lee S. J., Kwon K. Y., Choi S. Y., Lee K. U., Park J. Y., Moon I. J., Park J. G., Yoshizumi M., Ouchi Y., Lee I. K. (2002) Circ. Res. 90, 1325–1332 [DOI] [PubMed] [Google Scholar]
- 34. Edlin R. S., Tsai S., Yamanouchi D., Wang C., Liu B., Kent K. C. (2009) J. Vasc. Surg. 49, 1289–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kavurma M. M., Khachigian L. M. (2003) J. Cell Biochem. 89, 289–300 [DOI] [PubMed] [Google Scholar]
- 36. Sakakibara K., Kubota K., Worku B., Ryer E. J., Miller J. P., Koff A., Kent K. C., Liu B. (2005) J. Biol. Chem. 280, 25470–25477 [DOI] [PubMed] [Google Scholar]
- 37. Friedman R. C., Farh K. K., Burge C. B., Bartel D. P. (2009) Genome Res. 19, 92–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Griffiths-Jones S., Grocock R. J., van Dongen S., Bateman A., Enright A. J. (2006) Nucleic Acids Res. 34, D140-D144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Benson D. A., Karsch-Mizrachi I., Lipman D. J., Ostell J., Sayers E. W. (2011) Nucleic Acids Res. 39, D32-D37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu X., Sempere L. F., Ouyang H., Memoli V. A., Andrew A. S., Luo Y., Demidenko E., Korc M., Shi W., Preis M., Dragnev K. H., Li H., Direnzo J., Bak M., Freemantle S. J., Kauppinen S., Dmitrovsky E. (2010) J. Clin. Invest. 120, 1298–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aprelikova O., Yu X., Palla J., Wei B. R., John S., Yi M., Stephens R., Simpson R. M., Risinger J. I., Jazaeri A., Niederhuber J. (2010) Cell Cycle 9, 4387–4398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cottonham C. L., Kaneko S., Xu L. (2010) J. Biol. Chem. 285, 35293–35302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pedrioli D. M., Karpanen T., Dabouras V., Jurisic G., van de Hoek G., Shin J. W., Marino D., Kälin R. E., Leidel S., Cinelli P., Schulte-Merker S., Brändli A. W., Detmar M. (2010) Mol. Cell Biol. 30, 3620–3634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shen J., Yang X., Xie B., Chen Y., Swaim M., Hackett S. F., Campochiaro P. A. (2008) Mol. Ther. 16, 1208–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mardaryev A. N., Ahmed M. I., Vlahov N. V., Fessing M. Y., Gill J. H., Sharov A. A., Botchkareva N. V. (2010) FASEB J. 24, 3869–3881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Suárez Y., Wang C., Manes T. D., Pober J. S. (2010) J. Immunol. 184, 21–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Creighton C. J., Fountain M. D., Yu Z., Nagaraja A. K., Zhu H., Khan M., Olokpa E., Zariff A., Gunaratne P. H., Matzuk M. M., Anderson M. L. (2010) Cancer Res. 70, 1906–1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen G., Wu L., Deng C. Q. (2011) J. Ethnopharmacol. 135, 7–14 [DOI] [PubMed] [Google Scholar]
- 49. Lu R., Wang X., Chen Z. F., Sun D. F., Tian X. Q., Fang J. Y. (2007) J. Biol. Chem. 282, 12249–12259 [DOI] [PubMed] [Google Scholar]
- 50. Qin S., Zhang C. (2011) J. Cardiovasc. Pharmacol. 57, 8–12 [DOI] [PMC free article] [PubMed] [Google Scholar]