

Background: Growing resistance to influenza antivirals calls for novel therapeutics.
Results: Cholesterol conjugates of HA-derived peptides inhibit influenza infection. HA refolding is trapped at an intermediate stage, arresting fusion.
Conclusion: Cholesterol-conjugated peptides, which potently block fusion of extracellularly fusing viruses, are now shown to block fusion of the intracellularly fusing influenza virus.
Significance: We described a new potential anti-influenza strategy.
Keywords: Animal Viruses, Antiviral Agents, Cholesterol, Electron Microscopy (EM), Endosomes, Membrane Fusion, RNA Viruses, Virus Entry
Abstract
We previously described fusion-inhibitory peptides that are targeted to the cell membrane by cholesterol conjugation and potently inhibit enveloped viruses that fuse at the cell surface, including HIV, parainfluenza, and henipaviruses. However, for viruses that fuse inside of intracellular compartments, fusion-inhibitory peptides have exhibited very low antiviral activity. We propose that for these viruses, too, membrane targeting via cholesterol conjugation may yield potent compounds. Here we compare the activity of fusion-inhibitory peptides derived from the influenza hemagglutinin (HA) and show that although the unconjugated peptides are inactive, the cholesterol-conjugated compounds are effective inhibitors of infectivity and membrane fusion. We hypothesize that the cholesterol moiety, by localizing the peptides to the target cell membrane, allows the peptides to follow the virus to the intracellular site of fusion. The cholesterol-conjugated peptides trap HA in a transient intermediate state after fusion is triggered but before completion of the refolding steps that drive the merging of the viral and cellular membranes. These results provide proof of concept for an antiviral strategy that is applicable to intracellularly fusing viruses, including known and emerging viral pathogens.
Introduction
The impact of influenza infection is felt each year when approximately 20% of the population of the world falls ill. In the United States, influenza infections occur in epidemics each winter. Although vaccination is the primary strategy for influenza prevention, there are a number of likely scenarios for which vaccination is inadequate, and effective antiviral agents would be of utmost importance (1). During any influenza season, antigenic drift in the virus may occur after formulation of the vaccine of the year has taken place, rendering the vaccine less protective so that outbreaks can occur more easily. Vaccine production by current methods cannot be carried out with the speed required to halt the progress of a new strain of influenza virus. Antiviral agents form an important part of the approach to epidemic and pandemic influenza (2).
Although two classes of anti-influenza compounds are available, the neuraminidase inhibitor class of compounds are currently the only option in most clinical settings because of the high level of resistance to the amantadine class of antivirals (2). As predicted by molecular studies, however, resistance to the neuraminidase inhibitors is now emerging rapidly (3). Several mutations in the neuraminidase (NA)4 active site that prevent the molecular rearrangements necessary for oseltamivir to fit, but do not affect NA activity, lead to resistance (3). These influenza isolates are generally still sensitive to the inhaled agent zanamivir. However, emergence of resistance to this last option is of great concern, leaving humans with no antiviral agents for treatment of influenza.
Peptides that correspond to a number of viral fusion proteins can bind to conformational intermediates during viral entry and inhibit infection. For HIV, one such clinically effective peptide is enfuvirtide. Enfuvirtide is derived from the C-terminal region of HIV gp41. It binds in trans to the prehairpin (extended) intermediate state in which the N-terminal fusion peptide of gp41 has inserted into the target host membrane, whereas the C-terminal transmembrane domain of gp41 remains anchored in the viral envelope. Enfuvirtide binding inhibits the conversion of gp41 from the prehairpin to the post-fusion, hairpin (folded) state where the two membranes would be drawn together. For several other viruses that utilize class I fusion proteins, this approach has been hindered by the relatively low potency and short half-life in vivo of the corresponding peptides. Recently, we showed that cholesterol-conjugated, membrane-targeted peptides derived from the fusion protein of enveloped viruses are potent inhibitors of infection and also gain improved pharmacokinetic properties. In particular, we showed that a cholesterol-conjugated inhibitor of HIV is 50–400-fold (depending on the viral strain) more potent than enfuvirtide (4) and that cholesterol-conjugated inhibitors of human parainfluenza virus type 3 are effective inhibitors of both human parainfluenza virus type 3 and henipavirus infection in vitro (5–8) and for Nipah virus are effective antivirals in vivo (9). We have suggested previously that cholesterol conjugation of antiviral peptides could also be useful for viruses that fuse in the endosomal compartment, including influenza virus, by endowing the inhibitory peptides with the ability to be trafficked, along with virus, to the intracellular sites of membrane fusion (8).
Here we demonstrate that a cholesterol-conjugated peptide derived from the influenza virus HA, the receptor binding/fusion protein, inhibits influenza infection. This targeted peptide traps HA in a transient intermediate state, thus preventing the progression of fusion.
EXPERIMENTAL PROCEDURES
Transient Expression of HA, Junin GP NiV G/F, and VSV G Genes
Transfections were performed according to the Lipofectamine and Plus or the Lipofectamine 2000 manufacturer's protocols (Invitrogen).
Cells and Viruses
293T (human kidney epithelial), Vero (African green monkey kidney cells) and CV1 cells were grown in DMEM (Mediatech, Cellgro) supplemented with 10% FBS and antibiotics in 5% CO2. The effect of peptides on H3N2 influenza (from ATCC, VR-544, lot 57899071) plaque number was assessed by a plaque reduction assay. Briefly, CV-1 cell monolayers were inoculated with 100–200 plaque-forming units of influenza H3N2 in the presence of various concentrations of peptides. After 120 min, 2× minimal essential medium containing 10% FBS was mixed with 1% methylcellulose and added to the dishes. The plates were then incubated at 37 °C for 4 days. After removing the medium overlay, the cells were stained for plaque detection. The number of plaques in the control (no peptide, HPIV3-derived peptides) and experimental wells were counted under a dissecting stereoscope. The IC50 (concentrations required for a 50% decrease in plaque number) were determined by plotting the percent of decrease in plaque number versus inhibitor concentration.
β-Gal Complementation-based Fusion Assay
We previously adapted a fusion assay on the basis of α complementation of β-gal (10, 11). Receptor-bearing cells expressing the ω peptide of β-gal are mixed with 1918 HA-expressing cells that also express the α peptide of β-gal, in the presence of or absence of inhibitory peptide. Cell fusion leads to complementation. Fusion was stopped by lysing the cells and, after addition of the substrate, fusion was quantitated on a Spectramax M5 microplate reader.
Labeling RBCs with DiO
0.5% RBCs in CO2-independent medium (Invitrogen, catalog no. 18045) were labeled using VybrantTM DiO (Molecular Probes, catalog no. V22889) cell labeling solution at 37 °C for 30 min. The RBCs were washed and then resuspended to 2% final RBC concentration.
RBC Fusion with HA-expressing Cells
293T cells expressing 1918 HA were treated with cycloheximide and 40 milliunit neuraminidase for 1 h. They were then overlaid with DiO-labeled RBCs at room temperature for 30 min. The cells were then washed with CO2-independent medium and treated with CO2-independent medium at pH 4.9 or 7.4 in the absence or presence of 50 mm of the indicated peptide. After 30 min at room temperature, pictures were taken using a fluorescent microscope.
Pseudotyped Virus Infection Assay
The VSV-ΔG-RFP is a recombinant VSV derived from the cDNA of VSV Indiana, in which the G gene is replaced with the Ds-Red (RFP) gene. Pseudotypes with 1918 HA were generated as described (12, 13). Briefly, 293T cells were transfected with plasmids encoding either VSV-G, 1918 HA, Junin GP, or NiV G/F. 24 h post-transfection, the dishes were washed and infected (multiplicity of infection of 1) with VSV-ΔG-RFP complemented with VSV G. Supernatant fluids containing pseudotyped virus (1918 HA, Junin GP, NiV G/F, or VSV-G) were collected 24 h post-infection and stored at −80 °C. For infection assays, 1918 HA and NiV G/F or VSV-G pseudotypes (controls) were used at a multiplicity of infection of 0.25 to infect 293T cells. Peptides were added at various concentrations. RFP production at 96 h was analyzed by fluorescence microscopy (14), Spectramax M5 analysis and FACS analysis (BD Biosciences FACSCalibur).
Plasmids
The genes of NiV WT G and WT F were codon-optimized and synthesized by GeneArt (Germany) and subcloned into the mammalian expression vector pCAGGS using EcoRI or XhoI and BglII. The 1918 HA was codon-optimized and synthesized by EpochBiolabs and subcloned into the mammalian expression vector pCAGGS. Junin envelope glycoprotein (GP-C) in pCAGGS was generously provided by Thomas Briese.
Peptide Synthesis
All peptides were produced by standard Fmoc-solid phase methods. The cholesterol moiety was attached to the peptide via reaction with a bromoacetyl derivative of cholesterol, as described previously (4, 8, 9).
Virus and Liposome Preparation for Fluorescence Spectroscopy
Gradient-purified X31 (H3N2) influenza A virus grown in embryonated eggs was purchased from Charles River Laboratories. Virus stocks were first centrifuged at 2,320 relative centrifugal force for 5 min to remove precipitates formed by egg proteins. Then the virion supernatant was concentrated to 5–6 mg/ml by centrifugation at 21,000 rcf and stored in 250 mm NaCl, 10 mm HEPES, 50 mm sodium citrate (pH 7.5) buffer. Labeling with the lipophilic dye DiD (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine) was performed by adding 500 μl of approximately 1 mg/ml X31 virus-diluted solutions to 5 μl of DiD Vybrant solution (Invitrogen) followed by a 2-h incubation at 37 °C with gentle rocking. The dye-labeled virus was harvested by ultracentrifugation and again resuspended to 5–6 mg/ml X31 virus solution, then stored at 4 °C. Liposomes composed of either pure DOPC or DOPC and cholesterol (Avanti Polar Lipids) in 4:1 w/w ratios were performed as described previously with a few modifications (15). The buffer used for resuspension of dried lipids was 250 mm NaCl, 10 mm HEPES, 50 mm sodium citrate (pH 7.5) containing 25 mm sulforhodamine B (SRB) fluorophore (Invitrogen). Resuspended lipid was subjected to five sequential freeze/thaw cycles in liquid nitrogen followed by 25 extrusions through polycarbonate filters with 200-nm pore size (Avanti Polar Lipids). The dye-encapsulating liposomes eluted as a single band from PD-10 gel filtration columns (GE Healthcare) and were stored in the same pH 7.5 buffer used for virus storage.
Fluorescence Spectroscopy
Prior to fluorescence experiments, the cholesterol-tagged P155–185-chol peptide was incubated with SRB-containing DOPC and DOPC:cholesterol liposomes at 1, 3, and 5 μm final concentrations for 30 min at 25 °C. In control experiments with P155–185 peptide lacking the cholesterol tag, an identical procedure was followed. DiD-labeled virus and inhibitor-decorated liposomes were mixed in a ratio of 1:10 with liposomes in excess. Parallel experiments in which the cholesterol-tagged inhibitor was incubated first with virus and then mixed with inhibitor-naïve liposomes were also carried out. To initiate the fusion reactions, the pH was dropped to 5.0 or 5.25 by the addition of aliquots of 250 mm NaCl, 10 mm HEPES, 50 mm sodium citrate (pH 3.0) buffer. Fluorescence spectroscopy was carried out in a Varian Cary Eclipse spectrophotometer using excitation/emission spectral pairs: λ excitation, 565 nm; λ emission, 585 nm (SRB) and λ excitation, 644 nm and λ emission, 665 nm (DiD) with 2.5 nm slit widths. The fluorescence cuvette was thermostated at 25 °C. In all experiments, the extent of liposome content leakage (SRB dequenching) and lipid mixing (DiD dequenching) was determined relative to the value of fully dequenched fluorescence exhibited following Triton X-100 solubilization of liposomes and virus (15).
Negative Staining and Electron Microscopy
The cholesterol-tagged P155–185-chol inhibitor and DOPC or DOPC:cholesterol liposomes were first mixed together at 5 μm inhibitor concentration and incubated at 25 °C for 30 min. Approximately 10-fold greater concentrations of liposomes and virus were used compared with the conditions in the fluorescence experiments to produce a reasonable density of particles in the field of view. After 30 min of incubation at 25 °C and pH 5.0, 5.25, or 5.5, 3 μl of each sample was transferred onto glow-discharged, carbon-coated electron microscope grids and stained with either nano-W (methylaminetungstate, NanoProbes) or nano-van (methylaminevanadate, NanoProbes). Imaging was performed using an FEI Company Tecnai G2 transmission electron microscope operating at 200 kV coupled to a Gatan Ultrascan 1000 charge-coupled device.
Detection of HA Conformational Change
293T cell monolayers were transiently transfected with 1918 HA (tagged at the C terminus with a GFP-derived epitope) and incubated overnight in DMEM supplemented with 10% FBS. The transfected cells were washed with Optimem and incubated for 1 h at 37 °C in Optimem supplemented with 100 mg/ml cyclohexamide to prevent de novo protein synthesis (16) and 40 milliunits of Clostridium perfringens neuraminidase (Sigma). The cells were treated with trypsin 5 mg/ml at 37 °C for 15 min and then incubated at room temperature in medium at the indicated pH with or without 50 mm of P155–185-chol-peptide. The cells were then lysed in DH buffer (50 mm HEPES, 100 mm NaCl, 5 mg/ml dodecyl maltoside, complete protease inhibitor mixture (Roche)). HA was immunoprecipitated with LC89 antibody (obtained from Judith M. White with permission from John J. Skehel) or with fusion peptide serum (from Judith M. White) (17). As a control, HA was also immunoprecipitated with anti-GFP antibody-conjugated agarose beads (Santa Cruz Biotechnology, Inc.). The immunoprecipitates were then subjected to SDS-PAGE and Western blot analysis with anti-GFP polyclonal antibodies conjugated to HRP.
Cytotoxicity Assay
293T cells were incubated in Optimem with the indicated concentration of peptide or vehicle (dimethyl sulfoxide) for 48 h. 10% v/v Alamar Blue (Invitrogen) was added according to the manufacturer's instructions. Fluorescence was read at an excitation of 570 nm and emission of 590 nm using a microplate fluorescence reader (Spectramax M5).
RESULTS
Design of the Peptide Inhibitors
The 16 subtypes of influenza HA can be divided in two major groups: group 1 (with two clades including H3) and group 2 (with 3 clades including H1) (18). On the basis of the post-fusion structure of the X-31 H3 HA ectodomain of the HA2 subunit of HA, EHA2 (19) (PDB code 1QU1), we generated three peptides corresponding to residues 155–185, containing 31, 27, or 21 residues (Fig. 1, A and B). These peptides were synthesized either with or without a cholesterol moiety linked via a thioether bond to a cysteine residue added C-terminally to the peptide (“cholesterol-tagged”). The 155–185 domain of HA, which is immediately upstream of the transmembrane domain, was chosen for the inhibitory peptide because it packs against the inner coiled-coil of HA as identified in the post-fusion, low-pH structure of the protein (19, 20). According to the commonly accepted model for viral fusion driven by class I proteins (21), formation of a complex between region 155–185 and the inner coiled-coil is central to the energetics of fusion (22). Within this domain, residues 176–185 participate in the “N-cap” motif, which forms an extensive set of interactions that terminates the central coiled-coil and fixes the N- and C-terminal regions of the molecule together at one extreme end of the rod-like structure (19). We compare inhibitors with or without this subdomain. Finally, residues 182–185, visible only in one of the monomers of the crystal structure, occupy a position between the end of the N-capped coiled-coil and transmembrane domain, similar to the role of the membrane proximal external region of HIV gp41 fusion protein (23). In HIV, this region rich in tryptophan and aromatic residues (24) interacts with the membrane and, in particular, with cholesterol (25). The role of the HIV membrane proximal external region in facilitating the generation of membrane curvature and increasing the stability of the post-fusion structure has been highlighted recently (26). For influenza HA, the presence of Trp-185 and Tyr-182 in this short stretch may suggest a similar role. If so, cholesterol conjugation may interfere with the natural function of this region and be detrimental to inhibitory activity, as we found for HIV (4). We therefore compare cholesterol-tagged inhibitors with or without the 182–185 region.
FIGURE 1.

Cholesterol-conjugated peptides derived from influenza HA are effective influenza virus entry inhibitors. A, structure of the influenza virus hemagglutinin in the low-pH post-fusion conformation, highlighting the region corresponding to the cholesterol-conjugated fusion inhibitors. Only one of the three units of the trimer is shown for clarity. The region corresponding to the cholesterol-tagged inhibitors and their controls is highlighted in yellow, violet and red. Yellow, amino acids 155–175, common to all peptides; violet, amino acids 176–181; red, amino acids 182–185. The linker GSGSG and the cholesterol group are C-terminal to the last HA2 residue included in the inhibitor. B, table showing the 6 HA derived peptides and their IC50 versus influenza H3N2 live virus in a plaque reduction assay. The amino acids from the HA sequence are shown in red.
Cholesterol-conjugated Peptides Inhibit Infection
We hypothesized that, as for the parainfluenza virus (8) and HIV (4), cholesterol conjugation would result in trafficking of the inhibitor peptide to the membrane compartment where the fusion protein is activated. For influenza, this would be the membrane of the endosome, where acidification drives HA to its fusion-ready state. At this point, the fusion inhibitory peptides would prevent progression of virus-cell membrane fusion by blocking the key HA folding steps required for the membrane merger (15, 27).
Accordingly, we found that the cholesterol-tagged peptides are effective inhibitors of infection with influenza A/H3N2 in cell culture. The longer peptide we designed (P155–185-chol) inhibits viral entry with an IC50 of 0.4 μm (Fig. 1B). Deletion of the C-terminal four residues resulted in an increase in IC50 to 2 μm, whereas truncation of residues 176–185, involved in the N-Cap motif in the HA2 hairpin structure, further reduces efficacy with IC50 > 10 μm (Fig. 1). The same peptides without a cholesterol tag did not significantly inhibit infection.
The Cholesterol-conjugated Inhibitor Displays Cross-subtype Antiviral Activity
Highly conserved sites on influenza virus HA proteins have been recently identified, and several groups have proposed that targeting these conserved sites may allow for broad-spectrum anti-influenza agents (28). One target is a highly conserved helical region in the stem of HA, and broadly cross-subtype neutralizing antibodies binding to this region have been independently identified, which neutralize the virus by blocking the conformational rearrangements associated with HA-mediated membrane fusion (29–32). We therefore asked whether our H3-derived fusion inhibitory peptides would likewise display cross-subtype neutralization or whether the inhibition would be restricted to homotypic HA. To this end, we tested the efficacy of our H3-derived peptide versus the H1 subtype 1918 HA. For these experiments, we adapted the pseudotyping strategy that we employ for highly pathogenic viruses (5, 34). For the experiment in Fig. 2, HA was pseudotyped onto a recombinant vesicular stomatitis virus (VSV) that expresses red fluorescent protein (RFP) but lacks its attachment protein, G (33, 34). The resulting pseudotyped virus bears the influenza HA. Supplemental Fig. 1A shows the hemagglutination titer of the H1 HA-pseudotyped virus. Infection of target 293T cells in 96-well plates by pseudotyped virus in the presence of various concentrations of P155–185-chol and P155–185, lacking the cholesterol anchor, was quantified by assessing the production of red fluorescence. The infected plates were incubated at 37 °C for 48 h, and the fluorescence in each well was measured. The cholesterol-conjugated peptide completely blocks H1 virus infection at a concentration of 20 μm, whereas the untagged peptide is only weakly active at the same concentration. To ensure that inhibition was specific to the HA-pseudotyped virus, we tested P155–185-chol on Junin GPC-pseudotyped virus (35) and found no significant activity (supplemental Fig. 1B). As a control for the effect of cholesterol tagging, a previously described HIV-derived cholesterol-tagged peptide (4) was also included in the experiment shown in Fig. 2. The HIV-derived cholesterol peptide has negligible activity at 20 μm and no inhibitory activity at 10 μm, at which concentration P155–185-chol peptide showed approximately 85% inhibition of viral entry. This minimal antiviral activity of the HIV-derived cholesterol-tagged peptide may be attributable to the toxic effect that this specific peptide exhibits in our cell viability assay at the same concentration (supplemental Fig. 1C). The influenza derived P155–185-chol peptide instead does not alter cell viability (supplemental Fig. 1C).
FIGURE 2.

H3 HA derived cholesterol conjugated peptides inhibit 1918 influenza HA mediated infection. Inhibition of viral entry by P155–185-chol (●), P155–185 (■), and HIV-derived cholesterol tagged peptide (▴) using 1918 HA-pseudotyped VSV-ΔG-RFP (vesicular stomatitis virus engineered to express red fluorescent protein and lacking its own G envelope protein, substituted by the 1918 HA).
The Cholesterol-conjugated Peptide Blocks Fusion by Preventing Merger of the Virus and Cell Membranes
We then asked whether P155–185-chol blocks HA-mediated fusion by trapping HA in a transient intermediate state and preventing it from progressing through the folding steps that lead to membrane merger. To test this idea, we used 200 nm of DOPC or DOPC:cholesterol (4:1 w/w) liposomes containing 25 mm sulforhodamine B fluorophore. The liposomes were incubated with 1, 3, 5, and 10 μm P155–185-chol for 30 min at 25 °C (pH 7.5). The inhibitor-decorated liposomes were then combined at pH 7.5 with X-31 H3N2 (A/Aichi/68) virus that had been labeled with the lipophilic dye DiD at an approximately 5- to 10-fold excess of liposome to virus particle. The fusion reaction was initiated by adding aliquots of predetermined volumes of 250 mm NaCl, 10 mm HEPES, 50 mm Na-Citrate (pH 3.0) to achieve a final pH of 5.0, 5.25, or 5.5. Fluorescence spectroscopy monitoring both SRB leakage and DiD transfer as the fusion reaction progressed was performed at 25 °C (15). In the experimental results shown in Fig. 3, in the presence of P155–185-chol, both liposome leakage (SRB fluorescence dequenching) and lipid mixing (DiD fluorescence dequenching) were inhibited, and inhibition was concentration-dependent. In identical experiments carried out with P155–185 lacking the cholesterol anchor, there was negligible inhibition (Fig. 4A). Likewise, when P155–185-chol was incubated with the virus particles rather than with the liposomes prior to acidification, minimal inhibition was observed, indicating that the inhibitor must be presented on the target membrane to be effective (Fig. 4B). From these data, we conclude that the cholesterol-anchored inhibitor must be presented on the target liposomal membrane rather than freely diffusing in solution or bound to the viral envelope. Lastly, although overall fusogenicity of pure DOPC liposomes was lower than DOPC:cholesterol liposomes, both showed similar degrees of inhibition of leakage and lipid mixing (not shown), suggesting that the inhibitor had a similar effect regardless of the target membrane composition.
FIGURE 3.

H3 HA-derived cholesterol conjugated peptides block influenza virus fusion. Fluorescence-monitored assays of fusion and content leakage used cholesterol-tagged inhibitor (P155–185-chol) added directly to preformed liposomes (4:1 w/w DOPC:cholesterol, 200-nm diameter). A water-soluble fluorophore, sulforhodamine B, was encapsulated in the aqueous lumen of the liposomes at self-quenching concentrations. The lipophilic fluorophore, DiD, was embedded in the viral membrane, also at self-quenching concentrations. Inhibitor-decorated liposomes were incubated with X-31 H3N2 virus prior to acidification to pH 5.25 (blue traces) or 5.0 (black traces). A and B, the fluorescence signals increased as a result of fluorophore dilution and fluorescence dequenching. Fluorescence assay at pH 5.25, exhibited inhibitor concentration-dependent inhibition of sulforhodamine B leakage (A) as well as inhibition of lipid mixing (B). 0, 1, 3, and 5 μm inhibitor reactions were performed. Only 0 μm (traces labeled with circles) and 5 μm (traces labeled with squares) data are shown here for clarity. 1 and 3 μm cases were intermediate in the degree of inhibition. pH 5.0 experiments exhibited the same concentration-dependent inhibition trends with faster kinetics.
FIGURE 4.

Fusion inhibition depends on the cholesterol anchor and the specific membrane in which the antiviral peptide is anchored. (A) and (B), fluorescence-monitored fusion reactions with sulforhodamine B fluorescence (encapsulated within the liposomes), which reports liposome permeabilization (top panels), and DiD fluorescence (embedded in the viral membrane), which reports lipid mixing (bottom panels). (A), if P155–185 is not anchored to a membrane by conjugation to cholesterol, inhibition is abolished. P155–185 was incubated at 5 μm (blue traces) with 4:1 w/w DOPC:cholesterol liposomes prior to mixing with virus and acidifying to pH 5.0. The fluorescence-dequenching traces reporting on liposome content leakage (top panel) and lipid mixing (bottom panel) are essentially superimposable with 0 μm inhibitor controls (black traces). (B), P155–185-chol was incubated at 10 μm only with virus prior to mixing with DOPC:cholesterol liposomes and acidification to pH 5.0 to initiate the fusion reaction. Even with 10 μm peptide preincubated with viral particles (red traces), inhibition of liposome leakage (top panel) and lipid mixing (bottom panel) is minimal, and the fluorescence-dequenching traces closely follow the control reactions with no inhibitor present (black traces).
To image complexes of virus and liposomes, negative-stain transmission electron microscopy was used. As shown in Fig. 5, liposomes that were incubated with 5 μm P155–185-chol were unable to fuse with virus (Fig. 5, A–D), even though under identical fusogenic conditions the same liposomes without inhibitor exhibited significant post-fusion complexes with completely merged virus and liposome membranes, which appear flattened on the grid, and a clear transfer of viral glycoproteins (HA and NA) onto the merged membrane surface (Fig. 5, E–G). In the contact zones between virus and the P155–185-chol decorated liposomes, the liposomal surface is highly coordinated by continuous swaths of viral glycoproteins, presumably HA, to the extent that some liposomes wrap around a virus particle. HA spikes remain in the interstitial space, bridging the two membrane surfaces, and the membranes are not in direct apposition with each other. This indicates that HAs have yet to complete their acid-triggered refolding, which would colocalize the viral transmembrane anchor and fusion peptides and draw the membranes together.
FIGURE 5.

Trapping the HA in a transient intermediate state with cholesterol-conjugated peptides. Negative-stain transmission electron microscopy, comparing liposome-virus complexes with 5 μm inhibitor at pH 5.25 (A and B) and pH 5.5 (C and D). In the presence of the inhibitor, the virus is closely associated with liposomes (suggesting fusion-peptide mediated interaction occurring in the absence of sialic acid influenza receptor present), the membranes have not joined, and there is no evidence of dispersed glycoproteins on liposomes. In the samples without the inhibitor present after incubation at pH 5.25 (E) or pH 5.5 (F and G), the virus and liposomes show significant merging of membranes, viral glycoprotein spikes have dispersed across the liposomal surface, and the liposomes appear flattened and more permeable to stain, suggesting a loss of bilayer integrity. Scale bars = 100 nm.
The Cholesterol-conjugated Peptide Does Not Block the Activation of Viral Fusion
To identify the stage of conformational change at which HA is arrested by the inhibitor, we used an antibody-based assay that has been shown to detect HA activation (17, 36, 37). For the experiment shown in Fig. 6, cells expressing 1918 H1 HA were treated with trypsin and incubated with 50 μm cholesterol-tagged peptide at either pH 4.9 or pH 7.4 for 30 min at room temperature. Under these conditions, syncytia formation and fusion measured by β-gal complementation assay are inhibited by P155–185-chol (supplemental Fig. 2, A and B). The HA was then immunoprecipitated with either monoclonal antibody LC89 or anti-fusion peptide (FP) serum (17) that recognize the low-pH conformation of HA (38, 39). A Western blot analysis of immunoprecipitated HA shows that in the presence of the cholesterol-tagged peptide, both FP serum and LC89-immunoprecipitated HA from the sample incubated at pH 4.9 but not from the sample incubated at pH 7.4. These results confirm that the P155–185-chol peptide does not prevent HA activation. We also conclude that the peptide activity does not induce premature activation of HA because the anti-fusion peptide antibody and serum do not react with HA in the presence of inhibitor at pH 7.4 as well as on the basis of the observations that the preincubation of virus with peptide does not result in fusion inhibition (Fig. 4B).
FIGURE 6.
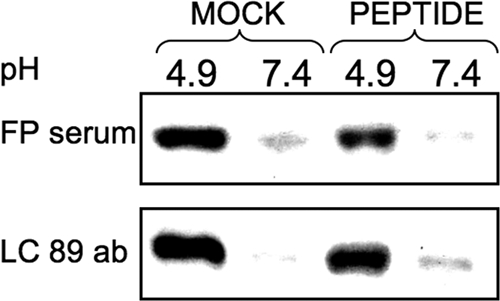
Cholesterol-conjugated peptides do not prevent pH-induced HA activation. Monolayers of cells expressing HA were incubated for 30 min in media at pH 4.9 or pH 7.4 with or without 50 μm P155–185-chol peptide as indicated. HA conformation was analyzed by immunoprecipitation with the conformation-specific antibodies (ab) LC89 and anti-FP antiserum. Representative Western blots show HA detected by polyclonal anti-GFP-HRP-conjugated antibodies. We note that the fusion peptide sequence recognized by the FP serum is highly conserved across serotypes. The LC89 antibody was originally raised against X-31 HA that had been treated with acidic pH. The HA2 residues recognized by LC89, 106–112, are HTIDLT in X-31. In 1918 H1N1, residues 106–112 are RTLDFH. The sequence specificity of LC89 binding is linked primarily to having a threonine at position 107. If this is mutated, LC89 binding is greatly reduced (38, 39). Given the conservation of Thr-107 in X-31 and H1N1 HA2, the LC89 antibody may be able to still recognize the conformational epitope in our experiments in with HA from 1918 H1N1. In terms of the pattern of ionizable, polar, and apolar residues, the two sequences are also similar.
DISCUSSION
We report here the first example of a peptide fusion inhibitor for influenza virus. Viruses such as influenza that fuse in intracellular compartments have been considered a difficult target for fusion inhibitors because the peptide needs to be internalized into host cells to have access to transient intermediate conformations of the viral fusion proteins, which become populated only following cell entry (21, 40). Our results demonstrate that the addition of a cholesterol group confers antiviral activity to the anti-influenza peptide. Because influenza cell entry and infection occurs via endocytosis, we interpret the inhibition that we observe as a reflection of the peptide being enriched first at the cellular membrane, because of the cholesterol anchor, and then being internalized together with the virus in the endosomes, where acidification triggers the fusogenic activity of HA and converts it to the conformation recognized by the inhibitor peptide.
Most fusion-inhibitory peptides that have been described are helical either in isolation or once bound to their target (4, 9, 21, 27, 41–43). Their proposed mechanism of action is to prevent the natural refolding of the fusion protein into a 6-helix bundle. For influenza HA, a short 6-helix bundle is located at the membrane distal end of the protein, whereas the membrane proximal region has been portrayed as a “leash” that packs into the grooves of the N-terminal helical regions (22). Mutations that alter the proper interaction between the leash and the groove decreased fusion capability (22). Our inhibitory peptides were designed on the basis of this leash region, and we propose that they prevent fusion by competing with the intramolecular binding of the C-terminal segment of HA2 to the groove. The HA-derived peptides may block fusion by arresting the refolding of the fusion protein at an intermediate stage. The trapped conformation is possibly similar to that populated by leash mutants such as a previously reported one with five alanines in position 171–175, which undergoes the acid pH-induced helix-to-loop transition to a state recognized by antibody LC89 but is unable to mediate lipid mixing (22). A non-helical fusion inhibitory peptide has also recently been described for human T-cell leukemia virus (44).
Because the peptide fusion inhibitors are on the basis of a relatively conserved region of the HA2 subunit, they are expected to exhibit reasonably broad-spectrum inhibition, consistent with our observations of similar efficacy in the infectivity assays with the H1 and H3 HA-bearing virus. For the next generation of inhibitors, we are refining the analysis of HA sequence alignments of this region to design consensus peptides with an even broader spectrum of activity. Additionally, although reasonably potent, P155–185-chol peptides are not expected to display in vivo inhibitory activity. On the basis of our experience with HIV and paramyxoviruses, to achieve this, we would need to achieve single-digit nanomolar to subnanomolar in vitro IC50. It is encouraging though that the fusion-inhibitory activity with liposomes seen in Fig. 3, A and B, correlates with the entry inhibition seen in Fig. 2, indicating that the limited potency results from inefficient peptide binding to the transient intermediate structure populated prior to final hairpin formation. Numerous examples in the literature suggest that this feature may be improved by suitable mutagenesis of the peptide binding interface (5, 9, 45, 46), and we are currently pursuing this strategy.
Electron microscopy of virus-liposome complexes in Fig. 5 shows liposomes bound to HA-bearing viral particles. Because no sialic acid receptor is present on the liposomes, their coordination by the viral particles either results from the HA fusion peptides grappling to the liposomal membrane or from binding of the cholesterol-anchored inhibitor peptide to HA spikes. This may be occurring without significant insertion of the fusion peptides into the liposomal outer leaflet. We note that SRB leakage, like lipid mixing detected by DiD dequenching, is inhibited in a concentration-dependent manner (Fig. 3), which may be consistent with a scenario in which the fusion peptides are prevented from binding to and destabilizing the liposomal bilayer, which would allow content leakage. For paramyxovirus fusion proteins we have recently shown that cholesterol-conjugated peptides trap a fusion intermediate before insertion of the fusion peptide into the host cell membrane (9). One possibility is that the anti-influenza P155–185 peptide segment is bound to activated HA, which bears an extended N-terminal trimeric helical bundle with fusion peptides presented at the end, but they may be held out of reach of or prevented from inserting sufficiently into the target membrane by the hexapeptide-PEG4 linker that joins P155–185 and the cholesterol anchor. This possibility may be tested by further varying the linker in future iterations of peptide design. Lower levels of liposome content leakage could also result from the inhibition of target membrane bending that we have observed previously in the absence of fusion inhibitors (15). Indeed, in Fig. 5, A–D, liposome and virus membranes appear intact without acute protrusions or pinched features that might be focal points for membrane permeability. Our combined results from electron microscopy, fluorescence, and antibody binding studies indicate that in the presence of P155–185-chol, the hemagglutinin spikes are trapped in a transitional conformation with exposed fusion peptides, but they are not capable of destabilizing membranes to induce leakage, and they have not completed the conformational refolding that would thrust the two membranes together to induce fusion. Cryo-electron tomography has recently been used to image native snapshots of HA-mediated fusion pore formation (15). Similar analysis of the peptide trapped HA intermediate may enable characterization of hemagglutinin spikes and the detailed nature of membrane deformation in the fusion-arrested state.
The finding that liposomes with P155–185-chol appear to arrest HA fusion at an intermediate stage also suggests a potential new, general approach to influenza vaccines as well as vaccines for other viral pathogens. Specifically, vaccination with trapped fusion intermediates, which expose normally inaccessible or only transiently accessible conserved epitopes to the immune system, may elicit potent immunity with broad coverage of viral isolates from different strains.
The cholesterol-tagging strategy presented here is broadly applicable, like the parallel approach of conjugating an inhibitory peptide to an arginine-rich sequence, recently described for a fusion inhibitor of Ebola virus (47). Conjugation to cholesterol also provides the critical advantage of endowing the peptide with an improved in vivo half-life, as we demonstrated in closely related systems (4, 49), a common issue for peptide inhibitors.
In conclusion, the results reported here have broad implications for antiviral development with application to a wide range of known and emerging viruses that fuse with intracellular compartments of infected cells. Applicable targets include influenza A and B viruses as well as arenaviruses (Junin), human metapneumoviruses, filoviruses (Ebola), flavivirus, and coronaviruses (severe acute respiratory syndrome) (8, 9, 33, 48).
Supplementary Material
Acknowledgments
We thank Dan and Nancy Paduano for support of innovative research projects, Ashton Kutcher and Jonathan Ledecky for their support, and the Friedman Family Foundation for renovation of our laboratories at Weill Cornell Medical College. We also thank Judith M. White and John J. Skehel for helpful discussion and providing essential reagents. We thank GTx, Inc. for the kind gift of VSV-ΔG-RFP VSV-G. We also thank Sergei Rudchenko for flow cytometry support in the Flow Cytometry Facility of the Hospital for Special Surgery/Weill Cornell Medical College. We thank the Northeast Center of Excellence for Bio-defense and Emerging Infections Disease Research Proteomics Core for peptide synthesis and purification.
This work was supported, in whole or in part, by NIAD, National Institutes of Health Northeast Center of Excellence for Biodefense and Emerging Infectious Disease Research Grant U54AI057158 (to M. P.), Principal Investigator of Center of Excellence Grant W. I. Lipkin (to A. M.), National Institutes of Health Grants R01AI076335, R01AI31971, and R21AI090354 (to A. M.); R21EBO11707 (to M. P.); and R00GM080352 (to K. K. L.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2.
- NA
- neuraminidase
- DiO
- 3,3′-dioctadecyloxa carbocyanine perchlorate
- DiD
- 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine
- SRB
- sulforhodamine B
- VSV
- vesicular stomatitis virus
- RFP
- red fluorescent protein
- FP
- fusion peptide
- DOPC
- 1,2-dioleoyl-sn-glycero-3-phosphocholine.
REFERENCES
- 1. Moscona A. (2008) Annu. Rev. Med. 59, 397–413 [DOI] [PubMed] [Google Scholar]
- 2. Moscona A. (2005) N. Engl. J. Med. 353, 1363–1373 [DOI] [PubMed] [Google Scholar]
- 3. Moscona A. (2009) N. Engl. J. Med. 360, 953–956 [DOI] [PubMed] [Google Scholar]
- 4. Ingallinella P., Bianchi E., Ladwa N. A., Wang Y. J., Hrin R., Veneziano M., Bonelli F., Ketas T. J., Moore J. P., Miller M. D., Pessi A. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 5801–5806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Porotto M., Carta P., Deng Y., Kellogg G. E., Whitt M., Lu M., Mungall B. A., Moscona A. (2007) J. Virol. 81, 10567–10574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Porotto M., Doctor L., Carta P., Fornabaio M., Greengard O., Kellogg G. E., Moscona A. (2006) J. Virol. 80, 9837–9849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Porotto M., Yokoyama C. C., Orefice G., Kim H. S., Aljofan M., Mungall B. A., Moscona A. (2009) J. Virol. 83, 6947–6951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Porotto M., Yokoyama C. C., Palermo L. M., Mungall B., Aljofan M., Cortese R., Pessi A., Moscona A. (2010) J. Virology 84, 6760–6768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Porotto M., Rockx B., Yokoyama C. C., Talekar A., Devito I., Palermo L. M., Liu J., Cortese R., Lu M., Feldmann H., Pessi A., Moscona A. (2010) PLoS Pathog. 6, e1001168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moosmann P., Rusconi S. (1996) Nucleic Acids Res. 24, 1171–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Porotto M., Fornabaio M., Kellogg G. E., Moscona A. (2007) J. Virol. 81, 3216–3228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Takada A., Robison C., Goto H., Sanchez A., Murti K. G., Whitt M. A., Kawaoka Y. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 14764–14769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Negrete O. A., Levroney E. L., Aguilar H. C., Bertolotti-Ciarlet A., Nazarian R., Tajyar S., Lee B. (2005) Nature 436, 401–405 [DOI] [PubMed] [Google Scholar]
- 14. Porotto M., Murrell M., Greengard O., Lawrence M. C., McKimm-Breschkin J. L., Moscona A. (2004) J. Virol. 78, 13911–13919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee K. K. (2010) EMBO J. 29, 1299–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Obrig T. G., Culp W. J., McKeehan W. L., Hardesty B. (1971) J. Biol. Chem. 246, 174–181 [PubMed] [Google Scholar]
- 17. Leikina E., Ramos C., Markovic I., Zimmerberg J., Chernomordik L. V. (2002) EMBO J. 21, 5701–5710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Russell R. J., Kerry P. S., Stevens D. J., Steinhauer D. A., Martin S. R., Gamblin S. J., Skehel J. J. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 17736–17741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen J., Skehel J. J., Wiley D. C. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 8967–8972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen J., Lee K. H., Steinhauer D. A., Stevens D. J., Skehel J. J., Wiley D. C. (1998) Cell 95, 409–417 [DOI] [PubMed] [Google Scholar]
- 21. Eckert D. M., Kim P. S. (2001) Annu. Rev. Biochem. 70, 777–810 [DOI] [PubMed] [Google Scholar]
- 22. Park H. E., Gruenke J. A., White J. M. (2003) Nat. Struct. Biol. 10, 1048–1053 [DOI] [PubMed] [Google Scholar]
- 23. Noah E., Biron Z., Naider F., Arshava B., Anglister J. (2008) Biochemistry 47, 6782–6792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Salzwedel K., West J. T., Hunter E. (1999) J. Virol. 73, 2469–2480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Veiga A. S., Castanho M. A. (2007) FEBS J. 274, 5096–5104 [DOI] [PubMed] [Google Scholar]
- 26. Buzon V., Natrajan G., Schibli D., Campelo F., Kozlov M. M., Weissenhorn W. (2010) PLoS Pathog. 6, e1000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Harrison S. C. (2008) Nat. Struct. Mol. Biol. 15, 690–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Harrison C. (2009) Nat. Rev. Drug Discov. 8, 276. [DOI] [PubMed] [Google Scholar]
- 29. Corti D., Voss J., Gamblin S. J., Codoni G., Macagno A., Jarrossay D., Vachieri S. G., Pinna D., Minola A., Vanzetta F., Silacci C., Fernandez-Rodriguez B. M., Agatic G., Bianchi S., Giacchetto-Sasselli I., Calder L., Sallusto F., Collins P., Haire L. F., Temperton N., Langedijk J. P., Skehel J. J., Lanzavecchia A. (2011) Science 333, 850–856 [DOI] [PubMed] [Google Scholar]
- 30. Ekiert D. C., Bhabha G., Elsliger M. A., Friesen R. H., Jongeneelen M., Throsby M., Goudsmit J., Wilson I. A. (2009) Science 324, 246–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sui J., Hwang W. C., Perez S., Wei G., Aird D., Chen L. M., Santelli E., Stec B., Cadwell G., Ali M., Wan H., Murakami A., Yammanuru A., Han T., Cox N. J., Bankston L. A., Donis R. O., Liddington R. C., Marasco W. A. (2009) Nat. Struct. Mol. Biol. 16, 265–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ekiert D. C., Friesen R. H., Bhabha G., Kwaks T., Jongeneelen M., Yu W., Ophorst C., Cox F., Korse H. J., Brandenburg B., Vogels R., Brakenhoff J. P., Kompier R., Koldijk M. H., Cornelissen L. A., Poon L. L., Peiris M., Koudstaal W., Wilson I. A., Goudsmit J. (2011) Science 333, 843–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schmidt A. G., Yang P. L., Harrison S. C. (2010) J. Virol. 84, 12549–12554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Porotto M., Orefice G., Yokoyama C. C., Mungall B. A., Realubit R., Sganga M. L., Aljofan M., Whitt M., Glickman F., Moscona A. (2009) J. Virol. 83, 5148–5155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rojek J. M., Kunz S. (2008) Cell. Microbiol. 10, 828–835 [DOI] [PubMed] [Google Scholar]
- 36. White J. M., Wilson I. A. (1987) J. Cell Biology 105, 2887–2896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chernomordik L. V., Leikina E., Kozlov M. M., Frolov V. A., Zimmerberg J. (1999) Mol. Membr. Biol. 16, 33–42 [DOI] [PubMed] [Google Scholar]
- 38. Daniels R. S., Douglas A. R., Skehel J. J., Wiley D. C. (1983) J. Gen. Virol. 64, 1657–1662 [DOI] [PubMed] [Google Scholar]
- 39. Wharton S. A., Calder L. J., Ruigrok R. W., Skehel J. J., Steinhauer D. A., Wiley D. C. (1995) EMBO J. 14, 240–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Skehel J. J., Wiley D. C. (2000) Annu. Rev. Biochem. 69, 531–569 [DOI] [PubMed] [Google Scholar]
- 41. Lamb R. A., Paterson R. G., Jardetzky T. S. (2006) Virology 344, 30–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yin H. S., Wen X., Paterson R. G., Lamb R. A., Jardetzky T. S. (2006) Nature 439, 38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lambert D. M., Barney S., Lambert A. L., Guthrie K., Medinas R., Davis D. E., Bucy T., Erickson J., Merutka G., Petteway S. R., Jr. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 2186–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mirsaliotis A., Lamb D., Brighty D. W. (2008) J. Virol. 82, 4965–4973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Welch B. D., Francis J. N., Redman J. S., Paul S., Weinstock M. T., Reeves J. D., Lie Y. S., Whitby F. G., Eckert D. M., Hill C. P., Root M. J., Kay M. S. (2010) J. Virol. 84, 11235–11244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kahle K. M., Steger H. K., Root M. J. (2009) PLoS Pathog. 5, e1000674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Miller E. H., Harrison J. S., Radoshitzky S. R., Higgins C. D., Chi X., Dong L., Kuhn J. H., Bavari S., Lai J. R., Chandran K. (2011) J. Biol. Chem. 286, 15854–15861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schmidt A. G., Yang P. L., Harrison S. C. (2010) PLoS Pathog. 6, e1000851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Porotto M., Rockx B., Yokoyama C. C., Talekar A., Devito I., Palermo L. M., Liu J., Cortese R., Lu M., Feldmann H., Pessi A., Moscona A. (2010) PLoS Pathog. 6, e1001168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.