

Background: Arginine-rich salmon protamine does not condense DNA in sperm as densely as expected from the number of arginines.
Results: Inserting neutral and negatively charged amino acids into model hexa-arginine peptides decreases attraction between DNA helices.
Conclusion: DNA packing efficiency in sperm nuclei depends on amino acid composition.
Significance: Mispackaging because of protamine chemistry has strong biological implications for DNA damage in sperm.
Keywords: Biophysics, DNA-binding Protein, DNA-Protein Interaction, Protein-Nucleic Acid Interaction, Spermatogenesis, X-ray Scattering, DNA Condensation, Protein-Nucleic Acid Interaction, Sperm Packaging, Intermolecular Forces
Abstract
In spermatogenesis, chromatin histones are replaced by arginine-rich protamines to densely compact DNA in sperm heads. Tight packaging is considered necessary to protect the DNA from damage. To better understand the nature of the forces condensing protamine-DNA assemblies and their dependence on amino acid content, the effect of neutral and negatively charged amino acids on DNA-DNA intermolecular forces was studied using model peptides containing six arginines. We have previously observed that the neutral amino acids in salmon protamine decrease the net attraction between protamine-DNA helices compared with the equivalent homo-arginine peptide. Using osmotic stress coupled with x-ray scattering, we have investigated the component attractive and repulsive forces that determine the net attraction and equilibrium interhelical distance as a function of the chemistry, position, and number of the amino acid inserted. Neutral amino acids inserted into hexa-arginine increase the short range repulsion while only slightly affecting longer range attraction. The amino acid content alone of salmon protamine is enough to rationalize the forces that package DNA in sperm heads. Inserting a negatively charged amino acid into hexa-arginine dramatically weakens the net attraction. Both of these observations have biological implications for protamine-DNA packaging in sperm heads.
Introduction
In cells, DNA primarily exists in a highly compact state despite its high charge density. Understanding the physical basis of DNA packaging is a necessary first step toward elucidating how nature both generates and employs condensates to store and protect genetic information in vivo. During vertebrate spermatogenesis, chromatin is dramatically reorganized in developing spermatids via histone replacement with protamines to achieve an even more compacted state for efficient genetic delivery and DNA protection (1–4). Dense packaging of DNA in sperm nuclei is considered important to protect the DNA against damage by mutagens or oxidative species (1, 5–7). Protamines are small, arginine-rich, nuclear proteins that condense the spermatid genome into a genetically inactive state. Salmon protamine is a 32-amino acid peptide with 21 arginines and 11 neutral amino acids (PR4S3RPVR5PRVSR6G2R4). Unlike histone-compacted DNA, the physical properties of reconstituted protamine-DNA assemblies closely resemble those of DNA condensed by multivalent cations that are much smaller, have much less charge, and have been well characterized such as Mn2+, Co(NH3)63+, spermidine, spermine, and several arginine oligopeptides (8–17). DNA is packaged by protamine to densities within the range seen for DNA condensed with the smaller multivalent ions (12, 14–16, 18–21). Under conditions of low DNA concentration, protamine will condense DNA into toroids that are on the same order of size as DNA condensed with the small multivalent cations (1, 8, 22–25). Most importantly, the forces measured for protamine-DNA arrays by the osmotic stress technique show very similar characteristics to DNA precipitated by multivalent ions (15, 26). The forces underlying DNA compaction by protamines can be investigated using the same techniques as used for other, simpler condensing ions. We can begin to understand the dependence of DNA packing density in sperm nuclei on specific protamine properties.
The physical origins of the forces acting to compact multivalent ion-DNA assemblies are still debated. Experimental studies (8–15, 26, 27) have aimed to elucidate the fundamental physical mechanisms responsible for DNA condensation. In vitro experiments have shown that DNA condensation from bulk solution critically depends on the valence of the counterions and that typically a net charge of +3 or larger in water is typically required to overcome the inherently large repulsion between the like-charged polyelectrolytes, although some +2 metal ions will condense DNA. Upon condensation, the resulting compacted structures have well defined equilibrium surface separations of the DNA double helices of 7–15 Å, depending on the identity of the condensing ion. The finite separation of helices indicates a delicate balancing of a short range repulsive force with a longer range attraction (15, 26, 27).
Using combined osmotic stress and single molecule tweezing experiments (27), we have previously characterized the distance dependence of the two component forces. The attractive force varies with DNA-DNA spacing as a 4–5-Å decay length exponential, whereas the repulsive force is a 2–2.5-Å decay length exponential. Using this constraint, we can effectively separate the attractive and repulsive contributions to the force-distance curves. The amplitudes of the two forces were extracted from the force curves for a set of homologous arginine peptides, R1 through R6 and polyarginine (26). The magnitude of the attractive force depends sensitively on the length of the arginine peptide. The repulsive force amplitude, in contrast, is nearly independent of arginine peptide length. The equilibrium spacing between DNA helices in reconstituted +21 charged salmon protamine-DNA arrays is much larger than expected from the arginine peptide results. This is surprising because it is generally considered that cation charge is the dominating determinant of DNA compaction. The separation of the forces for salmon protamine-DNA assemblies (26) indicated that the attractive force amplitude was very close to that expected for 21 arginines but that the repulsive force had a significantly larger amplitude than observed for the arginine peptide series. This additional repulsive force therefore is the primary reason for the observed lower packaging efficiency of protamine compared with that expected from the arginine peptide series. Salmon protamine is long enough that the extra repulsion could be due to defects in its binding along the DNA helix. To determine whether this extra repulsion is a general property of including neutral amino acids in arginine-based peptides or whether it is something peculiar to protamine, we here investigate the effect on forces of inserting neutral amino acids into model hexa-arginine (R6) peptides.
We report that neutral amino acids inserted into R6 significantly increase the amplitude of the short range repulsive force. We observe a smaller decrease in the amplitude of the long range attraction. The effect on force amplitudes of incorporating increasing numbers of alanines into the middle of R6 is additive at least through four. There is relatively little difference in inserting alanine, serine, proline, or isoleucine. Indeed, the increase in the short repulsive force for salmon protamine condensed DNA is in reasonable quantitative agreement with its neutral amino acid content. Mammalian protamines typically have a significantly higher fraction of neutral amino acids than salmon protamine, which would suggest less attraction and looser DNA packing and perhaps greater accessibility of mutagens and oxidizing species to DNA. This may be the reason why mammals reinforce tight packaging of DNA by forming protamine-protamine disulfide bridges that are absent in fish (1, 3).
We also investigated the effect on forces of incorporating a single negative charge, glutamate or phosphorylated serine, into R6. Protamine-DNA net attraction is substantially weakened, much more than simply reducing the net charge by one. Indeed, the equilibrium spacing is significantly greater than that seen with R3. The short range repulsion is dramatically increased, and the longer range attraction moderately decreased. This has biological implications for the initial in vivo binding of phosphorylated protamine to DNA. DNA tight packaging is hypothesized to be correlated with the removal of the phosphates from protamine (1, 3, 5, 6).
EXPERIMENTAL PROCEDURES
Materials
All of the arginine peptides were custom synthesized and purified (>98%) by GenScript Corporation. The peptides were neutralized with Tris base and used without further purification. Bioultra grade PEG, average molecular weight of 8,000, was purchased from Fluka Chemical Company. All chemicals were used without further purification. High molecular weight DNA (molecular weight > 5 × 106) was prepared and purified from adult chicken whole blood as described previously (15) and dialyzed against 10 mm Tris-Cl (pH 7.5), 1 mm EDTA. Salmon sperm nuclei were purchased from Sigma, rehydrated in buffer, and used without further purification.
Osmotic Stress
The method for direct force measurements by osmotic stress has been previously described in detail (15, 28). In brief, condensed macromolecular arrays, such as DNA, are equilibrated against a bathing polymer solution of known osmotic pressure. The polymer, typically PEG, is too large to enter the condensed DNA phase, thus applying a direct osmotic pressure on the condensate. Water, salt, and other small solutes are free to exchange between the PEG and condensed DNA phases. After equilibration, the osmotic pressures in both phases are the same. The interhelical spacing, Dint, can be determined from Bragg scattering of x-rays as a function of the applied PEG osmotic pressure.
Arginine peptide-compacted DNA samples used osmotic stress and for x-ray scattering were prepared in two ways. The arginine-based peptides were titrated into 200 μl of 1 mg/ml chicken erythrocyte DNA (2 mm bp) in 10 mm Tris-Cl (pH 7.5) until some precipitation was noted. The samples were thoroughly mixed before slowly adding more arginine peptide. Precipitation occurred when the arginine and DNA phosphate concentrations were about equal. Alternatively, 1 ml of arginine peptide solution in 10 mm Tris (pH 7.5) was added to DNA pellets (∼200 μg) obtained from ethanol precipitation from 0.3 m sodium acetate. The monomer arginine concentration of the arginine peptide was ∼1 mm. There was a slight expansion of the DNA pellet as the arginine peptides bind to DNA before recompacting. The condensed samples were centrifuged, and the DNA pellets transferred to PEG solutions (1–1.5 ml) containing at least 100 μm arginine peptide and 10 mm Tris (pH 7.5) in screw cap microtubes. Incubating with 50 or 200 μm arginine peptide did not affect interhelical spacings. The samples were equilibrated for at least several days with occasional vigorous mixing before transferring to fresh solutions. The samples were considered equilibrated after 1–2 weeks of incubation. No change in the x-ray scattering pattern is observed after 6 months of storage. X-ray scattering profiles did not depend on the method used to prepare the initial DNA pellet or the pellet size. PEG osmotic pressures were measured directly using a Wescor Vapro Vapor Pressure Osmometer (model 5520).
X-ray Scattering
Nickel-filtered Cu-Kα radiation from an UltraBright microfocus x-ray source from Oxford Instruments equipped with polycapillary focusing x-ray optics was used for the small angle x-ray scattering experiments. The primary beam was also collimated by a set of slits. The samples were sealed with a bath of equilibrating solution in the sample cell and then mounted into a temperature-controlled holder at 20 °C (29). The flight path between the sample and detector was helium-filled. Typical exposure times were ∼45 min. Diffraction patterns were recorded by direct exposure of Fujifilm BAS image plates and digitized with a Fujifilm FLA 3000 scanner. The images were analyzed using FIT2D (AP Hammersley, ESRF) and SigmaPlot 10.01 (SPSS) software programs. The sample to image plate distance was calibrated using silver behenate and found to be ∼16.7 cm. Mean pixel intensities between scattering radii r − 0.05 mm and r + 0.05 mm averaged over all angles of the powder pattern diffraction, <I(r)>, were used to calculate integrated radial intensity profiles, 2πr<I(r)>. The strong scattering peaks correspond to interaxial Bragg diffraction from DNA helices packed in a hexagonal array. The Bragg spacing, DBr, and the actual distance between helices, Dint, are related by Dint = 2DBr/. For different samples equilibrated at the same PEG concentration, interaxial spacings are reproducible to within ∼0.1 Å.
Force Analysis
Many polyvalent counterions, including all the R6 derivatives discussed in this work, cause DNA to spontaneously condense in vitro, resulting in a finite equilibrium separation between the hexagonally packed DNA helices. Thermodynamic forces between polycation condensed DNA helices can be investigated by the osmotic stress technique. Previous results indicated that DNA-DNA forces can be described by two exponentials at close interaxial spacings, the last 20 Å of surface-to-surface separation (15, 26, 27). We fit the osmotic pressure Π versus spacing D curves to a double exponential equation with variable pre-exponential factors A and R:
or equivalently
with the decay length λ fixed at 4.8 Å. This form and decay length constraint is the result of experiments combining osmotic stress measurements with single molecule, magnetic tweezer experiments to separate the attractive, and repulsive free energies at the equilibrium spacing for several commonly used condensing agents (27). Equation 2 with λ = 4.8 Å gives very good fits for the arginine peptide-DNA complexes previously examined (26). The results are only slightly dependent on the decay length λ over the range of approximately ± 0.3 Å. For cations that spontaneously assemble DNA, the coefficients R and A are connected through the equilibrium interaxial spacing Deq because Π (Deq) = 0, giving a fitting equation with only a single variable R.
The repulsive and attractive free energy contributions from the two individual forces per DNA base pair at a spacing D can be calculated by integrating Π dV for each exponential from ∞ to D assuming hexagonal packing,
and
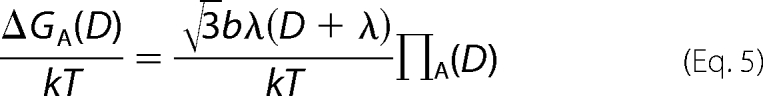 |
where b is the linear spacing between DNA base pairs, 3.4 Å.
RESULTS
Fig. 1 shows osmotic stress force curves, the log of the osmotic pressure of the excluded PEG shown as dependent on the interhelical spacing, for DNA condensed by R6, R3, salmon protamine, and R3A3R3. The arrows show the interaxial spacing in the absence of applied osmotic pressure. The solid lines are fits of the data to Equation 3 that separates the net force into its repulsive and attractive components. As we reported previously (26), the high pressure data for R6 and R3 converge even though the equilibrium spacings are very different. The primary effect of increasing the number of arginines in the RN series is on the magnitude of the 4.8 Å decay length attraction. The 2.4 Å decay length repulsion that is dominant at the high osmotic pressures is only slightly dependent on the number of arginines. DNA condensed with salmon protamine shows a significantly greater repulsive force at high osmotic pressures than R6 and R3. The separation of forces indicated that the long range attraction for salmon protamine-DNA arrays is consistent with its 21 arginines but that the short range repulsion is significantly larger than for the homo-arginine RN series (26). The high osmotic pressure amplitude for DNA condensed with the synthetic peptide that has neutral amino acids inserted, R3A3R3, is quite close to the salmon protamine data, indicating that the 2.4 Å decay length repulsions are more similar than for R6 or R3 condensed DNA. The large difference in the equilibrium spacings between protamine and R3A3R3 would indicate, however, that the long range attractions for the two are quite different.
FIGURE 1.

Osmotic stress force curves are shown for DNA condensed by R6, R3 salmon protamine, and R3A3R3. Π is the osmotic pressure of PEG that is excluded from the DNA phase. DNA interaxial spacings are measured by x-ray scattering. The arrows indicate the equilibrium spacing in the absence of PEG. The solid lines are fits of the data to the double exponential function given by Equation 3, separating the overall force into its repulsive and attraction components. Note the convergence of the R6 and R3 data at high osmotic pressures indicating a similar 2.4 Å decay length repulsion. The different equilibrium spacings for R6 and R3 indicate different attractive force amplitudes. Similarly the high osmotic pressure data for salmon protamine and R3A3R3 are similar, indicating similar short range repulsions. Again the difference in equilibrium spacing for these two indicates different attractive force amplitudes. In addition to varied concentrations of PEG, the bathing solutions in equilibrium with the DNA phase contained 10 mm Tris-Cl (pH 7.5) and 100 μm of peptide or salmon protamine.
Fig. 2 shows the osmotic stress force curves for a set of R6 peptides with alanines inserted in the middle, R3ANR3, where N varies from 0 to 4. The solid lines are the fits to Equation 3. Even though all of the peptides have a charge of +6, the equilibrium spacing between helices increases by ∼0.7 Å/alanine, indicating that either the amplitude of the 2.4 Å decay length force (R) increases or the amplitude of the 4.8 Å decay length attraction (A) decreases (or both) in Equation 1. The high osmotic pressure data indicate that the repulsive force increases with the increasing number of alanines.
FIGURE 2.

Osmotic stress force curves are shown for DNA condensed by the peptide series R3ANR3. The arrows indicate the equilibrium spacing in the absence of applied PEG osmotic pressure. The solid lines are fits of the data to Equation 3. Note the progressive increase in equilibrium spacing as the number of alanines is increased. Note also the increase in the amplitude at high pressures with an increasing number of alanines, indicating the short range repulsive force is increasing. In addition to varied concentrations of PEG, the bathing solutions in equilibrium with the DNA phase contained 10 mm Tris-Cl (pH 7.5) and 100 μm of peptide.
Fig. 3A shows the dependence of the fitted values for the osmotic pressure components of the two forces at 25 Å, ΠR(25 Å) and ΠA(25 Å), on the number of inserted alanines, N. Within experimental error, both ΠA and ΠR vary linearly with N. The 2.4 Å decay length repulsion increases and the 4.8 Å decay length attraction decreases as the number of alanines increases. The effect of the inserted alanines is primarily on the short range repulsion. The value of ΠR increases by ∼50% between n = 0 and 4, whereas ΠA decreases by only ∼18% over the same range. The slope dΠR/dN is +2.45 × 107 erg/ala-cm3, but only −4.4 × 106 erg/ala-cm3 for dΠA/dN. Fig. 3B shows essentially the same data, but now as free energy contributions from attraction and repulsion at 25 Å in units of kT/base pair.
FIGURE 3.

The intermolecular forces for DNA condensed by the series R3ANR3 are separated into their repulsive and attractive components. In A, the osmotic pressure contributions from the 2.4 Å decay length repulsion, ΠR, at 25 Å and from the 4.8 Å decay length attraction, ΠA, at 25 Å are shown as dependent on the number of alanines inserted. In B, the dependence of the free energy contributions, ΔGR and ΔGA, at 25 Å, calculated from Equations 4 and 5, on the number of alanines is shown.
Fig. 4 shows a comparison of osmotic stress curves for DNA condensates with different placements of the A3 insertion: R3A3R3, A3R6, and R6A3. DNA force curves with R3A2R3 and R3A4R3 are shown for comparison. Equilibrium interaxial spacings at Π = 0 that represent the balance between the attractive and repulsive interhelical forces are indicated by arrows in the figure. The R6A3-DNA data shows a transition to smaller spacings at low osmotic pressures. The solid lines are fits of Equation 3 to the R3A3R3, A3R6, and R6A3 data. The asterisk shows the best fitting post-transition equilibrium spacing for the R6A3 data. Although the actual, pre-transition equilibrium spacing at Π = 0 for R6A3 is larger than for R3A3R3, the apparent post-transition spacing is smaller. The high osmotic pressure data for A3R6 and R6A3 more closely resemble that for R3A2R3 than R3A3R3. The osmotic pressure contributions at 25 Å ΠR(25 Å) and ΠA(25 Å) and the free energy contributions, ΔGR(25 Å) and ΔGA(25 Å), for these peptides are given in Table 1. Although placement of the A3 insertion does make some difference, it is less than the difference between R3A2R3 and R3A4R3.
FIGURE 4.

Osmotic stress force curves are shown for DNA condensed by A3R6, R3A3R3, and R6A3. Force curves for DNA condensed by R3A2R3 and R3A4R3 are also shown for comparison. The arrows indicate equilibrium spacings in the absence of applied osmotic pressure for A3R6, R3A3R3, and R6A3 condensed DNA. The solid lines are the best fits to Equation 3. Placement of A3 makes little difference to the observed forces. The data with R6A3 show a transition at low pressures. The asterisk indicates the best fitting equilibrium spacing after the transition. Osmotic pressure and free energy contributions from the repulsive and attractive force components calculated from the fitting parameters are given in Table 1. The DNA pellet bathing solutions contained various concentrations of PEG, 10 mm Tris-Cl (pH 7.5), and 100 μm peptide.
TABLE 1.
Contributions from the attractive and repulsive force components to DNA condensation
The equilibrium interhelical spacings (± 0.1 Å) from direct x-ray measurements and repulsive and attractive force component contributions to osmotic pressures (± 5%) and free energies (± 5%) at 25 Å calculated from fits to force curves are shown for DNA condensed by various hexa-arginine peptides.
| Peptide | Deq | ΠR(25 Å)/108 erg/cm3 | −ΠA(25 Å)/108 erg/cm3 | ΔGR(25 Å)/kT/base pair | −ΔGA(25 Å)/kT/base pair |
|---|---|---|---|---|---|
| Å | |||||
| R6 | 28.6 | 2.03 | 0.965 | 1.94 | 2.01 |
| R3 | 30.15 | 1.68 | 0.54 | 1.61 | 1.12 |
| Salmon protamine | 29.26 | 3.02 | 1.33 | 2.89 | 2.35 |
| R3A3R3 | 30.7 | 2.67 | 0.815 | 2.56 | 1.70 |
| A3R6 | 30.0 | 2.535 | 0.89 | 2.43 | 1.85 |
| R6A3 | 30.4 | 2.58 | 0.83 | 2.47 | 1.73 |
| R3AR3 | 29.25 | 2.20 | 0.905 | 2.20 | 1.89 |
| R3SR3 | 29.05 | 2.33 | 1.00 | 2.23 | 2.08 |
| R3PR3 | 29.45 | 2.48 | 0.98 | 2.38 | 2.04 |
| R3IR3 | 29.4 | 2.28 | 0.915 | 2.18 | 1.91 |
| R3ER3 | 31.5 | 2.825 | 0.73 | 2.70 | 1.52 |
| R3S(PO4−)R3 | 33.25 | 4.455 | 0.80 | 4.26 | 1.77 |
Fig. 5 shows the effect of varying the nature of the neutral amino acid incorporated in R3XR3 peptides on osmotic stress force curves. The inserted amino acids examined span a range of physical and chemical properties: alanine, hydrophilic serine, hydrophobic isoleucine, and bent proline. Although there are differences among the amino acids, all the curves fall between R6 and R3A2R3. The fitted values for the osmotic pressure contributions at 25 Å, ΠR(25 Å) and ΠA(25 Å), and the free energy contributions, ΔGR(25 Å) and ΔGA(25 Å), of the two exponential components are given in Table 1 for these peptides. Again, the short range repulsion is affected significantly more than the long range attraction. The identity of the inserted neutral amino acid affects forces only slightly.
FIGURE 5.

Osmotic stress forces curves are shown for DNA condensed by R3AR3, R3SR3, R3IR3, and R3PR3. Force curves are also shown for DNA arrays with R6 and R3A2R3 for comparison. The arrows indicate equilibrium spacings in the absence of applied osmotic pressure. The solid lines are fits of the data to Equation 3. The identity of the neutral amino acid makes little difference to measured forces. Osmotic pressure and free energy contributions from the repulsive and attractive components of the force calculated from the fitting parameters are given in Table 1. The DNA pellet bathing solutions contained various concentrations of PEG, 10 mm Tris-Cl (pH 7.5), and 100 μm peptide.
Inserting a negative charge into R6 has a profound effect on forces. Fig. 6 shows osmotic stress force curves for DNA condensed with glutamic acid peptide R3ER3 and with the phosphorylated serine peptide R3(S-PO4−)R3. The net attraction is much weaker than seen for R3-DNA even though the two peptides have a net charge of +5. Two exponentials with 2.4 and 4.8 Å decay lengths still fit the data relatively well, although the fit to the phosphorylated serine peptide is somewhat worse than for the others shown here and in previous work (15, 26). The magnitude of the attractive force for these two peptides is decreased significantly more than for the inserted neutral amino acids, but the increase in the magnitude of the repulsive force is comparatively even greater. Tabulated values for ΠR, ΠA, ΔGR, and ΔGA at 25 Å are given in Table 1.
FIGURE 6.

Osmotic stress force curves are shown for DNA condensed by R3ER3 and R3S(PO4−)R3. For comparison, force curves for DNA assemblies with R6, R3SR3, and R3 are shown. The arrows indicate equilibrium spacings in the absence of applied osmotic pressure. The solid lines are the best fits of the data to Equation 3. Inserting one negative charge into R6 greatly reduces the net attraction. Osmotic pressure and free energy contribution from the repulsive and attraction force components are given in Table 1. The DNA pellet bathing solutions contained various concentrations of PEG, 10 mm Tris-Cl (pH 7.5), and 100 μm peptide.
DISCUSSION
X-ray scattering and cryo-electron microscopy indicate that the vast majority of DNA in sperm nuclei is packed in hexagonal arrays with a spacing between nearest neighbor helices of ∼30 Å (18, 21). This is closely similar to the arrangement of DNA condensed by much simpler multivalent ions in vitro (8, 10, 14, 15, 22, 25, 26). The specific placement of protamine within the DNA arrays has led to several models (1, 30–33). Most experimental data, however, are consistent with protamine binding in the grooves of DNA (31, 34). We have previously shown that the same double exponential fit can be used to describe the force curves for DNA condensed by a wide variety of cations, including protamine (15, 26, 27). The similarity of force curves indicates an underlying universality of interaction. This commonality is further amplified in Fig. 7 that directly compares osmotic stress forces curves for reconstituted salmon protamine-DNA assemblies and for intact salmon nuclei. The striking similarities of the curves suggest that in vivo packaging of DNA in sperm nuclei is governed by the same fundamental physical principles that underlie in vitro DNA condensation and that measurement of forces in reconstituted assemblies is fully relevant to understanding DNA compaction in sperm.
FIGURE 7.

Osmotic stress forces curves for reconstituted salmon-protamine DNA and for isolated salmon sperm nuclei are compared. The similarities in force curves between reconstituted samples and in vivo salmon nuclei packaging are striking. The equilibrium spacings at zero applied pressure (indicated by the arrow) are the same within experimental error. Subtle differences are observed at high pressure that may indicate more uniform binding of protamine to DNA in sperm nuclei. The DNA pellet bathing solutions contained various concentrations of PEG, 10 mm Tris-Cl (pH 7.5), and 100 μm peptide or protamine.
The distance between neighboring helices in these condensed arrays is determined by the balance between short range repulsion and a longer range attractive force. Our previous work with arginine homopeptides indicated that the attractive force increases with the number of arginines, whereas the repulsion is very weakly dependent on peptide length. As a consequence, the interaxial spacing between helices decreases as the number of arginines in the homopeptide increases. The interaxial spacing, or equivalently packaging efficiency, for DNA condensed by salmon protamine with 21 arginines, however, was only about the same as penta-arginine. Separation of the force components for salmon protamine-DNA condensed arrays indicated that the attractive force was consistent with the 21 arginines present but that the repulsion was much greater than for the arginine homopeptides. The results here indicate that the neutral amino acids found in salmon protamine are responsible for this increased repulsion. Non-arginine amino acids incorporated into R6 significantly affect the condensation forces (Figs. 2 and 3). Inserting non-arginine amino acids into R6 simultaneously increases the repulsive force and decreases the attractive force. The increase in amplitude of the short range repulsive force is significantly larger than the decrease in the attractive force amplitude. The increase in the osmotic pressure contribution from the short range repulsion, ΠR, with an increasing number N of alanines, dΠR/dN (Fig. 3B), is ∼5–6-fold greater than the decrease with N in ΠA. The incorporation of even four alanines into R6 only lowers the attractive force to a value comparable with R5. The contribution of the repulsive force, however, results in an equilibrium interaxial spacing for R3A4R3 that is significantly larger than for R3. The amplitude of the short range repulsive force for DNA condensed by salmon protamine is between that for DNA condensed by R3A3R3 and R3A4R3. The fraction neutral amino acids are quite similar for salmon protamine (34%) and the two peptides. This would strongly suggest that neutral amino acid content alone is sufficient to explain the increased short range repulsion of salmon protamine.
There are several differences between mammalian and piscine protamines. Most but not all mammals have two protamines, P1 and P2, which are both longer than piscine protamines. The average fraction arginine of mammalian protamines, 50–60%, is significantly smaller than for fish, 65–70% (1, 3). On the basis of the results here, the increase in neutral amino acid content would be expected to further increase the repulsive force, increasing the equilibrium spacing between helices in mammalian sperm. Extrapolating the results of Figs. 1 and 2, we would predict that the equilibrium interhelical spacing of R3A6R3 condensed DNA (50% arginine) would be 33 Å and that R3A7R3 (46% arginine) would not be able to condense DNA without added osmotic pressure. DNA damage caused by mutagens and reactive oxidizing species is thought related to DNA mispackaging (1, 5–7, 35). A looser packaging of DNA caused by an increased fraction of neutral amino acids would increase the accessibility of small molecules to the DNA. This might explain another difference between mammalian and piscine protamine-DNA packaging. Disulfide bridges between protamines in mammalian sperm are required for tight packaging. Reduction of the disulfide bridges is required for in vitro decondensation of mammalian sperm nuclei (21, 36). Many piscine protamines, on the other hand, do not have cysteines.
Perhaps not surprisingly, the incorporation of a negatively charged amino acid into R6 has a large effect on forces. Again, however, the effect is mainly to increase the amplitude of the short range repulsive force dramatically, a 2-fold larger ΠR(25 Å) for R3(S-PO4−)R3-DNA compared with R3AR3-DNA. The long range attractive force is changed to a much lesser extent, only a 12% decrease in ΠA(25 Å). The amplitudes of the attractive force for R3ER3-DNA and R3(S-PO4−)R3-DNA with net charges of +5 are actually very close to that measured for R5-DNA. The large increase in the repulsive force, however, causes the equilibrium interhelical spacings for both R3ER3 and R3(S-PO4−)R3 condensed DNA to be much greater than for R3. The replacement of histones by protamines occurs in several steps (1, 5). First, acetylated histones are replaced by transition proteins. Second, serine-phosphorylated protamines then replace the transition proteins. It is only after removing the phosphate groups that DNA is tightly packed. Incomplete dephosphorylation has been suggested to be a cause of increased DNA damage and male infertility (5). The results here show that phosphorylation has a much larger effect on weakening the net attraction between helices than simply reducing the protamine charge would suggest.
Understanding the cause of the much larger changes in the amplitude of the repulsive force compared with attraction caused by inclusion of non-arginine amino acids is problematic. Our previous work on condensed DNA (15, 26) showed that their osmotic stress force curves can be well described by the double exponential fit of Equation 3 with a 2.4 Å decay length short range repulsion and a 4.8 Å decay length attraction. Both of these forces can be rationalized within either an electrostatic or hydration force framework. Within an electrostatic framework, the longer decay length force is due to the direct interaction of charges on apposing helixes, which can be either attractive or repulsive depending on the number of bound charges and the interhelical correlation of charges, whereas the shorter λ/2 decay length force in an image charge repulsion is due to the interaction of charges on one DNA helix with the low dielectric DNA core of another. Equivalently, from a hydration force perspective, the longer decay length force is due to direct interaction of surface hydration structures on apposing helices (37–39). This force can be either attractive or repulsive depending on the correlation of complementary water structures. The short range repulsive force is the hydration equivalent of the image charge repulsion. The hydration atmosphere extending out from one helix and stabilizing the surface hydration structure is disrupted by the presence of a second DNA helix. The decay length of this image charge-like force is necessarily half that of the direct force (15). We have emphasized hydration interactions because we have seen similar forces between uncharged macromolecules, between charged salts and uncharged macromolecules, and between neutral solutes and uncharged or charged macromolecular surfaces where the only commonality is the presence of water in all of these systems (37, 38).
The theoretical framework of Korneyshev et al. (40) and Korneyshev and Leikin (41) provides an attractive model for discussing correlation and attraction. The binding of cationic charges in the major, in particular, or the minor grooves can naturally lead to attractive interhelical correlations between the bound positive charges and the phosphate backbone. Based on fiber diffraction (42) and Raman spectroscopy (31), a model of the complex of polyarginine with DNA has been developed. The peptides seem to form ordered structures, perhaps an extended helix, in the major groove of DNA, with ∼1 arginine/phosphate. Protamines seem to bind in a somewhat similar way. There are some differences, however, in Raman peaks and x-ray scattering intensities between polyarginine- and protamine-bound DNA (31, 42). All of the DNA charges seem to be neutralized by arginines in protamine-DNA arrays despite the additional neutral amino acids. Observed force magnitudes of protamine-DNA arrays are not sensitive to the presence of tetramethyl ammonium+ or Mg2+ that would have given very different force magnitudes if bound as a neutralizing ion.2 To achieve charge neutralization with oligoarginine peptides that have incorporated non-arginine amino acids, either the neutral amino acids must loop out if binding is restricted to the major groove or additional peptides bind elsewhere, e.g. the minor groove. We see no indication that an additional steric repulsion of looped out neutral amino acids modifies the forces seen at high pressures. This would be particularly evident for R6A4R6 that would project out from the DNA by ∼5 Å if DNA was neutralized by arginines binding in the major groove only. The small loss of attraction with included neutral amino acids could be due to minor groove binding of the additional peptide needed to neutralize DNA. Minor groove binding is expected to result in less attraction than binding in the major groove within the Korneyshev-Leikin formalism. Fluctuations in the positioning of the bound neutral amino acids inserted into the middle of R6 may also perturb the correlation of charges on apposing helices. The amplitudes of the attractive force are somewhat greater for A3R6 and R6A3 than for R3A3R3, suggesting that the contiguous six arginines may be better correlated, but the effect is small. There is a low osmotic pressure transition observed for R6A3. This peptide has a C-terminal, negatively charged carboxylate that is well separated from the positively charged arginines. An osmotic pressure-dependent change in the DNA binding conformation of the C-terminal end of the peptide may account for the observed transition. This low osmotic pressure transition is not observed with DNA condensed with A3R6.
Filling the minor groove with peptide to neutralize DNA charge would also increase the short range repulsive image force both for electrostatic and hydration interactions simply because water is displaced. This indirect effect of neutral amino acids would contrast with a direct interaction of these amino acids with neighboring DNA helices. In addition to alanine, we examined hydrophobic, hydrophilic, and rigidly bent amino acids incorporated into R6 (Fig. 4). Although there is some variation in force amplitudes with the identity of the neutral amino acid, the variation observed is less than incorporating a second alanine. This is consistent with a more indirect effect of these amino acids on forces. The individual changes in the force amplitudes are generally close to the limits of our fitting errors. There is no additional attraction because of hydrophobic interactions of correlated isoleucines on opposing helices. In salmon protamine, 3 of the 32 amino acids are prolines that may help protamines bend around DNA in the major or minor groove. There is no strong effect on forces, however, resulting from the incorporation of proline in R6.
Because most DNA repair mechanisms are turned off within sperm cells, proper packaging is likely critical for maintaining DNA integrity. Currently experiments are underway to investigate differences in packaging between piscine and mammalian sperm nuclei as well as probing interconnections between protamine phosphorylation and both male infertility and birth defects because of DNA mutations caused by mispackaging.
Footnotes
This work was supported, in whole or in part, by National Institutes of Health Eunice Kennedy Shriver Intramural Research Program.
J. E. DeRouchey and D. C. Rau, unpublished observation.
REFERENCES
- 1. Balhorn R. (2007) Genome Biol. 8, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Green G. R., Balhorn R., Poccia D. L., Hecht N. B. (1994) Mol. Reprod. Dev. 37, 255–263 [DOI] [PubMed] [Google Scholar]
- 3. Lewis J. D., Song Y., de Jong M. E., Bagha S. M., Ausió J. (2003) Chromosoma 111, 473–482 [DOI] [PubMed] [Google Scholar]
- 4. Braun R. E. (2001) Nat. Genet. 28, 10–12 [DOI] [PubMed] [Google Scholar]
- 5. Oliva R. (2006) Hum. Reprod. Update 12, 417–435 [DOI] [PubMed] [Google Scholar]
- 6. Andrabi S. M. (2007) J. Assist. Reprod. Genet. 24, 561–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Muratori M., Marchiani S., Maggi M., Forti G., Baldi E. (2006) Front. Biosci. 11, 1491–1499 [DOI] [PubMed] [Google Scholar]
- 8. Bloomfield V. A. (1996) Curr. Opin. Struct. Biol. 6, 334–341 [DOI] [PubMed] [Google Scholar]
- 9. Bloomfield V. A. (1997) Biopolymers 44, 269–282 [DOI] [PubMed] [Google Scholar]
- 10. DeRouchey J., Netz R. R., Rädler J. O. (2005) Eur. Phys. J. E 16, 17–28 [DOI] [PubMed] [Google Scholar]
- 11. Pelta J., Livolant F., Sikorav J. L. (1996) J. Biol. Chem. 271, 5656–5662 [DOI] [PubMed] [Google Scholar]
- 12. Qiu X., Andresen K., Lamb J. S., Kwok L. W., Pollack L. (2008) Phys. Rev. Lett. 101, 228101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Raspaud E., Olverade la Cruz M., Sikorav J. L., Livolant F. (1998) Biophys. J. 74, 381–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Raspaud E., Durand D., Livolant F. (2005) Biophys. J. 88, 392–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rau D. C., Parsegian V. A. (1992) Biophys. J. 61, 246–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schellman J. A., Parthasarathy N. (1984) J. Mol. Biol. 175, 313–329 [DOI] [PubMed] [Google Scholar]
- 17. Widom J., Baldwin R. L. (1983) Biopolymers 22, 1595–1620 [DOI] [PubMed] [Google Scholar]
- 18. Feughelman M., Langridge R., Seeds W. E., Stokes A. R., Wilson H. R., Hooper C. W., Wilkins M. H., Barclay R. K., Hamilton L. D. (1955) Nature 175, 834–838 [PubMed] [Google Scholar]
- 19. Suau P., Subirana J. A. (1977) J. Mol. Biol. 117, 909–926 [DOI] [PubMed] [Google Scholar]
- 20. Suwalsky M., Traub W. (1972) Biopolymers 11, 2223–2231 [DOI] [PubMed] [Google Scholar]
- 21. Blanc N. S., Senn A., Leforestier A., Livolant F., Dubochet J. (2001) J. Struct. Biol. 134, 76–81 [DOI] [PubMed] [Google Scholar]
- 22. Brewer L. R. (2011) Integr. Biol. (Camb.) 3, 540–547 [DOI] [PubMed] [Google Scholar]
- 23. Balhorn R., Brewer L., Corzett M. (2000) Mol. Reprod. Dev. 56, 230–234 [DOI] [PubMed] [Google Scholar]
- 24. Conwell C. C., Vilfan I. D., Hud N. V. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 9296–9301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hud N. V., Downing K. H. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 14925–14930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. DeRouchey J., Parsegian V. A., Rau D. C. (2010) Biophys. J. 99, 2608–2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Todd B. A., Parsegian V. A., Shirahata A., Thomas T. J., Rau D. C. (2008) Biophys. J. 94, 4775–4782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Parsegian V. A., Rand R. P., Fuller N. L., Rau D. C. (1986) Methods Enzymol. 127, 400–416 [DOI] [PubMed] [Google Scholar]
- 29. Mudd C. P., Tipton H., Parsegian A. V., Rau D. (1987) Rev. Sci. Instrum. 58, 2110–2114 [Google Scholar]
- 30. Raukas E., Mikelsaar R. H. (1999) Bioessays 21, 440–448 [DOI] [PubMed] [Google Scholar]
- 31. Hud N. V., Milanovich F. P., Balhorn R. (1994) Biochemistry 33, 7528–7535 [DOI] [PubMed] [Google Scholar]
- 32. Teif V. B. (2005) Biophys. J. 89, 2574–2587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vilfan I. D., Conwell C. C., Hud N. V. (2004) J. Biol. Chem. 279, 20088–20095 [DOI] [PubMed] [Google Scholar]
- 34. Prieto M. C., Maki A. H., Balhorn R. (1997) Biochemistry 36, 11944–11951 [DOI] [PubMed] [Google Scholar]
- 35. Castillo J., Simon L., de Mateo S., Lewis S., Oliva R. (2011) J. Androl. 32, 324–332 [DOI] [PubMed] [Google Scholar]
- 36. Perreault S. D., Wolff R. A., Zirkin B. R. (1984) Dev. Biol. 101, 160–167 [DOI] [PubMed] [Google Scholar]
- 37. Todd B. A., Sidorova N. Y., Rau D. C., Stanley C., Begley T. P. (2007) In Wiley Encyclopedia of Chemical Biology, John Wiley & Sons, Inc [Google Scholar]
- 38. Stanley C., Rau D. C. (2011) Curr. Opin. Colloid Interface Sci., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Leikin S., Parsegian V. A., Rau D. C., Rand R. P. (1993) Annu. Rev. Phys. Chem. 44, 369–395 [DOI] [PubMed] [Google Scholar]
- 40. Kornyshev A. A., Lee D. J., Leikin S., Wynveen A. (2007) Rev. Mod. Phys. 79, 943–996 [Google Scholar]
- 41. Kornyshev A. A., Leikin S. (1999) Phys. Rev. Lett. 82, 4138–4141 [Google Scholar]
- 42. Fita I., Campos J. L., Puigjaner L. C., Subirana J. A. (1983) J. Mol. Biol. 167, 157–177 [DOI] [PubMed] [Google Scholar]