

Abstract

We report a new method for the highly regio-, diastereo-, and enantioselective palladium-catalyzed allylic alkylation of 2-substituted pyridines that allows for the formation of homoallylic stereocenters containing alkyl, aryl, heteroaryl, and nitrogen substituents. When the reaction is conducted with asymmetric acyclic electrophiles, both linear and branched products may be obtained exclusively by selecting the appropriate regioisomeric starting material and ligand, an example of the “memory effect.” Deuterium-labeling studies reveal that though no such phenomenon occurs with racemic cyclic electrophiles, the chiral ligand employed reacts kinetically faster with the enantiomer of substrate for which it is “matched,” and yet eventually converts all “mismatched” substrate to product.
The development of palladium-catalyzed asymmetric allylic alkylations (AAAs) continues to be productive area of research.1 Still, there are only a few examples of such reactions that form an adjacent stereocenter diastereoselectively, and these are almost exclusively limited to enolate nucleophiles.2 This narrow substrate scope reflects the inherent challenge of simultaneously forming vicinal stereocenters selectively. After reporting a method for employing 2-methylpyridines in palladium-catalyzed AAAs,3 we wondered whether an analogous reaction with higher-order 2-substituted pyridines could be effected in a diastereo- and enantioselective fashion (Scheme 1, A). We hypothesized that, upon coordination of the pyridyl nitrogen atom with BF3, benzylic deprotonation would provide a nucleophile that would exist as a single geometric isomer because of the steric demands imposed by the Lewis acid. If such control were possible, alkylation products might be obtained with both high diastereo- and enantiocontrol.
Scheme 1.

Reaction hypothesis and initial result.
Unfortunately, when 2-ethylpyridine (1) was reacted with allylic carbonate 2 under the previously optimized conditions,3 1H NMR revealed no desired product and partial decomposition of 2 (Scheme 1, B). Replacing the tert-butyl carbonate ester with the more robust pivalate ester provided an electrophile that was completely stable to the reaction conditions, and with this substrate the desired alkylation product (3a) could be obtained in 15% yield, >19:1 dr, and 95% ee. Although the reaction conversion was low, the excellent diastero- and enantioselectivity validated our hypothesis. Disappointingly, higher yields could not be obtained by heating the reaction or by using other strong bases (e.g. LiTMP, t-BuOK, KHMDS, or n-BuLi).
The breakthrough came when a single equivalent of n-BuLi was added to the nucleophilic complex generated with three equivalents of LiHMDS; under these conditions, 3a was now isolated in 85% yield, >19:1 dr, and 94% ee. Presumably, the introduction of n-BuLi quantitatively deprotonates the equivalent of HMDS formed in the initial deprotonation event, driving the reaction to completion.
Other 2-substituted pyridines that underwent reaction with cyclohex-2-enyl pivalate were identified (Table 1). A range of 2-alkyl- and 2-aryl-substituted pyridines performed well, as did the corresponding five-membered ring electrophile (see 3b). Large substituents generally provided the highest level of stereocontrol; for example, 2-(naphthalene-2-ylmethyl)- pyridine gave 3g in 90% yield, >19:1 dr, and 98% ee.4 Heteroaryl substitution was also tolerated (3h), as was an aryl bromide (3i), which did not undergo oxidative addition by palladium(0) or lithium-halogen exchange with n-BuLi. Finally, a nitrogen-containing stereocenter could also be established (3j).5 The relative and absolute stereochemistry of the products was assigned by analogy to 3d, which was bromocyclized to provide single crystals of the corresponding pyridinium cation suitable for X-ray diffraction (Scheme 2).6
Table 1.
AAA Reactions with 2-Substituted Pyridyl Nucleophilesa
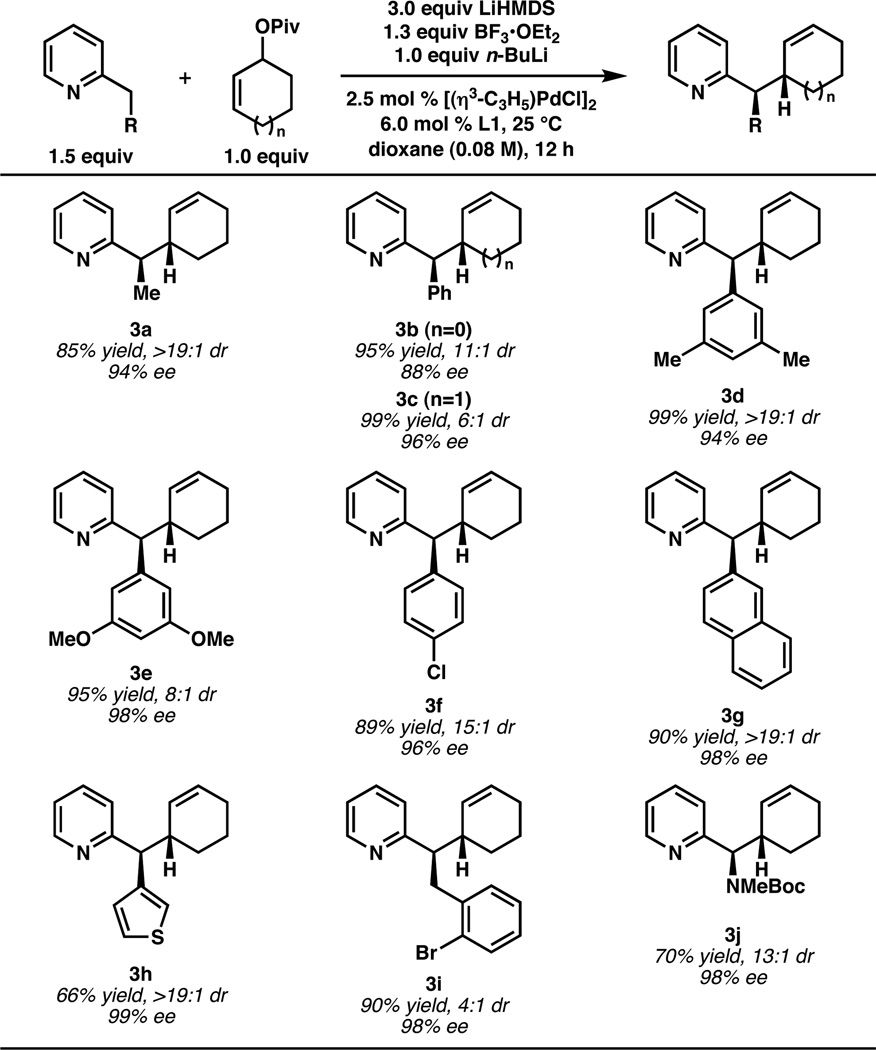 |
Yield reflects combined isolated yield of both diastereomers; dr and ee determined by 1H NMR analysis of the crude reaction mixture and chiral HPLC, respectively.
Scheme 2.

Bromocyclization of 3d.
The alkylation is sensitive to the steric nature of the nucleophile. When additional substituents were placed either at the benzylic (e.g. 2-isopropylpyridine) or homobenzylic (e.g. 2-neopentylpyridine) positions, no desired product was observed. Conversely, if the 2-pyridyl substituent is insufficiently sterically demanding for the two possible geometric configurations of the nucleophile to be adequately differentiated, the product is formed with low diastereocontrol (e.g. when 2-(methoxymethyl)pyridine was employed, the product was obtained in 78% yield but 3:2 dr).
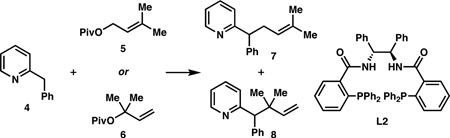
The reaction is not limited to cyclic electrophiles. Indeed, when 2-benzylpyridine (4) was reacted with pivalate 5, linear product 7 was obtained in 74% yield and 89% ee (Table 2, entry 1). Unexpectedly, when the reaction was instead conducted with regioisomeric pivalate 6, a mixture of linear and branched products (7 and 8) was formed (entry 2). This is an example of the “memory effect,” in which the regiochemistry of the electrophile influences the regiochemistry of the product.7 When the reaction was performed with 5 in benzene with L2, the linear product was again formed, but with little enantiocontrol (entry 3). However, with 6, the branched product could be obtained in 64% yield and 63% ee8 (entry 4). Thus either regioisomeric product may be obtained exclusively by choosing the appropriate allylic ester and ligand.
Table 2.
“Memory Effect” Observed with Linear Electrophilesa
 | ||||||
|---|---|---|---|---|---|---|
| entry | substrate | ligand | solvent | 7:8b | yield (%)c | ee (%)d |
| 1 | 5 | L1 | dioxane | >19:1 | 74 | 89 |
| 2 | 6 | L1 | dioxane | 1:1.6 | 71 | 7:64 |
| 8:17 | ||||||
| 3 | 5 | L2 | PhH | >19:1 | 32 | 3.6 |
| 4 | 6 | L2 | PhH | <1:19 | 64 | 63 |
Reactions run under the standard conditions.
Determined by 1H NMR of the crude reaction mixture.
Combined isolated yield of both regioisomers.
Determined by chiral HPLC.
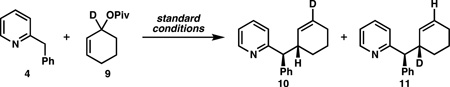
To assess whether such an effect was occurring with cyclic electrophiles, deuterated substrate 9 was prepared (Table 3). When the reaction was conducted with 4 and racemic ligand, regioisomer 11 was obtained with high selectivity (entry 1), while nonracemic ligands gave an equal mixture of 10 and 11 (entries 2 and 3). These results show: (1) no “memory effect” is operative, since no bias for nucleophilic attack is observed with the (S,S)- or (R,R)-ligand alone, (2) when placed in competition, each ligand is able to perform a near-perfect kinetic resolution of the electrophile, since each reacts fastest with the enantiomer of 9 for which it is “matched” (providing 11 as the common product of “matched” ionization pathways), and (3) despite this high degree of chiral recognition, a single enantiomer of ligand is capable of converting both “matched” and “mismatched” substrate to product.
Table 3.
AAA Reactions with Deuterated Substrate 9
 | ||||||
|---|---|---|---|---|---|---|
| entry | ligand | 10:11a | yield (%)b | drc | ee (%)d | |
| 1 | (S,S)- + (R,R)-L1 | 1:19 | 97 | 9:1 | - | |
| 2 | (S,S)-L1 | 1:1 | 93 | 9:1 | +90 | |
| 3 | (R,R)-L1 | 1:1 | 99 | 9:1 | −91 | |
Determined by 1H NMR of the product mixture.
Combined isolated yield of both regioisomers.
Determined by 1H NMR of the crude reaction mixture.
Determined by chiral HPLC.
In summary, we have reported a new method for the highly regio-, diastereo-, and enantioselective allylic alkylation of 2-substituted pyridines. Investigations of the reaction mechanism, the role of lithium aggregates, and applications of this strategy to other nucleophilic classes are ongoing.
Supplementary Material
Acknowledgements
We thank the National Science Foundation and the National Institutes of Health (GM13598) for supporting our programs and Johnson Matthey for palladium salts. Dr. Victor G. Young, Jr. (X-Ray Crystallographic Laboratory, University of Minnesota) is gratefully acknowledged for XRD analysis.
Footnotes
Supporting Information Available. Experimental procedures and analytical data for all new compounds are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Trost BM, Crawley ML. Chem. Rev. 2003;103:2921. doi: 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]; (b) Lu Z, Ma S. Angew. Chem. Int. Ed. 2008;47:258. doi: 10.1002/anie.200605113. [DOI] [PubMed] [Google Scholar]
- 2.Ketones: Braun B, Laicher F, Meier T. Angew. Chem. 2000;112:3637. doi: 10.1002/1521-3773(20001002)39:19<3494::aid-anie3494>3.0.co;2-4. Trost BM, Schroeder GM. Chem. Eur. J. 2004;11:174. doi: 10.1002/chem.200400666. Trost BM, Xu J, Reichle M. J. Am. Chem. Soc. 2007;129:282. doi: 10.1021/ja067342a. Zheng WH, Zheng BH, Zhang Y, Hou XL. J. Am. Chem. Soc. 2007;129:7718. doi: 10.1021/ja071098l. Trost BM, Xu J, Schmidt T. J. Am. Chem. Soc. 2008;130:11852. doi: 10.1021/ja8038954. β-keto esters: Trost BM, Sacchi KL, Schroeder GM, Asakawa N. Org. Lett. 2002;4:3427. doi: 10.1021/ol0265766. Imino esters: Baldwin IC, Williams JMJ. Tetrahedron: Asymmetry. 1995;6:1515. Nakoji M, Kanayama T, Okino T, Takemoto Y. J. Org. Chem. 2002;67:7418. doi: 10.1021/jo0260645. Azalactones: Trost BM, Ariza X. Angew. Chem. Int. Ed. Engl. 1997;36:2635. Trost BM, Lee CB. J. Am. Chem. Soc. 1998;120:6818. Nitroalkanes: Trost BM, Surivet J-P. J. Am. Chem. Soc. 2000;122:6291.
- 3.Trost BM, Thaisrivongs DA. J. Am. Chem. Soc. 2008;130:14092. doi: 10.1021/ja806781u. [DOI] [PubMed] [Google Scholar]
- 4.Interestingly, the 1-naphthyl analog of 3g could be obtained in 100% conversion by 1H NMR but only 2:1 dr.
- 5.No reaction was observed with the unmethylated carbamate or the corresponding phthalimide-protected aminopyridine.
- 6.(a) Szychowski J, Wróbel JT, Leniewski A. Bull. Acad. Pol. Sci. Ser. Sci. Chim. 1980;28:9. [Google Scholar]; (b) Waetzig SR, Tunge JA. J. Am. Chem. Soc. 2007;129:4138. doi: 10.1021/ja077070r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Trost BM, Bunt RC. J. Am. Chem. Soc. 1998;120:70. [Google Scholar]; (b) Lloyd-Jones GC, Stephen SC. Chem. Eur. J. 1998;4:2539. [Google Scholar]
- 8.No racemization was observed under the reaction conditions.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.