

Abstract
Glutamatergic systems, including AMPA receptors (AMPARs), are involved in opiate-induced neuronal and behavioral plasticity, although the mechanisms underlying these effects are not fully understood. In the present study, we investigated the effects of repeated morphine administration on AMPAR expression, synaptic plasticity, and context-dependent behavioral sensitization to morphine. We found that morphine treatment produced changes of synaptic AMPAR expression in the hippocampus, a brain area that is critically involved in learning and memory. These changes could be observed 1 week after the treatment, but only when mice developed context-dependent behavioral sensitization to morphine in which morphine treatment was associated with drug administration environment. Context-dependent behavioral sensitization to morphine was also associated with increased basal synaptic transmission and disrupted hippocampal long-term potentiation (LTP), whereas these effects were less robust when morphine administration was not paired with the drug administration environment. Interestingly, some effects may be related to the prior history of morphine exposure in the drug-associated environment, since alterations of AMPAR expression, basal synaptic transmission, and LTP were observed in mice that received a saline challenge 1 week after discontinuation of morphine treatment. Furthermore, we demonstrated that phosphorylation of GluA1 AMPAR subunit plays a critical role in the acquisition and expression of context-dependent behavioral sensitization, as this behavior is blocked by a viral vector that disrupts GluA1 phosphorylation. These data provide evidence that glutamatergic signaling in the hippocampus plays an important role in context-dependent sensitization to morphine and supports further investigation of glutamate-based strategies for treating opiate addiction.
Introduction
It has been suggested that drugs of abuse affect the neural substrates of learning and memory, which plays an important role in addiction (Vorel et al., 2001; Weiss, 2005). Rodent models of drug addiction have demonstrated a role of drug-associated contexts in drug reward and relapse (Weiss, 2005; Shen et al., 2006; Koya et al., 2009; Rothwell et al., 2010; Bossert et al., 2011). Moreover, it has been shown that rodents exhibit increased locomotor activity when repeated drug administration is paired with a context (context-dependent behavioral sensitization). This effect is less robust when drug administration is not paired with an environment (Koya et al., 2009; Liang et al., 2011a,b). Previous studies have found that contextual conditioning with either cocaine or amphetamine requires the hippocampus (Meyers et al., 2006; Shen et al., 2006). However, it remains unknown whether context-dependent behavioral sensitization to morphine is hippocampus dependent.
AMPA receptors (AMPARs) are a subclass of glutamate receptors that mediate the majority of fast excitatory transmission in the brain. In the adult hippocampus, the GluA1 and GluA2 subunits play a key role in activity-dependent synaptic trafficking of AMPARs (Wang et al., 2005). Drug-induced changes to synaptic AMPAR expression can alter spine morphology and lead to changes in synaptic strength (Russo et al., 2010). We have previously observed increased insertion of GluA2-lacking AMPARs in hippocampal synapses 12 h after repeated morphine administration. This was mainly due to increased GluA1 and GluA3 levels at the postsynaptic density (PSD), with total levels of GluA2 being unaltered (Billa et al., 2010). However, the cellular mechanisms underlying these effects remain unknown, and it is unclear whether alterations of AMPAR expression can be observed at later time points when animals are exposed to the drug-associated environment and whether the observed biochemical changes are responsible for morphine-induced behavioral adaptations such as context-dependent behavioral sensitization.
The present study uses a multifaceted approach to examine the underlying neural substrates of context-dependent behavioral sensitization. We provide evidence that repeated morphine treatment produces changes in AMPAR trafficking and synaptic expression in the hippocampus. Morphine-induced alterations of synaptic AMPAR expression can be observed 1 week later, following context-dependent behavioral sensitization, and they are associated with increased basal synaptic transmission and disrupted long-term potentiation (LTP). These alterations are reduced or absent when morphine treatment is not paired with a context. In addition, our findings indicate that the administration of morphine in the drug-associated environment is sufficient to promote changes in AMPAR expression and function, since alterations in AMPAR expression, synaptic transmission, and plasticity are observed in mice that received a saline challenge 1 week after discontinuation of morphine treatment. Finally, we demonstrate that changes in synaptic AMPAR expression and trafficking that occur during repeated morphine treatment are required for context-dependent behavioral sensitization, as this behavior is blocked when hippocampal GluA1 phosphorylation is disrupted. These data provide evidence that AMPAR composition and function in the hippocampus play an important role in context-dependent behavioral sensitization to morphine.
Materials and Methods
Animals and morphine treatment.
Experimental protocols involving animals were approved by the Institutional Animal Care and Use Committee at Columbia University, according to the U.S. Department of Agriculture Animal Welfare Act (Public Law 89-544) and the National Institutes of Health's Guide for the Care and Use of the Laboratory Animals (Department of Health, Education, and Welfare Publication No. 85-23, revised 1985). Six- to 8-week-old male C57BL/6 mice (20–25 g) (Harlan) were maintained on a 12 h light/dark cycle and allowed to acclimatize to their environment for 1 week before drug administration. Morphine sulfate was dissolved in saline at the concentration of 1 mg/ml. For the experiments in which we examined the effects of repeated morphine administration, mice were injected intraperitoneally with escalating doses of morphine (5, 8, 10, and 15 mg/kg) or equivalent volume of saline in their home cages every 12 h as described previously (Morón et al., 2007; Billa et al., 2010). Animals were killed either at 12 h or 1 week after the last injection. The hippocampi were dissected and stored at −80°C until use.
Context-dependent behavioral sensitization.
In a first set of experiments, we assessed context-dependent behavioral sensitization to morphine using four identical chambers (41.5 × 41.5 × 30 cm) equipped with photobeams that detected locomotor activity (AccuScan Instruments). Mice were randomly assigned to three groups (n = 6–8 per group; for a schematic of the behavioral experiments, see Fig. 4A). Mice received four training sessions in the locomotor activity chambers (once every 12 h) and were administered saline or escalating doses of morphine (5, 8, 10, and 15 mg/kg) during training. Seven days later, locomotor activity was assessed following a challenge dose of saline or 5 mg/kg morphine. All sessions began with a habituation period in which mice were treated with saline and allowed to explore the locomotor activity chambers for 60 min. Following habituation, mice received an injection of saline or morphine in the locomotor activity chambers and activity was measured for 90 min. All mice were killed by decapitation 90 min after receiving the challenge injection, and hippocampi were dissected for use in electrophysiology or immunoblotting experiments described below.
Figure 4.
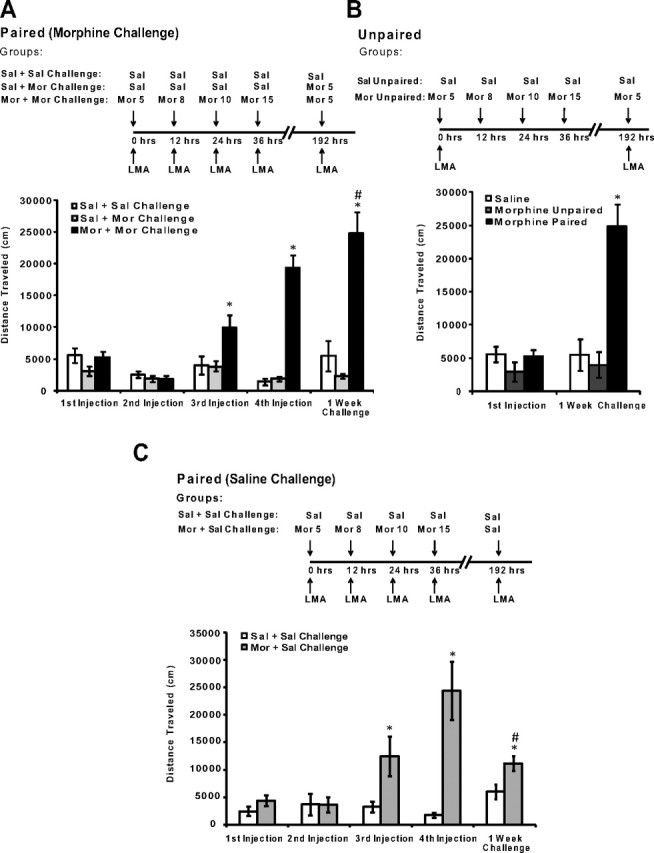
Behavioral sensitization following treatment with escalating doses of morphine is selectively expressed in a drug-associated environment. A–C, A schematic of the design is shown for either context-dependent behavioral sensitization or the unpaired group. LMA indicates that locomotor activity was recorded immediately following injection. A, The effects of escalating doses of morphine on context-dependent behavioral sensitization. Mice treated with escalating doses of morphine exhibited increased context-dependent locomotor activity when compared with saline-treated mice and with mice that only received 5 mg/kg morphine on the challenge day (*). Furthermore, mice treated with escalating doses of morphine had increased locomotor activity during the 1 week challenge of 5 mg/kg morphine when compared with the initial effects of 5 mg/kg morphine on locomotor activity (#). B, In contrast, behavioral sensitization to the effects of 5 mg/kg morphine was not observed when morphine administration was not consistently paired with the locomotor activity chambers (morphine unpaired). C, Mice treated with a saline challenge after context-dependent morphine treatment exhibited a conditioned response when compared with saline-treated mice (*) and had increased locomotor activity after the 1 week saline challenge when compared with the initial effects of the first 5 mg/kg injection of morphine (#). N = 8. *,#p < 0.05, one-way ANOVA with Tukey's post hoc comparisons or unpaired t tests. Error bars indicate SEM.
In a second set of experiments, we investigated whether the effects of morphine on locomotor activity were context dependent. Mice were assigned to one of three groups (n = 6–8 per group; see Fig. 4B). Mice assigned to the morphine unpaired condition received the initial injection of 5 mg/kg morphine in the locomotor activity chambers and received the remaining doses of morphine (8, 10, and 15 mg/kg) in their home cage. Seven days later, morphine unpaired mice received a 5 mg/kg morphine or saline challenge and locomotor activity was assessed for 90 min. Mice in the morphine-paired and saline-paired groups received all injections in the locomotor activity chambers as described in the previous experiment.
A third set of experiments investigated whether the above-described procedure of morphine administration in the drug-associated environment could trigger a conditioned response 1 week later when animals receive a challenge injection of saline (n = 8 per group; see Fig. 4C). During training, mice were treated with escalating doses of morphine (5, 8, 10, and 15 mg/kg) in the locomotor activity chambers in the same manner as described in the first behavioral experiment. One week later, mice received a saline injection before a 60 min habituation period and were treated with a second saline injection in the locomotor activity chambers, and activity was recorded for 90 min. Control mice in this experiment received saline injections throughout the experiment. Following all behavioral experiments, mice were killed by decapitation, and hippocampi were dissected for electrophysiology or immunoblotting experiments described below.
Surgeries and viral gene transfer.
We used a viral vector that expressed a mutated version of the GluA1 protein in which the S845 phosphorylation site is no longer functional (S845A). Specifically, the S845A GluA1 protein has a serine-to-alanine substitution in position 845 of the GluA1 protein that disrupts phosphorylation at the S845 site that can be observed 3 d after infection (see Fig. 7B). This viral vector was characterized and validated in previous studies (Carlezon et al., 2000; Christoffel et al., 2011). Mice (n = 6–8 per group) were anesthetized with isoflurane (5% for induction, 2.5% for maintenance) and were placed in a stereotaxic apparatus (VWR Instruments). The skull was exposed and aligned so that the height of lambda and bregma was identical. Four holes were drilled into the skull, and a Hamilton syringe (Hamilton Co.) was used to bilaterally infuse 0.5 μl of herpes simplex virus (HSV) expressing the S845A GluA1 protein (HSV-S845A), a vector expressing the control protein lacZ (HSV-lacZ), or expressing a control green fluorescent protein (HSV-GFP). Viral vectors were infused into the hippocampus at the following sites: −1.7 mm anterior/posterior (AP), ±1.5 mm medial/lateral (ML), −1.8 mm dorsal/ventral (DV); −3.0 mm AP, ±3.0 mm ML, −2.2 mm DV; −3.0 mm AP, ±3.0 mm ML, −4.1 mm DV (all coordinates posterior to bregma; see Fig. 7A). Mice infused with HSV-GFP were killed 3 d later and hippocampal slices were visualized under a fluorescent microscope to determine the spread of HSV-GFP in the hippocampus (see Fig. 7A). For behavioral experiments, mice were trained and tested in context-dependent behavioral sensitization in the same manner as described above; training began 3 d after the viral vector infusion. Mice that received HSV-S845A were treated with escalating doses of morphine, whereas mice that received HSV-lacZ were administered saline or escalating doses of morphine during context-dependent behavioral sensitization. Infusion of HSV vectors produces an increase in protein expression that peaks ∼3 d after infusion and returns to basal levels 6 d after infusion (Barrot et al., 2002). Therefore, the acquisition of context-dependent behavioral sensitization occurred during a peak period of protein overexpression (3–4 d after infection), whereas expression of behavioral sensitization was measured 11 d after infection during which protein expression has likely returned to basal levels.
Figure 7.
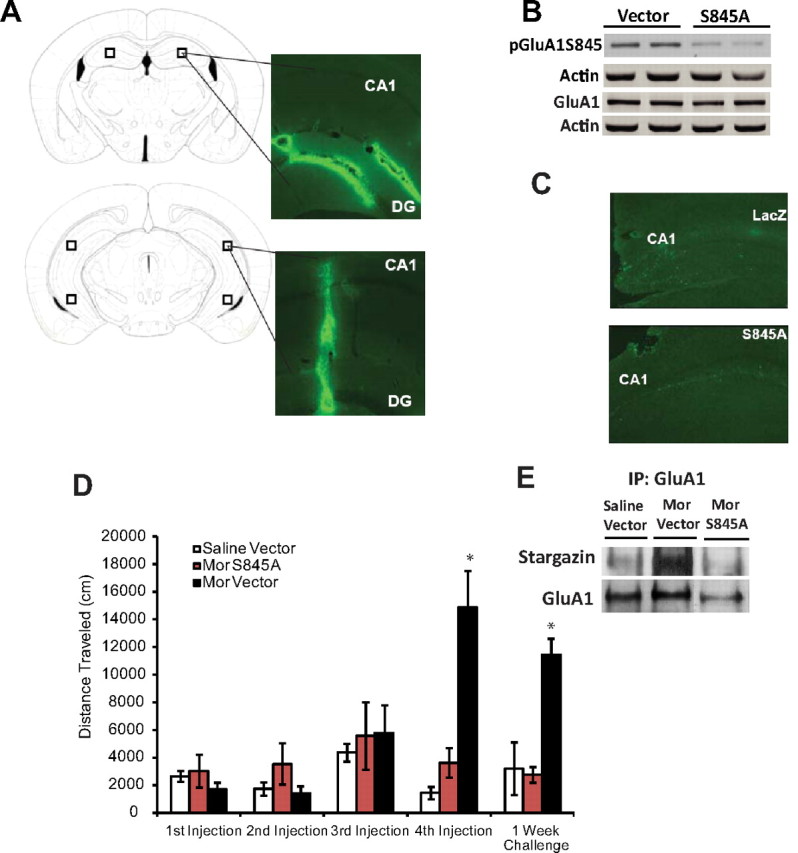
GluA1 S845 phosphorylation in the hippocampus is required for context-dependent behavioral sensitization to morphine. A, Representative examples of HSV infection in the hippocampus. Mice received hippocampal infusions of HSV-GFP, and tissue was collected 3 d after infection. The area of infection is indicated by HSV-GFP-labeled neurons, and diagrams from a mouse brain atlas (Franklin and Paxinos, 2007) indicate the hippocampal regions targeted for viral infusion. B, Infusions of HSV-S845A into the hippocampus decreased S845 GluA1 phosphorylation (3 d after infection) in naive mice but did not alter total levels of GluA1. C, Representative hippocampal slices stained with TUNEL, a marker of DNA fragmentation. Overexpression of S845A did not produce any observable changes in neurotoxicity in the hippocampus 5 d after HSV-S845A infusion. D, Mice treated with escalating doses of morphine that received a HSV-lacZ control protein exhibited increased context-dependent locomotor activity when compared with all other groups, whereas morphine-treated mice that received hippocampal infusions of HSV-S845A did not exhibit context-dependent behavioral sensitization to morphine. E, Infusion of HSV-S845A disrupted the association between GluA1 and stargazin that is observed following context-dependent sensitization to morphine. N = 6–7. *p < 0.05, one-way ANOVA with Tukey's post hoc comparisons or unpaired t tests. Error bars indicate SEM.
TUNEL staining.
We used TUNEL staining to detect DNA fragmentation that could be related to the overexpression of GluA1 S845A by viral vector infusion in the hippocampus. In situ labeling of fragmented DNA was performed with TUNEL staining (green fluorescence) 5 d after viral infusion using a commercially available in situ cell death detection kit (Roche) according to the instructions provided by the manufacturer as previously described (Lee et al., 2004).
Subcellular fractionation.
Subcellular fractionation to obtain the PSD was performed as previously described (Morón et al., 2007; Billa et al., 2009, 2010). Briefly, hippocampi from individual mice were homogenized in 0.32 m sucrose solution containing 0.1 mm CaCl2, protease and phosphatase inhibitors (Sigma-Aldrich). The homogenate was then adjusted to a final concentration of 1.25 m sucrose by adding 2 m sucrose and 0.1 mm CaCl2. The homogenate was placed in an ultracentrifuge tube, overlaid with 1 m sucrose, and centrifuged at 100,000 × g for 3 h at 4°C. The synaptosomal fraction was collected at the 1.25 m/1 m sucrose interface and then solubilized in 20 mm Tris-HCl, pH 6.0, buffer containing 1% Triton X-100 (TX-100) for 20 min at 4°C with gentle rotation. The synaptosomal fraction was then centrifuged at 40,000 × g for 30 min at 4°C. The resulting pellet, containing isolated synaptic junctions, was resuspended in 20 mm Tris-HCl, pH 8, 1% TX-100, and 0.1 mm CaCl2, and incubated at 4°C for 20 min to separate the presynaptic fraction. After centrifuging at 40,000 × g for 30 min at 4°C, the insoluble pellet containing the PSD fraction was collected, dissolved in 1% SDS, and kept at −80°C until use.
The synaptosomal fraction was obtained as previously described (Morón et al., 2002, 2007). Briefly, hippocampi were homogenized in 10 mm Tris-HCl buffer, pH 7.4, containing 0.320 m sucrose, 1 mm EDTA, and protease and phosphatase inhibitors (Sigma-Aldrich). The homogenate was centrifuged at 1000 × g for 5 min, and the supernatant was collected and centrifuged again at 16,000 × g for 15 min. After washing once in the lysis buffer, the resulting pellet was dissolved in 0.1% Triton-Tris buffer with gentle mixing at 4°C for 20 min and then subjected to centrifugation at 20,000 × g for 30 min. The pellet was collected as the synaptosomal membrane fraction.
Immunoblotting.
Equal amounts of protein (10 μg) were loaded and separated in 10% SDS-polyacrylamide gels and then transferred to nitrocellulose membranes. Membranes were incubated overnight at 4°C with selective antibodies to N-ter GluA1 (1:1000), phospho-GluA1S845 (1:1000), N-ter GluA2 (1:1000), phospho-GluA2Y876 (1:1000), PSD95 (1:1000), GRIP1 (1:1000), PICK1 (1:1000), and stargazin (1:1000). All the antibodies used in this study were from Millipore Bioscience Research Reagents unless otherwise indicated. The membranes were then incubated with either HRP-conjugated or fluorescent secondary antibody. After extensive washes, blots were visualized by either enhanced chemiluminescence (ECLplus; GE Healthcare) according to the manufacturer's instruction or scanned directly by the Odyssey infrared fluorescent imaging system (LI-COR Biosciences). To ensure equal protein loading, membranes were reprobed with an antibody selective to actin (Millipore Bioscience Research Reagents). Actin was chosen as a loading control because it is highly concentrated in the PSD (Matus et al., 1982). Actin levels did not change across any drug treatment for all experiments. Quantification was performed by comparing the intensity of the band with protein-specific antibodies to actin band intensity to control for variation in loading and transfer. These values were normalized to saline control values.
Coimmunoprecipitation.
Coimmunoprecipitation was performed as previously described (Morón et al., 2007; Billa et al., 2010) with some modifications. In brief, 4–5 μg of GluA1, GluA2, stargazin, PSD95, or clathrin antibodies were incubated with 20 μl of 50% protein A agarose slurry (Thermo Fisher Scientific) for 4 h at 4°C. The beads bound with antibody were pelleted by centrifugation at 1000 × g for 2 min and washed three times with 0.1% Tris-buffered saline (TBS)/Triton X-100. PSD fractions (100 μg) were added to 500 μl of 0.1% TBS/Triton X-100 and incubated with the antibody-conjugated beads overnight with gentle rocking at 4°C. The immunoprecipitates were then collected by centrifugation at 1000 × g for 2 min to pellet the agarose-bound antibody. After washing three times with ice-cold Tris-buffered saline containing 0.1% Tween 20 (TBST), the immunoprecipitates were resuspended in 2 × Laemmli sample buffer and boiled for 5 min to dissociate the proteins from beads. After removal of the beads by centrifugation at 2000 × g for 2 min, the supernatant was subjected to SDS-PAGE separation and the target proteins were detected by immunoblotting.
Protein cross-linking using BS3.
Levels of cell surface and intracellular AMPAR subunit proteins were analyzed using a protein cross-linking assay as previously described (Boudreau and Wolf, 2005). Briefly, slices of 400 μm thickness were prepared from fresh hippocampi and allowed to recover in 95% O2/5% CO2 saturated artificial CSF (ACSF) (117 mm NaCl, 2.5 mm KCl, 1.3 mm NaH2PO4, 1.2 mm MgSO4, 2.0 mm CaCl2, 26 mm NaHCO3, and 12 mm glucose) for 10 min. Next, hippocampal slices were incubated with 2 mm BS3 in ACSF for 30 min at 4°C with gentle agitation. The reaction was quenched by adding 100 mm glycine. After brief washing in cold ACSF, the slices were collected and the synaptosomal fraction was obtained as described above. Unmodified (intracellular) and cross-linked (cell surface) AMPAR subunits were separated by SDS-PAGE and detected as described above.
Electrophysiology.
Mice were killed by decapitation immediately following context-dependent behavioral sensitization, and brains were removed rapidly in an ice-cold cutting solution containing 175 mm sucrose, 7 mm MgCl2, 2.5 mm KCl, 1.2 mm NaH2PO4, 0.5 mm CaCl2, 26 mm NaHCO3, and 25 mm glucose. Brains were mounted on a platform and sliced at 350–400 μm thickness while submerged in an ice-cold cutting solution. Slices were transferred to beakers that contained ACSF and incubated for at least 30 min. After incubation, slices were transferred to the recording chamber. The temperature of both the incubating and recording chambers was maintained at 30 ± 1°C, and 95% O2/5% CO2 was bubbled continuously during dissection, incubation, and recording.
A concentric bipolar stimulating electrode placed in the Schaffer collateral pathway was used to evoke synaptic responses, and a glass Ag/AgCl electrode filled with ACSF was used to record extracellular field EPSPs (fEPSPs) in the stratum radiatum of CA1. At the start of each experiment, a stimulus–response curve was constructed by recording fEPSPs after stimulating the slice with increasing intensities. Following a 20 min recording of baseline synaptic responses, LTP was induced by two high-frequency stimulation trains (100 Hz, 1 s duration) separated by a 20 s intertrain interval. During LTP recording, pulses at an intensity eliciting 50% of a maximum slope were used.
Visualized whole-cell patch-clamp recordings were made from the CA1 area of the hippocampus with recording pipettes containing the following (in mm): 130 Cs-methylsulfonate, 10 Na-methylsulfonate, 10 EGTA, 1 CaCl2, 10 HEPES, 5 QX-314 · Cl, 0.1 spermine tetrahydrochloride, 2 Mg2+-ATP. Osmolarity was adjusted to 280–290 osmol L−1 and pH to 7.35–7.4 with CsOH. Open pipette resistance was 2–4 MΩ, and access resistance during recordings was <20 MΩ. Evoked EPSCs were recorded (Axopatch 200B; Molecular Devices) in neurons voltage clamped at −70 mV holding potentials. To isolate AMPA-mediated responses, the experiments were performed in the presence of NMDA, GABAA, and GABAB receptor blockers: 50 mm d-APV (2-amino-5-phosphonovalerate), 100 mm picrotoxin, and 2 mm CGP55845 [(2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl](phenylmethyl)phosphinic acid hydrochloride]. The records were filtered at 1 kHz.
Data acquisition and analysis were performed using Clampex and Clampfit 8.2 (Molecular Devices).
Data analysis.
Results are expressed as the mean ± SEM. For experiments with two groups, statistical comparisons were made using two-tailed unpaired t tests, whereas data from experiments with three or more groups were analyzed using one-way ANOVAs followed by Tukey's post hoc comparisons. Linear regression was used to compare the slopes of fiber volley (FV)–fEPSP ratios. Significance was set at p < 0.05. Statistical analyses of data were generated by using Prism software (version 5.0; GraphPad Software).
Results
Administration of escalating doses of morphine in the home cage increases the phosphorylation levels of both GluA1 and GluA2 subunits
It is thought that phosphorylation of AMPAR subunits controls synaptic targeting and function of AMPARs (Ehlers, 2000; Lee et al., 2000). We examined the effects of morphine on the phosphorylation levels of GluA1 at the S845 site, which is known to be phosphorylated by PKA (Colledge et al., 2000) and can ultimately lead to receptor insertion in the membrane (Ehlers, 2000; Lee et al., 2000). Figure 1 shows that, 12 h after treatment of mice with escalating doses of morphine in the home cage, there was a significant increase in GluA1 S845 phosphorylation in the total homogenate (Fig. 1A; n = 3–4 per group; t(5) = 2.97; p = 0.03), in the crude synaptosomal fraction (Fig. 1B; n = 4 per group; t(6) = 4.58; p = 0.004), and in the PSD (Fig. 1C; n = 3–4 per group; t(5) = 3.68; p = 0.01). These data indicate that escalating doses of morphine increase GluA1 S845 phosphorylation, which could affect synaptic trafficking of GluA1-containing AMPARs.
Figure 1.

A–C, Administration of escalating doses of morphine in the home cage alters phosphorylation levels of GluA1 and GluA2 in the hippocampus. Twelve hours after treatment with escalating doses of morphine in the home cage, pGluA1 was increased in the homogenate, synaptosomal fraction, and the PSD relative to saline-treated mice, whereas pGluA2 was increased only in the total homogenate and PSD. Furthermore, repeated morphine treatment differentially alters synaptic targeting of GluA1 and GluA2 AMPAR subunits in the homogenate. D, The association between stargazin and GluA1 and the association between PSD95 and GluA1 were increased 12 h after treatment with escalating doses of morphine. E, The association between GRIP1 and GluA2 decreased 12 h after repeated morphine treatment, whereas no changes were observed in the association between PICK and GluA2. Inset, Representative blot showing stronger association of stargazin with GluA1 than with the GluA2 subunit. N = 3–4. *p < 0.05, morphine versus saline, unpaired t test. Error bars indicate SEM.
Next, we examined the effects of repeated morphine administration on the Y876 phosphorylation site of the GluA2 subunit, a site phosphorylated by Src family tyrosine kinases (Hayashi and Huganir, 2004; Wang et al., 2005). Figure 1 shows that Y876 phosphorylation increased in the total homogenate (Fig. 1A; n = 4 per group; t(6) = 4.22; p = 0.006) and the PSD (Fig. 1C; n = 3–4 per group; t(5) = 3.04; p = 0.03) but remained unchanged in the synaptosomal fraction (Fig. 1B) after morphine treatment. It is important to note that, unlike GluA1 S845 phosphorylation, which could be partially responsible for the synaptic insertion of the subunit (Wolf et al., 2004; Jiang et al., 2006), studies that have investigated phosphorylation of GluA2 at Y876 have yielded mixed results. Specifically, Hayashi and Huganir (2004) reported that Y876 phosphorylation leads to a removal of the GluA2 subunit from the synapse, whereas Moult et al. (2006) have found that tyrosine dephosphorylation may lead to synaptic removal of GluA2-containing AMPARs. Together, these data suggest that repeated morphine administration triggers an increase in overall GluA2-lacking receptor expression at the synapse by simultaneously increasing synaptic insertion of GluA1 subunits and possibly a removal of GluA2 subunits from the synapse.
The interaction between GluA1 and GluA2 and their respective PDZ domain-containing proteins at the synapse are differentially affected by the administration of escalating doses of morphine
The targeting of AMPAR subunits to and from the synapse is regulated by several PDZ domain-containing proteins such as stargazin, GRIP1, and PICK1 (Díaz, 2010). As repeated morphine administration alters GluA1 and GluA2 phosphorylation at the synapse (Fig. 1A–C), it is possible that morphine may alter the synaptic expression of AMPAR subunits by regulating the interactions between the receptor subunits and PDZ domain-containing proteins. Therefore, we performed coimmunoprecipitation experiments in hippocampal PSD fractions that directly analyzed the effects of morphine on the association between GluA1 and stargazin/PSD95 and between GluA2 and GRIP1/PICK1.
Interestingly, the association between stargazin and GluA1 in the PSD was significantly enhanced 12 h following morphine administration in the home cage (Fig. 1D; n = 3–4 per group; t(5) = 4.11; p = 0.009). Furthermore, the association between PSD95 and stargazin was increased in morphine-treated mice (Fig. 1D; n = 3–4 per group; t(5) = 4.10; p = 0.009), suggesting that morphine administration triggers the formation of a GluA1/stargazin/PSD95 complex at the PSD, which would enhance the synaptic insertion and immobilization of GluA1. We also investigated possible interactions between GluA2 and stargazin, as stargazin can interact with all AMPAR subunits (Díaz, 2010; Kato et al., 2010). However, stargazin was absent in the immunoprecipitate obtained with antibody for GluA2 (data not shown). To confirm the lack of an interaction between stargazin and GluA2 in the PSD, we examined the levels of stargazin in the unbound fraction, which contains proteins that do not precipitate with the antibody–beads complex. As expected, levels of stargazin remaining in the unbound fraction after immunoprecipitation with antibody to GluA2 were higher than those observed after immunoprecipitation with the antibody for GluA1 (Fig. 1D, inset; n = 3 per group; t(4) = 4.85; p = 0.008). This suggests that, in the PSD, stargazin is preferentially associated to the GluA1 subunit in the hippocampus.
Next, we examined the effects of morphine on the association between GluA2 and GRIP1/PICK1. We observed that repeated morphine administration significantly disrupted the interaction between GluA2 and GRIP1, whereas the association between GluA2 and PICK1 remained unaltered (Fig. 1E; n = 3–4 per group; t(5) = 2.66; p = 0.04). These data are in agreement with previous studies showing that phosphorylation of GluA2 at Y876 disrupts GRIP1/GluA2 interactions (Hayashi and Huganir, 2004; Wang et al., 2005). Overall, these data indicate that repeated morphine administration alters protein–protein interactions between AMPAR subunits and their regulatory proteins at the synapse, which could in turn affect the synaptic insertion of these receptors. These alterations of AMPAR synaptic targeting by morphine may lead to changes in neuroplasticity at later time points that could lead to dysfunctional changes in behavior.
Administration of escalating doses of morphine promotes internalization of GluA2 in the synaptosomal fraction
The data shown above suggest that morphine affects synaptic insertion of AMPARs by altering the interaction between the different receptor subunits and their regulatory proteins. To directly examine this, we analyzed the effects of morphine on cell surface expression of AMPAR subunits using a protein cross-linking assay with BS3 (Boudreau and Wolf, 2005). Figure 2A shows that there were no significant changes in the cell surface levels of GluA1 in the synaptosomal fraction 12 h after morphine administration in the home cage. It is possible that the observed increase in phosphorylation of GluA1 in the synaptosomal fraction and at the PSD (Fig. 1B,C) is not due to increased cell surface expression. However, it is also possible that the observed increase in GluA1 insertion is produced locally at the PSD, and therefore cannot be detected using this cross-linking protocol. However, Figure 2B shows that repeated morphine administration led to a significant decrease of GluA2 levels in the cell surface (Fig. 2B; n = 3 per group; t(4) = 4.36; p = 0.01) and in the S/I ratio (Fig. 2B; n = 3 per group; t(4) = 3.28; p = 0.03), suggesting that morphine causes internalization of GluA2 subunits in the synaptosomal fraction. Because AMPARs are known to undergo clathrin-dependent endocytosis (Man et al., 2000), we next investigated the effect of morphine on the association of GluA2 with clathrin in the synaptosomal fraction. Levels of clathrin associated with GluA2 were significantly increased following morphine treatment (Fig. 2C; n = 3–4 per group; t(5) = 2.65; p = 0.04). Therefore, these data indicate that repeated morphine administration decreases cell surface expression of GluA2 in the synaptosomal fraction by triggering its internalization via a clathrin-dependent mechanism.
Figure 2.
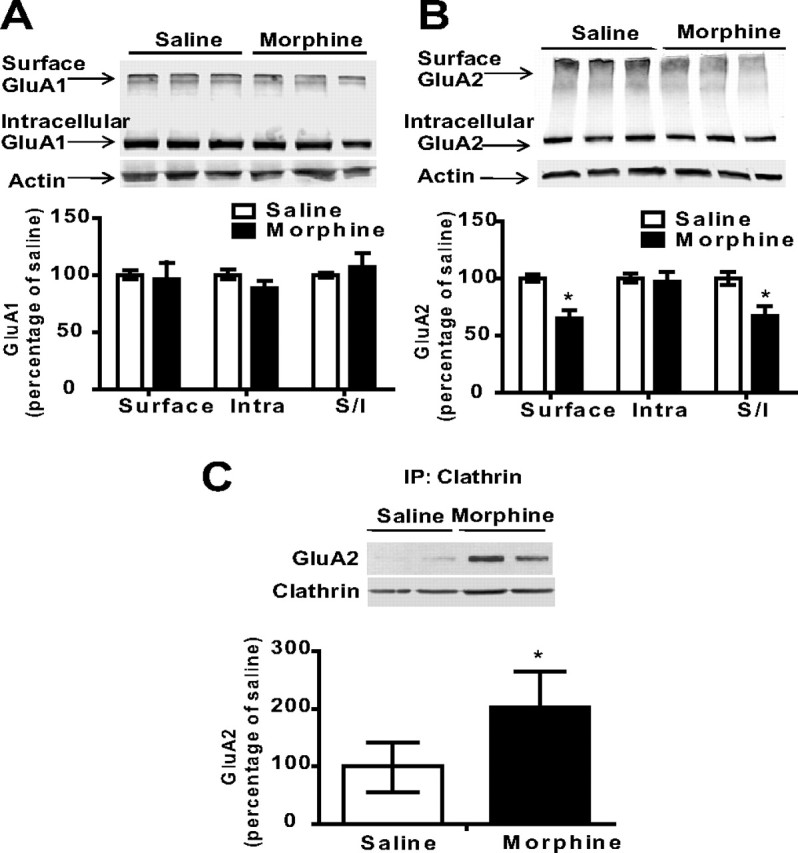
Administration of escalating doses of morphine in the home cage triggers internalization of the GluA2 subunit of AMPA receptors. A, B, Using a protein cross-linking assay with BS3, we found no changes in cell surface expression of GluA1 in the synaptosomal fraction, but GluA2 was decreased on the cell surface and in the surface/intracellular ratio 12 h after repeated morphine treatment. C, The association between clathrin and GluA2 was increased 12 h after repeated morphine treatment, suggesting that repeated morphine administration leads to an increased internalization of GluA2 subunits and thus decreases cell surface expression of GluA2. N = 3–4. *p < 0.05, morphine versus saline, unpaired t test. Error bars indicate SEM.
Together, these data demonstrate that the repeated administration of morphine in the home cage produces short-term changes in hippocampal GluA1 and GluA2 phosphorylation and alters the interaction between PDZ domain-containing proteins and AMPAR subunits. Next, we examined whether these alterations in the expression and phosphorylation of AMPAR subunits could be observed 1 week later. Mice were treated in the home cage with escalating doses of morphine as described above, but in this case they were killed 1 week after the last morphine injection. Figure 3 shows that there were no changes in either the expression or the phosphorylation of GluA1 and GluA2 AMPAR subunits in the total homogenate or in the PSD. Overall, these results show that repeated morphine treatment in the home cage can lead to short-term alterations of GluA1 and GluA2 AMPAR subunit expression, but these changes are no longer present after 1 week.
Figure 3.
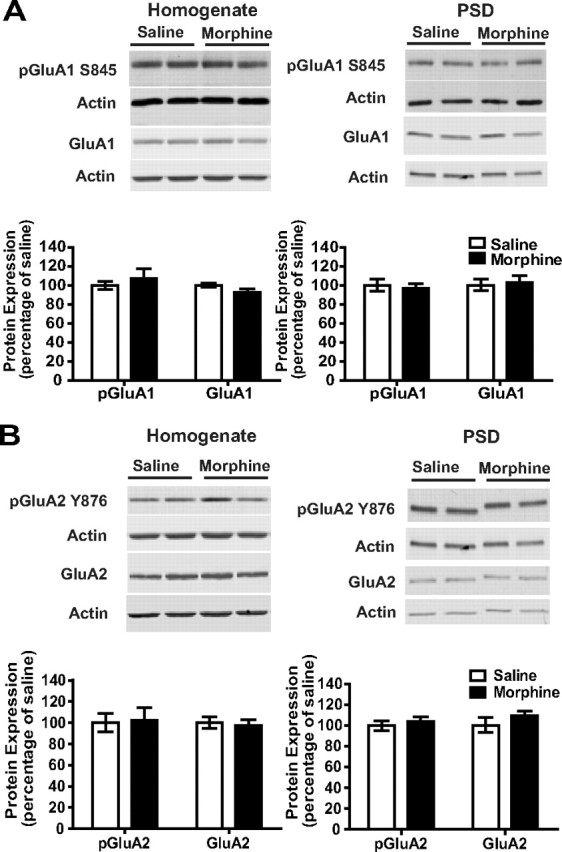
AMPAR expression was not altered 1 week after repeated morphine administration in the home cage. A, B, One week after the administration of escalating doses of morphine, no changes in GluA1 or GluA2 AMPAR subunits were observed in either the homogenate or the PSD, suggesting that alterations of AMPAR expression are not observed if morphine administration occurred in the home cage. Error bars indicate SEM.
Behavioral sensitization following treatment with escalating doses of morphine is selectively expressed in a drug-associated environment
Morphine-induced changes in AMPARs were not observed 1 week later when morphine was administered in the home cage. However, a question that remains unresolved is whether the drug-associated environment triggers alterations to behavior, synaptic plasticity, and AMPAR function in the hippocampus. We used context-dependent behavioral sensitization to investigate learned associations between morphine and the drug administration environment. Mice were treated with escalating doses of morphine (5, 8, 10, and 15 mg/kg) in a novel context every 12 h and locomotor activity was recorded (for schematic, see Fig. 4A). Enhanced locomotor activity was observed following treatment with 10 (Fig. 4A; n = 8 per group; F(2,19) = 8.62; p = 0.002) and 15 mg/kg morphine (Fig. 4A; n = 8 per group; F(2,19) = 80.42; p < 0.0001). One week later, a challenge of 5 mg/kg morphine produced sensitized locomotor activity in the drug-associated context (Fig. 4A; n = 8 per group; F(2,19) = 37.08; p < 0.0001) compared with mice treated with saline. Furthermore, morphine-treated mice exhibited sensitization to 5 mg/kg morphine relative to the initial effects of 5 mg/kg morphine (Fig. 4A; n = 8 per group; t(14) = 6.05; p < 0.0001).
It is possible that the effect of escalating doses of morphine on locomotor activity may not be due to associations between morphine and the context, but rather to other processes such as changes in motor function. Therefore, we treated mice with escalating doses of morphine using an unpaired design in which morphine was administered in different drug administration environments (for schematic, see Fig. 4B). In these conditions, mice showed no increase in locomotor activity following treatment with a 5 mg/kg challenge dose of morphine (p > 0.05), whereas mice that received context-dependent morphine treatment exhibited behavioral sensitization (Fig. 4B; n = 8 per group; F(2,22) = 12.31; p = 0.0003). These data indicate that associations between repeated morphine treatment and the drug administration environment can produce robust behavioral adaptations that may model drug-seeking behavior (Robinson and Berridge, 1993). Furthermore, behavioral sensitization to morphine was selectively expressed in a drug-associated environment, suggesting that this effect was due to the association between morphine and the drug administration environment, and not due to other processes such as motor function.
Mice that received context-dependent morphine treatment showed sensitization to a challenge dose of morphine, but it is unknown whether context-dependent treatment with escalating doses of morphine is sufficient to trigger a conditioned response 1 week later (for schematic, see Fig. 4C). Thus, we performed experiments in which mice received escalating doses of morphine using a paired design and then were treated with a saline challenge 1 week later. Treatment with 10 mg/kg (Fig. 4C; n = 8 per group; t(14) = 2.64; p = 0.02) and 15 mg/kg (Fig. 4C; n = 8 per group; t(14) = 4.55; p = 0.0005) morphine enhanced locomotor activity. One week later, saline was administered in the context and a significant enhancement in locomotor activity was observed relative to control mice that received saline during both the training and the challenge (Fig. 4C; n = 8 per group; t(14) = 2.95; p = 0.01). However, this effect was less robust than that observed after the administration of a morphine challenge (Fig. 4A). Locomotor activity in morphine-treated mice that received a saline challenge was also enhanced relative to the initial effects of 5 mg/kg morphine (Fig. 4C; n = 8 per group; t(14) = 4.57; p = 0.0005). Therefore, these data show that context-dependent morphine treatment was sufficient to produce a conditioned response (i.e., increased locomotor activity) and that this effect was magnified when animals received a drug challenge injection 1 week after discontinuation of morphine treatment.
Phosphorylation of the GluA1 subunit is increased following context-dependent behavioral sensitization
We have shown that escalating doses of morphine in the home cage can produce alterations of synaptic AMPAR expression (Fig. 1); however, these effects are no longer observed when animals are killed 1 week after morphine administration (Fig. 3). Therefore, it is possible that morphine-induced changes to AMPAR expression may depend on whether morphine administration is associated with the drug administration environment. To test this, hippocampi were collected following context-dependent behavioral sensitization and changes in synaptic AMPAR expression were assessed. The results from immunoblotting experiments showed changes in the expression and synaptic redistribution of AMPARs following context-dependent behavioral sensitization. We found that morphine-treated mice that exhibited context-dependent behavioral sensitization following a 5 mg/kg morphine challenge 1 week later had increased pGluA1 (Fig. 5A; n = 4 per group; F(2,9) = 5.92; p = 0.02) and GluA1 levels (Fig. 5A; n = 4 per group; F(2,9) = 7.00; p = 0.01) in the total homogenate compared with saline-treated mice. Furthermore, the expression of context-dependent behavioral sensitization to morphine was associated with an increase in pGluA1 (Fig. 5A; n = 3–4 per group; F(2,8) = 9.56; p = 0.008) and a decrease in GluA2 levels (Fig. 5A; n = 3–4 per group; F(2,9) = 7.85; p = 0.01) in the PSD. Interestingly, no changes in AMPAR expression or phosphorylation were observed in mice that were repeatedly treated with saline and then received an acute challenge of morphine. These data indicate that the observed changes following context-dependent sensitization were not due to the acute challenge that mice received 90 min before these biochemical analyses.
Figure 5.

Alterations in synaptic distribution of AMPARs following context-dependent behavioral sensitization to morphine. A, Hippocampal tissue from saline- and morphine-treated mice was collected after context-dependent behavioral sensitization followed by a morphine challenge 7 d later (paired group). Context-dependent treatment of morphine produced an increase in pGluA1 and GluA1 in the total homogenate, whereas no effects were observed in pGluA2 and GluA2 levels. Furthermore, context-dependent treatment of morphine produced an increase in pGluA1 and decreased GluA2 levels in the PSD. B, Hippocampal tissue from saline- and morphine-treated mice was collected after context-dependent behavioral sensitization followed by a saline challenge 7 d later (paired group). Mice that received a saline challenge 1 week after context-dependent morphine treatment showed increased GluA1 and decreased pGluA2 in the PSD, whereas pGluA1 was decreased in the total homogenate. C, In contrast, no changes in synaptic GluA1 or GluA2 were observed when mice were treated with escalating doses of morphine in an unpaired design, but pGluA2 levels were increased in the homogenate. S+S, Saline plus Saline Challenge; S+M, Saline plus Morphine Challenge; M+M, Morphine plus Morphine Challenge; M+S, Morphine plus Saline Challenge. N = 3–4. *p < 0.05, one-way ANOVA with Tukey's post hoc comparisons or unpaired t test. Error bars indicate SEM.
Changes in synaptic AMPAR expression were only observed when mice were treated with escalating doses of morphine using a paired design, as mice that received repeated morphine treatment in an unpaired design showed no changes in behavior upon receiving a morphine challenge (Fig. 4B) and no changes in synaptic AMPAR expression (Fig. 5C; p > 0.05). However, pGluA2 was increased in the total homogenate in mice that received unpaired morphine treatment (Fig. 5C; n = 3–4 per group; t(6) = 6.06; p = 0.0009). Together, these data suggest that the effects of repeated morphine treatment on synaptic AMPAR expression can be observed 1 week later only if morphine treatment is paired with a context and these changes are associated with the expression of behavioral sensitization when mice receive a morphine challenge 1 week later. In contrast, changes in synaptic AMPAR expression are not observed when morphine administration is not consistently associated with the drug administration environment; this is in agreement with the lack of behavioral sensitization observed in these mice upon the administration of the morphine challenge.
As shown in Figure 4C, context-dependent treatment with escalating doses of morphine was sufficient to produce a conditioned response. Therefore, changes in AMPAR expression were examined in mice that received context-dependent morphine treatment followed by a saline challenge 1 week later. We found that morphine-treated mice that exhibited a conditioned response after a saline challenge had increased GluA1 and decreased pGluA2 levels in the PSD (Fig. 5B; n = 3–4 per group; t(5) = 3.83, p = 0.01, and t(5) = 2.78, p = 0.04, respectively), whereas pGluA1 levels were decreased in the total homogenate (Fig. 5B; n = 3–4 per group; t(5) = 2.80; p = 0.02). These data suggest that changes in AMPAR expression observed 1 week after drug treatment depend upon two factors: the conditions in which escalating doses of morphine were administered (paired vs unpaired administration) and whether a challenge dose of morphine or saline was given 1 week after the context-dependent morphine treatment.
Basal synaptic transmission is increased but hippocampal LTP is impaired following context-dependent behavioral sensitization
We have previously reported that repeated morphine administration acutely increases basal synaptic transmission in the hippocampus (Billa et al., 2010). Therefore, we next investigated whether context-dependent behavioral sensitization to morphine could lead to changes in the stimulus–response curves of fEPSPs in the hippocampus. Mice that received context-dependent morphine treatment exhibited a significantly greater response than saline-treated mice when slices were stimulated with 5–8 V (Fig. 6A; n = 4 per group; 5 V, F(2,8) = 6.09, p = 0.02; 6 V, F(2,8) = 33.45, p = 0.0001; 7 V, F(2,8) = 35.23, p = 0.0001; 8 V, F(2,8) = 41.89, p < 0.0001). In contrast, mice that received morphine in an unpaired design exhibited an intermediate stimulus–response slope that was significantly different from both morphine-paired mice and saline-treated mice following stimulation with 6, 7, or 8 V (Fig. 6A). To test whether the differences in synaptic transmission were due to differences in the positioning of the stimulating electrode, we plotted fEPSP slope against FV amplitude. Figure 6A, inset, shows that paired morphine treatment significantly increased fEPSP slopes at similar presynaptic FV amplitudes compared with saline treatment or to the unpaired group (Fig. 6A, inset; n = 4 per group; linear regression comparisons; Morphine plus Morphine Challenge vs Saline plus Morphine Challenge: F(1,38) = 35.09, p < 0.0001; Morphine plus Morphine Challenge vs Unpaired Morphine Treatment: F(1,38) = 25.74, p < 0.0001). The Unpaired Morphine Treatment versus Saline plus Morphine Challenge comparison was also significant (F(1,44) = 6.42; p = 0.01). Thus, context-dependent morphine treatment can produce changes in basal synaptic transmission.
Figure 6.
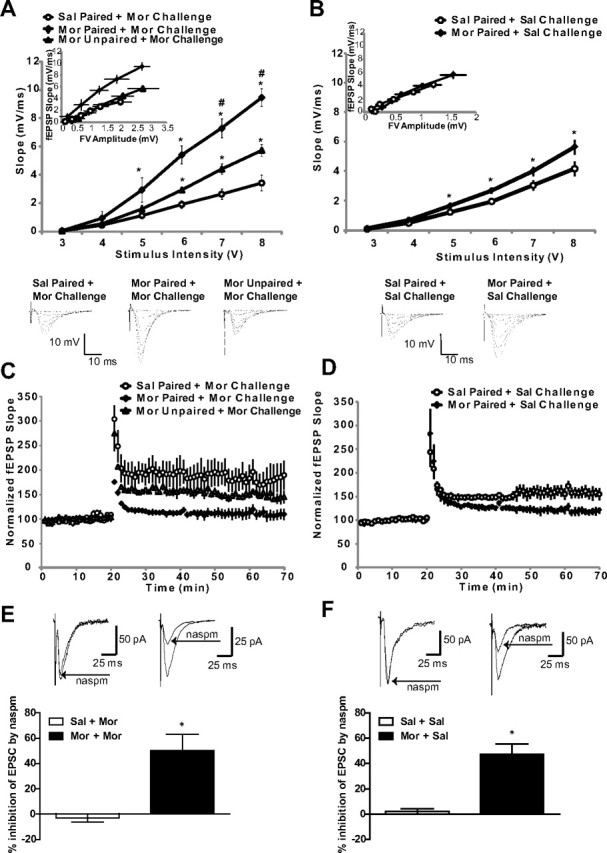
Increased synaptic transmission and disrupted LTP following context-dependent behavioral sensitization to morphine. A, Mice treated with morphine in a paired design that received a morphine challenge showed a significantly greater fEPSP slope relative to saline-treated mice (*). Mice that were administered morphine in an unpaired design exhibited an intermediate stimulus–response slope that was significantly different from both morphine-paired mice (#) and saline-treated mice (*). B, Mice treated with morphine in a paired design that received a saline challenge exhibited a significantly enhanced fEPSP slope compared with saline-treated mice (*). The insets show fEPSP–FV relationships. C, Mice that received a morphine challenge 7 d after context-dependent morphine treatment showed disrupted LTP when compared with saline-treated mice, whereas mice that received unpaired morphine showed an intermediate effect relative to morphine-paired and saline-treated mice. D, Mice that received a saline challenge 7 d after context-dependent morphine treatment showed impaired LTP when compared with saline-treated mice. E, F, The histograms show an average percentage inhibition (mean ± SEM) of EPSCs recorded from hippocampal slices. Sample traces (averages of 20 trials) were recorded before and after naspm application (100 μm, 5 min). Naspm reduced the amplitude of EPSCs in mice that received context-dependent morphine treatment (*), regardless of whether a saline or morphine challenge was administered 1 week later. N = 4–6. *,#p < 0.05, one-way ANOVA with Tukey's post hoc comparisons or unpaired t tests. Error bars indicate SEM.
Context-dependent administration of morphine followed by a saline challenge 1 week later also triggered an increase in basal synaptic transmission (Fig. 6B; n = 4–6 per group; 5 V, t(8) = 3.00, p = 0.01; 6 V, t(8) = 3.41, p = 0.009; 7 V, t(8) = 2.51, p = 0.03; 8 V, t(8) = 2.44, p = 0.04), although this effect was less robust than that observed when animals received a morphine challenge (Fig. 6A), and FV–fEPSP relationships were not significantly different between the groups (Fig. 6B, inset; p > 0.05), suggesting that differences in electrode positioning partly account for the observed changes. It is possible that prior exposure to morphine in the drug-associated environment may elicit changes in basal synaptic transmission 1 week after morphine treatment, but these changes are further enhanced when animals receive a morphine challenge. These data, along with the biochemical data shown above, suggest that context-dependent treatment with escalating doses of morphine can alter AMPAR expression and basal synaptic transmission, but these effects are enhanced if mice receive a morphine challenge 1 week after context-dependent treatment.
In animal models, repeated morphine administration can alter hippocampal LTP (Pu et al., 2002) and can affect cell signaling molecules involved in hippocampal synaptic plasticity, such as AMPARs (Billa et al., 2009, 2010; Morón et al., 2010). However, it is unknown whether context-dependent morphine treatment can produce changes in the expression of AMPARs and alter hippocampal LTP. To address this, we examined hippocampal LTP following context-dependent behavioral sensitization. When comparing the three groups during the final 10 min of recording (60–70 min after baseline recording commenced), morphine-treated mice that exhibited context-dependent behavioral sensitization followed by a morphine challenge 1 week later had significantly impaired hippocampal LTP relative to all other groups (Fig. 6C; n = 3–5 per group; F(2,27) = 321.3; p < 0.0001). In contrast, mice that received morphine in an unpaired design exhibited an intermediate change in LTP that was decreased compared with saline-treated mice, but was enhanced compared with morphine-paired animals (Fig. 6C). Finally, context-dependent administration of morphine followed by a saline challenge 1 week later led to an impairment of LTP relative to mice that received saline throughout the behavioral experiment (Fig. 6D; n = 4–6 per group; t(18) = 21.02; p < 0.0001). Together, the results from fEPSP recordings suggest that a learned association between morphine and the drug administration environment could saturate excitatory synaptic transmission in the hippocampus, resulting in an impairment of LTP. This impairment of LTP can be observed 1 week later, regardless of whether a saline or morphine challenge is administered.
A selective blocker of GluA2-lacking receptors inhibits evoked EPSCs following context-dependent sensitization to morphine
To confirm that the effects observed in biochemical and behavioral experiments were mediated by an increased insertion of GluA2-lacking receptors at the synapse, we performed whole-cell patch-clamp recordings from the CA1 area of hippocampal slices obtained from mice that received context-dependent morphine treatment followed by a morphine challenge 1 week later (Fig. 6E). Bath application of 1-naphthylacetylsperimine (naspm), a selective blocker of GluA2-lacking AMPARs, decreased the amplitude of EPSCs in slices from morphine-paired animals by 50.2 ± 13% (Fig. 6E; n = 3–4; t(5) = 4.61; p = 0.006). In saline-paired mice, the blocker had no effect (n = 4). Similarly, we found that naspm reduced the amplitude of EPSCs by 48.3 ± 8.2% in mice that received context-dependent morphine treatment followed by a saline challenge 1 week later (Fig. 6F; n = 3–4; t(5) = 4.58; p = 0.006). Therefore, these data confirm that associations between the environment and morphine administration selectively increase the insertion of GluA2-lacking AMPARs. This effect was not due to the acute administration of morphine challenge that the animals received 90 min before the recordings, but instead was due to prior context-dependent treatment with morphine.
Context-dependent behavioral sensitization to morphine requires phosphorylation of the GluA1 AMPAR subunit in the hippocampus
The present results demonstrate that repeated morphine administration can produce increases in hippocampal S845 GluA1 phosphorylation. One week later, this effect can be only observed in animals with a prior history of morphine exposure in the drug-associated environment that show a sensitized response upon receiving a drug challenge (Fig. 5). It is possible that the acquisition and expression of context-dependent behavioral sensitization to morphine depends upon the initial increases in synaptic GluA1 phosphorylation in the hippocampus. To determine whether increased GluA1 phosphorylation is required for context-dependent behavioral sensitization to morphine, we infused a HSV vector into the hippocampus that contained a mutated version of the GluA1 subunit (S845A) in which phosphorylation at the S845 site was disrupted while levels of GluA1 remained unaltered (Fig. 7A,B). Even though we did not observe a significant increase in the expression levels of GluA1 upon infusion of the HSV-S845A construct, we performed TUNEL staining to exclude any possible neurotoxic effect due to overexpression of the S845A mutant. We found no differences in TUNEL staining between mice that were infused with S845A or the lacZ control protein (Fig. 7C). These data are consistent with a previous study that reported that infusion of S845A did not lead to increased membrane expression of homomeric GluA1 AMPARs (Choi et al., 2011), and suggest that S845A infusion does not have a neurotoxic effect or leads to overexpression of GluA1 homomers.
To examine the role of S845 GluA1 phosphorylation in context-dependent behavioral sensitization to morphine, mice were trained 3 d after S845A infusion and were challenged with 5 mg/kg morphine 1 week later. Infusion of HSV vectors produces a peak in protein expression ∼3–4 d after viral infusion (Barrot et al., 2002), and the data in Figure 7B show significant disruption of GluA1 phosphorylation at the S845 site 3 d after infusion. Therefore, it is likely that phosphorylation levels of S845 are disrupted during the entire training of context-dependent behavioral sensitization, since HSV-mediated expression returns to basal levels 6 d after infusion (Barrot et al., 2002). Mice that received an HSV vector expressing the control protein lacZ showed increased locomotor activity after treatment with 15 mg/kg morphine (Fig. 7D; n = 6–7 per group; F(2,16) = 23.10; p < 0.0001) and exhibited behavioral sensitization to morphine 1 week later, following treatment with 5 mg/kg morphine (Fig. 7D; n = 6–7 per group; F(2,16) = 15.70; p = 0.0002). In contrast, the infusion of S845A into the hippocampus blocked the acquisition and expression of context-dependent behavioral sensitization to morphine (Fig. 7D). Together, these data demonstrate that associations between morphine and the drug administration environment that elicit a sensitized response upon the administration of a drug challenge critically depend upon GluA1 S845 phosphorylation.
Finally, to test whether the blockade of context-dependent behavioral sensitization to morphine by the infusion of S845A was associated with changes in the interactions between GluA1 and PDZ domain-containing proteins, we performed coimmunoprecipitation studies to directly examine the association between GluA1 and stargazin in the PSD. Stargazin regulates AMPAR trafficking through its delivery of AMPARs to the surface membrane and through its interaction with PSD-95 and other PDZ-domain containing proteins (Chen et al., 2000). The interaction between stargazin and PSD-95 is disrupted by PKA phosphorylation of stargazin (Choi et al., 2002; Stein and Chetkovich, 2010). The association of stargazin with GluA1 was quantified by normalizing the levels of stargazin to the amount of GluA1 in the immunoprecipitate. We found that mice that received a HSV vector containing the control protein lacZ and exhibited context-dependent behavioral sensitization upon receiving a morphine challenge showed a 120% increase in the association between GluA1 and stargazin at the PSD fraction when compared with saline-treated mice (Fig. 7E). However, this association was decreased 40% by the infusion of HSV-S845A (Fig. 7E). Interestingly, this effect correlates with the lack of context-dependent sensitization to morphine observed after infusion with the HSV-S845A construct and suggests that disruption of GluA1 S845 phosphorylation could impair the interaction between the GluA1 subunit and stargazin, which in turn could lead to alterations of this morphine-induced behavior.
Discussion
The present study demonstrates that alterations of synaptic AMPAR expression in the hippocampus are required for morphine-induced context-dependent sensitization. We showed that the initial morphine exposure altered AMPAR expression by differentially modulating the synaptic targeting of GluA1 and GluA2 subunits. One week later, changes in synaptic AMPAR expression were only observed when repeated morphine administration was paired with the environment, regardless of whether mice received a saline or morphine challenge. Furthermore, these changes were required for the learned association between the effects of morphine and the drug administration environment, since targeted disruption of hippocampal GluA1 AMPAR subunit phosphorylation by viral transfection blocked context-dependent behavioral sensitization to morphine.
Our data highlight an importance of early morphine-induced changes in neuroplasticity, as these drug-induced changes may lead to neuronal and behavioral consequences when morphine administration is paired with a specific environment. We have previously reported that the administration of escalating doses of morphine increases the insertion of Ca2+-permeable (GluA2-lacking) AMPARs and enhances hippocampal excitatory transmission (Billa et al., 2010). In the present study, we demonstrated that this morphine-induced increase in GluA2-lacking receptors was due to both the synaptic insertion of GluA1-contaning receptors and synaptic removal of GluA2-containing receptors. These changes in synaptic AMPAR expression were mediated by interactions between AMPAR subunits and the synaptic regulatory proteins stargazin and GRIP1. Furthermore, the initial effects of morphine on synaptic AMPAR expression were crucial for behavioral adaptations that occurred 1 week later, as disruption of GluA1 phosphorylation by viral gene transfer prevented both the acquisition and expression of context-dependent behavioral sensitization. These data are supported by studies reporting that the initial effects of morphine can lead to neurobehavioral adaptations that can be observed at later time points (Vanderschuren et al., 2001; Borgkvist et al., 2008) and suggest that pharmacological interventions that block these initial morphine-induced changes could reduce the likelihood of dependence.
Hippocampal GluA1-containing AMPARs are critically involved in mediating context-dependent behavioral sensitization to morphine
The development of behavioral sensitization may model the intensification of drug craving in human addicts (Robinson and Berridge, 1993), and therefore, this is a well characterized model of drug-induced behavioral plasticity (Boudreau and Wolf, 2005; Shen et al., 2006; Ferrario et al., 2010). In the present study, we found that associations between morphine treatment and contextual stimuli could lead to changes in synaptic AMPAR expression in the hippocampus, and that these changes were selectively expressed when morphine was associated with the context. When mice received context-dependent morphine treatment and were treated with a morphine challenge, we observed behavioral sensitization to morphine, whereas treatment with a saline challenge produced a conditioned response, although this effect was less robust than that observed after a morphine challenge. Thus, context-dependent morphine treatment altered AMPAR expression at the PSD, but the pattern of changes in AMPAR expression depended upon whether a saline or morphine challenge was administered. If mice received a saline challenge after context-dependent morphine treatment, an increase in GluA1 was observed at the PSD, whereas the administration of a morphine challenge enhanced synaptic GluA1 phosphorylation. These data suggest that context-dependent morphine treatment promotes redistribution of GluA1 at the PSD, whereas the morphine challenge triggers an increase in synaptic GluA1 phosphorylation that could be required for context-dependent behavioral sensitization. In support, we demonstrated that the disruption of GluA1 phosphorylation at S845 in the hippocampus, which is thought to modulate insertion of this receptor subunit at the PSD (Colledge et al., 2000; Lee et al., 2000), prevented the acquisition and the expression of behavioral sensitization. This effect may also be due to a disruption of protein–protein interactions between the GluA1 subunit and stargazin, a regulatory protein responsible for targeting GluA1 to the synapse (Díaz, 2010). We found that the administration of a saline or morphine challenge produced opposing changes in GluA2 expression at the PSD, with decreased pGluA2 expression at the PSD following a saline challenge (suggesting an increase in GluA2 cell surface expression), and decreased synaptic GluA2 expression observed in the PSD after treatment with a morphine challenge. However, our results from patch-clamp experiments indicate that EPSC amplitude in both groups was decreased after application of naspm, a selective blocker of GluA2-lacking AMPARs. Thus, the data from biochemistry and patch-clamp experiments suggest that an overall increase in GluA2-lacking receptor insertion occurs after administration of a saline or morphine challenge.
Although changes in behavior and AMPAR expression were observed following context-dependent morphine treatment, unpaired morphine treatment did not produce behavioral sensitization and produced few changes in AMPAR expression (i.e., increased GluA2 phosphorylation in the homogenate). An increase in GluA2 phosphorylation could argue against the idea that changes in AMPAR expression were only observed if morphine was paired with the context. However, at the PSD there were no changes in AMPAR expression following unpaired morphine treatment. Overall, these data suggest that associations between contextual stimuli and morphine use can facilitate the development of morphine addiction by enhancing cravings for morphine or through increased drug-seeking behavior. This is supported by studies that reported that exposure to drug-associated stimuli or environments can facilitate relapse (Crombag et al., 2008; Van den Oever et al., 2008), underscoring the importance of drug-associated contexts in the development and maintenance of addiction.
Hippocampal synaptic plasticity is altered following context-dependent behavioral sensitization to morphine
In the present study, we found an enhancement of hippocampal basal synaptic transmission and impairment of LTP 1 week after context-dependent morphine treatment when animals received either a saline or a morphine challenge. The effect of the morphine challenge on basal synaptic transmission was much greater relative to mice that received a saline challenge after context-dependent morphine treatment. A disruption of LTP was observed regardless of whether a saline or morphine challenge were administered, suggesting that this effect is related to prior history of morphine exposure in the environment. Together, these results suggest that prior history of morphine exposure in the context is sufficient to trigger functional changes that may be related to alterations of AMPARs. For instance, the observed alterations in synaptic transmission and plasticity may be due to an increase in synaptic insertion of GluA2-lacking AMPARs induced by repeated morphine treatment. An increase in phosphorylation of GluA1 at S845 can lead to increased channel conductance (Roche et al., 1996). Thus, an increase of pGluA1 levels after a morphine challenge could lead to increased basal synaptic transmission as we observed in our experiments. In addition, it is likely that decreased synaptic levels of GluA2 could magnify this effect since GluA2-containing AMPARs possess lower channel conductance (Swanson et al., 1997). It has been shown that a spike timing-dependent plasticity protocol produces LTP in naive or saline-treated mice but does not induce LTP in cocaine-treated mice, and that this effect of cocaine may be due to a drug-induced saturation of AMPARs at the synapse (Ungless et al., 2001; Luu and Malenka, 2008). Similar mechanisms may be responsible for LTP impairment in morphine-treated mice.
Although the experimental design in the present study does not allow us to conclude that the molecular changes observed 12 h after morphine administration continuously persist over time, the data obtained in this study indicate that the administration of a saline or drug challenge 1 week after context-dependent morphine treatment triggers changes in AMPAR expression, synaptic expression, and behavior, while no changes in AMPAR expression or function can be observed 1 week later, if mice receive initial morphine treatment in the home cage. In addition, the data collected after intrahippocampal infusion of the S845A GluA1 viral vector provide evidence that the initial molecular effects triggered by context-dependent administration of morphine are critical for the expression of behavioral sensitization 1 week later. Further studies may be needed to ascertain whether the administration of the challenge and/or the presentation of the drug-paired environment by themselves may have any additional effects to those triggered by the initial administration of morphine in the paired context.
Conclusions
In conclusion, we find that context-dependent behavioral sensitization to morphine is associated with changes in synaptic AMPAR expression and function that could ultimately trigger alterations in basal synaptic transmission and plasticity in the hippocampus, and that the GluA1 subunit plays a key role in mediating these sensitized responses that occur after drug administration. Several studies have reported that changes in AMPAR distribution are associated with sensitization to psychostimulants (Boudreau and Wolf, 2005; Boudreau et al., 2007; Ghasemzadeh et al., 2009), although the majority of these studies have been performed in brain areas related to the reward pathway, such as the NAc and the VTA. In addition, previous studies reported that withdrawal from cocaine leads to potentiation of accumbal synapses through the insertion of AMPARs (Kourrich et al., 2007; Conrad et al., 2008). These data, along with the results of the present study, suggest that the behavioral effects of opiate and psychostimulant drugs may partially depend on drug-induced alterations in glutamatergic signaling. The targeted disruption of these changes to AMPAR function may prevent the development of morphine-induced context-dependent sensitization and therefore provide support for further investigations of glutamate-based strategies for the treatment of opiate addiction.
Footnotes
This work was supported by National Institutes of Health Grants DA025036 and DA027460 (J.A.M.). We thank Drs. Bruce Hope for helpful discussions and suggestions regarding this manuscript. We also thank Daniel Christoffel for his help with the surgeries and the viral gene transfer, and Dr. Agustin Zapata for technical assistance with the behavioral studies.
The authors declare no competing financial interests.
References
- Barrot M, Olivier JD, Perrotti LI, DiLeone RJ, Berton O, Eisch AJ, Impey S, Storm DR, Neve RL, Yin JC, Zachariou V, Nestler EJ. CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc Natl Acad Sci U S A. 2002;99:11435–11440. doi: 10.1073/pnas.172091899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billa SK, Sinha N, Rudrabhatla SR, Morón JA. Extinction of morphine-dependent conditioned behavior is associated with increased phosphorylation of the GluR1 subunit of AMPA receptors at hippocampal synapses. Eur J Neurosci. 2009;29:55–64. doi: 10.1111/j.1460-9568.2008.06560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billa SK, Liu J, Bjorklund NL, Sinha N, Fu Y, Shinnick-Gallagher P, Morón JA. Increased insertion of glutamate receptor 2-lacking alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors at hippocampal synapses upon repeated morphine administration. Mol Pharmacol. 2010;77:874–883. doi: 10.1124/mol.109.060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgkvist A, Valjent E, Santini E, Hervé D, Girault JA, Fisone G. Delayed, context- and dopamine D1 receptor-dependent activation of ERK in morphine-sensitized mice. Neuropharmacology. 2008;55:230–237. doi: 10.1016/j.neuropharm.2008.05.028. [DOI] [PubMed] [Google Scholar]
- Bossert JM, Stern AL, Theberge FR, Cifani C, Koya E, Hope BT, Shaham Y. Ventral medial prefrontal cortex neuronal ensembles mediate context-induced relapse to heroin. Nat Neurosci. 2011;14:420–422. doi: 10.1038/nn.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Wolf ME. Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci. 2005;25:9144–9151. doi: 10.1523/JNEUROSCI.2252-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Reimers JM, Milovanovic M, Wolf ME. Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci. 2007;27:10621–10635. doi: 10.1523/JNEUROSCI.2163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Haile CN, Coppersmith R, Hayashi Y, Malinow R, Neve RL, Nestler EJ. Distinct sites of opiate reward and aversion within the midbrain identified using a herpes simplex virus vector expressing GluR1. J Neurosci. 2000;20:RC62. doi: 10.1523/JNEUROSCI.20-05-j0002.2000. (1–5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- Choi J, Ko J, Park E, Lee JR, Yoon J, Lim S, Kim E. Phosphorylation of stargazin by protein kinase A regulates its interaction with PSD-95. J Biol Chem. 2002;277:12359–12363. doi: 10.1074/jbc.M200528200. [DOI] [PubMed] [Google Scholar]
- Choi KH, Edwards S, Graham DL, Larson EB, Whisler KN, Simmons D, Friedman AK, Walsh JJ, Rahman Z, Monteggia LM, Eisch AJ, Neve RL, Nestler EJ, Han MH, Self DW. Reinforcement-related regulation of AMPA glutamate receptor subunits in the ventral tegmental area enhances motivation for cocaine. J Neurosci. 2011;31:7927–7937. doi: 10.1523/JNEUROSCI.6014-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffel DJ, Golden SA, Dumitriu D, Robison AJ, Janssen WG, Ahn HF, Krishnan V, Reyes CM, Han MH, Ables JL, Eisch AJ, Dietz DM, Ferguson D, Neve RL, Greengard P, Kim Y, Morrison JH, Russo SJ. IκB kinase regulates social defeat stress-induced synaptic and behavioral plasticity. J Neurosci. 2011;31:314–321. doi: 10.1523/JNEUROSCI.4763-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron. 2000;27:107–119. doi: 10.1016/s0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–121. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crombag HS, Bossert JM, Koya E, Shaham Y. Review. Context-induced relapse to drug seeking: a review. Philos Trans R Soc Lond B Biol Sci. 2008;363:3233–3243. doi: 10.1098/rstb.2008.0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz E. Regulation of AMPA receptors by transmembrane accessory proteins. Eur J Neurosci. 2010;32:261–268. doi: 10.1111/j.1460-9568.2010.07357.x. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Ferrario CR, Li X, Wang X, Reimers JM, Uejima JL, Wolf ME. The role of glutamate receptor redistribution in locomotor sensitization to cocaine. Neuropsychopharmacology. 2010;35:818–833. doi: 10.1038/npp.2009.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in sterotaxic coordinates. New York: Academic; 2007. [Google Scholar]
- Ghasemzadeh MB, Mueller C, Vasudevan P. Behavioral sensitization to cocaine is associated with increased glutamate receptor trafficking to the postsynaptic density after extended withdrawal period. Neuroscience. 2009;159:414–426. doi: 10.1016/j.neuroscience.2008.10.027. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Huganir RL. Tyrosine phosphorylation and regulation of the AMPA receptor by SRC family tyrosine kinases. J Neurosci. 2004;24:6152–6160. doi: 10.1523/JNEUROSCI.0799-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Suppiramaniam V, Wooten MW. Posttranslational modifications and receptor-associated proteins in AMPA receptor trafficking and synaptic plasticity. Neurosignals. 2006;15:266–282. doi: 10.1159/000105517. [DOI] [PubMed] [Google Scholar]
- Kato AS, Gill MB, Yu H, Nisenbaum ES, Bredt DS. TARPs differentially decorate AMPA receptors to specify neuropharmacology. Trends Neurosci. 2010;33:241–248. doi: 10.1016/j.tins.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Kourrich S, Rothwell PE, Klug JR, Thomas MJ. Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci. 2007;27:7921–7928. doi: 10.1523/JNEUROSCI.1859-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koya E, Golden SA, Harvey BK, Guez-Barber DH, Berkow A, Simmons DE, Bossert JM, Nair SG, Uejima JL, Marin MT, Mitchell TB, Farquhar D, Ghosh SC, Mattson BJ, Hope BT. Targeted disruption of cocaine-activated nucleus accumbens neurons prevents context-specific sensitization. Nat Neurosci. 2009;12:1069–1073. doi: 10.1038/nn.2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- Lee HT, Xu H, Nasr SH, Schnermann J, Emala CW. A1 adenosine receptor knockout mice exhibit increased renal injury following ischemia and reperfusion. Am J Physiol Renal Physiol. 2004;286:F298–F306. doi: 10.1152/ajprenal.00185.2003. [DOI] [PubMed] [Google Scholar]
- Liang J, Chen JH, Chen XH, Peng YH, Zheng XG. Gene expression of conditioned locomotion and context-specific locomotor sensitization controlled by morphine-associated environment. Behav Brain Res. 2011a;216:321–331. doi: 10.1016/j.bbr.2010.08.012. [DOI] [PubMed] [Google Scholar]
- Liang J, Zheng X, Chen J, Li Y, Xing X, Bai Y, Li Y. Roles of BDNF, dopamine D3 receptors, and their interactions in the expression of morphine-induced context-specific locomotor sensitization. Eur Neuropsychopharmacol. 2011b;21:825–834. doi: 10.1016/j.euroneuro.2010.12.006. [DOI] [PubMed] [Google Scholar]
- Luu P, Malenka RC. Spike timing-dependent long-term potentiation in ventral tegmental area dopamine cells requires PKC. J Neurophysiol. 2008;100:533–538. doi: 10.1152/jn.01384.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man HY, Lin JW, Ju WH, Ahmadian G, Liu L, Becker LE, Sheng M, Wang YT. Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron. 2000;25:649–662. doi: 10.1016/s0896-6273(00)81067-3. [DOI] [PubMed] [Google Scholar]
- Matus A, Ackermann M, Pehling G, Byers HR, Fujiwara K. High actin concentrations in brain dendritic spines and postsynaptic densities. Proc Natl Acad Sci U S A. 1982;79:7590–7594. doi: 10.1073/pnas.79.23.7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers RA, Zavala AR, Speer CM, Neisewander JL. Dorsal hippocampus inhibition disrupts acquisition and expression, but not consolidation, of cocaine conditioned place preference. Behav Neurosci. 2006;120:401–412. doi: 10.1037/0735-7044.120.2.401. [DOI] [PubMed] [Google Scholar]
- Morón JA, Brockington A, Wise RA, Rocha BA, Hope BT. Dopamine uptake through the norepinephrine transporter in brain regions with low levels of the dopamine transporter: evidence from knock-out mouse lines. J Neurosci. 2002;22:389–395. doi: 10.1523/JNEUROSCI.22-02-00389.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morón JA, Abul-Husn NS, Rozenfeld R, Dolios G, Wang R, Devi LA. Morphine administration alters the profile of hippocampal postsynaptic density-associated proteins: a proteomics study focusing on endocytic proteins. Mol Cell Proteomics. 2007;6:29–42. doi: 10.1074/mcp.M600184-MCP200. [DOI] [PubMed] [Google Scholar]
- Morón JA, Gullapalli S, Taylor C, Gupta A, Gomes I, Devi LA. Modulation of opiate-related signaling molecules in morphine-dependent conditioned behavior: conditioned place preference to morphine induces CREB phosphorylation. Neuropsychopharmacology. 2010;35:955–966. doi: 10.1038/npp.2009.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moult PR, Gladding CM, Sanderson TM, Fitzjohn SM, Bashir ZI, Molnar E, Collingridge GL. Tyrosine phosphatases regulate AMPA receptor trafficking during metabotropic glutamate receptor-mediated long-term depression. J Neurosci. 2006;26:2544–2554. doi: 10.1523/JNEUROSCI.4322-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu L, Bao GB, Xu NJ, Ma L, Pei G. Hippocampal long-term potentiation is reduced by chronic opiate treatment and can be restored by re-exposure to opiates. J Neurosci. 2002;22:1914–1921. doi: 10.1523/JNEUROSCI.22-05-01914.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-p. [DOI] [PubMed] [Google Scholar]
- Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- Rothwell PE, Gewirtz JC, Thomas MJ. Episodic withdrawal promotes psychomotor sensitization to morphine. Neuropsychopharmacology. 2010;35:2579–2589. doi: 10.1038/npp.2010.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo SJ, Dietz DM, Dumitriu D, Morrison JH, Malenka RC, Nestler EJ. The addicted synapse: mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci. 2010;33:267–276. doi: 10.1016/j.tins.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen F, Meredith GE, Napier TC. Amphetamine-induced place preference and conditioned motor sensitization requires activation of tyrosine kinase receptors in the hippocampus. J Neurosci. 2006;26:11041–11051. doi: 10.1523/JNEUROSCI.2898-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein EL, Chetkovich DM. Regulation of stargazin synaptic trafficking by C-terminal PDZ ligand phosphorylation in bidirectional synaptic plasticity. J Neurochem. 2010;113:42–53. doi: 10.1111/j.1471-4159.2009.06529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson GT, Kamboj SK, Cull-Candy SG. Single-channel properties of recombinant AMPA receptors depend on RNA editing, splice variation, and subunit composition. J Neurosci. 1997;17:58–69. doi: 10.1523/JNEUROSCI.17-01-00058.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Van den Oever MC, Goriounova NA, Li KW, Van der Schors RC, Binnekade R, Schoffelmeer AN, Mansvelder HD, Smit AB, Spijker S, De Vries TJ. Prefrontal cortex AMPA receptor plasticity is crucial for cue-induced relapse to heroin-seeking. Nat Neurosci. 2008;11:1053–1058. doi: 10.1038/nn.2165. [DOI] [PubMed] [Google Scholar]
- Vanderschuren LJ, De Vries TJ, Wardeh G, Hogenboom FA, Schoffelmeer AN. A single exposure to morphine induces long-lasting behavioural and neurochemical sensitization in rats. Eur J Neurosci. 2001;14:1533–1538. doi: 10.1046/j.0953-816x.2001.01775.x. [DOI] [PubMed] [Google Scholar]
- Vorel SR, Liu X, Hayes RJ, Spector JA, Gardner EL. Relapse to cocaine-seeking after hippocampal theta burst stimulation. Science. 2001;292:1175–1178. doi: 10.1126/science.1058043. [DOI] [PubMed] [Google Scholar]
- Wang JQ, Arora A, Yang L, Parelkar NK, Zhang G, Liu X, Choe ES, Mao L. Phosphorylation of AMPA receptors: mechanisms and synaptic plasticity. Mol Neurobiol. 2005;32:237–249. doi: 10.1385/MN:32:3:237. [DOI] [PubMed] [Google Scholar]
- Weiss F. Neurobiology of craving, conditioned reward and relapse. Curr Opin Pharmacol. 2005;5:9–19. doi: 10.1016/j.coph.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Wolf ME, Sun X, Mangiavacchi S, Chao SZ. Psychomotor stimulants and neuronal plasticity. Neuropharmacology. 2004;47(Suppl 1):61–79. doi: 10.1016/j.neuropharm.2004.07.006. [DOI] [PubMed] [Google Scholar]