

Abstract
A protocol for the enantioselective [3+2] cycloaddition of trimethylenemethane with electron-deficient olefins has been developed. The synthesis of novel phosphoramidite ligands was critical in this effort, and the preparation and reactivity of these ligands is detailed. The evolution of the ligand design, commencing with acyclic amine-derived phosphoramidites and leading to cyclic pyrrolidine and azetidine structures is discussed. The conditions developed to effect an asymmetric TMM reaction using 2-trimethylsilylmethyl allyl acetate were shown to be tolerant of a wide variety of alkene acceptors, providing the desired methylenecyclopentanes with high levels of enantioselectivity. The donor scope was also explored and substituted systems were tolerated, including one bearing a nitrile moiety. These donors were reactive with unsaturated acylpyrroles, giving the product cyclopentane rings bearing three stereocenters in high enantioselectivity and complete diastereoselectivity.
Introduction
The transition-metal catalyzed [3+2] trimethylenemethane (TMM) cycloaddition reaction (eq 1) is a versatile method for the chemo-, regio-, and diastereoselective construction of highly substituted five-membered rings.1 Utilization of Pd-TMM complexes derived from 3-acetoxy-2-trimethylsilylmethyl-1-propene (1) has resulted in the efficient catalytic syntheses of carbocycles2 and heterocycles.3 Although this methodology was disclosed over thirty years ago by our laboratory4, a general asymmetric variant of the cycloaddition remained elusive until 2006.5 In this full account, we will describe the development of the asymmetric TMM reaction and its application in the synthesis of complex carbocycles.
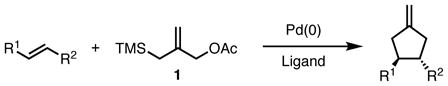 |
(1) |
The catalytic cycle for the TMM reaction of silyl allylic acetates (Scheme 1) was proposed early and has withstood the test of time.6 The zwitterionic Pd-TMM intermediate 2 is generated in situ by metal promoted ionization of the allylic acetate functionality in 1, followed by desilylation promoted by the displaced acetate. Complex 2 is nucleophilic in nature, and can add to electron-deficient systems, including olefins, aldehydes and imines. Addition of the nucleophilic Pd-TMM complex to the olefin gives intermediate 3, which is followed by collapse of the zwitterionic intermediate via intramolecular attack of the soft carbon nucleophile onto the π-allylpalladium. This affords the desired product 4, which can undergo a base-promoted isomerization of the exocyclic olefin to give the internal alkene depending on the reaction conditions.
Scheme 1.

Catalytic Cycle for the Pd-TMM Cycloaddition.
The mechanism in Scheme 1 illustrates the difficulty in the development of an asymmetric variant of the TMM reaction. Nucleophilic addition is the enantiodiscriminating step. The coordinated chiral ligands on the palladium are spatially remote to the reaction center, and this distance likely decreases their ability to transfer stereochemical information during the addition. Evidence for a distal approach was obtained in early mechanistic studies.7 Accordingly, prior to 2006, attempts to induce asymmetry into the TMM reaction had largely been limited to the use of chiral auxiliaries on the acceptor;8 chiral catalysis was largely unsuccessful. In fact, for several years, a single example of a catalytic, asymmetric TMM reaction stood alone in the literature: Hayashi disclosed an asymmetric [3+2] cycloaddition of 2-(sulfonylmethyl)-2-propenyl carbonate with simple esters and ketones in 1989 (Scheme 2).9 While the acceptor scope was limited to methyl vinyl ketone and methyl acrylate, and multiple equivalents were required, this represented an important advance in the development of the asymmetric reaction.
Scheme 2.

Hayashi’s Catalytic, Asymmetric Methylenecyclopentane Synthesis.
In the Trost group, work towards the development of an asymmetric reaction spanned more than two decades, and thus a comprehensive discussion is outside the scope of this account. It should be noted that the potential of phosphoramidite ligands in the asymmetric transformation was recognized by our group early after applications of this class of ligand began to appear in the literature. While conventional wisdom states that bidentate ligands provide the highest levels of reactivity and selectivity in asymmetric catalysis, an increasing body of literature demonstrates the utility of chiral monodentate phosphorus ligands.10 Indeed, in many asymmetric reactions, these structures have proven to be superior to bidentate ligands. Phosphoramidite ligands are one such class of privileged structures.11 Initially disclosed as a derivatization method for the determination of enantiomeric excesses of chiral amines,12 phosphoramidites were soon recognized as competent additives in catalytic, asymmetric conjugate additions13 and numerous other reactions.11 Due to the success of both phosphites and hexamethylphosphorus triamide as ligands in the achiral TMM reaction, we saw potential in the unique phosphoramidite ligand class containing both axial and point chiral elements. Our initial hit is shown in Scheme 3. Use of Feringa ligand L1 with the parent donor 1 provided the desired cycloadduct 6 of the unsaturated oxazolidinone 5 in 69% yield and 29% enantiomeric excess.14 After further experimentation, it became clear that ligand optimization would be required to reach the desired enantioselectivity level.
Scheme 3.

Initial Discovery of the Phosphoramidite-Catalyzed Asymmetric TMM Reaction.
Design and Application of A cyclic Amine Phosphoramidite Ligands to the Asymmetric Trimethylenemethane Cycloaddition Reaction
Of the diverse transformations promoted by phosphoramidites, a fairly substantial number rely on a few relatively similar ligand structures, specifically containing a BINOL backbone and secondary amine. This, in conjunction with a 2005 report from Alexakis which details the effects of structural changes to the amine segment of the phosphoramidite ligand on iridium-catalyzed asymmetric allylic alkylation,15 prompted us to study the effect of modifications of the amine on the TMM reaction.
For our initial studies (Scheme 4), we chose benzylidene acetone 7 as the TMM acceptor. Initially we chose a 4:1 ligand:palladium ratio, but quickly found that a 2:1 ratio gave the same product selectivity. Our standard reaction time was four hours, though the reaction was generally complete within 1–2 hours as determined by GC. As a basis for comparison, we first screened ligand L1. Under our standard conditions, we observed a 75% conversion and 33% enantiomeric excess at room temperature. We were quickly pleased to observe that small structural changes did in fact produce significant variation in reactivity and selectivity. Installation of a single ortho-methyl group (L2), 1-naphthyl (L3), or 2-naphthyl (L4) gave cyclopentane 8a with ee values comparable to the Feringa ligand but different levels of conversion. Replacing the ortho alkyl group with an alkoxy as in L5 or L6 led to a very quick reaction with high conversion but similar ee. We speculate that this may be due to a hemilabile coordination between the oxygen and palladium, providing a more active catalyst system. Increasing the steric bulk around both phenyl rings (L7) provided a nearly inactive catalyst system in contrast to the work of Alexakis.15 The conversion was restored with bis-methoxy ligand L8, perhaps due to a hemilabile coordination.
Scheme 4.

Initial Screen of Acyclic Amine Phosphoramidite Ligands.
While electronic effects of the system appear to be responsible for conversion, we speculated that steric factors may be the primary dictator of enantioselectivity. In order to test this, we first removed a methyl group entirely as in L9–L12. No reactivity was observed in any case and it became clear that catalyst decomposition with these ligands was very fast. Changing the relative stereochemistry of the chiral amine subunit as in L13 to L20 had a significant impact. Inversion of the stereocenter adjacent to the unsubstituted ring (L13) was not beneficial, but inversion of the stereocenter proximal to the substituted ring (L14–L18) substantially improved ee. The presence of a hemilabile alkoxy group again improved reactivity with L17 giving the product in 84% ee and 92% conversion at −25 °C. Interestingly, all of these ligands give product of the same absolute configuration, indicating that the BINOL is responsible for the overall product stereochemistry. Increasing the bulk closer to phosphorus (L19–L20) gave good conversion but decreased enantioselectivity.
In addition to the ligands shown in Scheme 4, other catalysts not containing the bis-benzylic and (R)-BINOL motifs were evaluated under the same conditions (Figure 1). In no case were satisfactory results obtained. While reasonable enantiomeric excess was observed with ligand L24 containing only a single stereocenter on the amine portion of the ligand, conversion was unacceptably low. Self-adapting catalyst L25, which proved useful in conjugate additions,16 showed no reactivity in the TMM cycloaddition. TADDOL-derived phosphoramidite L26 was also unreactive.
Figure 1.

Additional Acyclic Amine Ligands Evaluated in the [3+2] Cycloaddition.
Design and Application a Pyrrolidine Phosphoramidite Ligand to the Asymmetric Cycloaddition Reaction
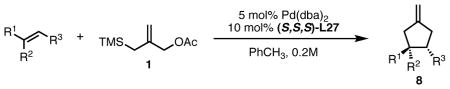
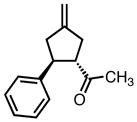
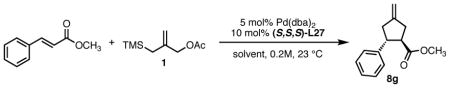
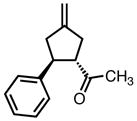
While modification of the acyclic amines led to respectable levels of enantioselectivity, we felt that more significant structural changes would be necessary to further elevate selectivity to the desired levels. As steric factors clearly play a significant role in both the conversion and enantioselectivity, we envisioned restricting the flexibility of amine fragment through constraint to a pyrrolidine, as this could eliminate conformations that lead to lower levels of selectivity. Additionally, the pyrrolidine is less sterically demanding. Since we previously observed that very bulky systems show decreased reactivity, this could provide a more active catalyst system. We first targeted diphenylpyrrolidine ligand L27 which had been synthesized previously and utilized in asymmetric conjugate addition reactions.17 We were gratified to observe an immediate jump in both reactivity and selectivity using this ligand, as compared to the majority of the acyclic amine-derived ligands. Under the standard conditions, the starting material was completely consumed within 30 minutes at room temperature to give the product in 76% isolated yield and 72% ee, which increased to 82% ee at −25 °C (Scheme 5).
Scheme 5.

Reaction of Benzylidene Acetone and TMS Propenyl Acetate Using Ligand (R,R,R)-L27.
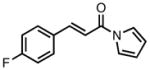
The pyrrolidine-based catalyst system L27 represented the most general and selective one for the cycloaddition to date. The substrate scope is shown in Table 1. To avoid confusion, it should be noted that these experiments were conducted using (S,S,S)-L27 unless otherwise stated. The absolute stereochemistry has been confirmed by analogy to a crystal structure obtained of a derivative of the cycloadduct of β-nitrostyrene with the parent donor.18 The reaction proved general for a variety of systems. Both aliphatic (entries 1–3) and aromatic (entries 4–6) ketones functioned in the reaction. As can be seen in entry 3, aliphatic groups on the olefin were also tolerated. The yields were good in all cases with enantioselectivities ranging from moderate to good in these systems. The reaction performed well at temperatures as low as −25 °C, with an increase in enantioselectivity of approximately 10% being observed upon lowering the temperature from 23 °C to −25 °C (entries 1–3). Esters (entry 7) and nitriles (entry 8) were tolerated, albeit with reduced enantioselectivity. Substituted tetralones proved to be excellent substrates for this reaction (entries 9–13) allowing the formation of an all-carbon quaternary center in enantioselectivities as high as 92%. We believe the increase in selectivity for these substrates has to do with their steric bulk, which most likely causes increased discrimination in the approach of the substrate to the palladium-ligand-donor complex, and therefore, the initial bond-forming event.
Table 1.
Initial Substrate Scope of the [3+2] Cycloaddition.a
 | |||||
|---|---|---|---|---|---|
| entry | substrate | product | temp (°C) | yield (%) | ee (%) |
| 1b |
|
 8a |
23 | 76 | 72 |
| −25 | 63 | 82 | |||
| 2 |
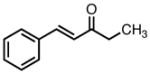
|
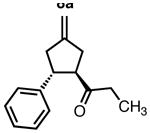 8b |
23 | 84 | 73 |
| −25 | 72 | 83 | |||
| 3 |
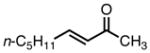
|
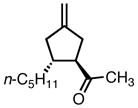 8c |
23 | 72 | 73 |
| −25 | 79 | 84 | |||
| 4 |
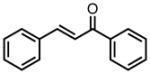
|
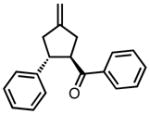 8d |
−25 | 83 | 80 |
| 5 |
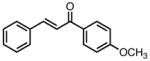
|
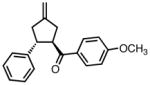 8e |
−25 | 73 | 72 |
| 6 |
|
 8f |
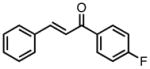
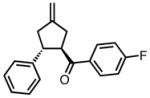
−25 | 80 | 74 |
| 7 |
|
 8g |
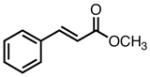
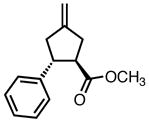
23 | 80 | 58 |
| 0 | 81 | 62 | |||
| 8 |
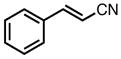
|
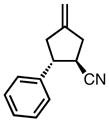 8h |
0 | 78 | 58 |
| 9 |
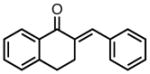
|
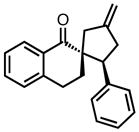 8i |
23 | 99 | 85 |
| −25 | 94 | 92 | |||
| 10 |
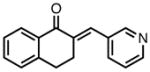
|
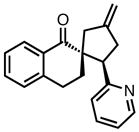 8j |
23 | 78 | 83 |
| −25 | 87 | 86 | |||
| 11 |
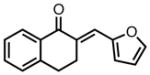
|
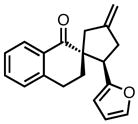 8k |
23 | 71 | 73 |
| −25 | 60 | 82 | |||
| 12 |
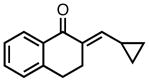
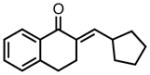
|
 8l |
23 | 95 | 79 |
| −25 | 70 | 87 | |||
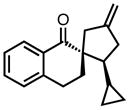
| 13 |
|
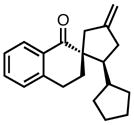 8m |
0 | 53 | 78 |
All reactions were performed at 0.2M in toluene with 5% Pd(dba)2, 10% ligand, 1.6 equiv donor 1, and stirred for 2–24 hours as monitored by GC for disappearance of the acceptor. Yields are isolated values; ee’s were determined by chiral HPLC or chiral GC.
Reaction performed with (R,R,R)-L27.
We also evaluated the effect of solvent on the asymmetric reaction (Table 2). Although toluene and THF have been the solvents of choice in the achiral reaction, we felt that an examination of solvent effect on yield and selectivity of this process using the new catalyst system was warranted. Reactions conducted in THF, diethyl ether, and DME gave good yields with modest ee values slightly lower than those observed in toluene (entries 2–4), while reactions in DCM, CH3CN, DMF, and dioxane gave low conversion to the cyclopentane product. Thus, toluene appears optimal.
Table 2.
Solvent Effects on Asymmetric [3+2] Cycloaddition.
 | |||
|---|---|---|---|
| entry | solvent | yield (%) | ee (%) |
| 1 | toluene | 80 | 58 |
| 2 | THF | 68 | 48 |
| 3 | DME | 80 | 53 |
| 4 | diethyl ether | 69 | 55 |
| 5 | DCM | 10 | 32 |
| 6 | CH3CN | 0 | 0 |
| 7 | DMF | 0 | 0 |
| 8 | dioxane | 35 | 39 |
Further Modification of the Pyrrolidine Phosphoramidite Ligand and Expanded Substrate Scope
With the discovery that the diphenylpyrrolidine moiety was very important for both selectivity and reactivity, we wanted to determine if modifications of the backbone could further improve the reaction. Ligands L28–L33 (Figure 2) were prepared and tested using benzylidene acetone as the acceptor under the standard conditions (5 mol% Pd(dba)2, 10 mol% ligand, 23 °C, toluene). Removal of the axial chirality element through use of a catechol (L28) or biphenyl (L29) backbone gave inactive catalysts. We tested a ligand containing a partially hydrogenated BINOL backbone (L30) with the hope that the increased bulk of the system would provide additional enantiodiscrimination and make the reaction more selective. Unfortunately, while full conversion was observed, the enantiomeric excess dropped to 61% from the 72% afforded by the fully unsaturated system. Under identical conditions, reversal of the BINOL enantiomer gave an inactive catalyst (L31), as did methyl and phenyl substitutions at the aryl moiety 3- and 3′-positions (L32–L33). We suspected that substitution at both the 3- and 3′-positions may increase the steric bulk to such an extent that substrate approach is prohibited.
Figure 2.

Pyrrolidine Phosphoramidite Ligands with Modified Backbones.
Due to our earlier success in improving the reaction selectivity by changes to the amine segment of the ligand, we hoped alteration of the pyrrolidine would also prove fruitful. Synthesis of the modified pyrrolidines was challenging as low solubility of intermediates prevented their preparation in a manner analogous to the method used to prepare the diphenylpyrrolidine.17 We decided to develop a new route to the desired 2,5-disubstituted pyrrolidines which would allow for maximum diversity, including preparation of unsymmetrically substituted compounds. In 2006, Campos disclosed a one-pot, sequential chiral lithiation, transmetallation, and Negishi coupling of N-Boc pyrrolidine which could be used to efficiently generate substituted pyrrolidines.19 While the authors include only a single example of the preparation of a disubstituted pyrrolidine, we hoped that we could make this a general process.
Our revised strategy for ligand synthesis is shown in Scheme 6. Carbamate protection of pyrrolidine proceeds in high yield and is followed by installation of the first aryl group. As shown in Table 3, the results are good, proceeding in high yields and enantiomeric excesses (>93%). This is not surprising as the enantiodiscriminating event is the lithiation step and thus the final value is independent of the identity of the aryl bromide utilized.
Scheme 6.

General Method for Synthesis of pyrrolidine Derived Phosphoramidite Ligands.
Table 3.
Synthesis of Mono- and Disubstituted Pyrrolidines.
| entry | R1 | ref | yield (%) | ee (%) | R2 | ref | yield (%) | dr |
|---|---|---|---|---|---|---|---|---|
| 1 | C6H5 | 11aa | 51 | 24:1 | ||||
| 2 | C6H5 | 10a | 81 | 93 | 2-MeOC6H4 | 11ab | 44 | 24:1 |
| 3 | 2-Naphthyl | 11ac | 45 | >50:1 | ||||
| 4 | 2-MeOC6H4 | 11ba | 37 | 16:1 | ||||
| 5 | 2-MeOC6H4 | 10b | 77 | 93 | C6H5 | 11ab | 33a | 21:1 |
| 6 | 2-Naphthyl | 11ca | 50b | >50:1 | ||||
| 7 | 2-Naphthyl | 10c | 75 | 95 | 1-Naphthyl | 11cb | 45b | >50:1 |
| 8 | 3,5-t-BuC6H3 | 11cc | 61b | >50:1 | ||||
| 9 | 3-Biphenyl | 10d | 86 | ND | 3-Biphenyl | 11d | 49b | >50:1 |
| 10 | 3,5-PhC6H3 | 10e | 70 | ND | 3,5-PhC6H3 | 11e | 48b | >50:1 |
| 11 | 4-Biphenyl | 10f | 82 | 94 | 4-Biphenyl | 11f | 23b,c | >50:1 |
| 12 | 1-Naphthyl | 10g | 83 | >93d | 1-Naphthyl | 11g | NDb,e | ND |
| 13 | 3,5-t-BuC6H3 | 10h | 54 | ND | 3,5-t-BuC6H3 | 11h | 0 | --- |
Reaction performed at 23 °C
With toluene cosolvent
After recrystallization from CH2Cl2:Pentane
Some overlap in HPLC trace
Could not completely purify by column chromatography, purified after deprotection.
Installation of the second aryl group proved more challenging (Table 3). While we were able to repeat the synthesis of diphenylpyrrolidine 11aa at room temperature (entry 1), the reaction failed when attempting to form ortho-methoxyphenyl pyrrolidine 11ab or 2-naphthyl pyrrolidine 11ac. In fact, significant reaction was only observed at ambient temperature in the synthesis of 11ab when the unsubstituted phenyl group was installed second (entry 5). In this case, the yield was only 33%. We suspected the low solubility of the organozinc intermediate in ethereal solvents in combination with the slower rate of oxidative addition of palladium to the more electron-rich aryl rings in the Negishi coupling might be the cause and accordingly performed the final step of the sequence at 60 °C. This immediately solved the lack of reactivity and produced the desired products (entries 1–4) in good yield. The diastereoselectivity was excellent, giving almost exclusively the desired trans-pyrrolidines. A second problem arose when we moved to more bulky substrates. We observed that the solubility of the anion in TBME was not very high as substantial precipitate was observed during the deprotonation step. Using our previous conditions, bis-2-naphthyl pyrrolidine 11ca was obtained in only 26% yield and many other disubstituted pyrrolidines did not form, even in trace quantities. To aid in the anion solubility, we added 10–30 vol% toluene relative to TBME and found that yields increased substantially with no loss of diastereoselectivity. Using these conditions, we were able to form sterically more bulky products including bis-2-naphthyl pyrrolidine 11ca (entry 6), mixed 1-naphthyl-2-naphthyl pyrrolidine 11cb (entry 7), mixed 2-naphthyl-3,5-tert-butylphenyl pyrrolidine 11cc (entry 8), bis-3-biphenyl pyrrolidine 11d (entry 9), and the very bulky bis-3,5-diphenylphenyl pyrrolidine 11e (entry 10), all in good yields and excellent diastereoselectivities. Significant quantities of the monosubstituted pyrrolidines could be recovered after the reaction, but the ee was considerably lower than that of the starting material, indicating competitive deprotonation between the disubstituted and more acidic (and more hindered) trisubstituted centers. Most likely, the transmetallation or Negishi coupling fails on the trisubstituted center for steric reasons, leading to nonselective protonation during workup and recovery of the pyrrolidine.
Having successfully generalized Campos’ lithiation strategy to form disubstituted pyrrolidines in high yields and selectivities, we wanted to further elaborate the structures to phosphoramidite ligands (Table 4). After deprotection (20 equivalents TFA/CH2Cl2, 4h, 23 °C), the free pyrrolidines 12a–g could easily be transformed into the desired ligands. A 0.5M solution of (R)-BINOL chlorophosphite 13 was added to the pyrrolidine, DMAP, and triethylamine in toluene to form the ligands. Monosubstituted pyrrolidine ligand L34 was prepared from the corresponding amine to determine if both substitutions are in fact necessary. The yields for the ligand synthesis were generally good regardless of the bulk of the pyrrolidine.
Table 4.
Deprotection of N-Boc Pyrrolidines and Phosphoramidite Synthesis.
| entry | N-Boc pyrrolidine | R1 | R2 | pyrrolidine | yield (%) | ligand | yield (%) |
|---|---|---|---|---|---|---|---|
| 1 | 10a | C6H5 | H | 12a | 92 | L34 | 38 |
| 2 | 11ab | C6H5 | 2-OMeC6H4 | 12ab | 97 | L35 | 81 |
| 3 | 11ac | C6H5 | 2-Naphthyl | 12ac | 100 | L36 | 82 |
| 4 | 11ba | 2-MeOC6H4 | 2-MeOC6H4 | 12ba | 71 | L37 | 41 |
| 5 | 11ca | 2-Naphthyl | 2-Naphthyl | 12ca | 98 | L38 | 84 |
| 6 | 11cb | 2-Naphthyl | 1-Naphthyl | 12cb | 100 | L39 | 77 |
| 7 | 11cc | 2-Naphthyl | 3,5-t-BuC6H3 | 12cc | 86 | L40 | 68 |
| 8 | 11d | 3-Biphenyl | 3-Biphenyl | 12d | 97 | L41 | 40 |
| 9 | 11e | 3,5-PhC6H3 | 3,5-PhC6H3 | 12e | 92 | L42 | 68 |
| 10 | 11f | 4-Biphenyl | 4-Biphenyl | 12f | 99 | L43 | 82 |
| 11 | 11g | 1-Naphthyl | 1-Naphthyl | 12g | 10a | L44 | 60 |
Isolated yield for two steps from 10g
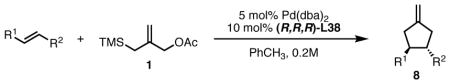
With the series of cyclic amine derived phosphoramidites in hand, we wanted to compare the levels of reactivity and selectivity with what we had observed in our previously explored catalyst systems (Scheme 7). We used identical conditions to those used to screen the acyclic systems. As was observed with the acyclic structures, ligands containing methylene centers adjacent to nitrogen as in L34 are unproductive in the cycloaddition. We immediately observed that the disubstituted cyclic systems were far superior to the acyclic systems in terms of reactivity. Even at −25 °C, complete conversion was observed in every case. The electronic nature of the aromatic rings proved to be less important than was previously observed in the acyclic systems. Installation of a single ortho-methoxy group (L35) showed essentially the same selectivity as the parent diphenyl ligand. A second methoxy substitution (L37) raised the selectivity by a small amount. Steric factors turned out to be differential in improving enantioselectivity. While the biphenyl system L43 gave only a small increase in ee, installation of a naphthyl group (L36) had a significant effect, increasing the ee to 84% at room temperature and 91% at −25 °C. This is the first time we had observed such a high selectivity in this system. The bis-1-naphthyl ligand L44 gave a 91% ee at room temperature and bis-2-naphthyl ligand showed the best results observed in the series, giving the cyclopentane product in 98% enantiomeric excess at room temperature. We also wanted to test the effect of ring size on reactivity and selectivity, and therefore screened the azetidine phosphoramidite L45. The lower steric demand led to a catalyst of higher reactivity, giving complete conversion in less than 15 minutes, but in only 52% ee. Clearly, steric factors are the most important in determining the enantioselectivity of these catalyst systems.
Scheme 7.

Selected Pyrrolidine and Azetidine Phosphoramidite Ligand Results.
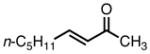
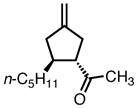
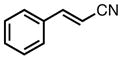
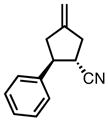
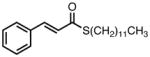
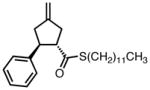
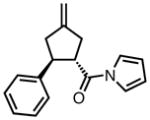
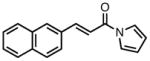
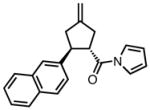
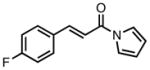
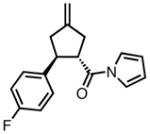
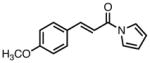
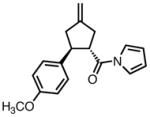
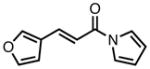
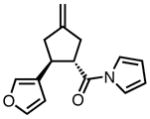
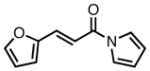
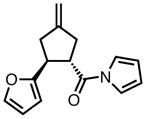
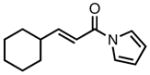
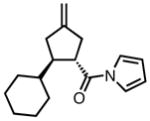
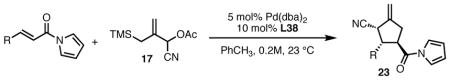
With ligand L38 in hand, we proceeded to examine the scope of the asymmetric TMM reaction. Gratifyingly, this ligand proved superior to those previously studied systems in terms of scope and selectivity. The substrate range was considerably broader and where comparisons were obtained, enantioselectivities were uniformly higher (Table 5). Both aliphatic and aromatic ketones performed well in the reaction (entries 1–3) providing the respective cyclopentanes in good yields and enantioselectivities of greater than 90%. In cases where conversion was low, the temperature of the reaction could be raised, generally with the effect of substantially increasing conversion with only a minor drop in enantioselectivity. The tolerance of this catalyst to elevated temperatures may explain the increased substrate scope using the naphthyl ligand. Esters were tolerated (entries 4–5) with ee values more than 30% higher than those obtained with the diphenyl ligand. An unsaturated nitrile gave good yield and selectivity (entry 6). Thioesters functioned well (entry 7) as did a Weinreb amide (entry 8). Conversion was reduced in the latter case, though the enantioselectivity remained high at temperatures as high as 75 °C. Oxazolidinones could be used in the reaction (entries 9–10), but the enantioselectivity dropped. Unfortunately, a cis-olefin gave no product (entry 11).
Table 5.
Substrate Scope of the [3+2] Cycloaddition with L38.a
 | |||||
|---|---|---|---|---|---|
| entry | substrate | product | temp (°C) | yield (%) | ee (%) |
| 1 |
|
 8a |
23 | 91 | 98 |
| 2 |
|
 8c |
23 | 80 | 95 |
| 3 |
|
 8f |
45 | 71 | 92 |
| 4 |
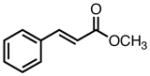
|
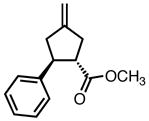 8g |
23 | 71b | 95 |
| 45 | 97 | 90 | |||
| 5 |
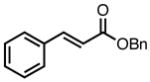
|
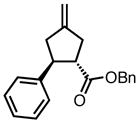 8n |
45 | 70 | 94c |
| 6 |
|
 8h |
23 | 28–50b | 95 |
| 45 | 98 | 92 | |||
| 7 |
|
 8o |
23 | 100 | 93 |
| 8 |
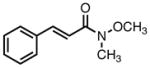
|
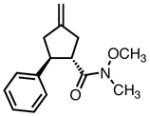 8p |
45 | 40 | 92 |
| 75 | 50 | 91 | |||
| 9 |
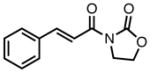
|
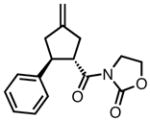 8q |
45 | 94 | 84 |
| 10 |
|
 8r |
45 | 86 | 77 |
| 11 |
|
 8u |
45 | 0 | – |
| 12 |
|
 8v |
45 | 80 | 91 |
| 13 |
|
 8w |
45 | 92 | 95 |
| 14 |
|
 8x |
23 | 99 | 93 |
| 15 |
|
 8y |
23 | 97 | 91 |
| 16 |
|
 8z |
23 | 99 | 86 |
| 17 |
|
 8aa |
23 | 99 | 66 |
| 18 |
|
 8ab |
75 | 100 | 91 |
All reactions were performed at 0.2M in toluene with 5% Pd(dba)2, 10% ligand, 1.6 equivalents donor 1, and stirred for 2–24 hours as monitored by GC for disappearance of the acceptor. Yields are isolated values; ee’s were determined by chiral HPLC or chiral GC.
Conversion.
ee obtained by conversion to the corresponding methyl ester using Otera’s catalyst (6 mol% catalyst, 60 equiv CH3OH, 0.1M PhCH3, 15h, 12% conversion).
Acylpyrroles represent a very interesting class of alkene acceptors as they are formally at the carboxylic acid oxidation state but have reactivity profiles similar to that of ketones. They are susceptible to nucleophilic attack and can be converted to a number of different products at various oxidation states. The chemistry of α,β-unsaturated acylpyrroles has been studied in conjugate additions,20 asymmetric epoxidations and cyclopropanations.21 A series of these compounds was prepared to examine the scope of this class of compounds in the TMM reaction. They were synthesized in the manner described by Shibasaki,22 and it was quickly discovered that they performed very well in the asymmetric cycloaddition reaction. Full conversion was obtained at room temperature and the reaction is tolerant of both aromatic (entries 12–17) and aliphatic (entry 18) groups. Steric bulk (entry 13), electron withdrawing groups (entry 14), electron donating groups (entry 15), and heterocyclic aromatic groups (entries 16–17) all gave high levels of conversion, although the ee dropped considerably with use of the 2-furyl substituent but not the 3-furyl system. A cyclohexyl substrate was tolerated (entry 18), and although conversion was low at 45 °C, the reaction went to completion at 75 °C without any deleterious effect on the enantioselectivity. Many of these products contain functional groups that can be further manipulated to generate cyclopentanes with high levels of molecular complexity.
Expansion of the Scope: Asymmetric Cycloadditions with Substituted TMM Donors
With our robust catalyst system in hand, we next turned our attention towards expansion of the TMM donor component of the reaction. Our studies of cycloadditions with substituted oxindoles and oxobenzofurans provided interesting insight into the TMM reaction and yielded a strategy for the facile construction of spirocyclic systems containing multiple adjacent stereogenic centers.23 To expand the scope of the reaction, we hoped to find a more general acceptor. We sought to find a system that, through subsequent synthetic manipulations, would allow access to an even wider range of cyclopentane containing structures. Our early studies focused on cyano donor 17, the synthesis of which is detailed in Scheme 8. Oxidation of key alcohol 15 with manganese dioxide gave aldehyde 16 and subsequent reaction with pyruvonitrile provided the donor 17 in a single step from the aldehyde in 58% yield.
Scheme 8.

Synthesis of Cyano-Substituted TMM donor 17.
We began to examine our cyano donor in the context of other electron deficient olefins. Unfortunately, our initial efforts were met with frustration with both benzylidene acetone and methyl cinnamate showing no reactivity. The tetralone substrates that had performed well with the diphenyl phosphoramidite ligand were also unreactive. Interestingly, GC-MS analysis of the crude reaction mixture seemed to indicate the presence of the cycloadduct between the dibenzylidene acetone ligand and the cyano donor. In light of this result, we began to look at the electronic properties of the substrates we were hoping to operate on. Specifically, we looked at orbital energies. In his studies of lanthanide catalyzed nucleophilic epoxidation reactions, Shibasaki observed a direct correlation between reaction rate and LUMO energy.22 He found that little, if any, conversion was observed with the morpholine amide 18 (<10% after 1h). The conversion increased substantially with decreasing LUMO energies and under identical conditions, conversions of approximately 40% and 60% were seen with the acylpyrrole 21 and chalcone 22 respectively. As Figure 3 shows, the LUMO energies of methyl cinnamate (19) and benzylidene acetone (7) are comparatively high (−1.72 and −1.88 eV respectively), whereas dibenzylidene acetone would be expected to lie considerably lower, near that of chalcone. The oxazolidinones, acylpyrroles and chalcones all have low-lying LUMOs. The acylpyrroles were the most attractive acceptors because of their versatility in further transformations and excellent performance with the parent TMM donor.
Figure 3.

LUMO Energies of α,β-Unsaturated Ketones and Carboxylic Acid Derivatives.
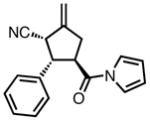
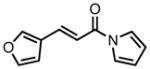
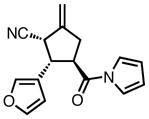
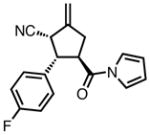
Gratifyingly, application of our standard reaction conditions to cyano donor 17 and acylpyrroles using ligand L38 gave us a single product in excellent yield and enantioselectivity (Table 6). The substrate scope was found to be very general. A diverse range of sterically and electronically different aryl and heteroaryl substituted acceptors performed well (entries 1–7). A diene acceptor reacted regioselectively with the 2,3-double bond only (entry 8). Substrates bearing saturated alkyl substituents were also reactive (entries 9–10) not requiring elevated temperatures as in the case of the parent donor (entry 10). Perhaps the most striking feature about this reaction was the diastereoselectivity. None of the trans, trans-diastereomer was observed in any case. In fact, the only evidence that small amounts may form was found in the case of 23c (entry 3). Trace quantities of the tautomerized product were obtained. We would expect the ee to remain unchanged if it were simply desired product isomerization. However, the observed ee was 84%, indicating that the isomerized material may potentially arise from the other diastereomer, which had lower ee, and that the rate of isomerization of this diastereomer is considerably higher, such that we do not observe any of the exocyclic olefin.
Table 6.
Substrate Scope of the TMM Reaction of Acylpyrroles with Nitrile Donor 17.
 | |||||
|---|---|---|---|---|---|
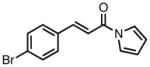
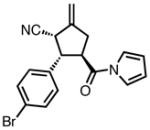
| entry | substrate | product | yield (%) | dr | ee (%) |
| 1b |
|
 23a |
87 | >20:1 | 92 |
| 2 |
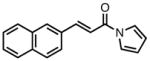
|
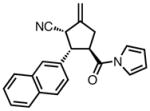 23b |
84 | >20:1 | 94 |
| 3 |
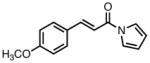
|
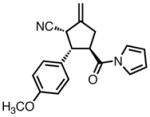 23c |
80 | >20:1 | 94 |
| 4 |
|
 23d |
98 | >20:1 | 95 |
| 5 |
|
 23e |
96 | >20:1 | 94 |
| 6 |
|
 23f |
100 | >20:1 | 95 |
| 7 |
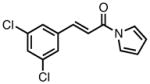
|
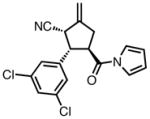 23g |
90 | > 20:1 | 97 |
| 8 |
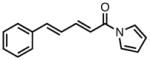
|
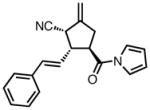 23h |
63 | > 20:1 | 95 |
| 9 |
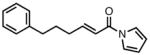
|
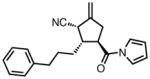 23i |
82 | > 20:1 | 94 |
| 10 |
|
 23j |
84 | > 20:1 | 94 |
All reactions were performed at 0.2M in toluene with 5% Pd(dba)2, 10% ligand L38, 1.5 equiv 17 and stirred for 3h; yields were combined isolated yields; ee’s were determined by HPLC with a chiral stationary phase column.
Reaction performed at 50 °C.
Another interesting feature of this reaction is the regioselectivity. Generation of the active Pd-TMM species occurs as shown in Scheme 9. Palladium mediated ionization of 17 gives π-allyl 24, which after acetate mediated desilylation gives TMM complex 25. While this initial ionization places the anion distal to the nitrile, the regioselectivity shown in the products above is derived exclusively from the donor species 26 in which the anion is adjacent to the nitrile. This anion is expected to be favored on both thermodynamic grounds, as the anion proximal to the nitrile is resonance-stabilized, and steric grounds, as this form places the bulk of the palladium-ligand complex distal to the donor substituent. The TMM species is dynamic in nature, and 25 and 26 are readily interchangeable by π-σ-π isomerization, which in this case occurs faster than attack of the anion to the olefin acceptor.24
Scheme 9.

Generation of Palladium-TMM Complex 26.
The relative and absolute stereochemistry were assigned by analogy to our previously disclosed acceptors,23 for which absolute stereochemistry has been confirmed by x-ray crystallography. Relative stereochemistry was also confirmed by NOE analysis of 23j (Figure 4). In addition, a geometry optimization of the palladium-ligand-TMM complex performed at the PM3 level using Spartan yielded the energy-minimized conformation shown in Figure 5. The structure indicates that one of the aryl rings of the BINOL could act as a wall such that the substrate approaches as depicted in Figure 5a; approach as depicted in Figure 5b would lead to significant steric interactions. While this model is not to be taken as definitive evidence in absence of a crystal structure, it does serve as a reasonable rationalization and accurately depicts the absolute and relative stereochemistry observed in this series, as well as that observed with oxindole, ketimine,25 and tropone26 acceptors.
Figure 4.
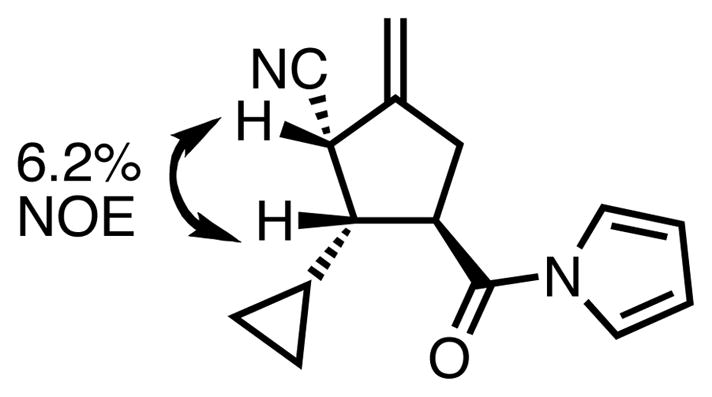
Confirmation of Relative Stereochemistry by NOE.
Figure 5.

Spartan Minimized (PM3) Palladium-TMM Complex.
The excellent reactivity of this specific substrate class in the TMM cycloaddition is very fortuitous. Because the nitrogen lone pair of an acylpyrrole is delocalized through the aromatic ring, there is little delocalization into the carbonyl. As a result, the compound, while formally at the carboxylic oxidation level, displays a reactivity profile similar to that of a ketone, but can be further transformed through synthetic manipulations common to the ester oxidation level. This leads to a wide range of accessible structures.
CONCLUSION
In summary, we have discovered and developed the first highly enantioselective asymmetric cycloadditions of trimethylenemethane. These transformations utilize a series of novel ligands which evolved from secondary amine-based phosphoramidites to a series of pyrrolidine-derived ligands during our studies. Sequential chiral lithiation, transmetallation, and Negishi coupling steps provided a general, efficient method that could be used in ligand preparation. Studies of the newly developed catalysts have culminated in the development of a robust synthetic method which can be used to prepare highly substituted cyclopentane scaffolds containing three contiguous stereocenters in high enantioselectivity and complete regio- and diastereoselectivity. The methodology is tolerant of a wide variety of electron-deficient olefins, including ketones, esters, nitriles, thioesters, and amides. Furthermore, the use of acylpyrrole substituents with substituted donors gives our method additional synthetic utility because it allows a good deal of flexibility in subsequent transformations. Applications of this novel methodology in the synthesis of complex molecules are highly anticipated.
EXPERIMENTAL SECTION
O,O′-(R)-(1,1′-dinaphthyl-2,2′-diyl)-N-((R,R)-2,5-di(2-naphthyl)pyrrolidine) phosphoramidite (L38)
(R)-tert-butyl 2-(naphthalen-2-yl)pyrrolidine-1-carboxylate (10c)
A solution of N-Boc pyrrolidine (4.9 g, 28.5 mmol, 1.2 eq) and (−)-sparteine (6.5 mL, 28.5 mmol, 1.2 eq) in tert-butyl methyl ether (60 mL) was cooled to −78 °C and s-BuLi (28.5 mmol, 1.2 eq, 27.1 mL of a 1.05M solution in cyclohexane) was added dropwise over 15 minutes. The pale yellow solution was stirred at this temperature for 3 hours, at which point ZnCl2 (17.1 mmol, 0.72 eq, 17.1 mL of a 1.0M solution in diethyl ether) was added dropwise slowly over 15 minutes. The solution was stirred at −78 °C for 30 minutes and then allowed to warm to room temperature and stir for an additional 30 minutes. 2-Bromonaphthalene (4.92 g, 23.75 mmol, 1.0 eq) was added followed by Pd(OAc)2 (256 mg, 1.14 mmol, 0.048 eq) and t-Bu3P-HBF4 (413 mg, 1.42 mmol, 0.06 eq), and the solution stirred overnight at room temperature. After 12 hours aqueous ammonia (2.5 mL) was added and the mixture stirred for one hour. At this time, the mixture was filtered through celite to remove salts and washed with tert-butyl methyl ether. The filtrate was washed with 1 NHCl and water, dried over MgSO4, filtered, and concentrated. The product was purified by flash chromatography on silica gel (90% methylene chloride in petroleum ether) to give the desired product as a white solid (5.3 g, 75%). 1H-NMR (400 MHz, CDCl3): 7.82-7.77 (m, 3H), 7.58 (s, 1H), 7.45 (br s, 2H), 7.31 (d, J = 8.4 Hz, 1H), 5.11 (s, 0.3H), 4.96 (s, 0.7H), 3.70-3.54 (m, 2H), 2.38 (br s, 1H), 1.99-1.87 (m, 3H), 1.50-1.43 (m, 3H), 1.14 (s, 6H); Chiral HPLC: Chiralcel® AD column, 1% isopropanol in heptane, 1.0 mL/min, λ = 254 nm, tR = 15.06 (major), 16.65 (minor).
(2R,5R)-tert-butyl 2,5-di(naphthalen-2-yl)pyrrolidine-1-carboxylate (11ca)
A solution of 10c (1.70 g, 5.7 mmol, 1.2 eq) and (−)-sparteine (1.31 mL, 5.7 mmol, 1.2 eq) in tert-butyl methyl ether (12 mL) was cooled to −78 °C. Toluene (3 mL) was added to aid in solubility. s-BuLi (5.7 mmol, 1.2 eq, 4.45 mL of a 1.28M solution in cyclohexane) was added dropwise over 15 minutes to give a dark yellow solution. The solution was stirred at this temperature for 3 hours, at which point ZnCl2 (3.42 mmol, 0.72 eq, 3.42 mL of a 1.0M solution in diethyl ether) was added dropwise slowly over 15 minutes. The solution was stirred at −78 °C for 30 minutes and then allowed to warm to room temperature and stir for an additional 30 minutes. 2-Bromonaphthalene (984 mg, 4.75 mmol, 1.0 eq) was added followed by Pd(OAc)2 (51 mg, 0.228 mmol, 0.048 eq) and t-Bu3P-HBF4 (83 mg, 0.285 mmol, 0.06 eq), and the solution stirred overnight at 60 °C. After 12 hours, the solution was cooled to room temperature and aqueous ammonia (500 μl) was added and the mixture stirred for one hour. At this time, the mixture was filtered through celite to remove salts and washed with tert-butyl methyl ether. The filtrate was washed with 1 NHCl and water, dried over MgSO4, filtered, and concentrated. The product was purified by flash chromatography on silica gel (10% ethyl acetate in petroleum ether) to give the desired product as a white solid (1.01 g, 50%). 1H-NMR (400 MHz, CDCl3): 7.86-7.82 (m, 6H), 7.69 (d, J = 9.2 Hz, 2H), 7.52-7.38 (m, 6H), 5.56 (d, J = 7.2 Hz, 1H), 5.41 (d, J = 7.6 Hz, 1H), 2.62-2.51 (m, 2H), 1.88-1.79 (m, 2H), 1.11 (s, 9H).
(2R,5R)-2,5-di(naphthalen-2-yl)pyrrolidine (12ca)
A solution of 11ca (1.06 g, 2.5 mmol, 1.0 eq) in CH2Cl2 (20 mL) was cooled to 0 °C and TFA (3.71 mL, 50 mmol, 20.0 eq) added. The mixture was stirred for 4 hours at room temperature and then concentrated. Ethyl acetate was added, followed by 2N NaOH (20 mL). Triethylamine (5 mL) was added. The organic phase was separated, dried over MgSO4, filtered, and concentrated in vacuo. Purification by flash chromatography on silica gel (10% ethyl acetate in pentane) gave the product as a white solid (795 mg, 98%); 1H-NMR (400 MHz, CDCl3): 7.88-7.82 (m, 8H), 7.58 (dd, J = 8.6, 1.6 Hz, 2H), 7.50-7.43 (m, 4H), 4.80 (t, J = 7.0 Hz, 2H), 2.58-2.47 (m, 2H), 2.10-2.00 (m, 2H); 13C-NMR (100 MHz, CDCl3): 143.2, 133.4, 132.6, 128.3, 127.8, 127.6, 126.0, 125.5, 125.0, 124.5, 62.5, 35.5.
O,O′-(R)-(1,1′-dinaphthyl-2,2′-diyl)-N-((R,R)-2,5-di(2-naphthyl)pyrrolidine) phosphoramidite (L38)
To a suspension of (R)-1,1′-binaphthyl-2,2′-diol (2.86 g, 10.0 mmol, 1.0 eq) in PCl3 (14.0 mL, 160 mmol, 16 eq) was added N-methylpyrrolidone (3 drops). The mixture was stirred at 60 °C for 15 minutes, cooled, and carefully concentrated to give a yellow, foamy solid. The residue was twice azeotroped with toluene (10 mL) and finally dissolved in toluene (20 mL) to make a 0.5M stock solution of chlorophosphite 13, which could be stored for several days at −15 °C. The intermediate was observed to be air sensitive, but the brief exposure during this procedure resulted in no more than 5% oxidation products (as judged by 31P NMR), which did not effect the subsequent reaction. 31P NMR (162 MHz, CDCl3): 179.0. A solution of BINOL chlorophosphite (1.56 mmol, 1.2 eq, 3.12 mL of a 0.5 M in solution in toluene) was added dropwise to a mixture of pyrrolidine 12ca (420 mg, 1.30 mmol, 1.0 eq), triethylamine (903 μl, 6.5 mmol, 5.0 eq), and DMAP (31.7 mg, 0.26 mmol, 0.2 eq) in toluene (8 mL) at 0 °C. The mixture was stirred overnight at room temperature and the ligand purified by flash chromatography on silica gel (25% dichloromethane in hexanes with 1% triethlyamine) to give the product as a white solid (694 mg, 84%) after trituration from hexanes. 1H-NMR (400 MHz, CDCl3): 7.94-7.80 (m, 9H), 7.57-7.52 (m, 5H), 7.43 (dd, J = 8.4, 2.0 Hz, 1H), 7.32-7.28 (m, 3H), 7.23-7.09 (m, 5H), 6.59 (d, J = 8.8 Hz, 1H), 5.86 (dd, J = 8.8, 0.8 Hz, 1H), 5.38 (d, J = 7.2 Hz, 2H), 2.59-2.48 (m, 2H), 1.84-1.76 (m, 2H); 31P NMR (162 MHz, CDCl3): 145.5; 13C-NMR (100 MHz, CDCl3): 149.4 (d, J = 9.8 Hz), 149.0, 143.3 (d, J = 4.5 Hz), 133.4, 132.8, 132.7, 132.1, 131.2, 130.2, 129.9, 128.5, 128.2, 128.1, 128.0, 127.9, 127.7, 127.1, 126.9, 126.2, 125.8, 125.7, 125.4, 125.3, 125.2, 124.6, 124.0, 122.0, 121.9, 120.8 (d, J = 3.1 Hz), 62.7 (d, J = 12.9 Hz), 33.2; IR (neat): 3054, 3008, 2970, 2933, 2870, 1619, 1591, 1507, 1463, 1326, 1230, 1068, 946, 818, 748; [a]24D = −117.8 (c 0.34, PhCH3), −1.3 (c 2.51, CHCl3); HRMS (ESI): calculated for C44H32NNaO2P [M+Na]+: 660.2068, found 660.2075.
General Procedure for Palladium-Catalyzed Asymmetric TMM Cycloaddition
A flask containing Pd(dba)2 (2.9 mg, 0.005 mmol, 0.05 eq), 2-naphthyl pyrrolidine phosphoramidite L38 (6.4 mg, 0.01 mmol, 0.10 eq), and (E)-3-(4-bromophenyl)-1-(1H-pyrrol-1-yl)prop-2-en-1-one (27.6 mg, 0.1 mmol, 1.0 eq) was evacuated and purged with argon. Toluene (0.5 ml) was added and the mixture stirred for 5 minutes at room temperature. Nitrile TMM donor 17 (35 μL, 0.15 mmol, 1.5 eq) was added and the solution was stirred at room temperature for 3 hours. The mixture was loaded directly onto a silica gel column and purified by flash chromatography on silica gel (15% ethyl acetate in hexanes) to give the desired product as a white solid (35.5 mg, quantitative).
23f
1H-NMR (400 MHz, CDCl3): 7.49 (dd, J = 8.8, 2.4 Hz, 2H), 7.30-7.22 (m, 2H), 7.19-7.15 (m, 2H), 6.32 (dd, J = 2.8, 2.4 Hz, 2H), 5.44-5.42 (m, 1H), 5.33-5.31 (m, 1H), 4.07-4.05 (m, 1H), 3.99-3.95 (m, 1H), 3.86-3.81 (m, 1H), 3.15-3.08 (m, 1H), 2.93-2.85 (m, 1H); 13C-NMR (100 MHz, CDCl3): 170.4, 142.9, 136.6, 132.2, 129.5, 122.3, 119.1, 117.8, 114.1, 112.6, 49.4, 47.5, 41.0, 35.6; IR (neat): 2961, 2916, 2849, 2243, 1704, 1665, 1467, 1261, 1070, 901, 800; [a]26D = 3.9 (c 0.41, CHCl3); HRMS (ESI): calculated for C18H15BrN2NaO [M+Na]+: 377.0265, found 377.0255; Chiral HPLC: Chiralpak® IA column, 5% isopropanol in heptane, 1.0 mL/min, λ = 254 nm, tR = 16.25 (minor), 29.06 (major).
Supplementary Material
Acknowledgments
We thank the NSF and NIH (Grant GM13598) for their generous support of our programs. S. M. S. thanks Eli Lilly and Roche for graduate fellowships. We thank Johnson-Matthey for their generous gifts of palladium salts.
Footnotes
Supporting Information. Detailed experimental details, compound characterization data, and spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Trost BM. Angew Chem Int Ed Engl. 1986;25:1. [Google Scholar]; (b) Trost BM. Pure Appl Chem. 1988;60:1615. [Google Scholar]; (c) Lautens M, Klute W, Tam W. Chem Rev. 1996;96:49. doi: 10.1021/cr950016l. [DOI] [PubMed] [Google Scholar]; (d) Yamago S, Nakamura E. Org React. 2002;61:1. [Google Scholar]
- 2.Chan DMT. Recent Advances in Palladium-Catalyzed Cycloadditions Involving Trimethylenemethane and its Analogs. In: Kobayashi S, Jorgensen KA, editors. Cycloaddition Reactions in Organic Synthesis. 1. Wiley-VCH; Weinheim, Germany: 2002. pp. 57–83. [Google Scholar]
- 3.(a) Trost BM, King SA, Schmidt T. J Am Chem Soc. 1989;111:5902. [Google Scholar]; (b) Trost BM, Marrs CM. J Am Chem Soc. 1993;115:6636. [Google Scholar]
- 4.Trost BM, Chan DMT. J Am Chem Soc. 1979;101:6429. [Google Scholar]
- 5.Trost BM, Stambuli JP, Silverman SM, Schwörer U. J Am Chem Soc. 2006;128:13328. doi: 10.1021/ja0640750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trost BM, Chan DMT. J Am Chem Soc. 1979;101:6432. [Google Scholar]
- 7.(a) Trost BM, Chan DMT. J Am Chem Soc. 1983;105:2326. [Google Scholar]; (b) Trost BM, Nanninga TN. J Am Chem Soc. 1985;107:1075. [Google Scholar]
- 8.(a) Chaigne F, Gotteland JP, Malacria M. Tetrahedron Lett. 1989;30:1803. [Google Scholar]; (b) Trost BM, Yang B, Miller M. J Am Chem Soc. 1989;111:6482. [Google Scholar]
- 9.Yamamoto A, Ito Y, Hayashi T. Tetrahedron Lett. 1989;30:375. [Google Scholar]
- 10.Börner A. Phosphorus Ligands in Asymmetric Catalysis: Synthesis and Applications. John Wiley & Sons; New York: 2008. [Google Scholar]
- 11.Teichert JF, Feringa BL. Angew Chem Int Ed. 2010;49:2486. doi: 10.1002/anie.200904948. [DOI] [PubMed] [Google Scholar]
- 12.Hulst R, de Vries NK, Feringa BL. Tetrahedron: Asymmetry. 1994;5:699. [Google Scholar]
- 13.de Vries AHM, Meetsma A, Feringa BL. Angew Chem Int Ed Engl. 1996;35:2374. [Google Scholar]
- 14.Trost BM, Yamano M. Unpublished results
- 15.Alexakis A, Polet D. Org Lett. 2005;7:1621. doi: 10.1021/ol050350w. [DOI] [PubMed] [Google Scholar]
- 16.Wakabayashi K, Aikawa K, Kawauchi S, Mikami K. J Am Chem Soc. 2008;130:5012. doi: 10.1021/ja710340n. [DOI] [PubMed] [Google Scholar]
- 17.Choi YH, Choi JY, Yang HY, Kim YH. Tetrahedron: Asymmetry. 2002;13:801. [Google Scholar]
- 18.Trost BM, Bringley DA. Unpublished results [Google Scholar]
- 19.Campos KR, Klapars A, Waldman JH, Dormer PG, Chen CY. J Am Chem Soc. 2006;128:3538–3539. doi: 10.1021/ja0605265. [DOI] [PubMed] [Google Scholar]
- 20.(a) Mita T, Sasaki K, Kanai M, Shibasaki M. J Am Chem Soc. 2005;127:514. doi: 10.1021/ja043424s. [DOI] [PubMed] [Google Scholar]; (b) Vakulya B, Varga S, Soos T. J Org Chem. 2008;73:3475. doi: 10.1021/jo702692a. [DOI] [PubMed] [Google Scholar]; (c) Shaghafi MB, Kohn BL, Jarvo ER. Org Lett. 2008;10:4743. doi: 10.1021/ol801830h. [DOI] [PubMed] [Google Scholar]
- 21.Kakei H, Sone T, Sohtome Y, Matsunaga S, Shibasaki M. J Am Chem Soc. 2007;129:13410. doi: 10.1021/ja076797c. [DOI] [PubMed] [Google Scholar]
- 22.Matsunaga S, Kinoshita T, Okada S, Harada S, Shibasaki M. J Am Chem Soc. 2004;126:7559. doi: 10.1021/ja0485917. [DOI] [PubMed] [Google Scholar]
- 23.Trost BM, Cramer N, Silverman SM. J Am Chem Soc. 2007;129:12396. doi: 10.1021/ja075335w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trost BM, Chan DMT. J Am Chem Soc. 1981;103:5972. [Google Scholar]
- 25.Trost BM, Silverman SM. J Am Chem Soc. 2010;132:8238. doi: 10.1021/ja102102d. [DOI] [PubMed] [Google Scholar]
- 26.Trost BM, McDougall PJ, Hartmann O, Wathen PT. J Am Chem Soc. 2008;130:14960. doi: 10.1021/ja806979b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.