

Abstract
Notch signaling and hepatocyte nuclear factor-6 (HNF-6) are two genetic factors known to affect lineage commitment in the bipotential hepatoblast progenitor cell (BHPC) population. A genetic interaction involving Notch signaling and HNF-6 in mice has been inferred through separate experiments showing that both affect BHPC specification and bile duct morphogenesis. To define the genetic interaction between HNF-6 and Notch signaling in an in vivo mouse model, we examined the effects of BHPC-specific loss of HNF-6 alone and within the background of BHPC-specific loss of RBP-J, the common DNA-binding partner of all Notch receptors. Isolated loss of HNF-6 in this mouse model fails to demonstrate a phenotypic variance in bile duct development compared to control. However, when HNF-6 loss is combined with RBP-J loss, a phenotype consisting of cholestasis, hepatic necrosis, and fibrosis is observed that is more severe than the phenotype seen with Notch signaling loss alone. This phenotype is associated with significant intrahepatic biliary system abnormalities, including an early decrease in biliary epithelial cells evolving to ductular proliferation and a decrease in the density of communicating peripheral bile duct branches. In this in vivo model, simultaneous loss of both HNF-6 and RBP-J results in down-regulation of both HNF-1β (hepatocyte nuclear factor-1β) and Sox9 (SRY-related HMG box transcription factor 9).
Conclusion
HNF-6 and Notch signaling interact in vivo to control expression of downstream mediators essential to the normal development of the intrahepatic biliary system. This study provides a model to investigate genetic interactions of factors important to intrahepatic bile duct development and their effect on cholestatic liver disease phenotypes.
Keywords: cholestasis, Albumin-Cre, Sox9, HNF-1β, RBP-J
Notch signaling is an intercellular signaling pathway required throughout embryonic development and adulthood for cell specification, lineage commitment, and maintenance of progenitor cells (1). In mammals, the canonical Notch pathway includes four receptors (Notch 1 [N1], N2, N3, N4) and two families of ligands (Jagged and Delta-like). Upon ligand binding, the Notch receptor undergoes sequential proteolysis, releasing the intracellular domain that translocates to the nucleus and associates with recombination signal binding protein immunoglobulin kappa J (RBP-J). Interaction with the Notch intracellular domain converts the RBP-J co-repressor complex into a co-activator complex and mediates gene transcription (2).
Human genetic disease and mutant mouse models have illustrated the importance of Notch signaling in the specification and remodeling of intrahepatic bile ducts (IHBDs). Alagille Syndrome (AGS) is an autosomal dominant disorder in humans caused by mutations in the Notch ligand Jagged1 (JAG1) and less commonly in the Notch receptor N2 (3-5). AGS is predominantly characterized by neonatal cholestasis due to paucity of IHBDs, although patients may also have significant cardiac, ocular, renal, pancreatic, and vascular defects (6). Mice heterozygous both for a null JAG1 allele and for a hypomorphic N2 mutation exhibit developmental defects similar to human AGS, including IHBD paucity (7). Recent studies in mice have further demonstrated the importance of JAG1 and N2 in causing IHBD remodeling defects (8-10), as well as identifying a role for Notch signaling in lineage commitment within the bipotential hepatoblast population. Mice with hepatoblast-specific deletion of RBP-J demonstrate a visible decrease in specified ductal cells contributing to ductal plate formation as well as a decrease in formed ductal structures (11, 12). These studies illustrate that Notch signaling is important in the process of IHBD formation in mice as well as humans.
Along with Notch signaling, HNF-6 (hepatocyte nuclear factor-6, Onecut-1) is one of the few known genetic factors that affect lineage commitment within the bipotential hepatoblast progenitor cell (BHPC) population in mice, directing differentiation into either hepatocyte or biliary epithelial cell (BEC) lineages in vitro (13). Mice with global loss of HNF-6 display disordered and delayed IHBD development during embryogenesis, which is associated with early post-natal cholestatic liver disease and a high rate of mortality. However, the mice that survived into adulthood showed no signs of overt hepatic defects (14), suggesting that HNF-6 functions primarily during embryonic development of the biliary tract and that other genetic factors direct continued IHBD development in the absence of HNF-6.
Experiments both in vitro and in vivo have shown that HNF-6 and Notch signaling modulate expression of various transcription factors, including HNF-1β (hepatocyte nuclear factor-1β) and Sox9 (SRY-related HMG box transcription factor 9) (12, 14-16). Inactivation of HNF-1β within the liver of mice results in severe cholestasis and paucity of IHBDs (17). Inactivation of Sox9 within the liver leads to a prolonged presence of asymmetrical primitive ductal structures during early ductal development (18). The ability of both HNF-6 and Notch signaling to modulate similar transcription factors suggests that similarities exist within their control of IHBD development. Given this possible overlap, we hypothesize that genetic defects in both factors will lead to an altered cholestatic liver disease phenotype.
To evaluate the role of HNF-6 during postnatal ductal development in an in vivo mouse model, we utilized a Cre-LoxP system to achieve genetic alteration specifically within the BHPC population. We studied the effect of HNF-6 removal as well as the effect of HNF-6 loss within the background of chronic loss of Notch signaling, achieved through deletion of RBP-J. Isolated hepatoblast-specific loss of HNF-6 fails to demonstrate a phenotypic variance in IHBD development compared to control. However, loss of HNF-6 in the setting of RBP-J loss results in extensive abnormalities in ductal development and intact IHBD structure, as well as cholestatic liver injury characterized by extensive hepatic necrosis and fibrosis. This phenotype was worse than that seen with RBP-J loss alone. These defects were associated with altered expression of transcription factors responsible for IHBD development, HNF-1β and Sox9, providing evidence of an interaction between HNF-6 and Notch signaling in vivo that leads to the modulation of transcription factors responsible for IHBD development. This provides a model to study the contribution of HNF-6 or associated transcription factors to the clinical severity of cholestatic liver injury in patients with IHBD defects related to Notch signaling defects.
MATERIALS AND METHODS
Mouse Lines
On a CD1 background, mice carrying conditional deletion alleles for HNF-6 (HNF-6flox/flox, HNF-6 KO) (19), RBP-J (RBPflox/flox, RBP KO) (20), or both HNF-6 and RBP-J (DKO) were crossed with mice carrying the Albumin-Cre (Alb-Cre) transgene (21). Further crosses were performed to obtain homozygous genotypes. Mouse and embryo genotypes were confirmed by polymerase chain reaction (PCR) analysis using previously published primer pairs. All breeding and experimental procedures were performed with approval from the Vanderbilt Institutional Animal Care and Use Committee. Infection with Helicobacter hepaticus was ruled out by PCR testing for bacterial DNA presence in mouse fecal samples.
Serum Chemistry
Blood was collected post-mortem from mice at postnatal day (P)60 and tested for serum alanine aminotransferase (ALT), alkaline phosphatase (ALP), total bilirubin (TB), and conjugated bilirubin by colorimetric endpoint assay (TecoDiagnostics, Anaheim, CA). Age-matched and littermate control mice without the Alb-Cre transgene were used for comparison.
Resin Casting and Tissue Clearance
Retrograde resin injection of the common bile duct was performed as previously published (11) with the following modifications. Following resin injection, the liver was placed in 4% paraformaldehyde for fixation at 4°C overnight. Sequential dehydration was performed with 1:1 methanol:PBS solution followed by 100% methanol at room temperature. Tissue clearance was achieved with 1:2 benzyl alcohol:benzyl benzoate (BABB) solution at room temperature. Liver lobes were photographed within BABB solution using a Leica MZ 16 FA stereoscope and QImaging RETIGA 4000R camera.
RNA Preparation and Quantitative Real-Time RT-PCR
Total liver RNA was prepared using TRIZOL (Invitrogen, Carlsbad, CA) and Turbo DNA-Free kit (Ambion, Austin, TX). Total RNA (2.5μg) was used for complementary DNA synthesis, performed with SuperScript III First-Strand (Invitrogen, Carlsbad, CA). Quantitative real-time RT-PCR was performed using the ABI Prism 7900 (Applied Biosystems, Foster City, CA). HNF-6, HNF-1β, Sox9, Onecut 2 (OC-2), and Hepatocyte nuclear factor-4 (HNF-4) messenger RNA (mRNA) was measured from 3 to 4 independent samples per genotype. Primer sequences are given in Supplemental Table 1.
Immunohistochemistry
Liver tissue was fixed overnight at 4°C in 4% paraformaldehyde, processed and embedded in paraffin. Embedded tissue was sectioned at 6μm. For CK19, wsCK, and DBA immunostaining, antigen retrieval was performed with slides incubated overnight at 55°C in 100mM Tris base solution, pH 10. For HNF-6 and HNF-1β immunostaining, antigen retrieval was performed with Proteinase K (Dako, Carpinteria, CA). Sections were incubated with primary antibody at 4°C overnight in blocking buffer (1% bovine serum albumin, 0.2% powdered skim milk, 0.3% Triton X-100 [Fisher BioReagents, Fair Lawn, NJ] in PBS) and then were incubated with appropriate secondary antibodies overnight at 4°C. Primary and secondary antibodies are listed in Supplemental Table 2. For biotin-SP-conjugated anti-immunoglobulin G (anti-IgG) secondary antibodies, a R.T.U. Vectastain Elite Universal ABC kit (Vector, Burlingame, CA) was developed using the substrate DAB (Vector) for chromogenic staining. Mayer’s hematoxylin was used as counterstain for chromogenic staining. For immunofluorescence, CY2 and CY3 secondary antibodies (Jackson ImmunoResearch, West Grove PA) were used with bisBenzimide counterstaining. Images were acquired either using an Axioplan2 microscope and QImaging RETIGA EXi camera or LSM510 Meta confocal microscope (Zeiss) at an optical depth of 1um. For Ki67 proliferation analysis, the total number of CK19-positive cells was counted (hilar and peripheral) from both control and DKO mice aged P3 (n=4 control; n=5 DKO), P15 (n=3 control; n=3 DKO), and P60 (n=3 control; n=5 DKO). Proliferation was determined based on the ratio of cells positive for both Ki67 and CK19 versus total cells positive for CK19. Apoptosis was investigated in control and DKO mice at P3 (n=1 control; n=4 DKO) with the use of ApopTag® Red In Situ Apoptosis Detection Kit (Millipore, Temecula, CA).
Statistical Analysis
Data was subjected to a two-tailed Student’s t-test and a P-value of ≤0.05 was considered statistically significant. Student’s t-test with Welch correction was applied to serum bilirubin measurements due to unequal variance amongst experimental groups.
RESULTS
Albumin-Cre mediates loss of HNF-6 within both BHPC lineages
HNF-6 is expressed within the liver and extrahepatic biliary system throughout embryonic development (22, 23). To investigate the consequence of HNF-6 deletion on BEC specification and IHBD development, we limited genetic alterations to the liver. We accomplished this through the use of Albumin-Cre (Alb-Cre) recombinase transgene. Alb-Cre activity is detected within the BHPC population prior to differentiation into hepatocytes and BECs (11). Hepatoblast-specific inactivation of HNF-6 by Alb-Cre was assessed by real-time RT-PCR of HNF-6 mRNA expression. With Alb-Cre directed recombination, HNF-6 mRNA was decreased compared to control at embryonic day (E)16.5 and this reached significance postnatally (Fig. 1A,B). To determine timing of HNF-6 protein loss directed by Alb-Cre recombination, immunostain analysis was performed at E16.5, E18.5, and postnatal day (P)0. At E16.5, HNF-6 protein was seen in a similar pattern in both control and HNF-6 KO mice, limited to periportal BECs (Fig. 1C,D). At E18.5, HNF-6 protein expression was visible in nearly all BECs and hepatocytes in control mice (Fig. 1E). However, by E18.5, HNF-6 protein expression was limited in HNF-6 KO mice to few isolated hepatocytes and few isolated BECs surrounding larger hilar portal veins (Fig. 1F, arrows). This pattern was consistent postnatally at P0 (Fig. 1G,H). These data indicate that Alb-Cre expression leads to inactivation of HNF-6 in both BHPC lineages by E18.5. The observed loss of HNF-6 protein in both hepatocytes and BECs is in agreement with previous ROSA26 reporter analysis, which has also demonstrated Alb-Cre directed recombination in cells contributing to both BHPC lineage derivatives (8, 11). Following this characterization, we then investigated the consequence of conditional loss of HNF-6 alone and in the setting of chronic cholestasis induced by the conditional loss of Notch signaling.
Fig. 1. Alb-Cre mediated recombination results in deletion of HNF-6.

(A-B) The relative expression ratio of HNF-6 mRNA was calculated using real-time RT-PCR and the deviation in cycle threshold (Ct) of control (without Alb-Cre), HNF-6 KO, RBP KO, and DKO samples and is expressed in comparison to GAPDH (Glyceraldehyde 3-phosphate dehydrogenase) at ages E16.5 and P60. A minimum of 3 RNA samples from separate mice were tested for each genotype. Statistical analysis was performed using a two-tailed Student’s t-test. Error bars represent standard error of the mean. *P<0.05, **P<0.01, ***P<0.001 (C-H) Immunostaining for HNF-6 protein in paraffin sections counterstained with Mayer’s hematoxylin in control and HNF-6 KO genotypes at ages E16.5, E18.5, and P0. (C-D) HNF-6 protein expression is similar in control and HNF-6 KO mice at E16.5, largely limited to periportal BECs. HNF-6 protein is visibly decreased compared to control by E18.5 (E-F) as well as P0 (G-H) in HNF-6 KO mice, with arrows representing the HNF-6 immuno-positive areas within this field limited to scattered periportal BECs surrounding larger hilar portal veins, confirming that Alb-Cre mediated recombination at the HNF-6 locus occurs prior to birth. (G) Scale bar = 100μm.
Cholestatic liver injury is enhanced by loss of HNF-6 in the setting of RBP-J loss
To study the effect of HNF-6 loss in a genetic model of chronic cholestasis, we utilized a described model of Notch signaling loss through Alb-Cre mediated deletion of RBP-J (11, 24). Because RBP-J is the DNA-binding partner required by all four Notch receptors to effect canonical target gene expression, this approach circumvents possible functional redundancy through different receptors. Liver-specific inactivation of both HNF-6 and RBP-J (hereafter referred to as DKO) results in elevation of both total bilirubin and alkaline phosphatase (Table 1) versus control, HNF-6 loss alone, and RBP-J loss alone (P<0.01). The fraction of conjugated bilirubin was similar between DKO and control genotypes (DKO: 56.4%±12.8; Control: 31.4%±13.4, p=0.22). However, accumulation of conjugated bilirubin in mutant DKO mice suggests that the elevation of total bilirubin observed is not due to defective hepatic conjugation, which is an enzymatic process allowing for normal hepatic excretion of bilirubin.
Table 1.
Serum was obtained from age P60 mice of indicated genotypes and analyzed for total bilirubin (TB), alkaline phosphatase (ALP), and alanine aminotransferase (ALT). Control mice age P60 without Alb-Cre transgene were used for comparison. Values are listed as mean ± standard error of the mean. IU/L: international units per liter, mg/dL: milligrams per deciliter.
| Genotype | n | TB (mg/dL) | n | ALP (IU/L) | n | ALT (IU/L) |
|---|---|---|---|---|---|---|
| Control | 15 | 0.80 ± 0.20 | 10 | 22.5 ± 2.6 | 8 | 44.0 ± 4.2 |
| HNF-6 KO | 9 | 1.03 ± 0.30 | 3 | 12.2 ± 5.7 | 7 | 48.2 ± 7.8 |
| RBP KO | 4 | 1.10 ± 0.50 | 4 | 14.1 ± 1.6 | 9 | 96.1 ± 15.4 ** |
| DKO | 33 | 11.27 ±2.36 ** | 9 | 71.5 ± 10.2 ** | 11 | 86.8 ± 5.6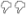
|
P values are calculated using two-tailed Student’s t-test.
P<0.01
P<0.0001
Both DKO and RBP KO mice demonstrate elevation in alanine aminotransferase (ALT) levels compared to control and HNF-6 KO mice (Table 1), indicative of hepatocellular injury. However, DKO mice also demonstrate extensive hepatic necrosis (Fig. 2D, arrowhead, Supplemental Fig. 1), as well as increased collagen deposition with areas of bridging fibrosis between portal tracts developing by age P60 (Fig. 2D, arrow). Isolated loss of either HNF-6 or RBP-J alone failed to show significant necrosis or collagen deposition compared to control at age P60 (Fig. 2A,B,C). With the observed elevation in total bilirubin and alkaline phosphatase demonstrating significant cholestasis, these data show that loss of HNF-6 in the setting of Notch signaling loss leads to enhanced cholestatic liver injury characterized by bridging hepatic fibrosis.
Fig. 2. Loss of HNF-6 and RBP-J results in hepatic fibrosis and necrosis.

(A-D) Representative paraffin sections from P60 mice stained for collagen with Gomori one-step trichrome stain. Genotypes are designated within figure. Age-matched control mice without Alb-Cre transgene were used for comparison. Images were obtained from the periphery of the left liver lobe. (A-C) Collagen deposition did not visibly differ compared to control with isolated loss of HNF-6 or RBP-J. (D) With loss of both HNF-6 and RBP-J, bridging fibrosis with collagen deposition (arrow) between portal veins, as well as extensive hepatic necrosis (arrowhead), was observed. PV, portal vein. (A) Scale bar = 100μm.
Loss of both HNF-6 and RBP-J results in early postnatal BEC paucity
To determine the intrahepatic ductal histopathology of mice with loss of HNF-6 alone and within the background of Notch signaling loss, we performed staining with wide-spectrum cytokeratin (wsCK) as a marker of BECs. Mice with isolated loss of HNF-6 showed no detectable phenotypic difference in IHBD wsCK staining compared to control (Fig. 3A,B,E,F,I,J). At age E16.5, RBP KO and DKO mice demonstrate hilar ductal plate formation of similar appearance to control mice (Fig. 3 A-D). This data agrees with previously published data, as mice with Alb-Cre or alpha-fetoprotein enhancer and albumin promoter Cre recombinase (AFP-Cre) mediated loss of RBP-J demonstrate ductal plate formation of normal appearance at age E16.5, but subsequently show a significant decrease in postnatal cytokeratin-positive BECs and formed IHBDs (11, 12). Consistent with this, at P3 there were visibly fewer wsCK-positive (+) cells associated with ductal plates and tubular structures in RBP KO mice (Fig. 3G). DKO mice also demonstrate a visible decrease in the number of wsCK+ cells at age P3 (Fig. 3H). At P15, a complete loss of all peripheral wsCK+ cells compared to control is observed (Fig. 3I,L). Cytokeratin-positive bile ducts in P15 DKO mice were only observed centrally within the hepatic lobe and co-stained positive with Dolichos biflorus agglutinin (DBA) (Supplemental Fig. 2). This was consistent among DKO mice examined at age P15 (n=5).
Fig. 3. Decrease in early postnatal wsCK+ BECs upon loss of both HNF-6 and RBP-J.

Paraffin sections from control (A,E,I), HNF-6 KO (B,F,J), RBP KO (C,G,K), and DKO (D,H,L) were immunostained for wsCK to mark BECs and counterstained with Mayer’s hematoxylin. HNF-6 KO mice showed no phenotypic variance in wsCK+ BECs compared to control at all ages evaluated (B,F,J). At E16.5, both RBP KO (C) and DKO (D) demonstrate wsCK+ cells (arrow) contributing to a ductal plate surrounding the portal vein similar to control (A). At P3, in RBP KO (G) and DKO mice (H), there are fewer wsCK+ BECs (arrow) compared to control (E) with DKO mice showing fewer formed ductal structures compared to RBP KO mice. At P15, while RBP KO mice show fewer formed ducts compared to control (K, arrow) there is complete absence of peripheral wsCK+ BECs in DKO mice (L, n=5) while control and HNF-6 KO mice show formed luminal ductal structures (I,J, arrow). In DKO mice at P15, portal veins were identified by surrounding mesenchymal cells, which are not present around central veins. Hepatic necrosis (L, arrowhead) was also present at P15 in DKO mice. PV, portal vein. Scale bar = 100μm.
To investigate the etiology of BEC paucity in DKO mice at P3 and P15, we analyzed both apoptosis and proliferation within BECs of DKO compared to age-matched controls. In DKO mice, there was no visible difference in apoptosis by TUNEL method within the wsCK+ BEC population compared to control at P3 (data not shown). Proliferation analysis performed by co-staining with cytokeratin-19 (CK19) and Ki67 (Supplemental Fig. 3A) showed no difference in the ratio of proliferative BECs in DKO mice at P3 and P15 when compared to age-matched controls (Supplemental Fig. 3B). These results indicate that the early postnatal BEC paucity observed in DKO mice is a consequence of abnormalities in BEC specification or a subsequent morphogenetic step.
Loss of HNF-6 and RBP-J results in alteration of intact communicating IHBD structure and ductular proliferation
Given the observed cholestatic phenotype and early postnatal IHBD paucity in DKO mice, we determined the effect of alterations in both HNF-6 and RBP-J on the intact communicating intrahepatic biliary system. To do this, we utilized an IHBD resin casting approach with tissue clearance to allow for direct visual analysis of the biliary cast within the left hepatic lobe. Figure 4 represents data obtained for all separate genotypes at age P60. Loss of RBP-J causes a decrease in the density of cast branches arising from major intrahepatic bile ducts (Fig. 4C), which correlates to a reduction in the number of bile ducts per portal vein (Fig. 4G) as previously quantified (24). While loss of HNF-6 alone failed to show an appreciable IHBD defect (Fig. 4B,F), loss of HNF-6 in the setting of RBP-J loss resulted in a further decrease in peripheral IHBD cast density as compared to RBP-J loss alone, with near complete loss of cast branching off main intrahepatic ducts (Fig. 4D). This phenotype was consistent among DKO mice (n=8).
Fig. 4. Severe decrease in communicating peripheral IHBDs with loss of HNF-6 and RBP-J.

Liquid resin/catalyst mix was retrograde injected into the common bile duct to obtain a cast of the communicating intrahepatic biliary architecture (A-D) in P60 mice, with images representative of entire left lobe cleared with BABB. (E-H) Corresponding paraffin sections from mice of equal genotype and age, immunostained for CK19 to mark BECs and counterstained with Mayer’s hematoxylin. Images obtained from periphery of left lobe. (A) Control (littermates without Alb-Cre transgene, n=3). (B) HNF-6 KO (n=5). (C) RBP KO (n=6). (D) DKO (n=8). While loss of RBP-J leads to a decrease in communicating peripheral IHBDs cast branches (C) and peripheral bile duct paucity (G), loss of both HNF-6 and RBP-J leads to a more severe decrease in communicating peripheral IHBD cast branches (D) yet an increase in the number of CK19+ BECs surrounding portal veins at P60 (H). (A) Scale bar = 1 mm. (E) Scale bar = 100 μm. (E-G) Arrow, CK19+ ductal structures.
Analysis of histopathology at this age revealed an unexpected yet consistent ductular reaction in DKO mice (Fig. 4H, n=10), defined as disorganized CK19+ BECs surrounding portal veins. These cells were present throughout peripheral periportal regions of the liver parenchyma and were not communicating with the intrahepatic biliary system based on resin cast analysis (Fig. 4D). Cytokeratin-positive BECs in P60 DKO animals had a higher proliferative index compared to BECs in age-matched control mice (Fig. 5A), as determined by proliferation analysis co-staining with CK19 and Ki67 (Fig. 5B,C). Importantly, these reactive CK19+ cells did not represent BECs that escaped Alb-Cre mediated HNF-6 deletion. HNF-6 protein expression was not visible in reactive peripheral ductular cells on immunostain analysis compared to control (Fig. 5D,E). In DKO mice at P60, only limited hilar BECs remained positive for HNF-6 protein compared to control (Fig. 5F,G), a pattern similar to that seen at earlier time points in HNF-6 KO mice (Fig. 1F,H).
Fig. 5. A proliferative cytokeratin-positive reactive population arises in adult mice with loss of both HNF-6 and RBP-J.

(A) The ratio of double Ki67/CK19+ to total CK19+ cells counted per mouse. A minimum of 354 total CK19+ cells were counted from each control mouse (n=3), with a total of 1390 CK19+ cell counted for control. A minimum of 531 total CK19+ cells were counted from each DKO mouse (n=5), with a total of 4126 CK19+ cells counted for DKO. CK19+ BECs in DKO mice have a significantly increased proliferation ratio compared to control mice. Statistical analysis was performed using a two-tailed Student’s t-test comparing proliferative ratios of separate mice. * P < 0.05. (B-C) Representative paraffin sections from the periphery of the left lobe in P60 control and DKO mice stained for Ki67 protein as a cellular marker for proliferation and CK19 as a marker for BECs. (B) Scale bar = 10 μm. (C) Arrow, dual labeled cell for Ki67 and CK19. (D-G) Paraffin sections from the left lobe in P60 Control and DKO mice stained for HNF-6 and CK19. (D-E) Periphery of left lobe. (F-G) Hilum of left lobe. Reactive peripheral ductal cells in DKO mice (E) remain negative for HNF-6 protein, while only limited hilar cells in DKO mice (G) stain positive for HNF-6 protein compared to control (D,F). (D) Scale bar = 20 μm.
Loss of HNF-6 and RBP-J alters expression of HNF-1β and Sox9
Loss of HNF-6 in the setting of RBP-J loss leads to more severe cholestatic liver injury and IHBD abnormalities compared to RBP-J loss alone. These findings indicate a possible genetic interaction between HNF-6 and Notch signaling during IHBD development. To molecularly characterize this interaction, we analyzed the expression of hepatic transcription factors by quantitative real-time RT-PCR analysis of total liver mRNA. Given that both HNF-6 and Notch signaling modulate expression of HNF-1β and Sox9 (12, 14-16) and that IHBD defects are observed with conditional loss of these genetic factors (17, 18), we hypothesized that either HNF-1β, Sox9, or both may be a common downstream mediator lost in DKO mice. Indeed, HNF-1β and Sox9 mRNA expression was decreased at E16.5 in DKO mice (Fig. 6A,B). Neither HNF-1β nor Sox9 were down regulated in HNF-6 KO mice and expression of both was increased in RBP KO mice at E16.5.
Fig. 6. Alteration in HNF-1β and Sox9 mRNA expression with loss of HNF-6 and RBP-J.

The relative expression ratio of HNF-1β (A,C,E) and Sox9 (B,D,F) mRNA was calculated using real-time RT-PCR. The deviation in cycle threshold (Ct) of experimental (HNF-6 KO, RBP KO, and DKO mice) versus control (without Alb-Cre) total liver samples are expressed in comparison to GAPDH (Glyceraldehyde 3-phosphate dehydrogenase) at ages E16.5 (A,B), P3 (C,D), and P60 (E,F). (A,B) At E16.5, expression of both HNF-1β and Sox9 is decreased only in DKO mice. (C,D) Expression of both factors remains decreased at P3 in DKO mice. HNF-1β and Sox9 expression are also decreased in RBP KO mice at P3. (E,F) At P60, Sox9 expression remains decreased in DKO mice while HNF-1β expression does not differ significantly from control. A minimum of 3 samples from separate mice were analyzed for each genotype at each age group. Statistical analysis was performed using a two-tailed Student’s t-test. Error bars represent standard error of the mean. *P<0.05, **P<0.01, ***P<0.001.
At P3, Sox9 expression was decreased in both RBP KO and DKO mice (Fig. 6D). While Sox9 expression remained decreased in DKO mice at P60, its expression did not differ significantly from control in RBP KO mice (Fig. 6F). At P3, HNF-1β expression was decreased in both RBP KO and DKO mice (Fig. 6C,D). At P60, RBP-J loss was associated with a continued decrease in HNF-1β expression while HNF-6 loss was associated with an increase in HNF-1β expression. Interestingly, HNF-1β expression did not differ compared to control at P60 in DKO mice (Fig. 6E). Overall, this pattern was consistent with immunostain analysis of HNF-1β protein expression (Fig. 7A-L). While expression of both HNF-1β and Sox9 was decreased at E16.5 and P3, expression of other transcription factors including HNF-4 and OC-2 were unchanged in DKO mice at these ages compared to control mice (data not shown). These observed modulations of HNF-1β and Sox9 expression during both embryonic and postnatal time points coincide with detectable alterations in the complex process of IHBD formation in DKO mice due to the loss of both HNF-6 and Notch signaling.
Fig. 7. Alteration in HNF-1β protein expression with loss of HNF-6 and RBP-J.

Representative paraffin sections from the left lobe of control (A,E,I), HNF-6 KO (B,F,J), RBP KO (C,G,K), and DKO mice (D,H,L) immunofluorescent stained for wsCK and HNF-1β. HNF-1β protein expression generally mirrored that of RNA expression profile. HNF-1β protein localization was visible in HNF-6 KO (B) and RBP KO (C), albeit lower in DKO (D) compared to control (A) at age E16.5. At P3, HNF-1β protein was visibly decreased in both RBP KO (G) and DKO mice (H) compared to control (E) peripheral ducts. At P60, HNF-1β was present in some ducts although there are fewer ducts in RBP KO mice (K). In DKO mice (L), HNF-1β protein staining appeared similar to control (I) at P60. HNF-6 KO mice (J) showed an increase in HNF-1β protein compared to control. (A) Scale bar = 10μm.
DISCUSSION
This study describes the modulation of postnatal IHBD development and cholestatic liver disease phenotype by HNF-6 and Notch signaling. RNA expression analysis of liver transcription factors presented in this study suggests that a direct in vivo genetic interaction between HNF-6 and Notch signaling exists. To date, no in vitro or in vivo studies have described the genetic interaction of these two factors in combination.
Independent genetic loss of HNF-1β or Sox9 leads to abnormalities IHBD development (17, 18). With loss of both HNF-6 and RBP-J, the expression of both HNF-1β and Sox9 was down regulated at E16.5 (Fig. 6A,B) and at P3 (Fig. 6C,D). Alb-Cre mediated recombination of the RBP-J locus begins at E14.5 (24). HNF-6 mRNA expression was decreased in HNF-6 KO mice and reached significance in DKO animals at E16.5 (Fig. 1A) with a visible decrease in HNF-6 protein expression by E18.5 in HNF-6 KO mice (Fig. 1F). During early postnatal time periods, DKO mice also demonstrated significant BEC paucity worse than that seen with RBP-J loss alone. This was not associated with changes in BEC apoptosis or proliferation (Supplemental Fig. 3, data not shown). Thus, in the setting of diminished HNF-6 and Notch signaling, there is a decreased expression of both Sox9 and HNF-1β during continued hepatoblast specification and IHBD morphogenesis. The observed decrease in BECs in DKO mice may be secondary to these changes in genetic factors essential for normal IHBD development, leading to a phenotype of severe IHBD paucity and cholestatic liver disease.
The control of HNF-1β and Sox9 expression by HNF-6 and Notch signaling may occur either in a parallel or an epistatic relationship, however we propose that this interaction occurs largely along parallel pathways. HNF-1β expression was increased in both single KO models at E16.5 (Fig. 6A,7B,C), suggesting possible compensation from the alternate parallel arm of either HNF-6 or Notch signaling. Within our model, hepatoblast-specific deletion of RBP-J alone results in an increase in Sox9 expression at E16.5 (Fig. 6B). This may be related to an observed 6-7 fold increase in HNF-6 mRNA and protein expression at E16.5 in RBP KO embryos (Fig. 1A, Supplemental Fig. 4B). With Notch signaling loss, this increase in HNF-6 is likely compensatory and may contribute to the observed increase in Sox9 expression. An alternate possibility would be an epistatic model in which Notch signaling occurs upstream of HNF-6, acting as an attenuator of HNF-6. However, previous experimental models have shown that constitutive Notch activation does not down-regulate expression of HNF-6 (12, 15). The possibility of HNF-6 occurring upstream of Notch signaling is also unlikely, given that Sox9 is a Notch target (12) and isolated hepatoblast-specific loss of HNF-6 did not result in any changes in Sox9 at ages E16.5 and P3 (Fig. 6B,D). The etiology of the decrease in Sox9 expression and increase in HNF-1β expression in HNF-6 KO mice compared to control at age P60 (Fig. 6E,F) is unknown. However, taken together these data suggest that control of factors essential for early IHBD development occurs along parallel mechanisms through HNF-6 and Notch signaling.
The pattern of HNF-1β and Sox9 expression in our model of conditional BHPC-specific loss of HNF-6 does not necessarily contradict previously published data describing a decrease in both Sox9 and HNF-1β expression with global HNF-6 loss. Initial regulation of both HNF-1β and Sox9 by HNF-6 appears to occur during early embryonic time points, with expression of both factors approaching or equaling control mice by E17.5 in a HNF-6 global loss model (14, 18). Given that HNF-6 protein expression is decreased compared to control by E18.5 (Fig. 1E,F), conditional deletion of HNF-6 by Alb-Cre may not occur early enough to affect the initial control of HNF-1β and Sox9 expression. However, our results do indicate a role for HNF-6, uncovered by the loss Notch signaling, in the continued control of downstream factor expression. We hypothesize that this role occurs in parallel with Notch signaling.
Interestingly, while Sox9 expression remains decreased in DKO animals at P60, the expression of HNF-1β is not decreased significantly compared to control mice at P60 (Fig. 6E,F, Fig. 7I,L). A ductular proliferative response is seen as well at this age, with multiple disorganized CK19+ BECs seen throughout the peripheral periportal regions of DKO livers (Fig. 4H, Fig. 5A,C). The etiology of this ductular response, as well as the restoration of HNF-1β during this adult time period, is unknown. Down-regulation of HNF-6 expression has been described to be an important factor in the ductular proliferative response seen in cholestatic liver injury resulting from bile duct ligation (25), and HNF-6 protein expression continues to remain decreased in the DKO ductular response (Fig. 5E). HNF-6 loss may thus contribute to the ductular response in the setting of cholestatic liver injury seen in DKO mice. However, this explanation by itself would not account for the restoration of HNF-1β expression. An alternate pathway or effector may thus drive the ductular proliferative response, and may or may not do so through improved expression of HNF-1β. Candidates include separate pathways such as Wnt or Hedgehog signaling (26, 27). Still, the observed abnormalities in both IHBD cast structure and cytokeratin-positive BECs indicate a severe defect in the formation of the communicating intrahepatic biliary system with loss of both HNF-6 and Notch signaling.
Associated with severe abnormalities in IHBD development in DKO mice, there was a worsening of cholestatic liver disease. This was evidenced by an increase in total bilirubin and alkaline phosphatase compared to RBP-J loss alone (Table 1), as well as extensive hepatic necrosis and bridging fibrosis (Fig. 2D). To date, HNF-6 polymorphisms have not been described in AGS patients. However, given that loss of HNF-6 in the setting of Notch signaling loss causes a clear phenotypic worsening of cholestatic liver disease, HNF-6 or downstream effectors may contribute to the clinical variability in disease phenotype that can be seen in AGS patients (6, 28).
In summary, our data indicate that HNF-6 and Notch signaling interact to affect expression of similar downstream mediators that are important in normal intrahepatic biliary development. We suggest that this interaction occurs along a parallel course, and loss of both HNF-6 and Notch signaling leads to subsequent loss of HNF-1β and Sox9 expression. This continued requirement for HNF-6 in the postnatal expression of genetic effectors essential to IHBD development has not been described previously. The complex regulatory profile of HNF-1β and Sox9 presented in this study does not prove that alterations in expression of these molecules are responsible for the abnormalities in IHBD development in DKO mice. However, alterations in their expression during periods of continued BEC specification and morphogenesis suggest that the observed worsened BEC paucity in DKO mice compared to RBP-J loss alone may be related to genetic loss of these important factors. The cholestatic livery injury observed in the adult mouse occurs with an associated ductular response. We hypothesize this may represent the involvement of a yet undefined alternate signaling mechanism that leads to the appearance of a proliferative cytokeratin-positive cell population and improvement in HNF-1β expression. This may serve as a model to identify alternate modulators of IHBD development and to study factors contributing to the cholestatic injury response, as well as further our understanding of clinical variability in patients with chronic cholestatic liver disease such as AGS.
Supplementary Material
Supplemental Fig. 1. Loss of both HNF-6 and RBP-J is associated with visible hepatic necrosis on gross examination of liver. (A-B) Representative images of the left lobe of the liver at age P15 in control mice (A) without Alb-Cre transgene and DKO mice (B). Multiple foci of hepatic necrosis (B, arrow) are visible on gross examination of DKO mice liver. These foci appear bile stained. (A) Scale bar = 1mm.
Supplemental Fig. 2. Formed IHBDs in DKO mice at P15 are located centrally within the hepatic lobe and stain positive for both DBA and CK19. Representative paraffin sections from the left lobe of P15 control (A) and DKO (B) mice stained for Dolichos biflorus agglutinin (DBA) and CK19 as a marker of BECs. In DKO mice (n=5), the only formed IHBDs were located centrally within the hepatic lobe and stained positive for both DBA and CK19. Peripheral CK19+/DBA− ducts and BECs were absent in DKO animals compared to control at age P15. (A) Scale bar = 10μm.
Supplemental Fig. 3. BECs present in DKO mice at P3 and P15 demonstrate similar proliferation ratio to BECs in control mice of same age. (A) Representative paraffin sections from the left lobe in P3 and P15 control (without Alb-Cre transgene, of equal age) and DKO mice stained for Ki67 protein as a proliferation maker and CK19 as a BEC marker. (A) Arrow, dual labeled cell for Ki67 and CK19. (A) Scale bar, 10um. (B) The ratio of dual labeled Ki67/CK19+ to total CK19+ cells counted separated by age and genotype. There was no statistical difference in P3 Control versus DKO (P = 0.10) and P15 Control versus DKO (P = 0.15) proliferation ratio. For control P3, a minimum of 85 total CK19+ cells were counted from each control mouse (n=4), with a total of 1099 CK19+ cells counted for P3 control. For DKO P3, a minimum of 61 total CK19+ cells were counted from each DKO mouse (n=5) with a total of 763 CK19+ cells counted for P3 DKO. For control P15, a minimum of 260 total CK19+ cells were counted from each control mouse (n=3), with a total of 1085 CK19+ cells counted for P15 control. For DKO P15, a minimum of 110 total CK19+ cells were counted from each DKO mouse (n=3), with a total of 421 CK19+ cells counted for P15 DKO. (B) Statistical analysis was performed using a two-tailed Student’s t-test comparing proliferative ratios of separate mice.
Supplemental Fig. 4. Alb-Cre mediated loss of RBP-J is associated with an increase in HNF-6 protein expression. (A,B) Representative paraffin sections from the left lobe immunostained for HNF-6 from E16.5 control (A) without Alb-Cre transgene and E16.5 RBP KO (B). At E16.5, RBP KO mice demonstrate an increase in HNF-6 protein expression, corresponding to an increase in HNF-6 mRNA expression demonstrated by quantitative RT-PCR analysis of total liver RNA also at E16.5 (Fig. 1A). (A) Scale bar, 100μm.
Acknowledgements
The authors thank Mark Magnuson and Tasuku Honjo for providing mice. The authors also thank Dong Hyun Lee and Kevin Song for help with genotyping mice; Teagan Walter, James Goldenring, Rick Peek, Louis Muglia, Lynette Gillis, D. Brent Polk, and Mark Magnuson for helpful comments and technical suggestions.
Financial Support:
These studies were supported by grants from the NIH to C.V. (T32-CA106183), from the NIH to M.G. (R01-DK065131), from the NIH to S.S.H (RO1-DK078640), the Vanderbilt Diabetes Research and Training Center (DK020593) and the Vanderbilt Digestive Disease Research Center (P30-DK058404) providing Core Services.
Abbreviations
- AGS
Alagille Syndrome
- RBP-J
recombination signal binding protein immunoglobulin kappa J
- IHBD
intrahepatic bile duct
- HNF-6
hepatocyte nuclear factor-6
- BHPC
bipotential hepatoblast progenitor cell
- BEC
biliary epithelial cell
- HNF-1β
hepatocyte nuclear factor-1β
- Sox9
SRY-related HMG box transcription factor 9
- OC-2
Onecut-2
- HNF-4
hepatocyte nuclear factor-4
- ALT
alanine aminotransferase
- ALP
alkaline phosphatase
- TB
total bilirubin
- BABB
benzyl alcohol: benzyl benzoate
- wsCK
wide-spectrum cytokeratin
- CK19
cytokeratin-19
- DBA
Dolichos biflorus agglutinin
- KO
knock-out
- DKO
double knock-out
References
- 1.Chiba S. Notch signaling in stem cell systems. Stem Cells. 2006;24:2437–2447. doi: 10.1634/stemcells.2005-0661. [DOI] [PubMed] [Google Scholar]
- 2.Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, Qi M, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16:243–251. doi: 10.1038/ng0797-243. [DOI] [PubMed] [Google Scholar]
- 4.Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, Piccoli DA, et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet. 1997;16:235–242. doi: 10.1038/ng0797-235. [DOI] [PubMed] [Google Scholar]
- 5.McDaniell R, Warthen DM, Sanchez-Lara PA, Pai A, Krantz ID, Piccoli DA, Spinner NB. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am J Hum Genet. 2006;79:169–173. doi: 10.1086/505332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Emerick KM, Rand EB, Goldmuntz E, Krantz ID, Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology. 1999;29:822–829. doi: 10.1002/hep.510290331. [DOI] [PubMed] [Google Scholar]
- 7.McCright B, Lozier J, Gridley T. A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development. 2002;129:1075–1082. doi: 10.1242/dev.129.4.1075. [DOI] [PubMed] [Google Scholar]
- 8.Geisler F, Nagl F, Mazur PK, Lee M, Zimber-Strobl U, Strobl LJ, Radtke F, et al. Liver-specific inactivation of Notch2, but not Notch1, compromises intrahepatic bile duct development in mice. Hepatology. 2008;48:607–616. doi: 10.1002/hep.22381. [DOI] [PubMed] [Google Scholar]
- 9.Lozier J, McCright B, Gridley T. Notch signaling regulates bile duct morphogenesis in mice. PLoS One. 2008;3:e1851. doi: 10.1371/journal.pone.0001851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hofmann JJ, Zovein AC, Koh H, Radtke F, Weinmaster G, Iruela-Arispe ML. Jagged1 in the portal vein mesenchyme regulates intrahepatic bile duct development: insights into Alagille syndrome. Development. 2010;137:4061–4072. doi: 10.1242/dev.052118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sparks EE, Huppert KA, Brown MA, Washington MK, Huppert SS. Notch signaling regulates formation of the three-dimensional architecture of intrahepatic bile ducts in mice. Hepatology. 2010;51:1391–1400. doi: 10.1002/hep.23431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zong Y, Panikkar A, Xu J, Antoniou A, Raynaud P, Lemaigre F, Stanger BZ. Notch signaling controls liver development by regulating biliary differentiation. Development. 2009;136:1727–1739. doi: 10.1242/dev.029140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clotman F, Lemaigre FP. Control of hepatic differentiation by activin/TGFbeta signaling. Cell Cycle. 2006;5:168–171. doi: 10.4161/cc.5.2.2341. [DOI] [PubMed] [Google Scholar]
- 14.Clotman F, Lannoy VJ, Reber M, Cereghini S, Cassiman D, Jacquemin P, Roskams T, et al. The onecut transcription factor HNF6 is required for normal development of the biliary tract. Development. 2002;129:1819–1828. doi: 10.1242/dev.129.8.1819. [DOI] [PubMed] [Google Scholar]
- 15.Tanimizu N, Miyajima A. Notch signaling controls hepatoblast differentiation by altering the expression of liver-enriched transcription factors. J Cell Sci. 2004;117:3165–3174. doi: 10.1242/jcs.01169. [DOI] [PubMed] [Google Scholar]
- 16.Matthews RP, Lorent K, Russo P, Pack M. The zebrafish onecut gene hnf-6 functions in an evolutionarily conserved genetic pathway that regulates vertebrate biliary development. Dev Biol. 2004;274:245–259. doi: 10.1016/j.ydbio.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 17.Coffinier C, Gresh L, Fiette L, Tronche F, Schutz G, Babinet C, Pontoglio M, et al. Bile system morphogenesis defects and liver dysfunction upon targeted deletion of HNF1beta. Development. 2002;129:1829–1838. doi: 10.1242/dev.129.8.1829. [DOI] [PubMed] [Google Scholar]
- 18.Antoniou A, Raynaud P, Cordi S, Zong Y, Tronche F, Stanger BZ, Jacquemin P, et al. Intrahepatic bile ducts develop according to a new mode of tubulogenesis regulated by the transcription factor SOX9. Gastroenterology. 2009;136:2325–2333. doi: 10.1053/j.gastro.2009.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang H, Ables ET, Pope CF, Washington MK, Hipkens S, Means AL, Path G, et al. Multiple, temporal-specific roles for HNF6 in pancreatic endocrine and ductal differentiation. Mech Dev. 2009;126:958–973. doi: 10.1016/j.mod.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han H, Tanigaki K, Yamamoto N, Kuroda K, Yoshimoto M, Nakahata T, Ikuta K, et al. Inducible gene knockout of transcription factor recombination signal binding protein-J reveals its essential role in T versus B lineage decision. Int Immunol. 2002;14:637–645. doi: 10.1093/intimm/dxf030. [DOI] [PubMed] [Google Scholar]
- 21.Postic C, Magnuson MA. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis. 2000;26:149–150. doi: 10.1002/(sici)1526-968x(200002)26:2<149::aid-gene16>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 22.Landry C, Clotman F, Hioki T, Oda H, Picard JJ, Lemaigre FP, Rousseau GG. HNF-6 is expressed in endoderm derivatives and nervous system of the mouse embryo and participates to the cross-regulatory network of liver-enriched transcription factors. Dev Biol. 1997;192:247–257. doi: 10.1006/dbio.1997.8757. [DOI] [PubMed] [Google Scholar]
- 23.Rausa F, Samadani U, Ye H, Lim L, Fletcher CF, Jenkins NA, Copeland NG, et al. The cut-homeodomain transcriptional activator HNF-6 is coexpressed with its target gene HNF-3 beta in the developing murine liver and pancreas. Dev Biol. 1997;192:228–246. doi: 10.1006/dbio.1997.8744. [DOI] [PubMed] [Google Scholar]
- 24.Sparks EE, Perrien DS, Huppert KA, Peterson TE, Huppert SS. Defects in hepatic Notch signaling result in disruption of the communicating intrahepatic bile duct network in mice. Dis Model Mech. 2011;4:359–367. doi: 10.1242/dmm.005793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holterman AX, Tan Y, Kim W, Yoo KW, Costa RH. Diminished hepatic expression of the HNF-6 transcription factor during bile duct obstruction. Hepatology. 2002;35:1392–1399. doi: 10.1053/jhep.2002.33680. [DOI] [PubMed] [Google Scholar]
- 26.Omenetti A, Popov Y, Jung Y, Choi SS, Witek RP, Yang L, Brown KD, et al. The hedgehog pathway regulates remodelling responses to biliary obstruction in rats. Gut. 2008;57:1275–1282. doi: 10.1136/gut.2008.148619. [DOI] [PubMed] [Google Scholar]
- 27.Sackett SD, Gao Y, Shin S, Esterson YB, Tsingalia A, Hurtt RS, Brondell K, et al. Foxl1 promotes liver repair following cholestatic injury in mice. Lab Invest. 2009;89:1387–1396. doi: 10.1038/labinvest.2009.103. [DOI] [PubMed] [Google Scholar]
- 28.Kamath BM, Bason L, Piccoli DA, Krantz ID, Spinner NB. Consequences of JAG1 mutations. J Med Genet. 2003;40:891–895. doi: 10.1136/jmg.40.12.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1. Loss of both HNF-6 and RBP-J is associated with visible hepatic necrosis on gross examination of liver. (A-B) Representative images of the left lobe of the liver at age P15 in control mice (A) without Alb-Cre transgene and DKO mice (B). Multiple foci of hepatic necrosis (B, arrow) are visible on gross examination of DKO mice liver. These foci appear bile stained. (A) Scale bar = 1mm.
Supplemental Fig. 2. Formed IHBDs in DKO mice at P15 are located centrally within the hepatic lobe and stain positive for both DBA and CK19. Representative paraffin sections from the left lobe of P15 control (A) and DKO (B) mice stained for Dolichos biflorus agglutinin (DBA) and CK19 as a marker of BECs. In DKO mice (n=5), the only formed IHBDs were located centrally within the hepatic lobe and stained positive for both DBA and CK19. Peripheral CK19+/DBA− ducts and BECs were absent in DKO animals compared to control at age P15. (A) Scale bar = 10μm.
Supplemental Fig. 3. BECs present in DKO mice at P3 and P15 demonstrate similar proliferation ratio to BECs in control mice of same age. (A) Representative paraffin sections from the left lobe in P3 and P15 control (without Alb-Cre transgene, of equal age) and DKO mice stained for Ki67 protein as a proliferation maker and CK19 as a BEC marker. (A) Arrow, dual labeled cell for Ki67 and CK19. (A) Scale bar, 10um. (B) The ratio of dual labeled Ki67/CK19+ to total CK19+ cells counted separated by age and genotype. There was no statistical difference in P3 Control versus DKO (P = 0.10) and P15 Control versus DKO (P = 0.15) proliferation ratio. For control P3, a minimum of 85 total CK19+ cells were counted from each control mouse (n=4), with a total of 1099 CK19+ cells counted for P3 control. For DKO P3, a minimum of 61 total CK19+ cells were counted from each DKO mouse (n=5) with a total of 763 CK19+ cells counted for P3 DKO. For control P15, a minimum of 260 total CK19+ cells were counted from each control mouse (n=3), with a total of 1085 CK19+ cells counted for P15 control. For DKO P15, a minimum of 110 total CK19+ cells were counted from each DKO mouse (n=3), with a total of 421 CK19+ cells counted for P15 DKO. (B) Statistical analysis was performed using a two-tailed Student’s t-test comparing proliferative ratios of separate mice.
Supplemental Fig. 4. Alb-Cre mediated loss of RBP-J is associated with an increase in HNF-6 protein expression. (A,B) Representative paraffin sections from the left lobe immunostained for HNF-6 from E16.5 control (A) without Alb-Cre transgene and E16.5 RBP KO (B). At E16.5, RBP KO mice demonstrate an increase in HNF-6 protein expression, corresponding to an increase in HNF-6 mRNA expression demonstrated by quantitative RT-PCR analysis of total liver RNA also at E16.5 (Fig. 1A). (A) Scale bar, 100μm.