

Abstract
Background
Chronic rhinosinusitis (CRS) is a heterogeneous disease characterized by local inflammation of the upper airways and sinuses and is frequently divided into polypoid CRS (CRSwNP) and non-polypoid CRS (CRSsNP). However, the mechanism of inflammation in CRS has still not been fully elucidated. The aim of the study was to investigate the role of IL-32, a recently discovered proinflammatory cytokine, in CRS.
Methods
We collected nasal epithelial cells and nasal tissue from patients with CRS and control subjects. We assayed mRNA for IL-32 by real-time PCR and measured IL-32 protein by ELISA, western blot and immunohistochemistry.
Results
The expression of mRNA for IL-32 was elevated in epithelial cells from uncinate tissue from patients with CRSsNP compared to patients with CRSwNP (p<0.05), control subjects (p=0.06) or epithelial cells from nasal polyp tissue (p<0.05). Production of IL-32 was induced by IFN-γ, TNF and dsRNA in primary airway epithelial cells. In whole tissue extracts, the expression of IL-32 protein was significantly elevated in patients with CRSwNP compared to patients with CRSsNP and control subjects. Immunohistochemistry data showed that IL-32 was detected in mucosal epithelial cells and inflammatory cells in the lamina propria. Levels of IL-32 were correlated with levels of CD3 and macrophage mannose receptor in nasal polyp tissue. Immunofluorescence data showed IL-32 co-localization with CD3 positive T cells and CD68 positive macrophages in nasal polyps.
Conclusion
Overproduction of IL-32 may be involved in the pathogenesis of CRS, although the role of IL-32 in the inflammation in CRSsNP and CRSwNP may be different.
Keywords: Chronic rhinosinusitis, Epithelial cells, Interleukin-32, Macrophages, T lymphocytes
Introduction
Chronic rhinosinusitis (CRS) is a chronic inflammatory disease of the sinuses and upper airways that affects over 30 million Americans (1). It is a significant cause of morbidity in the United States. Clinically, CRS is a heterogenous symptom complex, often resistant to medical therapy, that is typically characterized by two or more of the following: mucopurulent drainage, nasal obstruction, facial pain/pressure, and hyposmia/anosmia (1, 2). Current medications have variable efficacy and over 250,000 surgical procedures are performed annually in the United States on patients who failed maximal medical therapy. CRS is clinically classified into CRS with nasal polyps (CRSwNP) and CRS without nasal polyps (CRSsNP). In general, CRSwNP is associated more closely with the clinical complaints of nasal obstruction and smell loss, and CRSsNP is associated with the symptoms of facial pain/pressure and headaches (3). Although CRS has been characterized by chronic inflammation, the mechanisms of inflammation in CRSsNP and CRSwNP are generally different. CRSwNP, characteristically, has more Th2 polarization and a greater degree of tissue eosinophilia (4, 5). In contrast, CRSsNP demonstrates more Th1 polarization and collagen deposition in the nasal mucosa (4-6). However, the mechanism of inflammation in CRS has still not been fully elucidated.
Interleukin-32 (IL-32) is a recently described proinflammatory cytokine produced by T cells, NK cells, monocytes, endothelial cells, and epithelial cells (7, 8). It was cloned as a gene induced by IL-2 and initially named natural killer cell transcript 4, NK4, and its function remained unknown until 2005 (7, 9). There are 6 isoforms, α, β, γ, δ, ε and ζ, caused by mRNA splicing (8). There are potentially diverse roles for the different isoforms of IL-32; however, the functional differences are not well understood. Because the receptor for IL-32 and the ortholog of IL-32 in rodents have not been discovered, the function and regulation of IL-32 has been difficult to study. IL-32 is known to be induced by TNF, IL-1β, IFN-γ, dsRNA and LPS in monocytes, endothelial cells, epithelial cells, keratinocytes and fibroblasts (7, 10-15). IL-32 activates production of TNF and IL-8 through the activation of NF-κB and p38 MAP kinase (7). IL-32 synergizes with nucleotide oligomerization domain ligands to stimulate IL-1β and IL-6 release in a caspase-1-dependent manner (16). Recently, it has been reported that silencing endogenous IL-32 impairs IL-1β- or LPS-dependent production of TNF, IL-6 and IL-8 in endothelial cells and the monocytic cell line THP-1 cells (12, 17). Other groups have shown that IL-32 is a key endogenous factor in TCR- and IFN-γ-dependent apoptosis in T cells and keratinocytes (13, 18). Therefore, IL-32 is now recognized as a dual functional protein that acts as a proinflammatory cytokine and an endogenous regulator of cytokine production and apoptosis.
IL-32 is known to play an important role in host defense against microorganisms including Mycobacterium, HIV and influenza (10, 19-21). IL-32 has also been implicated in several inflammatory disorders including rheumatoid arthritis, Crohn's disease, atopic dermatitis, and chronic obstructive pulmonary disease (8, 13, 15, 22, 23). IL-32 expression has been found to correlate with disease severity in these conditions (13, 15, 24). Thus far, there have been no studies regarding IL-32 in sinus epithelial cells and in the disease states of CRS. In the present study, we investigated the expression of IL-32 and its potential role in the inflammatory response in CRSwNP and CRSsNP.
Methods
Patients and biopsies
CRS patients were recruited from the Allergy-Immunology clinic and the Otolaryngology clinic of the Northwestern Medical Faculty Foundation (NMFF) and the Northwestern Sinus Center at NMFF. Sinonasal and nasal polyp tissues were obtained from routine Functional Endoscopic Sinus Surgery in the patients with CRS. All subjects met the criteria for CRS as defined by the American Academy of Otolaryngology-Head and Neck Surgery Chronic Rhinosinusitis Task Force (1, 25). Patients with an established immunodeficiency, pregnancy, coagulation disorder, Churg-Strauss syndrome, diagnosis of classic allergic fungal sinusitis or cystic fibrosis were excluded from the study. Sinus tissues from inflammation-free control subjects undergoing endoscopic sinus surgery for surgical access to the anterior skull base were recruited from the Otolaryngology practice at NMFF. All subjects signed informed consent forms and the protocol governing procedures for this study has been approved by the Institutional Review Board of Northwestern University Feinberg School of Medicine. Details of subjects' characteristics are included in Table I and in Supporting information.
Table 1. Subject characteristics.
| Control | CRSsNP | CRSwNP | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Total no. of subjects | n=24 (13M/11F) | n=40 (15M/25F) | n=54 (42M/12F) | |||||||
| Age (y), median (range) | 42* (16-63)# | 37 (23-64) | 42 (26-74) | |||||||
| Y | N | U | Y | N | U | Y | N | U | ||
| Atopy | 1 | 19 | 4 | 20 | 11 | 9 | 24 | 17 | 13 | |
| Asthma | 0 | 23 | 1 | 5 | 30 | 5 | 23 | 29 | 2 | |
| Methodologies used | UT | UT | UT | NP | ||||||
| Nasal scraping cells | n=15 (11M/4F) | n=21 (8M/13F) | n=14 (13M/1F) | n=25 (23M/2F) | ||||||
| Age | 44 (27-63) | 36 (26-55) | 42 (26-61) | 43 (26-68) | ||||||
| Tissue RNA | n=9 (3M/6F) | n=18 (8M/10F) | n=19 (15M/4F) | n=26 (19M/7F) | ||||||
| Age | 39 (16-59) | 40 (23-64) | 41 (26-61) | 44 (26-74) | ||||||
| Tissue Extract | n=5 (1M/4F) | n=10 (3M/7F) | n=10 (9M/1F) | n=10 (9M/1F) | ||||||
| Age | 47 (25-63) | 36 (24-51) | 47 (30-74) | 42 (26-63) | ||||||
*median, #(range). Y; yes, N; no, U; unknown.
Cell culture, real-time PCR, western blot, immunohistochemistry and ELISA
The general methods for culture of primary nasal epithelial cells (PNEC) (26), culture of normal human bronchial epithelial cells (NHBE) (27), real-time PCR (28), western blot (29), immunohistochemistry (30) and ELISA (26) are described previously. The details of methods are described in Supporting information.
Statistics
All data are reported as the mean ± SEM unless otherwise noted. Differences between groups were analyzed using the Mann-Whitney U-test (in clinical samples) and the two-tailed paired Student's t test (in in vitro samples). Correlations were assessed by using the Spearman's rank correlation. A p value of less than 0.05 was considered significant.
Results
IL-32 Expression in nasal epithelial scraping cells in CRS
To determine the relevance of IL-32 expression in CRS, sinonasal and polyp tissues and nasal epithelial scraping cells were collected from 40 subjects with CRSsNP, 54 subjects with CRSwNP and 24 control subjects.
We assessed the expression of IL-32 in freshly isolated epithelial scraping cells from uncinate tissue (UT) from patients with CRSsNP, CRSwNP, and controls as well as epithelial scraping cells from nasal polyp (NP) tissue from patients with CRSwNP. IL-32 mRNA levels were increased in epithelial scraping cells from UT in patients with CRSsNP (n=21) compared to UT from controls (p=0.063, n=15), CRSwNP (p<0.05, n=14), and epithelial cells from NP tissue (p<0.05, n=25) (Fig 1).
Figure 1. Expression of IL-32 in epithelial scraping cells.
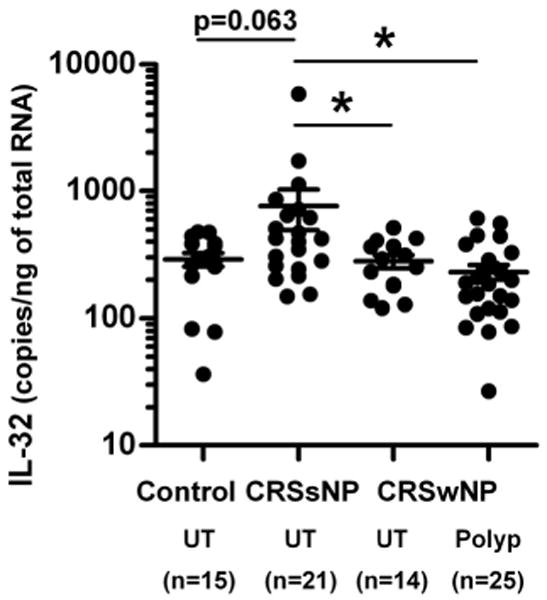
Total RNA was extracted from epithelial scraping cells from uncinate tissue (UT) and nasal polyps (Polyp) and expressions of IL-32 were analyzed using real-time PCR. IL-32 mRNA levels were increased in epithelial scraping cells from UT in patients with CRSsNP compared to UT from controls (p=0.063), CRSwNP (p<0.05), and nasal polyps (p<0.05, n=25). *p<0.05
Expression of IL-32 in primary human airway epithelial cells
Although we found upregulation of IL-32 in epithelial cells from patients with CRSsNP, regulation of IL-32 in primary cells has not been carefully studied. To our knowledge, no one has reported the regulation of IL-32 expression in primary airway epithelial cells. To study the regulation of IL-32 in primary nasal epithelial cells (PNEC) and primary normal human bronchial epithelial cells (NHBE), PNEC and NHBE were treated with known activating cytokines including TNF, IL-4, IL-17A, IFN-β and IFN-γ and TLR ligands including PGN (TLR2 ligand), dsRNA (TLR3 ligand) and LPS (TLR4 ligand) for 24 hours. Messenger RNA for IL-32 was significantly upregulated by stimulation with TNF (16-fold, 26-fold, p<0.05), IFN-γ (79-fold, 188-fold, p<0.05), and dsRNA (21-fold, 38-fold, p<0.05) and not affected by IL-4, IL-17A, IFN-β, PGN or LPS in both PNEC (n=7) and NHBE (n=5) (Fig 2A, 2B and data not shown). These suggest that the regulation of IL-32 in PNEC and NHBE is likely the same. We therefore focused on the regulation of IL-32 in NHBE for further studies.
Figure 2. Induction of IL-32 in human primary epithelial cells.
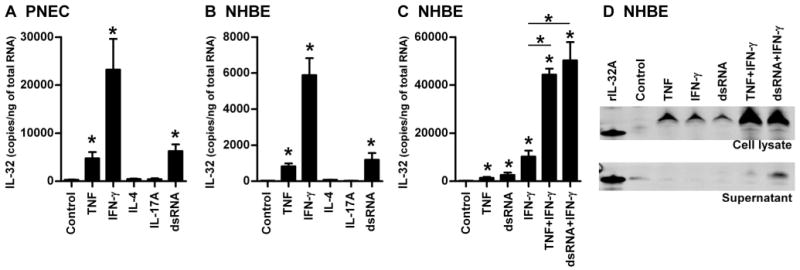
Primary nasal epithelial cells (PNEC, A) and primary normal human bronchial epithelial cells (NHBE, B and C) were incubated for 24 hours (A-C) with 100 ng/ml TNF, 10 ng/ml IFN-γ, 100 ng/ml IL-4, 100 ng/ml IL-17A, 5 μg/ml dsRNA. The expression of mRNAs for IL-32 was determined by real-time PCR (A-C). NHBE were stimulated with 50 ng/ml TNF, 10 ng/ml IFN-γ, 5 μg/ml dsRNA and their combinations for 48 hours and then protein expression in the cell lysates and supernatants was analyzed by western blot (D). Results shown are mean ± SEM of seven (A) and four (B and C) independent experiments. The results are representative of four separate experiments (D). *p<0.05.
It has been reported that stimulation of IL-32 mRNA is further enhanced by combining TNF and IFN-γ in intestinal epithelial cell lines (15). Therefore, we examined the effect of the combined stimulation of cytokines, TNF and IFN-γ, and the TLR3 ligand, dsRNA, on the expression of IL-32 in NHBE. We found that TNF and dsRNA synergistically and significantly enhanced IFN-γ-dependent IL-32 mRNA expression in NHBE (4.3-fold and 4.9 fold, n=3-4, p<0.05) (Fig 2C). We further confirmed this finding by western blot. IL-32 protein was found in the cell lysate after stimulation with TNF, IFN-γ and dsRNA (Fig 2D). In addition, expression of IL-32 in the cell lysates was further enhanced when NHBE were stimulated by the combination of IFN-γ and TNF or IFN-γ and dsRNA (Fig 2D). We detected IL-32 protein in the supernatants only when NHBE were stimulated by the combinations (Fig 2D). The IL-32 protein was detected as a 25 KDa band. This was larger than the recombinant IL-32A protein, indicating that the major isoforms in airway epithelial cells are less likely to be the α isoform.
IL-32 Expression in tissue samples in CRS
To further confirm at the protein level the elevation of IL-32 that we observed in nasal epithelial cells from UT from patients with CRSsNP, we used immunohistochemistry to detect IL-32 protein in UT and NP tissue. Immunohistochemistry demonstrated considerable IL-32 staining in the mucosal and the glandular epithelium as well as in submucosal cells (Fig 3A and data not shown). However, we did not observe more intense staining of IL-32 in epithelial cells from CRSsNP compared to the levels in control subjects and patients with CRSwNP. In contrast, we found that IL-32+ inflammatory cells were elevated in NP tissue from patients with CRSwNP (Fig 3A and data not shown). We therefore decided to determine the levels of IL-32 in sinus tissue. We assessed the expression of mRNA for IL-32 in UT from patients with CRSsNP, CRSwNP and controls, as well as NP tissue from CRSwNP. IL-32 mRNA was only significantly increased in NP tissue from patients with CRSwNP (p<0.001) in comparison to UT from patients with either form of CRS or control subjects (Fig 3B). To confirm this observation at the protein level, we made detergent extracts from homogenates of UT and NP tissues and then measured the IL-32 concentration by ELISA. We initially assayed the concentration of IL-32 in tissue extracts by a commercial IL-32α ELISA kit. However, the assay lacked adequate sensitivity to detect the IL-32α protein in tissue homogenates (data not shown). In addition, we found that the levels of expression of IL-32α were about 100-fold less than the levels of IL-32nonα in epithelial cells and in whole tissue extracts (Fig E2). This suggests that IL-32α is a minor isoform in nasal mucosa. We therefore decided to use an in-house ELISA system for pan-IL-32 isoforms. We found that the IL-32 protein was significantly increased in UT (n=10, 0.67 ± 0.16 ng/mg) and NP tissue (n=10, 0.46 ± 0.16 ng/mg) from patients with CRSwNP compared to UT from controls (n=5, 0.04 ± 0.07 ng/mg) (Fig 3C).
Figure 3. Expression of IL-32 in sinus tissue.
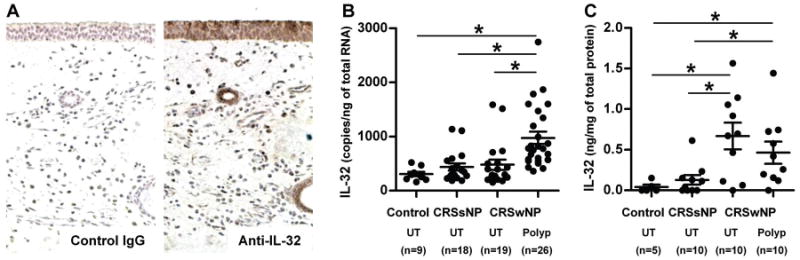
Representative immunostaining for IL-32 and control IgG1 in nasal polyp tissue from a patient with CRSwNP (A). IL-32 was detected in mucosal and glandular epithelium as well as in infiltrated inflammatory cells in nasal polyps. Total RNA was extracted from UT and nasal polyps (Polyp) and expression of IL-32nonA (IL-32) was analyzed by real-time PCR (B). IL-32 mRNA was significantly increased in nasal polyp tissue (p<0.001). The concentration of IL-32 in tissue homogenates of UT and Polyp was measured using ELISA (C). IL-32 protein was significantly increased in CRSwNP. IL-32 concentration was normalized to the concentration of total protein. *p<0.05.
Detection of IL-32 producing cells in nasal polyps
We found that IL-32+ cells were significantly elevated in the submucosal region of NP tissue and that IL-32 mRNA and protein were significantly elevated in NP tissue (Fig 3). We next proceeded to identify the IL-32 producing cells in the nasal mucosa of NP tissue. We first assessed whether the expression of IL-32 correlated with the levels of mRNA expression for markers of inflammatory cells in NP tissue by real-time PCR. We found that the levels of expression of IL-32 mRNA in NP tissue significantly and positively correlated with the expression of CD3 (r=0.789, p<0.001, n=26), macrophage mannose receptor (MMR, r=0.537, p=0.005), CD1c (r=0.481, p=0.013) and tryptase (r=0.489, p=0.011) but not with CD20, CD56, CD138 or ECP (Fig 4) indicating that IL-32 might be expressed in T cells, macrophages, dendritic cells and mast cells. To identify the IL-32 producing cells in the submucosal tissue of nasal polyps, we performed immunofluorescence analysis using an anti-IL-32 antibody and antibodies against markers of these cells including CD3 (T cells), CD68 (macrophages) and tryptase (mast cells). We found IL-32 co-localization with CD3+ T cells and CD68+ macrophages but not with mast cells in NP tissue (Fig 5).
Figure 4. Correlation of IL-32 with cell specific markers in nasal polyps.

Total RNA was extracted from nasal polyp tissue and the expression of IL-32 and cell specific markers was analyzed by real-time PCR (n=26). The correlations were assessed by using the Spearman rank correlation.
Figure 5. Identification of IL-32 producing cells by immunofluorescence assay.
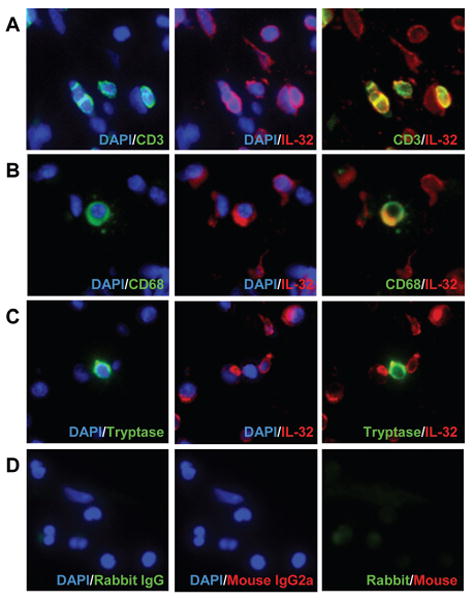
The immunofluorescence assay was performed using anti-human IL-32 mAb (red fluorescence), anti-CD3 mAb (green fluorescence) for T cells (A), anti-CD68 mAb (green fluorescence) for macrophages (B), anti-tryptase mAb (green fluorescence) for mast cells (C) and control IgG (D). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (blue fluorescence). IL-32 was detected in CD3+ T cells and CD68+ macrophages in nasal polyps as demonstrated by co-localization (yellow fluorescence) of the antibody staining. The results are representative of three separate patients.
Discussion
This study provides the first demonstration that IL-32 is increased in epithelial cells from patients with CRSsNP (Fig 1) and that the levels of IL-32 in primary airway epithelial cells are regulated by the Th1 cytokine IFN-γ, the proinflammatory cytokine TNF and the TLR3 ligand dsRNA (Fig 2). Significant levels of IL-32 mRNA and protein were found in whole tissue from patients with CRSwNP (Fig 3). The expression of IL-32 in nasal polyp tissue was positively correlated with levels of CD3 and MMR (Fig 4). IL-32 positive cells were more numerous in the submucosal tissue of nasal polyps from patients with CRSwNP, and they were co-localized with CD3 positive and CD68 positive cells indicating that T cells and macrophages are the major IL-32 producing cells in nasal polyps (Fig 3 and 5).
CRS is characterized by chronic inflammation of the paranasal sinuses and is frequently divided into polypoid CRS (CRSwNP) and non-polypoid CRS (CRSsNP). The lack of understanding of the mechanism for inflammation in CRS hinders the development of new treatments for this disease. IL-32 is a novel proinflammatory cytokine and also acts as an endogenous regulator of cytokine production and apoptosis. IL-32 is expressed in epithelial cells and the overexpression of IL-32 has been shown to be involved in Th1-related inflammatory disease including rheumatoid arthritis, Crohn's disease and chronic obstructive pulmonary disease (8, 15, 22, 23). In addition, there are very few studies of epithelial cells taken from patients with CRS and controls, and most of those studies focus on epithelial cells from patients with CRSwNP. We, therefore, sought to examine the expression of IL-32 in epithelial cells from patients with CRSsNP, which is characterized by Th1 polarized inflammation. In the current study, we report a very rare pattern of gene regulation in which expression is observed in epithelial cells from only patents with CRSsNP and not from those with CRSwNP. We demonstrated that mRNA encoding IL-32 was significantly increased in epithelial cells from patients with CRSsNP (Fig 1). Although IL-32 was found to be regulated by the Th1 cytokine IFN-γ in epithelial cell lines, to our knowledge, no one has shown the regulation of IL-32 in primary airway epithelial cells. We demonstrated that primary nasal and bronchial epithelial cells produced IL-32 when stimulated with IFN-γ, TNF, and dsRNA with a synergistic enhancement by the combination of IFN-γ with either TNF or dsRNA (Fig 2). Interestingly, there was no increased expression of IL-32 after treatment with the Th2 cytokine IL-4 or the Th17 cytokine IL-17A. Recently, Meyer et al demonstrated that Th1 cells but not Th2, Treg, or Th17 cells induced the expression of IL-32 in epidermal keratinocytes in an IFN-γ dependent manner (13). These results suggest that overexpression of IL-32 in epithelial cells from patients with CRSsNP may be induced by a Th1 environment.
We showed that IL-32 was upregulated in epithelial cells from patients with CRSsNP; however, it did not appear to be secreted, as demonstrated by low expression in nasal lavage fluids (data not shown). In in vitro studies, we did not see a substantial release of IL-32 protein in the culture supernatant of NHBE (Fig 2). Several groups have reported that IL-32 is detected in the cytoplasm of keratinocytes, endothelial cells, T cells and peripheral blood mononuclear cells but is not secreted from the cells (12, 13, 18, 19). Nold-Petry et al showed that silencing of endogenous IL-32 results in the reduction of constitutive as well as IL-1β-induced production of IL-1α, IL-6 and IL-8 in endothelial cells (12). Other groups have shown that silencing of endogenous IL-32 prevents apoptosis in HeLa cells and keratinocytes (13, 18). These results suggest that IL-32 may act as an endogenous regulator of cytokine production and apoptosis in nasal epithelial cells. Future studies will be required to investigate whether IL-32 acts as a controller of proinflammatory cytokine production in epithelial cells or an inducer of epithelial damage in patients with CRSsNP.
Although we found clear evidence that IL-32 was elevated in epithelial cells from CRSsNP, we could not find more intense staining of IL-32 in CRSsNP by immunohistochemistry. In addition, we could not obtain enough total protein from epithelial scraping cells from patients for our current ELISA (data not shown). It is recognized that immunohistochemistry is not a quantitative method. Future studies will be required to generate a high sensitivity assay system for detection of the IL-32 protein and to confirm the elevation of IL-32 protein in epithelial cells from patients with CRSsNP.
In contrast to epithelial cells, we found increased expression of IL-32 in whole tissue extracts from nasal polyps but not in whole tissue from patients with CRSsNP (Fig 3). We have recently reported a similar trend in the expression of S100A8/A9 in CRS. We showed that the expression of S100A8/A9 was decreased in epithelial cells from CRSwNP; however, the expression of S100A8/A9 was increased in whole tissue extracts from patients with CRSwNP (31). We also found that concentrations of S100A8/A9 were correlated with the concentration of neutrophil elastase suggesting that the elevation of S100A8/A9 in nasal polyp tissue was reflected by the infiltration of S100A8/A9 producing neutrophils in the tissue (31). In our current study, immunohistochemistry data clearly showed that IL-32 positive inflammatory cells were more abundant in nasal polyp tissue (Fig 3 and data not shown). This suggests that the infiltration of inflammatory cells may be responsible for the elevation of IL-32 in whole tissue from nasal polyps.
Nasal polyps are generally characterized by eosinophilic inflammation. In addition, it has been reported that lymphocytes, macrophages, dendritic cells and neutrophils also accumulate in the nasal mucosa of patients with CRSwNP (4, 5, 26, 30-33). Our immunofluorescence data clearly showed that IL-32 was detected in CD3+ T cells and CD68+ macrophages (Fig 5) and that the expression of IL-32 was positively correlated with their markers, CD3 and MMR, in nasal polyps (Fig 4). These results suggest that T cells and macrophages are the major IL-32 producing cells in the nasal mucosa of nasal polyps. Calabrese et al showed that IL-32 was elevated in the lungs of patients with chronic obstructive pulmonary disease and that IL-32 was detected in alveolar macrophages and CD8 positive T cells in addition to epithelial cells (23). These combined results suggest that macrophages and T cells are the major IL-32 producing inflammatory cells in airway inflammatory diseases.
Although there are 6 isoforms of IL-32, variations in the distribution and functional differences of those isoforms are not well described. IL-32ε and IL-32ζ were very minor isoforms and were found only in activated T cells (18). Alternative expression of the major isoform of IL-32 was reported in some cells; IL-32α was the major isoform in myofibroblasts, IL-32β was the major isoform in activated T cells and IL-32γ was the major isoform in epidermal keratinocytes (13, 14, 18). Collectively, these findings suggest that there are specific and subtle biological differences in the expression, and therefore likely the function, of the different isoforms. In the current study, we also detected IL-32α and the other isoforms separately. We showed that the levels of expression of IL-32α were 100-fold less than IL-32nonα isoforms in PNEC, NHBE, nasal scraping cells and nasal tissues, although the expression patterns were similar between the IL-32α and the IL-32nonα isoforms (Fig E1, E2 and data not shown). Western blot data showed that the molecular weight of IL-32 was 25 KDa in NHBE (Fig 2). This suggests that the major isoform was IL-32β and/or IL-32γ in NHBE. Choi et al demonstrated that IL-32β and IL-32γ have greater biological activity than IL-32α and IL-32δ in peripheral blood mononuclear cells (11). This suggests that the more active isoform of IL-32 is expressed in airway epithelial cells. Future studies will be required to determine the characterization of isoforms of IL-32 in CRS.
Although IL-32 is a proinflammatory cytokine that induces production of IL-1β, IL-6, TNF and IL-8 in monocytes and peripheral blood mononuclear cells, the mechanism of secretion of IL-32 is still unclear (7, 11). Goda et al showed that IL-32 was released from apoptotic T cells (18). In the present study, we showed that the release of IL-32 in primary epithelial cells occurred when cells were stimulated with the combination of IFN-γ and dsRNA, and combination of IFN-γ and dsRNA induced highest LDH release in NHBE (data not shown), indicating that epithelial cells might be able to release IL-32 only during cell death. Another source of IL-32 in nasal polyps could be macrophages. Bai et al demonstrated that IL-32 controlled pathogen-dependent cytokine production endogenously in the differentiated THP-1 macrophage line (10). However, it is still unknown whether primary macrophages release IL-32 or IL-32 acts intracellularly in cells contained within nasal polyps. Future studies will be required to determine whether IL-32 is released by activation or during cell death, and whether IL-32 acts extracellularly in patients with CRS.
In summary, we report here that IL-32 is elevated in epithelial cells from patients with CRSsNP and that IL-32 producing T cells and macrophages are elevated in nasal tissue from patients with CRSwNP. These results support the conclusion that the role of IL-32 in inflammation associated with CRSsNP and CRSwNP may be different. Future studies will center on the function of IL-32 in CRS to determine its role in inflammation and the immune system.
Supplementary Material
Acknowledgments
We thank Dr. David Conley, Dr. Leslie C. Grammer, Mr. James Norton and Ms. Kathleen E. Harris of Northwestern University Feinberg School of Medicine for valuable advice and sample collection from patients.
This research was supported in part by NIH grants, R01 HL078860, R01 AI072570 and R37 HL068546 and by the Ernest S. Bazley Trust.
Abbreviations
- CRS
Chronic rhinosinusitis
- CRSsNP
CRS without nasal polyps
- CRSwNP
CRS with nasal polyps
- IL-32
Interleukin-32
- GUSB
β-glucuronidase
- UT
uncinate tissue
- NP
nasal polyp
- MMR
macrophage mannose receptor
References
- 1.Meltzer EO, Hamilos DL, Hadley JA, Lanza DC, Marple BF, Nicklas RA, et al. Rhinosinusitis: establishing definitions for clinical research and patient care. J Allergy Clin Immunol. 2004;114(6 Suppl):155–212. doi: 10.1016/j.jaci.2004.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benninger MS, Ferguson BJ, Hadley JA, Hamilos DL, Jacobs M, Kennedy DW, et al. Adult chronic rhinosinusitis: definitions, diagnosis, epidemiology, and pathophysiology. Otolaryngol Head Neck Surg. 2003;129(3 Suppl):S1–32. doi: 10.1016/s0194-5998(03)01397-4. [DOI] [PubMed] [Google Scholar]
- 3.Banerji A, Piccirillo JF, Thawley SE, Levitt RG, Schechtman KB, Kramper MA, et al. Chronic rhinosinusitis patients with polyps or polypoid mucosa have a greater burden of illness. Am J Rhinol. 2007;21(1):19–26. doi: 10.2500/ajr.2007.21.2979. [DOI] [PubMed] [Google Scholar]
- 4.Van Zele T, Claeys S, Gevaert P, Van Maele G, Holtappels G, Van Cauwenberge P, et al. Differentiation of chronic sinus diseases by measurement of inflammatory mediators. Allergy. 2006;61(11):1280–1289. doi: 10.1111/j.1398-9995.2006.01225.x. [DOI] [PubMed] [Google Scholar]
- 5.Bachert C, Van Bruaene N, Toskala E, Zhang N, Olze H, Scadding G, et al. Important research questions in allergy and related diseases: 3-chronic rhinosinusitis and nasal polyposis - a GALEN study. Allergy. 2009;64(4):520–533. doi: 10.1111/j.1398-9995.2009.01964.x. [DOI] [PubMed] [Google Scholar]
- 6.Van Bruaene N, Derycke L, Perez-Novo CA, Gevaert P, Holtappels G, De Ruyck N, et al. TGF-beta signaling and collagen deposition in chronic rhinosinusitis. J Allergy Clin Immunol. 2009;124(2):253–259. 259 e251–252. doi: 10.1016/j.jaci.2009.04.013. [DOI] [PubMed] [Google Scholar]
- 7.Kim SH, Han SY, Azam T, Yoon DY, Dinarello CA. Interleukin-32: a cytokine and inducer of TNFalpha. Immunity. 2005;22(1):131–142. doi: 10.1016/j.immuni.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 8.Dinarello CA, Kim SH. IL-32, a novel cytokine with a possible role in disease. Ann Rheum Dis. 2006;65 3:iii61–64. doi: 10.1136/ard.2006.058511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dahl CA, Schall RP, He HL, Cairns JS. Identification of a novel gene expressed in activated natural killer cells and T cells. J Immunol. 1992;148(2):597–603. [PubMed] [Google Scholar]
- 10.Bai X, Kim SH, Azam T, McGibney MT, Huang H, Dinarello CA, et al. IL-32 is a host protective cytokine against Mycobacterium tuberculosis in differentiated THP-1 human macrophages. J Immunol. 2010;184(7):3830–3840. doi: 10.4049/jimmunol.0901913. [DOI] [PubMed] [Google Scholar]
- 11.Choi JD, Bae SY, Hong JW, Azam T, Dinarello CA, Her E, et al. Identification of the most active interleukin-32 isoform. Immunology. 2009;126(4):535–542. doi: 10.1111/j.1365-2567.2008.02917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nold-Petry CA, Nold MF, Zepp JA, Kim SH, Voelkel NF, Dinarello CA. IL-32-dependent effects of IL-1beta on endothelial cell functions. Proc Natl Acad Sci U S A. 2009;106(10):3883–3888. doi: 10.1073/pnas.0813334106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meyer N, Zimmermann M, Burgler S, Bassin C, Woehrl S, Moritz K, et al. IL-32 is expressed by human primary keratinocytes and modulates keratinocyte apoptosis in atopic dermatitis. J Allergy Clin Immunol. 2010;125(4):858–865. e810. doi: 10.1016/j.jaci.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 14.Nishida A, Andoh A, Shioya M, Kim-Mitsuyama S, Takayanagi A, Fujiyama Y. Phosphatidylinositol 3-kinase/Akt signaling mediates interleukin-32alpha induction in human pancreatic periacinar myofibroblasts. Am J Physiol Gastrointest Liver Physiol. 2008;294(3):G831–838. doi: 10.1152/ajpgi.00535.2007. [DOI] [PubMed] [Google Scholar]
- 15.Shioya M, Nishida A, Yagi Y, Ogawa A, Tsujikawa T, Kim-Mitsuyama S, et al. Epithelial overexpression of interleukin-32alpha in inflammatory bowel disease. Clin Exp Immunol. 2007;149(3):480–486. doi: 10.1111/j.1365-2249.2007.03439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Netea MG, Azam T, Ferwerda G, Girardin SE, Walsh M, Park JS, et al. IL-32 synergizes with nucleotide oligomerization domain (NOD) 1 and NOD2 ligands for IL-1beta and IL-6 production through a caspase 1-dependent mechanism. Proc Natl Acad Sci U S A. 2005;102(45):16309–16314. doi: 10.1073/pnas.0508237102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hong J, Bae S, Kang Y, Yoon D, Bai X, Chan ED, et al. Suppressing IL-32 in monocytes impairs the induction of the proinflammatory cytokines TNFalpha and IL-1beta. Cytokine. 2010;49(2):171–176. doi: 10.1016/j.cyto.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 18.Goda C, Kanaji T, Kanaji S, Tanaka G, Arima K, Ohno S, et al. Involvement of IL-32 in activation-induced cell death in T cells. Int Immunol. 2006;18(2):233–240. doi: 10.1093/intimm/dxh339. [DOI] [PubMed] [Google Scholar]
- 19.Netea MG, Azam T, Lewis EC, Joosten LA, Wang M, Langenberg D, et al. Mycobacterium tuberculosis induces interleukin-32 production through a caspase- 1/IL-18/interferon-gamma-dependent mechanism. PLoS Med. 2006;3(8):e277. doi: 10.1371/journal.pmed.0030277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nold MF, Nold-Petry CA, Pott GB, Zepp JA, Saavedra MT, Kim SH, et al. Endogenous IL-32 controls cytokine and HIV-1 production. J Immunol. 2008;181(1):557–565. doi: 10.4049/jimmunol.181.1.557. [DOI] [PubMed] [Google Scholar]
- 21.Li W, Sun W, Liu L, Yang F, Li Y, Chen Y, et al. IL-32: a host proinflammatory factor against influenza viral replication is upregulated by aberrant epigenetic modifications during influenza A virus infection. J Immunol. 2010;185(9):5056–5065. doi: 10.4049/jimmunol.0902667. [DOI] [PubMed] [Google Scholar]
- 22.Joosten LA, Netea MG, Kim SH, Yoon DY, Oppers-Walgreen B, Radstake TR, et al. IL-32, a proinflammatory cytokine in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2006;103(9):3298–3303. doi: 10.1073/pnas.0511233103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calabrese F, Baraldo S, Bazzan E, Lunardi F, Rea F, Maestrelli P, et al. IL-32, a novel proinflammatory cytokine in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;178(9):894–901. doi: 10.1164/rccm.200804-646OC. [DOI] [PubMed] [Google Scholar]
- 24.Cagnard N, Letourneur F, Essabbani A, Devauchelle V, Mistou S, Rapinat A, et al. Interleukin-32, CCL2, PF4F1 and GFD10 are the only cytokine/chemokine genes differentially expressed by in vitro cultured rheumatoid and osteoarthritis fibroblast-like synoviocytes. Eur Cytokine Netw. 2005;16(4):289–292. [PubMed] [Google Scholar]
- 25.Pearlman AN, Conley DB. Review of current guidelines related to the diagnosis and treatment of rhinosinusitis. Curr Opin Otolaryngol Head Neck Surg. 2008;16(3):226–230. doi: 10.1097/MOO.0b013e3282fdcc9a. [DOI] [PubMed] [Google Scholar]
- 26.Kato A, Peters A, Suh L, Carter R, Harris KE, Chandra R, et al. Evidence of a role for B cell-activating factor of the TNF family in the pathogenesis of chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2008;121(6):1385–1392. doi: 10.1016/j.jaci.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kato A, Favoreto S, Jr, Avila PC, Schleimer RP. TLR3- and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J Immunol. 2007;179(2):1080–1087. doi: 10.4049/jimmunol.179.2.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kato A, Truong-Tran AQ, Scott AL, Matsumoto K, Schleimer RP. Airway epithelial cells produce B cell-activating factor of TNF family by an IFN-beta-dependent mechanism. J Immunol. 2006;177(10):7164–7172. doi: 10.4049/jimmunol.177.10.7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chustz RT, Nagarkar DR, Poposki JA, Favoreto S, Jr, Avila PC, Schleimer RP, et al. Regulation and Function of the IL-1 Family Cytokine IL-1F9 in Human Bronchial Epithelial Cells. Am J Respir Cell Mol Biol. 2011;45(1):145–153. doi: 10.1165/rcmb.2010-0075OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poposki JA, Uzzaman A, Nagarkar DR, Chustz RT, Peters AT, Suh LA, et al. Increased expression of the chemokine CCL23 in eosinophilic chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2011;128(1):73–81. e74. doi: 10.1016/j.jaci.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tieu DD, Peters AT, Carter RT, Suh L, Conley DB, Chandra R, et al. Evidence for diminished levels of epithelial psoriasin and calprotectin in chronic rhinosinusitis. J Allergy Clin Immunol. 2010;125(3):667–675. doi: 10.1016/j.jaci.2009.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Claeys S, De Belder T, Holtappels G, Gevaert P, Verhasselt B, Van Cauwenberge P, et al. Macrophage mannose receptor in chronic sinus disease. Allergy. 2004;59(6):606–612. doi: 10.1111/j.1398-9995.2004.00471.x. [DOI] [PubMed] [Google Scholar]
- 33.Patadia M, Dixon J, Conley D, Chandra R, Peters A, Suh LA, et al. Evaluation of the presence of B-cell attractant chemokines in chronic rhinosinusitis. Am J Rhinol Allergy. 2010;24(1):11–16. doi: 10.2500/ajra.2010.24.3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.