

Abstract
APC is considered a gatekeeper for colorectal cancer (CRC). Cells with heterozygous APC mutations have altered expression profiles suggesting that the first APC hit may help set the stage for subsequent transformation. Therefore, we measured transformation efficiency following what we have designated as “simultaneous” versus “stepwise” Apc loss. We combined a conditional Apc allele (ApcCKO) with a Cre reporter gene and an out-of-frame Cre allele (Pms2cre) that stochastically becomes functional by a frameshift mutation in single cells. Loss of one Apc allele (ApcCKO/+) had little consequence, whereas simultaneous loss of both Apc alleles (ApcCKO/CKO) resulted in increased clonal expansion (crypt fission), consistent with the gatekeeper function of Apc. Interestingly, our analyses showed that most of the Apc-deficient crypts in ApcCKO/CKO mice appeared normal, with morphologic transformation, including β-catenin deregulation, occurring in only 17% of such crypts. To determine whether transformation efficiency was different following stepwise Apc loss, we combined ApcCKO with a germline mutant allele, either ApcMin or Apc1638N. Transformation efficiency following stepwise Apc loss (ApcMin/CKO or Apc1638N/CKO) was increased 5-fold and essentially all of the Apc-deficient cells were dysplastic. In summary, our data suggest that the gatekeeper function of Apc consists of two roles, clonal expansion and morphologic transformation, because simultaneous Apc loss frequently leads to occult clonal expansion without morphologic transformation, whereas stepwise Apc loss more often results in visible neoplasia. Finally, that Apc-deficient cells in certain scenarios can retain a normal phenotype is unexpected and may have clinical implications for surveillance strategies to prevent CRC.
Keywords: APC, cancer, simultaneous, stepwise, intestine
INTRODUCTION
Colorectal cancer (CRC) is thought to occur after the accumulation of mutations in multiple oncogenes and tumor suppressor genes. The order of these mutations is uncertain; however, APC is considered to have a gatekeeper role because its mutation is thought to initiate tumorigenesis (Fearon and Vogelstein, 1990). Germline APC mutations are associated with familial adenomatous polyposis (FAP), and APC mutations are found in the majority of adenomas (Forbes et al., 2010). Mice have been genetically manipulated with germline Apc mutations to mimic both FAP (Oshima et al., 1995; Sasai et al., 2000; Su et al., 1992) and attenuated FAP (Fodde et al., 1994). The gatekeeper function of Apc has been tested in murine models by combining a conditional Apc allele with an inducible Cre gene. Intestine-wide conditional loss of Apc using an epithelial-specific promoter resulted in morphologic hyperplasia within five days (Sansom et al., 2004). Loss of Apc in isolated Lgr5+ stem cells leads to highly efficient transformation, as evidenced by macroadenoma formation, within eight days following Cre induction (Barker et al., 2009), suggesting that loss of Apc is sufficient for transformation.
Here, we examine further the gatekeeping role of Apc by combining a conditional Apc allele (ApcCKO) (Kuraguchi et al., 2006) with an out-of-frame Cre allele (Pms2cre) that stochastically reverts back into frame (Miller et al., 2008). The Pms2cre mouse system used in this study is similar to cell type-specific conditional Cre/lox systems that model sporadic tumorigenesis by altering floxed genes in single cells surrounded by “normal” cells (Barker et al., 2009; Hung et al., 2010). However, in our system Cre activation occurs randomly throughout life without exogenous manipulations, therefore better mimicking the normal course of tumorigenesis. Moreover, multiple floxed alleles should be efficiently recombined within a single cell because the Cre allele should be constitutively expressed following frameshift mutation. The modified cell/cell lineage can be traced with a Cre-inducible marker gene, R26R (β-galactosidase or GFP), thus facilitating an assessment of the short- and longer-term consequences of target gene alteration. Because normal crypt stem cells are constantly replaced through neutral drift (Lopez-Garcia et al., 2010; Snippert et al., 2010), the mutated cell must compete with surrounding wild type stem cells to maintain crypt occupancy. By comparing clone sizes, it is possible to infer either negative selection (fewer or smaller β-gal+ foci) or positive selection (more or larger β-gal+ foci) conferred by specific mutation combinations even in the absence of morphologic changes.
The earliest phases of tumor progression are difficult to study because small tumors are easily overlooked, or may rapidly progress to larger tumors. Simplistically, loss of both Apc alleles should be sufficient to initiate intestinal tumorigenesis and lead to adenoma formation (Fearon and Vogelstein, 1990). In this study, combinations of different mutant Apc alleles reveal that the gatekeeper role of Apc is surprisingly complex because what we designate as “simultaneous” versus “stepwise” Apc loss yields different neoplastic phenotypes. Specifically, we have found that although simultaneous loss of both Apc alleles in otherwise Apc-normal intestines conferred a selective advantage (larger clone size), only a fraction of mutant cells became transformed. In contrast, we found that stepwise loss of Apc, in which Apc levels are compromised in the every cell of the mouse, resulted more often in overt transformation and adenoma formation.
RESULTS
Stochastic activation of Cre recombinase in the mouse small intestine
To study the consequences of targeted somatic mutation in single isolated cells, we developed a Cre-lox system in which recombination occurs in only a minority of intestinal crypts and is monitored by detection through expression of a Cre-reporter gene (Miller et al., 2008). Whole mount examination of Pms2cre/cre; Apc+/+; Rosa-β-gal mice at an average of 104 days revealed an average of 1845 β-gal+ foci in the proximal small intestine (Figure 1a). When sections were examined, we found that 2–3% of the crypts were β-gal+, with 24% of the β-gal+ foci involving more than one crypt, suggesting that only a minority of Cre reversion events occur during development in control mice (Figures 1c and d, 2a and c). Furthermore, because the overall frequency of β-gal+ crypts is low, β-gal+ foci involving more than one crypt likely represent a single Cre-activation event followed by crypt fission rather than Cre-activation in adjacent crypts (<0.1% chance of independent adjacent crypt activation).
Figure 1. Increased crypt fission in ApcCKO/CKO mice.

β-gal+ foci counted in whole mount (a, b), or sections of the proximal small intestine (c, d). (a) Note, there was no significant difference in the total number of β-gal+ foci between the three genotypes. (b) However, there was a significant increase in the number of “larger” β-gal+ foci, those involving more than three villi in ApcCKO/CKO mice (p=0.02). (c) In sections of the proximal small intestine, there was no significant difference in the percentage of β-gal+ foci. (d) In contrast, we did observe an increase in the percentage of β-gal+ foci involving more than one crypt in ApcCKO/CKO mice (p=0.001).
Figure 2. Increased number of larger β-gal+ foci in ApcCKO/CKO mice.

(a–d) Whole mount images of proximal small intestine of Apc+/+ and ApcCKO/CKO mice. Adenomas denoted with circles and larger normal β-gal+ foci with arrows. (b, d) Images showing examples of larger normal β-gal+ foci in ApcCKO/CKO mice. (e) Distribution of β-gal+ foci sizes in the proximal small intestine of Apc+/+, ApcCKO/+ and ApcCKO/CKO mice. ApcCKO/+ and ApcCKO/CKO mice show a significant increase in β-gal+ foci involving 3 villi (p=0.01). ApcCKO/CKO show a significant increase in β-gal+ foci involving 6 and 10 or more villi (p=0.02 and p=0.03, respectively).
Stochastic loss of a single Apc allele in individual crypts
To determine if stochastic inactivation of one Apc allele detectably alters intestinal homeostasis, we combined the Pms2cre allele with a conditional, floxed Apc allele, ApcCKO (Kuraguchi et al., 2006). No microadenomas or adenomas were seen in the Pms2cre/cre; ApcCKO/+ mice (average age of 116 days). There were no significant differences in the numbers or sizes of β-gal+ patches between ApcCKO/+ and Apc+/+ mice (p=0.97 and p=0.46) (Figures 1a and b). However, the numbers of β-gal+ foci with staining of 3 adjacent villi were increased in ApcCKO/+ mice compared to Apc+/+ mice (p=0.01) (Figure 2e). Because a single crypt can contribute cells to typically at most 3 villi (Lopez-Garcia et al., 2010; Miller et al., 2008), the proportions of β-gal+ foci with 3 adjacent stained villi can serve as an indicator of crypts harboring a majority of recombined cells. Therefore, these results suggest that loss of a single Apc allele confers a selective advantage over adjacent wild type cells, more often leading to dominance within the crypt. However, this selective advantage is limited because somatic loss of only a single Apc allele did not detectably increase crypt fission (Figure 1d) or lead to transformation.
“Simultaneous” loss of both Apc alleles within individual crypts
Next, we examined Pms2cre/cre; ApcCKO/CKO; Rosa-β-gal mice to determine whether the simultaneous loss of both Apc alleles altered intestinal homeostasis. We use the term “simultaneous” in a relative sense to distinguish such somatic loss of both alleles within a single cell lineage from “stepwise” loss, in which one defective Apc allele is inherited followed by stochastic somatic loss of the remaining allele (see below). We realize that Cre recombination at the two ApcCKO alleles in Pms2cre/cre; ApcCKO/CKO; Rosa-β-gal mice may occur over time in a particular cell lineage and not necessarily within the same cell cycle.
In contrast to Apc+/+ or ApcCKO/+ mice, ApcCKO/CKO mice developed anemia and were sacrificed at an average age of 114 days. Consistent with stochastic Cre-activation, β-gal+ foci were scattered throughout the intestines, with similar number of β-gal+ foci measured in both whole mount and sections when compared to either Apc+/+ or ApcCKO/+ mice (p=0.24 and p=0.97) (Figures 1a and c). However, the phenotypic consequences of simultaneous Apc loss were significantly different. As expected with complete Apc inactivation, β-gal+ macroscopic adenomas were now present, with an average of 90 adenomas in the small intestine. In whole mount, ~4% of spots were scored as adenomas; however, smaller changes such as microadenomas are difficult to score when examining the intestine in whole mount. Therefore, to determine the percentage of abnormal β-gal+ foci, we examined sections from the proximal small intestine. Interestingly, only 17% (22/129) of β-gal+ foci in the proximal small intestine of ApcCKO/CKO mice were morphologically abnormal (microadenoma or adenoma) (Figure 3). However, there were other phenotypic consequences of simultaneous Apc loss in the absence of morphological transformation. Notably, ApcCKO/CKO mice had a significantly higher proportion of β-gal+ foci involving either more than 3 villi or multiple adjacent crypts when compared to either Apc+/+ or ApcCKO/+ mice (p=0.02 and p=0.001) (Figures 1b and d, 2b and d). We note that the increased clonal expansion seen in ApcCKO/CKO, but not in ApcCKO/+ mice, supports efficient Cre recombination at both ApcCKO alleles. These results demonstrate that simultaneous loss of both Apc alleles, likely occurring during intestinal development, confers additional selective advantages over surrounding wild type cells, but separates clonal expansion from transformation. Clonal expansion, manifested by crypt fission (larger β-gal+ patches), without transformation was as common as clonal expansion with morphologic transformation.
Figure 3. Distribution of normal and adenomatous β-gal+ foci.

(a) Images of different representative β-gal+ foci illustrating a single β-gal+ cell, single β-gal+ crypt, multiple β-gal+ crypts, β-gal+ microadenoma and β-gal+ adenoma. (b) Percentage of the β-gal+ foci illustrated in (a).
Apc target alleles are efficiently recombined in β-gal+ foci
In principle, Cre expression following frameshift reversion should result in efficient recombination of multiple floxed alleles because Cre expression should persist. As pointed out above, the increased crypt fission in ApcCKO/CKO but not in ApcCKO/+ mice supports efficient recombination at both alleles. Nevertheless, the low percentage of β-gal+ foci in the Pms2cre/cre; ApcCKO/CKO; Rosa-β-gal mice showing morphologic transformation might be explained by inefficient recombination of both ApcCKO alleles. To verify Cre recombination efficiency, we used laser capture microdissection (LCM) on tissue cross sections to isolate β-gal+ cells from normal appearing crypts or adenomas (Figures 4a–c). Next, we used quantitative PCR to measure the relative proportions of the recombined versus unrecombined ApcCKO allele in microdissected samples enriched for β-gal+ cells (Figure 4d). As expected, β-gal+ adenomas showed significant enrichment for the recombined allele compared to β-gal− and ApcΔ/CKO samples (p<0.001). For morphologically normal β-gal+ foci, 76% (16/21) showed a significant 6–10 fold increase in signal of the recombined Apc allele over β-gal− cells and a significant 2–3 fold increase over ApcΔ/CKO cells (p<0.001 for both). Also, consistent with recombination of both Apc alleles in the normal appearing β-gal+ crypts, the enrichment of the recombined Apc allele in normal appearing β-gal+ tissue was similar to that found in adenoma tissue (p=0.85) (Figure 4e). These results show that ApcCKO is efficiently recombined in at least 75% of morphologically normal β-gal+ crypts suggesting that the majority of these normal appearing crypts are indeed Apc-deficient.
Figure 4. Apc is efficiently recombined in Pms2cre/cre; ApcCKO/CKO mice.

Images of (a) β-gal− crypts, (b) β-gal+ crypts and (c) adenomas from the proximal small intestine isolated on LCM CAPs. (d) Gel of PCR reactions for the different isolated cell types. CKO is the unrecombined allele and Δ is the recombined allele. Each gel was normalized to the signal obtained from an ApcΔ/CKO sample, which has a 1:1 ratio of recombined:unrecombined ApcCKO alleles. (e) Graph of normalized recombined:unrecombined ratios that illustrates that 76% (16/21) of normal appearing β-gal+ samples had a 6–10 fold increase in the recombined:unrecombined ratio over β-gal− cells (p<0.001), similar to the increase observed for the adenoma samples (p=0.85).
Characteristics of Cre-reporter+ foci in ApcCKO/CKO mice
To further examine morphologically normal β-gal+ or GFP+ crypts, most of which are Apc-deficient, we measured Wnt-dependent signaling, apoptosis and differentiation. Following Apc loss it is expected that: WNT signaling be activated as reported by β-catenin deregulation, apoptosis be increased as reported by TUNEL, and the villus cell marker alkaline phosphatase be eliminated (Sansom et al., 2004). As expected, all β-gal+ or GFP+ foci that were scored as adenomas or microadenomas showed deregulated β-catenin expression (Figures 5a, g–i), increased TUNEL staining (Figure 5b) and a lack of alkaline phosphatase (Figure 5c). However, these changes were not seen in morphologically normal β-gal+ or GFP+ crypts and villi. First, all normal appearing β-gal+ or GFP+ fociexamined (30/30) from ApcCKO/CKO mice showed normal levels and localization of β-catenin (Figures 5d, j-l). Second, only 7% (4/56) of normal appearing β-gal+ crypts had evidence of apoptosis, similar to the level of apoptosis in neighboring β-gal− crypts (54/1399) (Χ2=1.2, p=0.28) (Figure 5e). Finally, 10/10 β-gal+ villi examined expressed alkaline phosphatase, whereas all adenomas examined did not stain for alkaline phosphatase (Yate’s Χ2=7.9, p=0.005) (Figure 5f). These results illustrate further that when normal morphology is retained after simultaneous inactivation of both Apc alleles, the increased crypt dominance and fission is not accompanied by significant changes in β-catenin, apoptosis or differentiation.
Figure 5. Characteristics of Cre-reporter+ foci.
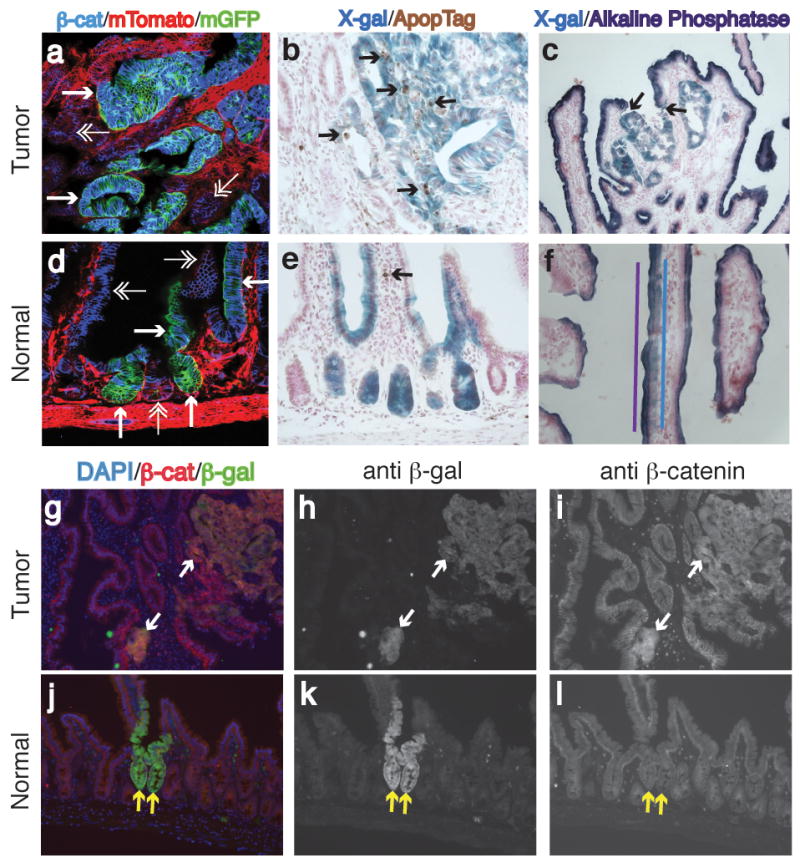
Characterization of Cre-Reporter+ tumor (a–c, g–i) and normal (d–f, j–l) tissue. (a, d) Show deregulated expression of β-catenin in GFP+ tumor tissue (single arrowheads), but normal β-catenin expression in normal GFP+ tissue (single arrowheads) (β-catenin:blue, mTomato:red[unrecombined], mGFP:green[recombined]). Double arrowheads point out unrecombined, epithelial tissue. (b, e) Show increased apoptosis in β-gal+ tumor tissue, but normal apoptosis levels in normal, β-gal+ tissue (β-gal+:blue, TUNEL:brown). Arrows denote TUNEL positive cells. (c, f) Loss of alkaline phosphatase staining in β-gal+ tumor tissue, but continued expression of alkaline phosphatase in normal, β-gal+ tissue (β-gal+:blue, alkaline phosphatase:purple). In c) arrows show demarcation of alkaline phosphatase and X-gal staining. In f) purple and blue lines illustrate the overlap of alkaline phosphatase and β-gal+ cells. (g, j) Merged images of immunofluorescence for β-catenin (red), β-gal (green) and DAPI (blue). Arrows point to the same β-gal+ regions. (h, k) Shown are images of only the β-gal (green) channel. (i, l) Images of only the β-catenin (red) channel. Note, deregulated β-catenin expression n the tumor, but normal β-catenin pattern in the normal tissue.
Attempts to generate Pms2cre/cre; Apc580S/580S mice
Previous studies by others suggested that isolated functional loss of both Apc alleles in mouse intestinal stem cells was sufficient for transformation (Akyol et al., 2008; Barker et al., 2009). However, these studies were performed with a different conditional Apc allele, Apc580S, from the ApcCKO allele used here in our study. For a more direct comparison of our findings with these other studies, we attempted to generate Pms2cre/cre; Apc580/580S mice. We first generated Pms2cre/+; Apc580S/580S and Pms2cre/+; ApcCKO/CKO mice. Next, we performed Pms2cre/+; Apc580S/580S × Pms2cre/+; Apc580S/580S and Pms2cre/+; ApcCKO/CKO × Pms2cre/+; ApcCKO/CKO matings, which are expected to yield 1 in 4 mice being Pms2cre/cre and either Apc580S/580S or ApcCKO/CKO. While we were able to generate mice homozygous for the ApcCKO allele in the expected Mendelian ratios (1:2:1) (15 Pms2+/+:35 Pms2cre/+:14 Pms2cre/cre) (Yate’s Χ2=0.35, p=0.84), we were unsuccessful in generating Pms2cre/cre; Apc580S/580S mice (9 Pms2+/+:22 Pms2cre/+:0 Pms2cre/cre) (Yate’s Χ2=9.9, p=0.007). In addition, other crosses designed to generate Pms2cre/cre; Apc580S/580S mice, albeit at a lower expected frequency, also failed to yield the desired genotype.
The ApcCKO allele is expressed at wild type levels
Previous studies have reported that the conditional Apc580S allele is hypomorphic, even prior to Cre recombination (Buchert et al., 2010; Shibata et al., 1997). The Apc allele used here, ApcCKO (Kuraguchi et al., 2006), when acted on by Cre results in the same deletion of exon 14 and truncation of Apc at amino acid 580. However, in the ApcCKO allele, the neomycin cassette was removed via FLP recombination, whereas the Apc580S allele retains the neomycin cassette, possibly accounting for its hypomorphic nature (Shibata et al., 1997). To determine the expression level of the ApcCKO allele, we measured Apc RNA levels by quantitative Real-Time PCR (qRT-PCR). While the Apc580S/580S small intestine expressed only 40% of wild type Apc RNA levels, in agreement with a previous estimate (Shibata et al., 1997), the ApcCKO/CKO small intestine expressed 101% of wild type Apc RNA levels (Table 1). In addition, whereas ApcCKO/Min mice are viable (see below), Apc580S/Min mice die in utero, presumably due to significantly reduced overall Apc levels (Buchert et al., 2010). We conclude that the inability to generate Pms2cre/cre; Apc580S/580S mice described above is also due to embryonic lethality.
Table 1.
Quantitative Real Time PCR for Apc expression levels in the small intestine
| Apc | Actin and GAPDH | Δ CT | ΔΔ CT | % Apc+/+ | |
|---|---|---|---|---|---|
| CT | CT | ||||
| Apc+/+ | 25.48 | 14.07 | 11.41 | 0 | 100 |
| ApcCKO/CKO | 24.11 | 12.71 | 11.40 | 0.01 | 101 |
| Apc580S/580S | 26.27 | 13.53 | 12.74 | −1.33 | 40 |
CT = cycle threshold (same threshold level of fluorescence for all samples)
ΔCT = CT (Apc) − CT (actin and GAPDH)
ΔΔCT = ΔCT (Apc+/+) − ΔCT (ApcCKO/CKO or Apc580S/580S)
%Apc+/+ = 2ΔΔCt × 100
“Stepwise” Apc loss results in more efficient transformation
The surprising finding that simultaneous Apc loss, when starting from normal Apc expression levels, frequently leads to clonal expansion (crypt fission) without morphologic transformation led us to use our system to determine the consequence of stepwise Apc loss, in which overall levels of Apc are reduced prior to complete Apc loss. We combined the ApcCKO allele with one of two germline Apc mutations, ApcMin or Apc1638N. The Min allele, expressing a protein truncated at amino acid 850, results in ~50 adenomas at 6 months of age (Moser et al., 1990) (data not shown). The 1638N allele results in truncation at amino acid 1638, is hypomorphic (~2% of wt) and predisposes to ~10 adenomas at 9 months of age (Smits et al., 1998) (data not shown). These two germline Apc alleles mimic the two different forms of FAP, normal and attenuated, based on differences in adenoma burden and onset of anemia.
Both ApcMin/CKO and Apc1638N/CKO mice became anemic at ~60 days of age, significantly more rapidly than the 114 days for ApcCKO/CKO mice (p<0.001) (Figure 6a). ApcMin/CKO mice had an average number of 171 adenomas in the small intestine, a significant increase (p=0.01) relative to ApcCKO/CKO mice. Apc1638N/CKO mice had an average of 225 adenomas in the small intestine, significantly more than ApcCKO/CKO mice (p=0.00002), but not significantly different from ApcMin/CKO mice (p=0.11) (Figure 6b). The percentage of β-gal+ cells in adenomas was similar between all three genotypes (ApcCKO/CKO, ApcMin/CKO and Apc1638N/CKO) (Χ2=4.2, p=0.4), but different from Apc1638N/+ mice (Χ2=132.4, p<0.001), suggesting that Cre recombination is responsible for adenoma formation (Figure 7). Examination of the proximal small intestine cross sections revealed that 82% (47/57) and 88% (50/57) of β-gal+ foci in the ApcMin/CKO and Apc1638N/CKO mice, respectively, were scored as either a microadenoma or adenoma, indicating a significant five-fold increase when compared to ApcCKO/CKO mice (Χ2=72.5, p<0.001 and Χ2=83.2, p<0.001) (Figure 3). Nearly all crypts (>80%) undergo transformation after stepwise loss of Apc, which is in marked contrast to the minority of crypts (<20%) that undergo transformation after simultaneous Apc loss, in which overall Apc levels are normal. We propose based on the contrasting “simultaneous” versus “stepwise” results reported here that the overall Apc landscape can influence the consequences of Apc loss.
Figure 6. Increased tumor formation in ApcMin/CKO and Apc1638N/CKO mice.

(a) Survival curve illustrating that ApcMin/CKO (red squares) and Apc1638N/CKO (green triangles) mice become anemic at an earlier age than ApcCKO/CKO (blue diamonds) mice (p<0.001). (b) Increased number of small intestinal adenomas in ApcMin/CKO and Apc1638N/CKO mice compared to ApcCKO/CKO mice (p=0.01).
Figure 7. Percentage of β-gal+ cells in proximal small intestine adenomas.
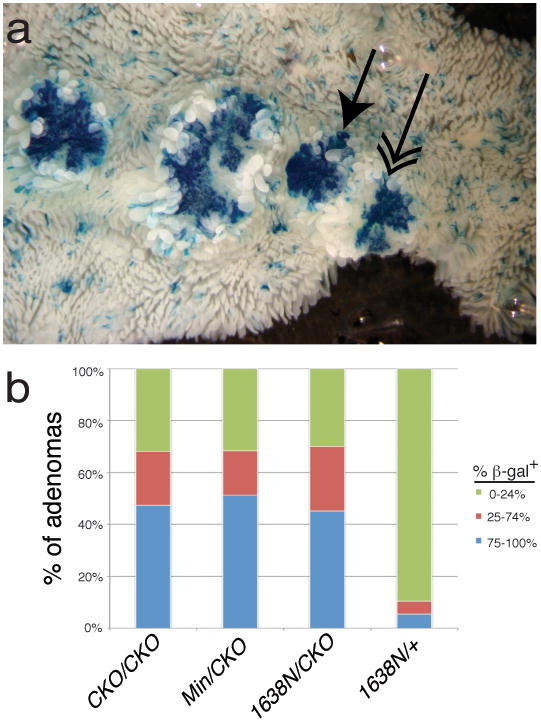
(a) Whole mount image of β-gal+ adenomas in ApcCKO/CKO mice. Single-headed arrow shows a tumor scored as 100% β-gal+. Double-headed arrow shows a tumor scored as 50% β-gal+. (b) Classification of adenomas into % β-gal+ in the proximal small intestine.
DISCUSSION
Progression to cancer is thought to occur through the acquisition of somatic changes associated with increasingly larger and more dysplastic precancerous lesions. Reconstruction of tumor progression is difficult because mutations can occur anywhere within the genome and serial observations are impractical. Human cancer genomes are difficult to interpret because they contain thousands of mutations. Most mutations are neutral “passenger” mutations and each cancer genome appears to be unique (Pleasance et al., 2010). For colorectal cancer, only a few genes (APC, TP53, KRAS) are mutated at frequencies above 15% (Attolini et al., 2010; Forbes et al., 2010; Sjoblom et al., 2006). Consistent with the hypothesis that APC is a gatekeeper gene (Kinzler and Vogelstein, 1996), APC mutations are found in many human colorectal cancers and are the most common mutation in adenomas (Forbes et al., 2010).
Mouse models facilitate testing of the specific roles of certain mutations, either in a familial, or germline, context, or with conditional models that create specific mutations sometime after birth. Here, we use the Pms2cre mouse system, which models several aspects of sporadic tumorigenesis, namely that mutations can occur anytime after conception, do not require external manipulations, and altered cells are initially isolated and surrounded by unaltered cells (Miller et al., 2008). In this system, Cre-mediated Apc deletion occurs stochastically in single isolated cells throughout life, induced by mutational slippage of an out-of-frame cre allele. The Pms2cre/cre mice used in this study are DNA mismatch repair (MMR) compromised, resulting in an increased rate of Cre reversion. The increased Cre reversion rate allowed us to achieve an optimal number of tumors by 4 months of age. Granted, MMR deficiency could have other consequences. However, because the control (those without conditional Apc alleles) and experimental mice (those with conditional Apc alleles) contain the same MMR defect, any consistent differences between the groups of mice should be due to the presence of the conditional alleles.
Using the Pms2cre system, we examined the consequences of “simultaneous” versus “stepwise” Apc loss. What we define as simultaneous Apc loss, in which Apc levels are normal prior to Apc loss, resulted in increased crypt fission, but only 17% of β-gal+ crypts exhibited phenotypic dysplasia, i.e. adenoma formation and deregulated β-catenin expression. In contrast, we found that stepwise Apc loss, in which partial Apc deficiency due to an inherited mutation was followed by stochastic somatic Apc loss, resulted in >80% phenotypic dysplasia, consistent with other studies (Akyol et al., 2008; Barker et al., 2009). Our findings reveal that the gatekeeper role of Apc is surprisingly complex because simultaneous Apc loss had different phenotypic consequences than stepwise Apc loss. One could say, based on our findings, that the gate can open in two steps and that the gatekeeper role of Apc can be divided into two parts---net cell proliferation and morphologic transformation.
A notable difference between ours and another mouse study, which found essentially 100% tumorigenesis after induced homozygous Apc loss (Barker et al., 2009), is that the conditional Apc allele, Apc580S, used in that study is hypomorphic, based on lower than wild type expression prior to Cre recombination (Buchert et al., 2010). In contrast, we have shown that the ApcCKO allele used here expresses Apc at a level similar to wild type. Therefore, the use of the ApcCKO allele allows us to uniquely engineer the loss of Apc in single, isolated cell lineages in mice with an otherwise normal Apc landscape. Our results show that inheriting an Apc defect (“one-hit”) fosters increased transformation upon somatic Apc loss, whereas simultaneous somatic Apc loss, in which overall Apc levels are normal prior to loss, frequently conferred clonal expansion through increased crypt fission, but did not necessarily result in morphologic transformation.
Although we believe that differences in overall Apc levels prior to Apc loss are important and can account for the contrasting results between our study and that of Barker et. al., the two Cre/lox systems employed do have some other relevant differences. The Lgr5-CreER mouse model (Barker et al., 2009) utilizes a single, low dose of tamoxifen to induce Cre recombination. Therefore, Apc loss occurs at a fixed point in time and likely within a limited time frame. Although both the Pms2cre and Lgr5-CreER systems resulted in a similar percentage of Apc-deficient (β-gal+) crypts, it is possible that the slow accumulation of Apc−/− cells occurring in Pms2cre mice, as opposed to cells becoming Apc−/− within a limited time frame in the Lgr5-CreER mice is another contributing factor. Regardless, it will be important to compare the two different, conditional Apc alleles using the Lgr5-CreER system.
It is commonly presumed that Apc−/− cells are absent when visible intestinal lesions are not present. However, the occult presence of Apc−/− cells with normal morphologies rather than lack of mutation may better explain why chemoprevention with Sulindac in FAP patients initially causes polyp regression, but polyps subsequently reappear, with progression even to cancer (Cruz-Correa et al., 2002; Lynch, 2010). Rather than eliminating Apc−/− cells, visible regression with Sulindac may instead represent metaplastic conversion back to a normal histology. The presence or persistence of normal appearing Apc−/− cells would complicate surveillance efforts to prevent CRC.
The additional alterations required for morphologic transformation following simultaneous homozygous Apc loss are uncertain, but such changes may evolve more readily after stepwise loss of the first Apc allele. Notably, although Apc+/− intestines appear morphologically normal, their circuitry is altered because expression and proteomic profiles differ between Apc+/+ and Apc+/− cells (Patel et al., 2011; Wang et al., 2010; Yeung et al., 2008). An acquired epigenetic mechanism or state present in Apc+/− cells, but not in Apc+/+ cells, could modulate Apc−/− cell phenotypic plasticity. Supporting a requisite epigenetic role in transformation, polyp formation in ApcMin mice is decreased when DNA methylation is inhibited (Eads et al., 2002; Laird et al., 1995), even though hypomethylation has been associated with both increased and decreased mutation rates (Chan et al., 2001; Chen et al., 1998). Alternatively Apc−/− cells may still arise in ApcMin mice, but more often retain normal morphologies when DNA methylation is inhibited.
Our findings with simultaneous Apc loss in mice suggest that Apc−/− cells frequently retained a normal phenotype, thus raising the possibility that Apc-deficient cells could exist in the human colon and go undetected by conventional screening techniques. But APC is almost certainly lost in a stepwise fashion in human CRC, thus normal appearing, APC-deficient cells might be uncommon in the human colon. However, the ApcMin and Apc1638N alleles used to determine the consequences of stepwise loss of Apc are constitutive mutants, thus not only is Apc reduced in the cell that will become transformed upon Apc loss, but Apc levels are also reduced throughout the mouse. Thus, partial loss of Apc activity may lead to a more transformation-prone state due to cell autonomous effects such as abnormal spindle orientation (Quyn et al., 2010). Alternatively, there could be cell non-autonomous effects such as cross-talk between myofibroblasts and the epithelium (Quante et al., 2011; Vermeulen et al., 2010). Studies with stochastic Apc loss targeting specific cell types should help distinguish between such explanations and elucidate further the important cell types involved in intestinal cancer.
The high frequency of APC mutations in human colorectal carcinomas indicates that APC function is usually lost during progression. The engineered isolated and simultaneous homozygous deletion of Apc in mice indicates that Apc loss can lead to clonal expansion but is usually insufficient for morphologic transformation. Our studies illustrate that the phenotypic plasticity observed in human cancers is also present very early in progression (Quintana et al., 2010). Further studies with different constellations of Cre-target genes can specifically test whether certain mutation combinations are sufficient for various stages of tumorigenesis. For example, using our system we found that Kras activation also resulted in clonal expansion without morphologic transformation (Miller et al., 2008). Potentially, the full malignant phenotype, including metastasis, can be engineered by Cre-mediated recombination of multiple oncogenes and/or tumor suppressor genes, thus facilitating specific testing of which combination(s) of pathway disruptions are necessary or sufficient for progression.
MATERIALS AND METHODS
Mice
Two different R26R (Rosa26 Reporter mice with either lox-stop-lox LacZ or pCAGG lox-mTomato-lox mGFP) alleles were used. Mice were housed in a specific pathogen free HEPA filtered room and were fed a diet of Purina PicoLab Rodent Diet 20.
Scoring of β-gal+ foci in whole mount intestine
At least 20 β-gal+ foci were counted for each third of the small intestine. Nearby β-gal+ foci were considered independent if not arising from the same crypt and surrounded by non-staining crypts. Adenomas, which involved multiple villi, were scored by whole mount and in cross sections. Microadenomas, involving a single villus, were determined by scoring cross sections.
Laser capture microdissection of intestinal tissue for PCR
Sections were examined using an ArcturusXT LCM instrument (Applied Biosystems) and β-gal+ or β-gal− crypts/villi were attached to a CapSure Macro LCM Cap (Applied Biosystems) with an infrared laser. Band intensity was used to measure the relative ratio of recombined to unrecombined DNA (Quantity One, Bio-Rad). A sample known to contain a 1:1 ratio of recombined to unrecombined, run on each gel, was used to normalize against unknown samples.
Immunofluorescence
Sections were incubated in primary antibody (rabbit anti-β-catenin H-102, SantaCruz; TRITC-conjugated mouse anti-β-catenin, BD Transduction Labs; or rabbit anti-β-galactosidase, Immunology Consultants Laboratory) diluted 1:100 overnight at 4°C, washed in TBST then incubated in secondary antibody (anti-rabbit AlexaFluor 488, 555 or 633, Invitrogen) diluted 1:100 for 1 hour at RT.
Statistics
Data were analyzed with StatPlus for Mac in Microsoft Excel. Fisher LSD post-hoc test was used after ANOVA test. Chi2 analysis was performed using a 2X2 or 2X3 Table.
Supplementary Material
Acknowledgments
We would like to thank Drs. James Stringer and Melissa Wong for critical reading of the manuscript. We also thank Drs. Raju Kucherlapati and Winfried Edelmann for the ApcCKO and Apc1638N mice, respectively, John Swain and Dr. Melissa Wong for ApcMin mice and Apc580S intestinal material and Dan Lioy and Drs. Gail Mandel and Paul Brehm for assistance with confocal microscopy. RML and DS were funded by NIH grant 2R01GM032741-28. JMF was funded by NIH training grant 5T32HD046420-05 and ACS postdoctoral fellow PF-11-067-01-TBE.
Footnotes
CONFLICT OF INTEREST: The authors declare no conflicts of interest.
References
- Akyol A, Hinoi T, Feng Y, Bommer GT, Glaser TM, Fearon ER. Generating somatic mosaicism with a Cre recombinase-microsatellite sequence transgene. Nat Methods. 2008;5:231–3. doi: 10.1038/NMETH.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attolini CS, Cheng YK, Beroukhim R, Getz G, Abdel-Wahab O, Levine RL, et al. A mathematical framework to determine the temporal sequence of somatic genetic events in cancer. Proc Natl Acad Sci U S A. 2010;107:17604–9. doi: 10.1073/pnas.1009117107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–11. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- Buchert M, Athineos D, Abud HE, Burke ZD, Faux MC, Samuel MS, et al. Genetic dissection of differential signaling threshold requirements for the Wnt/beta-catenin pathway in vivo. PLoS Genet. 2010;6:e1000816. doi: 10.1371/journal.pgen.1000816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan MF, van Amerongen R, Nijjar T, Cuppen E, Jones PA, Laird PW. Reduced rates of gene loss, gene silencing, and gene mutation in Dnmt1-deficient embryonic stem cells. Mol Cell Biol. 2001;21:7587–600. doi: 10.1128/MCB.21.22.7587-7600.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RZ, Pettersson U, Beard C, Jackson-Grusby L, Jaenisch R. DNA hypomethylation leads to elevated mutation rates. Nature. 1998;395:89–93. doi: 10.1038/25779. [DOI] [PubMed] [Google Scholar]
- Cruz-Correa M, Hylind LM, Romans KE, Booker SV, Giardiello FM. Long-term treatment with sulindac in familial adenomatous polyposis: a prospective cohort study. Gastroenterology. 2002;122:641–5. doi: 10.1053/gast.2002.31890. [DOI] [PubMed] [Google Scholar]
- Eads CA, Nickel AE, Laird PW. Complete genetic suppression of polyp formation and reduction of CpG-island hypermethylation in Apc(Min/+) Dnmt1-hypomorphic Mice. Cancer Res. 2002;62:1296–9. [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- Fodde R, Edelmann W, Yang K, van Leeuwen C, Carlson C, Renault B, et al. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc Natl Acad Sci U S A. 1994;91:8969–73. doi: 10.1073/pnas.91.19.8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Tang G, Bindal N, Bamford S, Dawson E, Cole C, et al. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 2010;38:D652–7. doi: 10.1093/nar/gkp995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung KE, Maricevich MA, Richard LG, Chen WY, Richardson MP, Kunin A, et al. Development of a mouse model for sporadic and metastatic colon tumors and its use in assessing drug treatment. Proc Natl Acad Sci U S A. 2010;107:1565–70. doi: 10.1073/pnas.0908682107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–70. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- Kuraguchi M, Wang XP, Bronson RT, Rothenberg R, Ohene-Baah NY, Lund JJ, et al. Adenomatous polyposis coli (APC) is required for normal development of skin and thymus. PLoS Genet. 2006;2:e146. doi: 10.1371/journal.pgen.0020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird PW, Jackson-Grusby L, Fazeli A, Dickinson SL, Jung WE, Li E, et al. Suppression of intestinal neoplasia by DNA hypomethylation. Cell. 1995;81:197–205. doi: 10.1016/0092-8674(95)90329-1. [DOI] [PubMed] [Google Scholar]
- Lopez-Garcia C, Klein AM, Simons BD, Winton DJ. Intestinal stem cell replacement follows a pattern of neutral drift. Science. 2010;330:822–5. doi: 10.1126/science.1196236. [DOI] [PubMed] [Google Scholar]
- Lynch PM. Pharmacotherapy for inherited colorectal cancer. Expert Opin Pharmacother. 2010;11:1101–8. doi: 10.1517/14656561003698123. [DOI] [PubMed] [Google Scholar]
- Miller AJ, Dudley SD, Tsao JL, Shibata D, Liskay RM. Tractable Cre-lox system for stochastic alteration of genes in mice. Nature Methods. 2008;5:227–229. doi: 10.1038/NMETH.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–4. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C, Taketo M. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc Natl Acad Sci U S A. 1995;92:4482–6. doi: 10.1073/pnas.92.10.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel BB, Li XM, Dixon MP, Blagoi EL, Nicolas E, Seeholzer SH, et al. APC +/− alters colonic fibroblast proteome in FAP. Oncotarget. 2011:2. doi: 10.18632/oncotarget.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–6. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19:257–72. doi: 10.1016/j.ccr.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana E, Shackleton M, Foster HR, Fullen DR, Sabel MS, Johnson TM, et al. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell. 2010;18:510–23. doi: 10.1016/j.ccr.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quyn AJ, Appleton PL, Carey FA, Steele RJ, Barker N, Clevers H, et al. Spindle orientation bias in gut epithelial stem cell compartments is lost in precancerous tissue. Cell Stem Cell. 2010;6:175–81. doi: 10.1016/j.stem.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Sansom OJ, Reed KR, Hayes AJ, Ireland H, Brinkmann H, Newton IP, et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004;18:1385–90. doi: 10.1101/gad.287404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasai H, Masaki M, Wakitani K. Suppression of polypogenesis in a new mouse strain with a truncated Apc(Delta474) by a novel COX-2 inhibitor, JTE-522. Carcinogenesis. 2000;21:953–8. doi: 10.1093/carcin/21.5.953. [DOI] [PubMed] [Google Scholar]
- Shibata H, Toyama K, Shioya H, Ito M, Hirota M, Hasegawa S, et al. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science. 1997;278:120–3. doi: 10.1126/science.278.5335.120. [DOI] [PubMed] [Google Scholar]
- Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- Smits R, van der Houven van Oordt W, Luz A, Zurcher C, Jagmohan-Changur S, Breukel C, et al. Apc1638N: a mouse model for familial adenomatous polyposis-associated desmoid tumors and cutaneous cysts. Gastroenterology. 1998;114:275–83. doi: 10.1016/s0016-5085(98)70478-0. [DOI] [PubMed] [Google Scholar]
- Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134–44. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256:668–70. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12:468–76. doi: 10.1038/ncb2048. [DOI] [PubMed] [Google Scholar]
- Wang D, Pezo RC, Corner G, Sison C, Lesser ML, Shenoy SM, et al. Altered dynamics of intestinal cell maturation in Apc1638N/+ mice. Cancer Res. 2010;70:5348–57. doi: 10.1158/0008-5472.CAN-09-4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung AT, Patel BB, Li X-M, Seeholzer SH, Coudry RA, Cooper HS, et al. One-Hit Effects in Cancer: Altered Proteome of Morphologically Normal Colon Crypts in Familial Adenomatous Polyposis. Cancer Res. 2008;68:7579–7586. doi: 10.1158/0008-5472.CAN-08-0856. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.