

Abstract
NMDA spikes are prominent in the basal dendrites of cortical pyramidal neurons and greatly expand their ability to integrate synaptic inputs. Calcium (Ca) signals during these spikes are important for synaptic plasticity and fundamentally depend on activation of NMDA receptors. However, the factors that shape the activation of these receptors and the initiation of NMDA spikes remain unclear. Here we examine the properties of NMDA spikes in the basal dendrites of layer 5 pyramidal neurons in the mouse prefrontal cortex. Using two-photon imaging, we demonstrate that NMDA spikes evoke large Ca signals in both postsynaptic spines and nearby dendrites. We find that the dendrite Ca signals depend on NMDA and AMPA receptors but not sodium (Na) or Ca channels. Using voltage-clamp recordings, we show that activation of dendrite NMDA receptors is enhanced by concerted synaptic activity. Blocking glutamate reuptake further increases activation of these receptors and promotes the initiation of NMDA spikes. We conclude that glutamate spillover and recruitment of extrasynaptic receptors contribute to the initiation of NMDA spikes. These results have important implications for how synaptic activity generates both electrical and biochemical signals in dendrites and spines.
Introduction
Dendritic spikes result from complex interactions between synaptic and voltage-sensitive conductances (Schiller et al., 1997; Stuart et al., 1997; Golding and Spruston, 1998). Initiating these spikes often requires concerted synaptic activity within individual dendritic branches (Losonczy and Magee, 2006). Local depolarization during dendritic spikes can activate both voltage-sensitive sodium (Na) and calcium (Ca) channels. The resulting Ca signals in dendrites and spines are important for synaptic plasticity in pyramidal neurons (Golding et al., 2002; Holthoff et al., 2004). The spatial confinement of these spikes also increases the complexity of synaptic integration and permits local computations (London and Häusser, 2005).
NMDA spikes are generated in the basal dendrites of cortical pyramidal neurons by extracellular stimulation (Schiller et al., 2000; Polsky et al., 2004, 2009; Milojkovic et al., 2007) or direct glutamate application (Schiller et al., 2000; Wei et al., 2001; Milojkovic et al., 2005; Major et al., 2008). Like other dendritic spikes, NMDA spikes are associated with supralinear increases in the amplitude and half-width of EPSPs. They are also responsible for large Ca signals restricted to tens of micrometers within individual dendritic branches. Unlike other dendritic spikes, however, NMDA spikes depend on the activation of NMDA receptors (NMDA-Rs) rather than Na and Ca channels.
Initiating NMDA spikes involves a positive feedback loop, in which local depolarization relieves the magnesium (Mg) block of NMDA-Rs (Schiller and Schiller, 2001). In the presence of bound glutamate, this enables NMDA-Rs to act like other voltage-sensitive channels. However, the conductance needed for NMDA spikes is extremely high (Rhodes, 2006), and the underlying mechanism remains unclear. One possibility is activation of silent synapses, but these are rare during the developmental periods when these spikes are studied (Durand et al., 1996; Busetto et al., 2008). Another possibility is glutamate spillover, which could activate extrasynaptic receptors on nearby spines and dendrites.
Although synaptic transmission is often viewed as a point-to-point process, glutamate can also escape the synaptic cleft (Kullmann et al., 1996; Asztely et al., 1997; Barbour and Häusser, 1997; Rusakov and Kullmann, 1998). Due to their high affinity, NMDA-Rs can sense this diffuse glutamate signal (Isaacson, 1999), which may pool from many active synapses (Arnth-Jensen et al., 2002), especially during high-frequency stimulation (Carter and Regehr, 2000). Glutamate binding to extrasynaptic NMDA-Rs could provide the high conductance required for NMDA spikes. It could also generate widespread Ca signals in dendrites and spines located further from the sites of glutamate release.
Here we examine the factors underlying NMDA spikes in layer 5 (L5) pyramidal neurons of the mouse prefrontal cortex (PFC). Using two-photon microscopy, we show that NMDA spikes generate Ca signals in spines and dendrites. The dendrite Ca signal requires NMDA-Rs and AMPA receptors (AMPA-Rs) but not Ca or Na channels. In voltage-clamp recordings, we show that dendrite Ca signals reflect activation of extrasynaptic NMDA-Rs. Blocking glutamate transporters enhances activation of these receptors and lowers the threshold for NMDA spikes. These results indicate that glutamate spillover promotes the generation of NMDA spikes, with important implications for synaptic plasticity and dendritic computation.
Materials and Methods
Preparation.
Recordings were made from L5 pyramidal neurons in the medial PFC of acute slices from postnatal day 21 to 28 Swiss-Webster mice of either sex, as previously described (Chalifoux and Carter, 2010, 2011). Mice were anesthetized with a lethal dose of ketamine/xylazine and perfused intracardially with ice-cold external solution containing the following (in mm): 65 sucrose, 75 NaCl, 25 NaHCO3, 1.25 NaH2PO4, 25 glucose, 2.5 KCl, 1 CaCl2, 5 MgCl2, 0.4 Na-ascorbate, 3 Na-pyruvate (295–305 mOsm), bubbled with 95% O2/5% CO2. Coronal slices (300 μm thick) were cut in ice-cold external solution and transferred to ACSF containing the following (in mm): 119 NaCl, 25 NaHCO3, 1.25 NaH2PO4, 21 glucose, 2.5 KCl, 2 CaCl2, 1 MgCl2, 0.4 Na-ascorbate, 3 Na-pyruvate (295–305 mOsm), bubbled with 95% O2/5% CO2. After 30 min at 35°C, slices were stored for 30 min at 24°C, after which experiments were conducted at 33−34°C.
In all experiments, the ACSF contained 10 μm gabazine to block GABAA-Rs and 10 μm d-serine to prevent NMDA-R run-down. We found no evidence for epileptic activity as long as slices were maintained at near-physiological temperatures. In voltage-clamp experiments, the ACSF contained 10 μm NBQX to block AMPA-Rs. In some experiments, one or more of the following drugs were added to the ACSF (in μm): 10 R-CPP, 10 NBQX, 30 cyclopiazonic acid (CPA), 10 mibefradil, 20 nimodipine, 1 ω-conotoxin MVIIC, 0.3 SNX-482, 10 TBOA (threo-β-benzyloxyaspartic acid). All chemicals were from Sigma or Tocris Bioscience, except for SNX-482 (Alomone Labs).
Physiology recordings.
Whole-cell recordings were obtained from L5 pyramidal neurons identified with IR-DIC at 450–550 μm from the pial surface. Borosilicate recording pipettes (2–4 MΩ) were filled with one of two internal solutions. Current-clamp recordings used (in mm): 135 K-gluconate, 7 KCl, 10 HEPES, 10 Na-phosphocreatine, 4 Mg2-ATP, 0.4 NaGTP, 290–295 mOsm, pH 7.35 with KOH. In some experiments, 5 mm QX-314 was added to this internal. Voltage-clamp recordings used (in mm): 135 Cs-gluconate, 10 HEPES, 10 Na-phosphocreatine, 4 Mg2-ATP, 0.4 NaGTP, 290–295 mOsm, pH 7.35 with CsOH. Internal solutions also contained 1000 μm Fluo-4FF to monitor Ca levels and 30 μm Alexa Fluor-594 to image neuronal morphology. Dye concentrations were chosen to ensure that Ca signals were in the linear range of the indicators. Neurons were filled via the patch electrode for at least 20 min before imaging. Recordings were made using a Multiclamp 700B amplifier, filtered at 5 kHz for current-clamp recordings and 2 kHz for voltage-clamp recordings, and sampled at 10 kHz.
Two-photon microscopy.
Intracellular Ca imaging was performed with a custom microscope, as previously described (Carter and Sabatini, 2004; Carter et al., 2007). For two-photon imaging, 810 nm light was used to excite Fluo-4FF (green) and Alexa Fluor-594 (red), to monitor Ca signals and spine morphology, respectively. Reference frame-scans were taken between each acquisition to correct for small spatial drift over time. Baseline fluorescence was monitored and recordings were discarded if an increase was detected, which would indicate photo-damage.
For most experiments, green and red fluorescence signals were collected during 500 Hz line-scans across a spine-dendrite pair. For the experiments in Figure 8 (see below), green and red fluorescence signals were collected as the scanning mirrors sequentially passed through the spine-dendrite and spine-spine pairs for 2 ms to generate 250 Hz line-scans. Ca signals were quantified as changes in green fluorescence to red fluorescence (ΔG/R), normalized to the maximal green fluorescence to red fluorescence (Gsat/R), giving ΔG/Gsat. The value of Gsat/R was measured after each recording using a thin-walled pipette, containing the internal solution with a saturating concentration of Ca. This pipette was positioned directly above the recorded cell and used at the same recording temperature (33−34°C), as described previously (Chalifoux and Carter, 2010).
Figure 8.
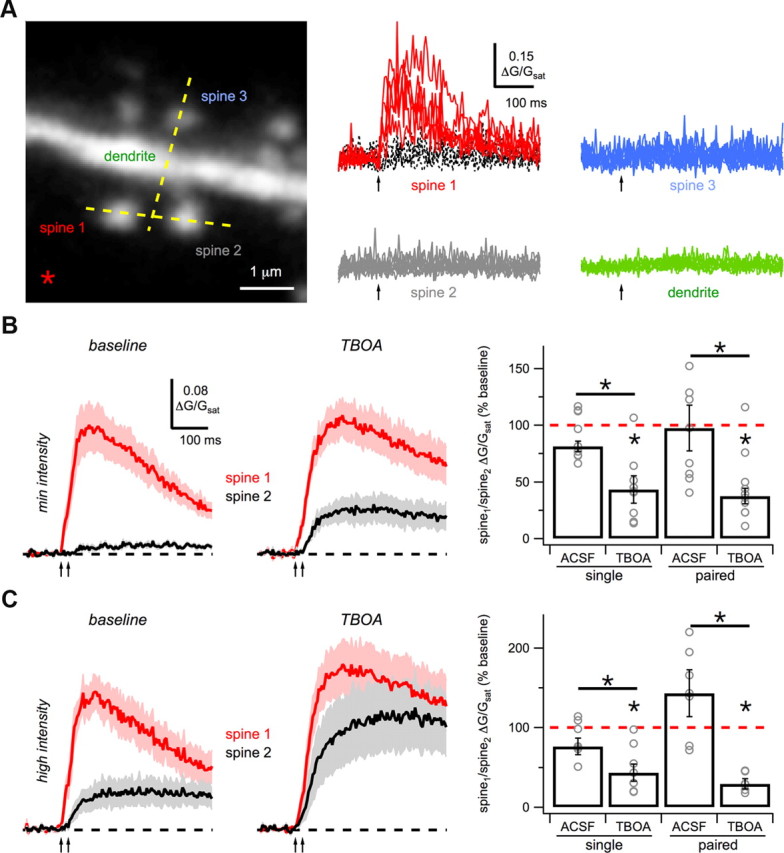
Glutamate spillover onto nearby spines. A, Left, Two-photon image stack of a basal dendrite showing line-scans (yellow dashed lines) through a spine-spine pair (spine 1 and spine 2) and spine-dendrite pair (spine 3 and dendrite), and the position of the theta-glass stimulation electrode (asterisk). Right, Single-pulse stimulation (arrow) evokes successes (red) and failures (black) in spine 1, but no Ca signals in nearby spines or dendrite. B, Average Ca signals in voltage-clamp with minimal-intensity paired-pulse stimulation (arrows) for spine 1 (red) and spine 2 (black) under baseline conditions (left) and after wash-in of 10 μm TBOA (middle). Right, Summary of SDR for single- and paired-pulse stimulation after wash-in of ACSF or TBOA. C, As in B, for high-intensity stimulation. Asterisks indicate significant difference from 100% or between ACSF and TBOA (p < 0.05).
Extracellular stimulation.
Excitatory synaptic inputs were stimulated with a theta-glass electrode (tip diameter = 5–20 μm) filled with the same external solution used for recording. Electrodes were bent to a 70° angle and placed in close proximity (5–15 μm) to a distal basal dendrite (>75 μm from the cell body). Stimulation was brief (0.2 ms) and paired-pulse stimulation was performed at 50 Hz [interstimulus interval (ISI) = 20 ms]. For current-clamp recordings (see Fig. 2), the stimulus intensity varied over a large range (5–100 μA). For current-clamp recordings in Figure 1 and in Figures 2–5 and 9 (below), intensities were defined as high (50–100 μA) and low (20–50 μA), based on their ability to generate a dendrite Ca signal, unless otherwise noted.
Figure 2.
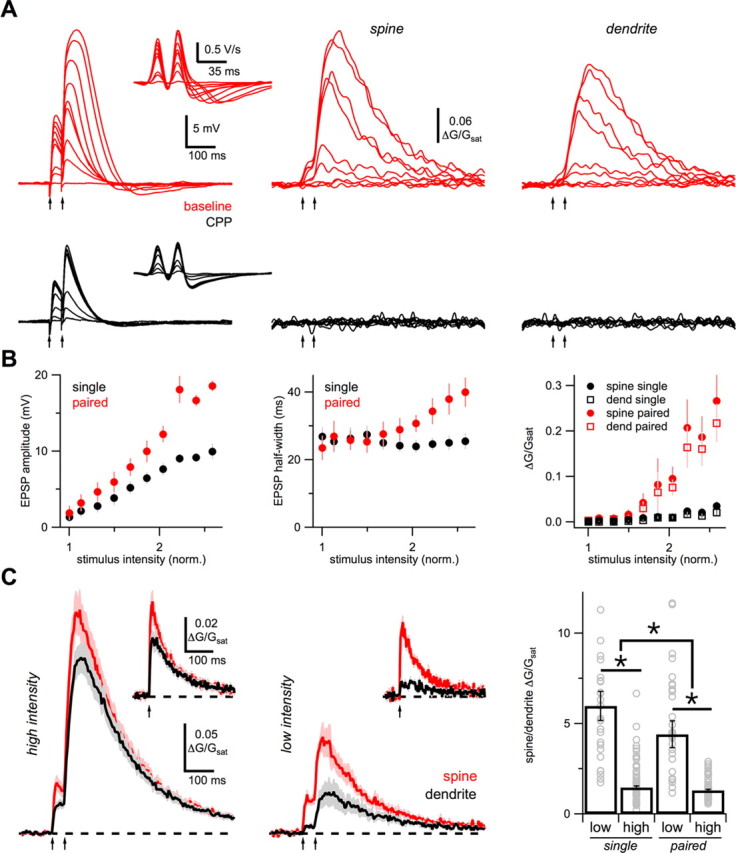
Concerted synaptic activity. A, Example experiment showing average EPSPs (left), first derivative of EPSPs (inset), and average Ca signals in spines (middle) and dendrites (right), for paired-pulse stimulation (arrows) at increasing stimulus intensities, in baseline conditions (red) and after wash-in of 10 μm CPP (black). B, Quantification of EPSP amplitude (left), EPSP half-width (middle), and spine (circles) and dendrite (squares) Ca signals (right), for single-pulse (black) and paired-pulse (red) stimuli, normalized to the minimal stimulus intensity. Individual points are means and vertical bars are SEMs. C, Average Ca signals in response to high-intensity (left) and low-intensity (middle) paired-pulse stimulation (arrows) in spines (red) and dendrites (black). Insets show spine (red) and dendrite (black) Ca signals in response to single-pulse stimulation. Right, Summary of SDR for low- and high-intensity, single- and paired-pulse stimulation. Bar graphs are median ± SE of the median. Gray open circles are individual experiments. Outliers are omitted for display purposes. Asterisks indicate significant difference between conditions (p < 0.05).
Figure 1.
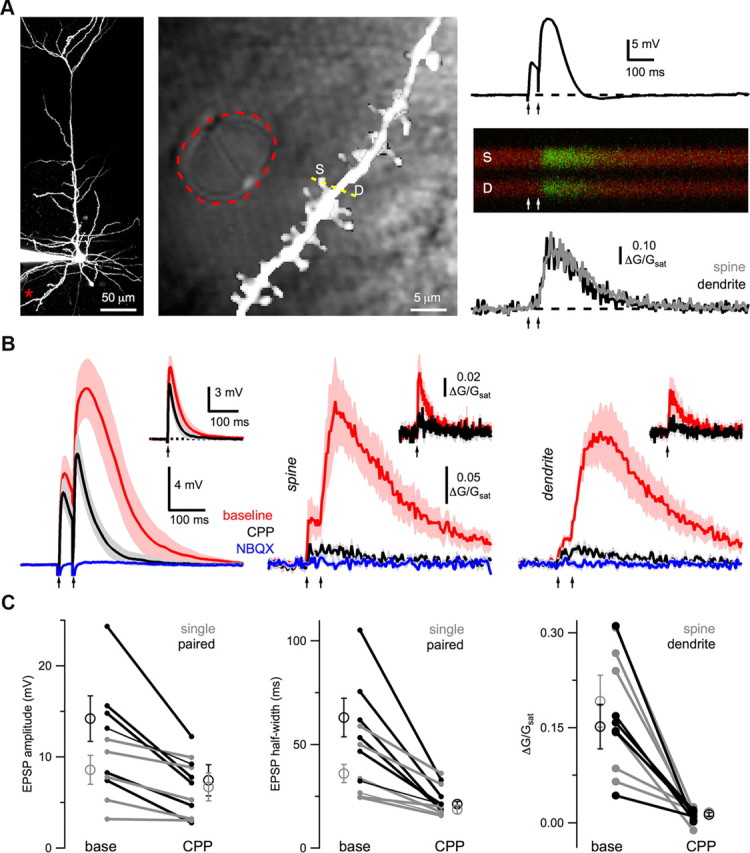
NMDA spike Ca signals. A, Left, Two-photon image of a L5 pyramidal neuron, showing the location of the theta-glass stimulating electrode (asterisk). Middle, High-magnification two-photon image overlaid on a laser-scanning DIC image, outlining the theta-glass stimulating electrode (red dashed line) and showing the line-scan (yellow dashed line) through a spine (S) and dendrite (D). Right, Paired-pulse stimulation (arrows) elicits an EPSP (top) and Ca signals (middle) that are quantified as ΔG/Gsat (bottom) in the spine (gray) and dendrite (black). B, Average EPSPs (left) and Ca signals in spines (middle) and dendrites (right) evoked by high-intensity, paired-pulse stimulation (arrows) in baseline conditions (red), after wash-in of 10 μm CPP (black), and subsequent wash-in of 10 μm NBQX (blue). The insets show the response to single-pulse stimulation in baseline conditions (red) and after wash-in of CPP (black). C, Summary of impact of CPP on EPSP amplitude (left), EPSP half-width (middle) and Ca signals (right) in spines (gray) and dendrites (black). Open circles are medians, error bars are SEs of the median, and connected filled circles are individual experiments.
Figure 3.

Diffusionally isolated Ca signals. A, Average high-intensity Ca signals in spines (left) and dendrites (right) in baseline conditions (red), after wash-in of 10 μm CPP (black), and subsequent wash-in of 10 μm NBQX (blue), using the low-affinity Ca indicator Fluo-5N (1000 μm). Insets show high-intensity single-pulse EPSPs and Ca signals. B, Summary of SDR using Ca indicators of four different Ca affinities and buffering capacities.
Figure 4.

Dendrite Ca sources. A, Average EPSPs (left) and Ca signals in spines (middle) and dendrites (right) in response to high-intensity, paired-pulse stimulation (arrows) before (red) and after (black) wash-in of 10 μm mibefradil (mib) and 20 μm nimodipine (nim). B, As in A, for wash-in of 30 μm CPA. C, Summary of impact of mib/nim and CPA on EPSP amplitude (left), EPSP half-width (middle), and Ca signals (right) in spines (open) and dendrites (hash). Asterisks indicate significant difference from 100% (p < 0.05).
Figure 5.

Postsynaptic depolarization. A, Average EPSPs (left) and Ca signals in spines (middle) and dendrites (right) in response to high-intensity, paired-pulse stimulation (arrows) in baseline conditions (red) and after wash-in of 10 μm NBQX (black). B, As in A, but with internal 5 mm QX-314 in baseline conditions (red), and sequential wash-in of 10 μm CPP (black) and 10 μm NBQX (blue). C, Summary of impact of CPP and NBQX on EPSP amplitude (left), EPSP half-width (middle), and Ca signals (right) in spines (open) and dendrites (hash), in the different baseline conditions. Asterisks indicate significant difference from 100% (p < 0.05).
Figure 9.
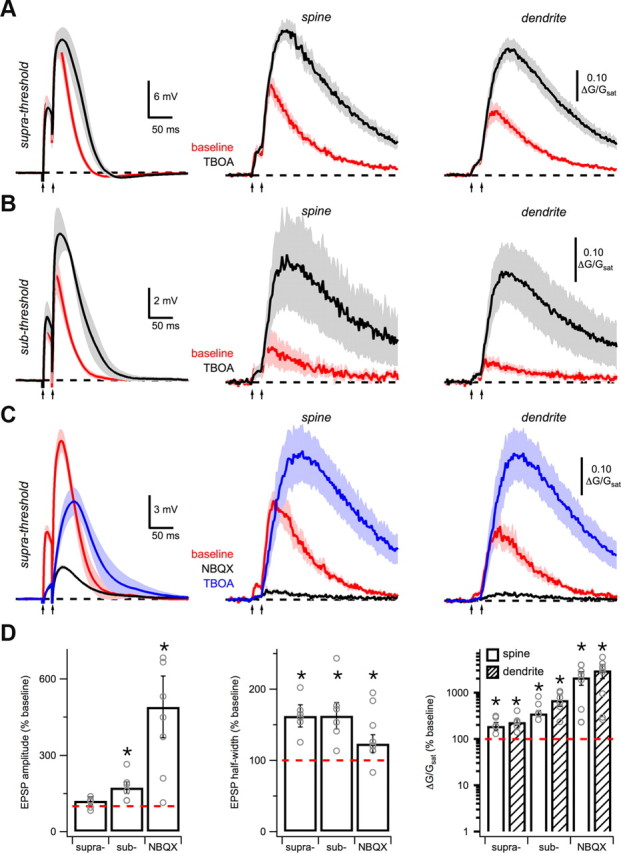
Glutamate spillover promotes NMDA spikes. A, Average EPSPs (left) and Ca signals in spines (middle) and dendrites (right) in response to suprathreshold intensity, paired-pulse stimulation (arrows) under baseline conditions (red) and after wash-in of 10 μm TBOA (black). B, As in A, for subthreshold intensity stimulation. C, Average EPSPs (left) and Ca signals in spines (middle) and dendrites (right) in response to suprathreshold intensity, paired-pulse stimulation under baseline conditions (red), after wash-in of 10 μm NBQX (black) and subsequent wash-in of 10 μm TBOA (blue). D, Summary of impact of TBOA on EPSP amplitude (left), half-width (middle), and Ca signals (right) in spines (open) and dendrites (hash). Asterisks indicate significant difference from 100%.
For voltage-clamp recordings, cells were held at the synaptic reversal potential (+15 to 30 mV), yielding zero net current. For recordings in Figures 6 to 8 (see below), the minimal intensities were small (1–20 μA) and characterized by their ability to generate spine Ca signals with successes and failures but no dendrite Ca signal with single pulses. Failures on individual trials were observed as previously described (Oertner et al., 2002; Chalifoux and Carter, 2010), and removed by calculating a threshold of two SDs above baseline noise. After finding a direct synaptic contact using minimal stimulation, the intensity was increased to investigate the Ca signals at nearby spines and dendrites. These high intensities were similar to those used to elicit NMDA spikes in current-clamp (50–100 μA) and the difference between minimal and high intensities was 30–80 μA (median = 30 ± 5 μA). In only one case did high intensity stimulation recruit a direct synaptic contact on another spine, characterized by its large amplitude and the presence of successes and failures, which we excluded from our analysis.
Figure 6.
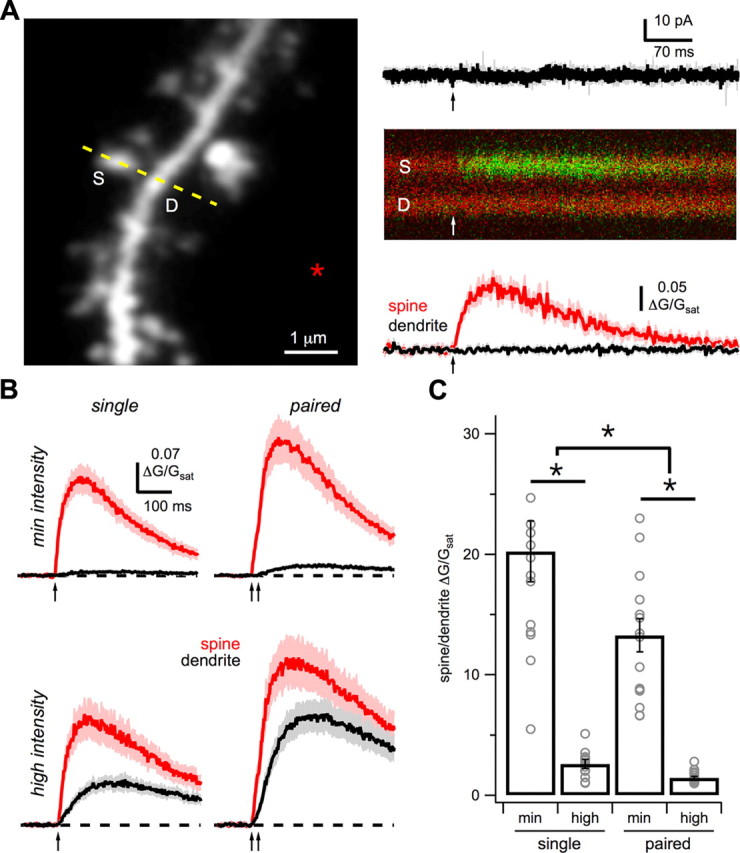
NMDA-R Ca signals. A, Left, Two-photon image stack of a basal dendrite with line-scan (yellow dashed line) through a spine (S) and adjacent dendrite (D), and the position of the theta-glass stimulation electrode (asterisk). Right, Single-pulse stimulation (arrow) in voltage clamp evokes no current while holding at +15 mV (top) but generates a Ca signal in the spine (middle), quantified as ΔG/Gsat (bottom) in the spine (red) and dendrite (black). B, Average Ca signals in spines (red) and dendrites (black) in response to single- (left) and paired-pulse (right) stimulation (arrows) at minimal (top) and high (bottom) intensities in voltage clamp. C, Summary of SDR under the different stimulation conditions. Asterisks indicate significant difference between conditions (p < 0.05).
Data acquisition and analysis.
Image and physiology data were acquired using National Instruments boards and custom software written in MATLAB (MathWorks). Off-line analysis was performed using custom routines written in Igor Pro (WaveMetrics). Ca signal amplitudes were calculated over a 10 ms window around the peak. EPSP amplitudes were calculated over a 5 ms window around the peak. Two-photon images were treated with a 1.5-pixel radius Gaussian filter for display purposes. Traces in Figure 2A were low-pass filtered for display purposes.
Electrophysiological and imaging data are reported in the text as median ± SE of the median. Traces in figures are the mean ± SEM. Summary data in figures are in paired-plot form, showing the median, SE of the median, and individual experiments. Summary data in some figures are shown in bar graph form (median, SE of the median, and individual experiments) because of the large number of N. The SE of the median was calculated as the SD of the medians computed from 100,000 bootstrapped samples from the data. Significance was defined as p < 0.05 and determined using the nonparametric two-tailed Wilcoxon–Mann–Whitney two-sample rank test or the two-tailed Wilcoxon signed rank test for paired data (when appropriate), neither of which make assumptions about the data distribution. For multiple comparisons, significance was determined with the Kruskal–Wallis test and post hoc multiple comparisons were performed with the Dunn–Holland–Wolfe test.
Results
NMDA spike Ca signals
We studied NMDA spikes in L5 pyramidal neurons from acute slices of mouse PFC, using a combination of whole-cell recordings and two-photon imaging. We evoked spikes with a theta-glass extracellular stimulus electrode (diameter = 5–20 μm) in close proximity (Δx = 5–15 μm) to the distal domain of the basal dendrites (median: 110 μm; range: 75–200 μm) (Fig. 1A). Paired-pulse stimulation (ISI = 20 ms) evoked prolonged EPSPs characteristic of NMDA spikes (Fig. 1B), as previously observed (Schiller et al., 2000; Polsky et al., 2004, 2009). The amplitude and half-width of the second EPSP (14.0 ± 2.5 mV; 63 ± 10 ms; n = 6) were significantly greater than the first EPSP (8.6 ± 1.6 mV, p = 0.03; 36 ± 6 ms, p = 0.03; Fig. 1C). Wash-in of the NMDA-R antagonist CPP (10 μm) significantly decreased both the first (amplitude: p = 0.03; half-width: p = 0.03) and second (amplitude: p = 0.03; half-width: p = 0.03) EPSPs (Fig. 1B,C). However, the impact of CPP was significantly greater for the second EPSP (amplitude: p = 0.002; half-width: p = 0.04). Subsequent wash-in of the AMPA-R antagonist NBQX (10 μm) completely blocked the remaining EPSPs (Fig. 1B), indicating that direct electrical stimulation did not occur.
In pyramidal neurons, NMDA spikes are associated with large Ca signals (Schiller et al., 2000; Wei et al., 2001), but their subcellular locations have not been explored. Given their dependence on NMDA-Rs and synaptic activity, we predicted that Ca signals during NMDA spikes would be restricted to postsynaptic spines. To test this, we used two-photon imaging in the same recordings to measure Ca signals with the low-affinity Ca indicator Fluo-4FF (1 mm) (Fig. 1A). Surprisingly, we found that paired-pulse stimulation evoked large Ca signals in both spines and nearby dendrites (Fig. 1B). As with EPSPs, wash-in of CPP eliminated most of these Ca signals (spine: p = 0.03; dendrite: p = 0.03; Fig. 1B,C), indicating a requirement for NMDA-Rs. Subsequent wash-in of NBQX completely blocked the remaining response, confirming a lack of direct electrical stimulation (Fig. 1B). In separate experiments, we found that wash-in of TTX (1 μm) blocked both EPSPs and Ca signals (n = 5: data not shown), further indicating they are not due to electroporation. These results indicate that NMDA spikes are associated with large Ca signals in both spines and adjacent dendrites.
Concerted synaptic activity
The triggering of NMDA spikes usually requires repetitive activation of multiple inputs to a local segment of dendrite (Schiller et al., 2000; Polsky et al., 2004, 2009). We next characterized how EPSPs and Ca signals during NMDA spikes depend on stimulus number and intensity (Fig. 2A). With increasing intensity, we found that the second EPSP gradually became larger and broader than the first. This boosting was reflected in the first derivative of these EPSPs, whose late negative component also increased. In the same recordings, we also found that local Ca signals were first undetectable but then grew after a threshold. This supralinear increase was only seen for paired stimuli and corresponded to the jump in amplitude and half-width of EPSPs (Fig. 2B). In all experiments (n = 6), the increases in both EPSPs and Ca signals were blocked by subsequent application of CPP. In separate experiments, we also found that no NMDA spikes occurred when the NMDA-R blocker MK-801 (1 mm) was added to the internal solution (n = 3; data not shown). Together, our results indicate that a combination of repetitive and synchronous synaptic activity promotes the generation of supralinear EPSPs and Ca signals during NMDA spikes.
We further quantified the relationship between spine and dendrite Ca signals as the spine/dendrite ratio (SDR) at different stimulus intensities and numbers (Fig. 2C). At high intensities (50–100 μA), paired-pulse stimulation yielded similar Ca signals in spines and dendrites, resulting in a relatively low SDR (1.5 ± 0.08; n = 65; Fig. 2C). At low intensities (20–50 μA), paired-pulse stimulation yielded much smaller Ca signals in dendrites than spines, leading to a significantly higher SDR (4.4 ± 0.7, p = 3.0 × 10−10; n = 33; Fig. 2C). At both high and low intensities, the SDRs were also significantly reduced for the second pulse compared with the first pulse (high: p = 2 × 10−5; low: p = 0.008; Fig. 2C). These results indicate that the dendrite Ca signal increases disproportionately relative to the spine Ca signal with increasing synaptic activity, suggesting a novel Ca source is engaged in the dendrites during NMDA spikes.
Diffusional isolation
Synaptic Ca signals are usually prevented from reaching the dendrite by the high spine neck resistance and Ca extrusion mechanisms (Sabatini et al., 2002; Grunditz et al., 2008). How is the dendrite Ca signal generated during NMDA spikes? One possibility is that the Ca indicator is simply diffusing from nearby spines. All Ca indicators act as mobile buffers, capable of shuttling Ca from spine to dendrite (Sabatini et al., 2002). Pooling in the dendrite should be minimal when using dyes with low Ca affinity (KD) and buffer capacity (KB), but still could occur if Ca signals are sufficiently high. To examine this possibility, we imaged spine and dendrite Ca signals with a range of Ca indicators with different Ca affinities and buffer capacities (Maravall et al., 2000), including 1000 μm Fluo-4FF (KD ∼ 10 μm; KB ∼ 67), 1000 μm Oregon-Green BAPTA-5N (KD ∼ 20 μm; KB ∼ 40), 1000 μm Fluo-5N (KD ∼ 90 μm; KB ∼ 11) and 200 μm Fluo-5N (KD ∼ 90 μm; KB ∼ 2) (Fig. 3A). We found the SDR was the same under all of these recording conditions, even with the lowest buffer capacity (p > 0.05, Kruskal–Wallis) (Fig. 3B). These results indicate that the dendrite Ca signal is unlikely due to Ca indicator diffusion, suggesting instead a novel Ca source in the dendrites themselves.
Dendrite Ca sources
The novel dendrite Ca signal during NMDA spikes could be mediated by a variety of Ca sources. Voltage-sensitive Ca channels (VSCCs) are located in spines and dendrites of pyramidal neurons and contribute to synaptic Ca signals (Bloodgood and Sabatini, 2007; Bloodgood et al., 2009; Higley and Sabatini, 2010). These channels can also mediate dendritic Ca spikes in the basal dendrites during suprathreshold synaptic activity (Kampa and Stuart, 2006). We found that wash-in of the T-type blocker mibefradil (10 μm) and L-type blocker nimodipine (20 μm) had no significant effect on EPSPs during NMDA spikes (amplitude: p = 0.09; half-width: p = 0.16; n = 6) (Fig. 4A,C). These blockers also caused a decrease in Ca signal amplitude in spines and dendrites (spine: 73 ± 9% of baseline, p = 0.03; dendrite: 64 ± 4% of baseline, p = 0.03), indicating that these channels are activated. However, these blockers had no effect on the SDR (p = 0.84), indicating that they are not required for the dendrite Ca signal. These effects are unlikely to have a presynaptic origin, as these blockers had no effect on the amplitude or paired-pulse ratio of EPSCs in voltage clamp (n = 5; data not shown). It was not possible to block additional VSCCs when using extracellular stimulation without greatly reducing glutamate release.
In addition to VSCCs, Ca release from internal stores contributes to synaptic Ca signals in some neurons (Emptage et al., 1999; Kovalchuk et al., 2000; Chalifoux and Carter, 2010) and may play a role in some dendritic spikes (Dudman et al., 2007). The endoplasmic reticulum (ER) has a high concentration of Ca and is intercalated throughout spines and dendrites (Spacek and Harris, 1997). Ca influx through spine NMDA-Rs could trigger Ca-induced Ca release (CICR) from the ER to trigger dendrite Ca signals. However, we found that wash-in of the sarcoendoplasmic reticulum Ca-ATPase (SERCA) inhibitor CPA (30 μm), which depletes internal Ca stores and blocks CICR, had no effect on EPSPs (amplitude: p = 0.94; half-width: p = 0.22; n = 7) or the SDR (p = 0.94) during NMDA spikes (Fig. 4B,C). However, CPA did cause a small increase in Ca signal amplitude in spines and dendrites (spine: 127 ± 17% of baseline, p = 0.10; dendrite: 128 ± 18% of baseline; p = 0.15), consistent with a role in clearance. These data confirm that SERCA helps remove synaptic Ca signals, but is not needed for the dendrite Ca signal that occurs during NMDA spikes.
Postsynaptic depolarization
Given that VSCCs and internal stores are not required for dendrite Ca signals, our results suggest that NMDA-Rs may be responsible. To generate NMDA spikes, a local depolarization must relieve the Mg block of these receptors (Schiller and Schiller, 2001). In principle, NMDA-Rs could act alone in a positive feedback loop to generate this depolarization (Rhodes, 2006). However, AMPA-Rs usually provide most of the postsynaptic depolarization and are known to influence synaptic Ca signals (Bloodgood et al., 2009). In support of a role for AMPA-Rs, we found that wash-in of NBQX (10 μm) significantly decreased the EPSP amplitude during NMDA spikes (p = 0.03; n = 6; Fig. 5A,C). Blocking AMPA-Rs also eliminated the Ca signals in both spines and dendrites (spine: p = 0.03; dendrite: p = 0.03; Fig. 5A,C). These results suggest that under our experimental conditions, AMPA-Rs help generate the local depolarization required to remove Mg block and elicit NMDA spikes.
While both AMPA-Rs and NMDA-Rs are clearly important, they are not the only depolarizing conductances that could help generate NMDA spikes. Voltage-sensitive Na channels (VSSCs) could also evoke a local depolarization to relieve Mg block of NMDA-Rs. These channels are present in the basal dendrites of L5 pyramidal neurons (Nevian et al., 2007; Acker and Antic, 2009) and can be activated during EPSPs (Araya et al., 2007). However, a requirement for VSSCs appears unlikely, as NMDA spikes were also elicited in the presence of the blocker QX-314 (5 mm) in the recording pipette (Fig. 5B,C). The SDR was similar in these experiments (1.7 ± 0.3, p = 0.27; n = 6), further indicating that the dendrite Ca signal does not depend on VSSC activation. These findings indicate that VSSCs are not required for NMDA spikes, and suggest that depolarization via AMPA-Rs may be sufficient to open NMDA-Rs.
Extrasynaptic NMDA-Rs
Our results suggest that activation of NMDA-Rs could generate the dendrite Ca signal during NMDA spikes. These receptors are likely to be extrasynaptic, as the vast majority of excitatory synapses are made onto spines. However, this activation is revealed only during local depolarization in current-clamp recordings, making it difficult to study. To avoid this problem, we voltage-clamped neurons at the reversal potential of NMDA-Rs (∼15 mV) and blocked AMPA-Rs with NBQX (10 μm). By using the low-affinity Ca indicator Fluo-4FF (1 mm), we were able to directly monitor NMDA-R activation in spines and dendrites (Fig. 6A).
With this approach, we found that single-pulse stimulation at minimal intensities evoked Ca signals in isolated spines but not adjacent dendrites (Fig. 6B). These results confirm that synaptic Ca signals are spatially restricted for tens of milliseconds, as previously observed (Oertner et al., 2002; Sabatini et al., 2002; Carter and Sabatini, 2004; Bloodgood and Sabatini, 2007; Chalifoux and Carter, 2010). In contrast, paired-pulse stimulation at minimal intensities also generated Ca signals in nearby dendrites (Fig. 6B). With higher pulse number, the dendrite Ca signal increased more than the spine Ca signal, yielding smaller SDRs (single: 20 ± 3; paired: 13 ± 1; p = 0.001; n = 16; Fig. 6C). Higher stimulation intensities also generated even larger Ca signals in both spines and dendrites (Fig. 6B). The dendrite Ca signal again increased disproportionately compared with the spine Ca signal, resulting in smaller SDRs (single: 2.5 ± 0.4; paired: 1.6 ± 0.1; p = 9 × 10−5; n = 16; Fig. 6C). In all conditions, spine and dendrite Ca signals were completely blocked by CPP (10 μm), indicating they are mediated by NMDA-Rs. These results demonstrate that the same stimuli that evoke NMDA spikes also allow glutamate to reach the dendrites and activate extrasynaptic NMDA-Rs.
A diffuse glutamate signal could selectively activate NR2B-containing NMDA-Rs, which may be uniquely present at extrasynaptic sites (Tovar and Westbrook, 1999). However, we found that the ifenprodil derivative Ro 25-6981 (1 μm), which preferentially blocks NR2B-containing receptors, reduced the NMDA-R Ca signals in both spines (single: 62 ± 10% of baseline; paired: 66 ± 13% of baseline; n = 9) and dendrites (single: 54 ± 16% of baseline; paired: 59 ± 10% of baseline) (data not shown). While the reduction was greater in dendrites than spines, this difference was not significant. These findings argue against the presence of two completely different populations of receptors in spines and dendrites. Instead, they are consistent with NR2B-containing NMDA-Rs sensing glutamate at both locations (Sobczyk et al., 2005).
Glutamate spillover
Our results suggest that glutamate spillover occurs during concerted synaptic activity and enables the activation of extrasynaptic receptors. Spillover from synapses is usually restricted by the presence of powerful glutamate reuptake mechanisms (Asztely et al., 1997; Isaacson, 1999; Carter and Regehr, 2000; Arnth-Jensen et al., 2002). If the dendrite Ca signal reflects spillover, we reasoned that blocking glutamate reuptake should further increase this response. At minimal intensities, we found that wash-in of a relatively low concentration of the glutamate transporter inhibitor TBOA (10 μm) enhanced the dendrite Ca signal (single: p = 0.02; paired: p = 0.01) and significantly decreased the SDR (single: p = 0.02; paired: p = 0.02; n = 8; Fig. 7A). At higher intensities, we observed similar changes in these Ca signals (single: p = 0.01; paired: p = 0.003) and SDR (single: p = 0.002; paired: p = 0.002; n = 10; Fig. 7B), which were not significantly different from those at minimal intensities. These results indicate that blocking glutamate transporters increases spillover to activate additional extrasynaptic NMDA-Rs on nearby dendrites.
Figure 7.

Glutamate spillover onto nearby dendrites. A, Average Ca signals in voltage clamp with minimal-intensity paired-pulse stimulation (arrows) for spines (red) and dendrites (black) under baseline conditions (left) and after wash-in of 10 μm TBOA (middle). Right, Summary of SDR for single- and paired-pulse stimulation after wash-in of ACSF or TBOA. B, As in A, for high-intensity stimulation. Asterisks indicate significant difference from 100% or between ACSF and TBOA (p < 0.05).
If the dendrite Ca signal is due to activation of extrasynaptic NMDA-Rs, we expected to observe similar responses in nearby spines. To investigate this possibility, we performed simultaneous line-scans through spine-spine and spine-dendrite pairs (see Materials and Methods) (Fig. 8A). Single-pulse stimulation at minimal intensities evoked Ca signals at isolated spines but not nearby spines or dendrites. As seen for dendrite Ca signals, higher stimulation intensities also evoked Ca signals at nearby spines, yielding a smaller spine/spine ratio (SSR). The Ca signals at nearby spines did not exhibit successes and failures, suggesting they are not generated by direct synaptic contacts. To determine whether these Ca signals could reflect glutamate spillover, we again examined the impact of blocking glutamate reuptake. At minimal intensities, we observed that wash-in of TBOA enhanced Ca signals at nearby spines (single: p = 0.08; paired: p = 0.005) and significantly decreased the SSR (single: p = 0.048; paired: p = 0.01; n = 10; Fig. 8B). At higher intensities, we found similar changes in these Ca signals (single: p = 0.02; paired: p = 0.001) and SSR (single: p = 0.02; paired: p = 0.02; n = 7; Fig. 8C), which were not significantly different from those at minimal intensities. Given the geometry of spines and dendrites, these results are difficult to explain by diffusion of the Ca indicator. Instead, they suggest that glutamate diffuses from nearby terminals to activate extrasynaptic NMDA-Rs on both spines and dendrites.
Enhancing NMDA spikes
Having established that glutamate spillover occurs onto spines and dendrites, we next examined the consequences for the initiation of NMDA spikes. We first determined the impact of blocking glutamate reuptake on NMDA spikes evoked by suprathreshold stimulation (Fig. 9A). Wash-in of TBOA enhanced NMDA spikes by broadening EPSPs (amplitude: p = 0.22; half-width: p = 0.03; n = 6) and increasing Ca signals (spine: p = 0.03; dendrite: p = 0.03; Fig. 9D). These results are consistent with enhanced NMDA-R activation promoting the regenerative feedback loop underlying NMDA spikes. We then asked whether increasing spillover could also generate NMDA spikes in response to subthreshold stimulation (Fig. 9B). In this case, wash-in of TBOA allowed subthreshold stimulation to elicit NMDA spikes, enhancing both EPSPs (amplitude: p = 0.03; half-width: p = 0.03; n = 6) and Ca signals (spine: p = 0.03; dendrite: p = 0.03; Fig. 9D). For both supra- and subthreshold synaptic activity, blocking glutamate transporters had only a minimal impact on synaptic responses evoked by single stimuli. Together, our results demonstrate that increasing glutamate spillover promotes the generation of NMDA spikes in the basal dendrites of these neurons.
While our results indicate that AMPA-Rs are usually needed for the generation of NMDA spikes in these neurons, a sufficiently large glutamate signal could activate enough extrasynaptic NMDA-Rs to eliminate this requirement (Rhodes, 2006). Finally, we tested whether increasing spillover could enable NMDA spikes even in the absence of AMPA-Rs (Fig. 9C). In the presence of NBQX, wash-in of TBOA converted subthreshold responses into NMDA spikes, enhancing both EPSPs (amplitude: p = 0.008; half-width: p = 0.04; n = 8) and Ca signals (spine: p = 0.007; dendrite: p = 0.007; Fig. 9D). In contrast, blocking glutamate transporters had no impact on synaptic responses evoked by single stimuli. These results demonstrate that spillover enables the generation of NMDA spikes even when AMPA-Rs are unavailable. Together, our findings indicate that the spatial profile of the glutamate signal is a crucial factor in the ability of synaptic inputs to generate nonlinear responses in the dendrites.
Discussion
We have shown that NMDA spikes generate large Ca signals in the basal dendrites of L5 pyramidal neurons in the mouse PFC. As predicted for active synaptic inputs, these spikes produce Ca signals in spines. Surprisingly, they also evoke Ca signals in nearby dendrites. These dendrite Ca signals have similar amplitudes and kinetics to spine Ca signals and do not reflect diffusion of the Ca indicator. While they depend on depolarization via AMPA-Rs, they do not require Na channels, Ca channels or internal Ca stores. Imaging Ca signals in voltage clamp reveals activation of extrasynaptic NMDA-Rs in the dendrite and nearby spines, which depends on both stimulation intensity and number. Consistent with glutamate spillover, blocking glutamate transporters increases the NMDA-R Ca signal found in nearby dendrites and spines. This increase in extrasynaptic NMDA-R activation promotes NMDA spikes and lowers their threshold, thereby reducing the requirement for AMPA-R depolarization.
Dendrite Ca signals
NMDA spikes have previously been shown to generate Ca signals in restricted segments of dendritic branches (Schiller et al., 2000; Wei et al., 2001; Milojkovic et al., 2007; Major et al., 2008). However, the location of these Ca signals was unknown at the subcellular level of individual spines and dendrites. In many neurons, excitatory inputs evoke NMDA-R Ca signals in spines but not adjacent dendrites (Koester and Sakmann, 1998; Yuste et al., 1999; Kovalchuk et al., 2000). Spines are able to isolate these signals due to their narrow, high resistance necks and powerful Ca extrusion mechanisms (Sabatini et al., 2002; Grunditz et al., 2008). Given their dependence on NMDA-Rs, and lack of requirement for other Ca sources, we expected that NMDA spikes would also generate Ca signals only in activated spines. We were thus surprised to discover that these spikes generate large Ca signals in both spines and adjacent dendrites.
Our initial explanation was that the dendrite Ca signal resulted from Ca indicator diffusing from the spine head to the dendrite. Four lines of evidence suggest this is not the case. First, we used multiple Ca indicators to show that dendrite Ca signals occurred even when the buffering capacity was extremely low. Under these conditions, there is essentially no shuttling of Ca ions from spines to neighboring dendrites (Sabatini et al., 2002). Second, we showed in voltage clamp that synaptic Ca signals were isolated to the spine head when using minimal intensity stimulation. These results are similar to previous demonstrations of spatially restricted Ca signals in activated spines (Oertner et al., 2002; Sabatini et al., 2002; Carter and Sabatini, 2004; Bloodgood and Sabatini, 2007; Chalifoux and Carter, 2010). Third, we also detected similar Ca signals in nearby spines that grew with increasing stimulus intensity and number. This signal is unlikely to be due to the diffusion of Ca indicator from a spine to the dendrite to another spine. Finally, blocking glutamate transporters should have no effect on the diffusion of Ca indicator from spine to dendrite. However, TBOA consistently increased the Ca signals mediated by extrasynaptic NMDA-Rs in dendrites and spines. Thus, our results indicate that the dendrite Ca signal does not reflect diffusion of the Ca indicator and instead is due to glutamate spillover.
The relief of Mg block is usually thought to allow NMDA-Rs to provide the regenerative depolarization needed for NMDA spikes (Schiller and Schiller, 2001). As expected, we found that both EPSPs and Ca signals require activation of NMDA-Rs under baseline conditions. We also found that these responses require activation of AMPA-Rs but not voltage-sensitive Na or Ca channels. These results suggest that AMPA-Rs contribute to the initial depolarization required to trigger NMDA spikes. They also agree with previous studies showing that high stimulus intensities are needed to generate NMDA spikes when AMPA-Rs are blocked (Polsky et al., 2009). Under these conditions, the NMDA-R conductance may become sufficiently large to evoke NMDA spikes (Rhodes, 2006). Together, our results suggest that glutamate spillover contributes to this high conductance, promoting the initiation of these spikes.
Glutamate spillover
While synaptic transmission is often considered as a point-to-point process, glutamate can also escape from the synaptic cleft and act diffusely (Kullmann et al., 1996; Asztely et al., 1997; Barbour and Häusser, 1997; Rusakov and Kullmann, 1998). In most cases, spillover is detected by changes in the amplitude, kinetics or pharmacology of somatic EPSCs due to an extended glutamate signal. For example, low concentrations of glutamate can preferentially activate high-affinity NMDA-Rs to generate slow EPSCs (Isaacson, 1999). Increasing stimulation intensity and number enhances spillover by activating many presynaptic terminals to generate prolonged EPSCs (Carter and Regehr, 2000). Moreover, blocking glutamate transporters enhances spillover by eliminating one of the primary clearance mechanisms for extrasynaptic glutamate (Arnth-Jensen et al., 2002). However, interpreting these effects is often complicated, as spillover is usually only indirectly inferred by changes in somatically recorded EPSCs.
In our experiments, we used two-photon microscopy to directly detect glutamate spillover as NMDA-R-mediated Ca signals. We used voltage clamp to eliminate contributions from voltage-sensitive ion channels and isolate NMDA-R responses. As in current-clamp, increasing the stimulation number and intensity produced Ca signals in dendrites. Increasing the amount of synchronous synaptic activity also generated graded Ca signals in nearby spines. These spine and dendrite Ca signals did not exhibit the stochastic failures and successes observed at direct synaptic connections (Oertner et al., 2002; Chalifoux and Carter, 2010). Instead, they reflect activation of extrasynaptic NMDA-Rs following glutamate spillover from nearby synapses. Activation of these receptors appears and builds at the same stimulus intensities and numbers needed to generate NMDA spikes. There is thus a close relationship between the stimuli required to elicit NMDA spikes and the activation of extrasynaptic NMDA-Rs by glutamate spillover.
The spatial and temporal extent of extracellular glutamate signals depends on diffusion, buffering and uptake (Barbour and Häusser, 1997; Rusakov and Kullmann, 1998). Dextrans have previously been used to reduce glutamate spillover by increasing viscosity and slowing diffusion (Min et al., 1998). However, we found that adding dextrans dramatically shrank our slices, moving both stimulus and recording pipettes. Glutamate scavengers have also been used to eliminate low concentrations of extrasynaptic glutamate (Min et al., 1998). However, these scavengers can also reduce peak glutamate signals (Turecek and Trussell, 2000), which can make their effects difficult to interpret. Blocking transporters has often been used to enhance glutamate spillover at synapses throughout the brain (Asztely et al., 1997; Isaacson, 1999; Carter and Regehr, 2000; Arnth-Jensen et al., 2002). We found that blocking transporters dramatically enhanced extrasynaptic NMDA-R activation on spines and dendrites. Ultimately, this NMDA-R activation is sufficient to allow pure NMDA spikes even in the absence of AMPA-R activation.
Functional implications
Dendritic spikes in pyramidal neurons result from complex interactions between synaptic inputs and voltage-sensitive conductances (Schiller et al., 1997; Stuart et al., 1997; Golding and Spruston, 1998). In many cases, voltage-sensitive Na or Ca channels mediate the regenerative depolarization during these spikes. The presence of these channels in both spines and dendrites enables these spikes to propagate along a dendritic branch (Losonczy and Magee, 2006). This restricted spread allows individual branches to act as distinct processing units during synaptic integration (Polsky et al., 2004). This compartmentalization of processing in turn enhances the computational capabilities of individual pyramidal neurons (London and Häusser, 2005).
NMDA spikes are unique in requiring both concerted synaptic activity and the sustained binding of glutamate to NMDA-Rs (Schiller et al., 2000). In this case, the depolarization is not due to Na or Ca channels, but instead involves relief of Mg block and opening of NMDA-Rs (Schiller and Schiller, 2001). This local feedback loop means that NMDA spikes can propagate only limited distances along a dendritic branch (Rhodes, 2006; Milojkovic et al., 2007; Major et al., 2008). In the absence of spillover, the need for bound glutamate also implies that NMDA spikes should only occur at postsynaptic spines. However, our results indicate that activation of extrasynaptic NMDA-Rs allows these spikes to spread to nearby spines and dendrites. Our findings suggest that the generation of NMDA spikes is influenced by both dendritic nonlinearities and the glutamate signal. Consequently, these spikes may be impacted by multiple factors that shape the glutamate signal, including local activity in both neuronal and glial networks.
The confinement of NMDA-R Ca signals to spines is often thought to underlie synapse specificity during synaptic plasticity (Zucker, 1999). The Ca signals evoked during NMDA spikes are important triggers of synaptic plasticity in the basal dendrites of cortical pyramidal neurons (Holthoff et al., 2004; Gordon et al., 2006). However, our results show that these Ca signals are not restricted to postsynaptic spines, and can also occur in nearby spines and dendrites. This suggests that glutamate spillover may increase the spatial extent of synaptic plasticity along a dendritic branch. Decreasing transporter activity could also reduce synapse specificity by further increasing the spread of glutamate. Thus, glutamate spillover is likely to be an important factor shaping synaptic plasticity and other Ca-dependent processes. In conclusion, the ability of glutamate spillover to promote the generation of NMDA spikes has important implications for electrical and biochemical signaling in the dendrites of pyramidal neurons.
Footnotes
This work was supported by NIH Grants F30 MH087409 (J.R.C.) and R01 MH085974 (A.G.C.). We thank members of the Carter laboratory, Michael Beierlein and Bernardo Sabatini, for helpful discussions and comments on the manuscript. We thank Adam Weiss for help with programming.
The authors declare no competing financial interests.
References
- Acker CD, Antic SD. Quantitative assessment of the distributions of membrane conductances involved in action potential backpropagation along basal dendrites. J Neurophysiol. 2009;101:1524–1541. doi: 10.1152/jn.00651.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya R, Nikolenko V, Eisenthal KB, Yuste R. Sodium channels amplify spine potentials. Proc Natl Acad Sci U S A. 2007;104:12347–12352. doi: 10.1073/pnas.0705282104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnth-Jensen N, Jabaudon D, Scanziani M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci. 2002;5:325–331. doi: 10.1038/nn825. [DOI] [PubMed] [Google Scholar]
- Asztely F, Erdemli G, Kullmann DM. Extrasynaptic glutamate spillover in the hippocampus: dependence on temperature and the role of active glutamate uptake. Neuron. 1997;18:281–293. doi: 10.1016/s0896-6273(00)80268-8. [DOI] [PubMed] [Google Scholar]
- Barbour B, Häusser M. Intersynaptic diffusion of neurotransmitter. Trends Neurosci. 1997;20:377–384. doi: 10.1016/s0166-2236(96)20050-5. [DOI] [PubMed] [Google Scholar]
- Bloodgood BL, Sabatini BL. Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron. 2007;53:249–260. doi: 10.1016/j.neuron.2006.12.017. [DOI] [PubMed] [Google Scholar]
- Bloodgood BL, Giessel AJ, Sabatini BL. Biphasic synaptic Ca influx arising from compartmentalized electrical signals in dendritic spines. PLoS Biol. 2009;7:e1000190. doi: 10.1371/journal.pbio.1000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busetto G, Higley MJ, Sabatini BL. Developmental presence and disappearance of postsynaptically silent synapses on dendritic spines of rat layer 2/3 pyramidal neurons. J Physiol. 2008;586:1519–1527. doi: 10.1113/jphysiol.2007.149336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci. 2000;20:4423–4434. doi: 10.1523/JNEUROSCI.20-12-04423.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, Sabatini BL. State-dependent calcium signaling in dendritic spines of striatal medium spiny neurons. Neuron. 2004;44:483–493. doi: 10.1016/j.neuron.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Carter AG, Soler-Llavina GJ, Sabatini BL. Timing and location of synaptic inputs determine modes of subthreshold integration in striatal medium spiny neurons. J Neurosci. 2007;27:8967–8977. doi: 10.1523/JNEUROSCI.2798-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalifoux JR, Carter AG. GABAB receptors modulate NMDA receptor calcium signals in dendritic spines. Neuron. 2010;66:101–113. doi: 10.1016/j.neuron.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalifoux JR, Carter AG. GABAB receptor modulation of voltage-sensitive calcium channels in spines and dendrites. J Neurosci. 2011;31:4221–4232. doi: 10.1523/JNEUROSCI.4561-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudman JT, Tsay D, Siegelbaum SA. A role for synaptic inputs at distal dendrites: instructive signals for hippocampal long-term plasticity. Neuron. 2007;56:866–879. doi: 10.1016/j.neuron.2007.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand GM, Kovalchuk Y, Konnerth A. Long-term potentiation and functional synapse induction in developing hippocampus. Nature. 1996;381:71–75. doi: 10.1038/381071a0. [DOI] [PubMed] [Google Scholar]
- Emptage N, Bliss TV, Fine A. Single synaptic events evoke NMDA receptor-mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron. 1999;22:115–124. doi: 10.1016/s0896-6273(00)80683-2. [DOI] [PubMed] [Google Scholar]
- Golding NL, Spruston N. Dendritic sodium spikes are variable triggers of axonal action potentials in hippocampal CA1 pyramidal neurons. Neuron. 1998;21:1189–1200. doi: 10.1016/s0896-6273(00)80635-2. [DOI] [PubMed] [Google Scholar]
- Golding NL, Staff NP, Spruston N. Dendritic spikes as a mechanism for cooperative long-term potentiation. Nature. 2002;418:326–331. doi: 10.1038/nature00854. [DOI] [PubMed] [Google Scholar]
- Gordon U, Polsky A, Schiller J. Plasticity compartments in basal dendrites of neocortical pyramidal neurons. J Neurosci. 2006;26:12717–12726. doi: 10.1523/JNEUROSCI.3502-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunditz A, Holbro N, Tian L, Zuo Y, Oertner TG. Spine neck plasticity controls postsynaptic calcium signals through electrical compartmentalization. J Neurosci. 2008;28:13457–13466. doi: 10.1523/JNEUROSCI.2702-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ, Sabatini BL. Competitive regulation of synaptic Ca2+ influx by D2 dopamine and A2A adenosine receptors. Nat Neurosci. 2010;13:958–966. doi: 10.1038/nn.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holthoff K, Kovalchuk Y, Yuste R, Konnerth A. Single-shock LTD by local dendritic spikes in pyramidal neurons of mouse visual cortex. J Physiol. 2004;560:27–36. doi: 10.1113/jphysiol.2004.072678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS. Glutamate spillover mediates excitatory transmission in the rat olfactory bulb. Neuron. 1999;23:377–384. doi: 10.1016/s0896-6273(00)80787-4. [DOI] [PubMed] [Google Scholar]
- Kampa BM, Stuart GJ. Calcium spikes in basal dendrites of layer 5 pyramidal neurons during action potential bursts. J Neurosci. 2006;26:7424–7432. doi: 10.1523/JNEUROSCI.3062-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koester HJ, Sakmann B. Calcium dynamics in single spines during coincident presynaptic and postsynaptic activity depend on relative timing of back-propagating action potentials and subthreshold excitatory postsynaptic potentials. Proc Natl Acad Sci U S A. 1998;95:9596–9601. doi: 10.1073/pnas.95.16.9596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalchuk Y, Eilers J, Lisman J, Konnerth A. NMDA receptor-mediated subthreshold Ca(2+) signals in spines of hippocampal neurons. J Neurosci. 2000;20:1791–1799. doi: 10.1523/JNEUROSCI.20-05-01791.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM, Erdemli G, Asztély F. LTP of AMPA and NMDA receptor-mediated signals: evidence for presynaptic expression and extrasynaptic glutamate spill-over. Neuron. 1996;17:461–474. doi: 10.1016/s0896-6273(00)80178-6. [DOI] [PubMed] [Google Scholar]
- London M, Häusser M. Dendritic computation. Annu Rev Neurosci. 2005;28:503–532. doi: 10.1146/annurev.neuro.28.061604.135703. [DOI] [PubMed] [Google Scholar]
- Losonczy A, Magee JC. Integrative properties of radial oblique dendrites in hippocampal CA1 pyramidal neurons. Neuron. 2006;50:291–307. doi: 10.1016/j.neuron.2006.03.016. [DOI] [PubMed] [Google Scholar]
- Major G, Polsky A, Denk W, Schiller J, Tank DW. Spatiotemporally graded NMDA spike/plateau potentials in basal dendrites of neocortical pyramidal neurons. J Neurophysiol. 2008;99:2584–2601. doi: 10.1152/jn.00011.2008. [DOI] [PubMed] [Google Scholar]
- Maravall M, Mainen ZF, Sabatini BL, Svoboda K. Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys J. 2000;78:2655–2667. doi: 10.1016/S0006-3495(00)76809-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milojkovic BA, Radojicic MS, Antic SD. A strict correlation between dendritic and somatic plateau depolarizations in the rat prefrontal cortex pyramidal neurons. J Neurosci. 2005;25:3940–3951. doi: 10.1523/JNEUROSCI.5314-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milojkovic BA, Zhou WL, Antic SD. Voltage and calcium transients in basal dendrites of the rat prefrontal cortex. J Physiol. 2007;585:447–468. doi: 10.1113/jphysiol.2007.142315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min MY, Rusakov DA, Kullmann DM. Activation of AMPA, kainate, and metabotropic receptors at hippocampal mossy fiber synapses: role of glutamate diffusion. Neuron. 1998;21:561–570. doi: 10.1016/s0896-6273(00)80566-8. [DOI] [PubMed] [Google Scholar]
- Nevian T, Larkum ME, Polsky A, Schiller J. Properties of basal dendrites of layer 5 pyramidal neurons: a direct patch-clamp recording study. Nat Neurosci. 2007;10:206–214. doi: 10.1038/nn1826. [DOI] [PubMed] [Google Scholar]
- Oertner TG, Sabatini BL, Nimchinsky EA, Svoboda K. Facilitation at single synapses probed with optical quantal analysis. Nat Neurosci. 2002;5:657–664. doi: 10.1038/nn867. [DOI] [PubMed] [Google Scholar]
- Polsky A, Mel BW, Schiller J. Computational subunits in thin dendrites of pyramidal cells. Nat Neurosci. 2004;7:621–627. doi: 10.1038/nn1253. [DOI] [PubMed] [Google Scholar]
- Polsky A, Mel B, Schiller J. Encoding and decoding bursts by NMDA spikes in basal dendrites of layer 5 pyramidal neurons. J Neurosci. 2009;29:11891–11903. doi: 10.1523/JNEUROSCI.5250-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes P. The properties and implications of NMDA spikes in neocortical pyramidal cells. J Neurosci. 2006;26:6704–6715. doi: 10.1523/JNEUROSCI.3791-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusakov DA, Kullmann DM. Extrasynaptic glutamate diffusion in the hippocampus: ultrastructural constraints, uptake, and receptor activation. J Neurosci. 1998;18:3158–3170. doi: 10.1523/JNEUROSCI.18-09-03158.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Oertner TG, Svoboda K. The life cycle of Ca(2+) ions in dendritic spines. Neuron. 2002;33:439–452. doi: 10.1016/s0896-6273(02)00573-1. [DOI] [PubMed] [Google Scholar]
- Schiller J, Schiller Y. NMDA receptor-mediated dendritic spikes and coincident signal amplification. Curr Opin Neurobiol. 2001;11:343–348. doi: 10.1016/s0959-4388(00)00217-8. [DOI] [PubMed] [Google Scholar]
- Schiller J, Schiller Y, Stuart G, Sakmann B. Calcium action potentials restricted to distal apical dendrites of rat neocortical pyramidal neurons. J Physiol. 1997;505:605–616. doi: 10.1111/j.1469-7793.1997.605ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller J, Major G, Koester HJ, Schiller Y. NMDA spikes in basal dendrites of cortical pyramidal neurons. Nature. 2000;404:285–289. doi: 10.1038/35005094. [DOI] [PubMed] [Google Scholar]
- Sobczyk A, Scheuss V, Svoboda K. NMDA receptor subunit-dependent [Ca2+] signaling in individual hippocampal dendritic spines. J Neurosci. 2005;25:6037–6046. doi: 10.1523/JNEUROSCI.1221-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spacek J, Harris KM. Three-dimensional organization of smooth endoplasmic reticulum in hippocampal CA1 dendrites and dendritic spines of the immature and mature rat. J Neurosci. 1997;17:190–203. doi: 10.1523/JNEUROSCI.17-01-00190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart G, Schiller J, Sakmann B. Action potential initiation and propagation in rat neocortical pyramidal neurons. J Physiol. 1997;505:617–632. doi: 10.1111/j.1469-7793.1997.617ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turecek R, Trussell LO. Control of synaptic depression by glutamate transporters. J Neurosci. 2000;20:2054–2063. doi: 10.1523/JNEUROSCI.20-05-02054.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei DS, Mei YA, Bagal A, Kao JP, Thompson SM, Tang CM. Compartmentalized and binary behavior of terminal dendrites in hippocampal pyramidal neurons. Science. 2001;293:2272–2275. doi: 10.1126/science.1061198. [DOI] [PubMed] [Google Scholar]
- Yuste R, Majewska A, Cash SS, Denk W. Mechanisms of calcium influx into hippocampal spines: heterogeneity among spines, coincidence detection by NMDA receptors, and optical quantal analysis. J Neurosci. 1999;19:1976–1987. doi: 10.1523/JNEUROSCI.19-06-01976.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Calcium- and activity-dependent synaptic plasticity. Curr Opin Neurobiol. 1999;9:305–313. doi: 10.1016/s0959-4388(99)80045-2. [DOI] [PubMed] [Google Scholar]