

Abstract
Objective
The purpose of this study was to examine the role of VE-cadherin in cellular processes underlying angiogenesis and the effects of VE-cadherin inhibition on retinal angiogenesis.
Methods
Retinal neovascularization was induced in newborn mice by exposure to 75% oxygen (day 7-12) followed by room air, and quantitatied from histological sections. Mice received daily intraperitoneal injections of either a VE-cadherin antagonist or a control peptide from day 12 to 17. In vitro cell migration, proliferation, and tubule formation was examined in the presence of the VE-cadherin antagonist. The effect of antagonist treatment on the integrity of established cell junctions was examined by FITC-dextran monolayer permeability and VE-cadherin immunocytochemistry.
Results
Treatment with the VE-cadherin antagonist significantly reduced retinal angiogenesis. Inhibition of VE-cadherin function suppressed tubule formation in endothelial cells. The antagonist treatment also decreased cell migration and proliferation. The antagonist treatment did not affect the integrity of existing cell junctions. Immunostaining for VE-cadherin and rates of monolayer permeability were comparable to untreated controls.
Conclusions
Our studies point to a pivotal role played by VE-cadherin in the angiogenic process.
Clinical Relevance
Inhibition of VE-cadherin might be an effective strategy for pharmacological inhibition in proliferative retinopathies.
Ocular angiogenesis, in response to tissue ischemia, is a leading cause of vision loss in numerous clinical conditions (1). Alterations in the retinal vasculature leading to new vessel formation is commonly seen in diabetic retinopathy, retinopathy of prematurity and retinal vein occlusion. The formation of new vessels in the eye and elsewhere is a multi-step process involving endothelial cell proliferation, migration, tubule formation and subsequent maturation of the newly formed vessels (2,3). These new vessels are formed by sprouting of endothelial cells from pre-existing vessels and their formation can be aided by the infiltration of bone marrow derived progenitor cells which differentiate into endothelial or microglial cells (4,5). The migration and assembly of endothelial cells into tube-like structures is a vital step in the morphogenesis of new blood vessels. The formation and stabilization of these vascular structures is mediated by cell-cell contact and the subsequent engagement of homotypic adhesion molecules on adjacent endothelial cells (6).
Endothelial cell junctions are populated by highly specialized families of cell adhesion molecules (7-9). The vascular endothelial cadherin (VE-cadherin), is localized at specialized cell junctions called adherens junctions and mediates calcium-dependent cell adhesion. The clustering of VE-cadherin ectodomains between adjacent endothelial cells begins the formation of stable adherens junctions between endothelial cells. In both normal and pathological angiogenesis, the formation and stabilization of cell-cell contact is a prerequisite to lumen formation, generation of basement membranes and inhibition of vascular regression (6, 10).
In addition to mediating cell adhesion, VE-cadherin has been implicated in a variety of cell signaling pathways crucial to the endothelium (11,12). These signaling functions depend upon the interaction of the VE-cadherin cytoplasmic domain with a number of accessory proteins including ß-catenin, plakoglogin and α-catenin. The interaction of the catenin-cadherin complex with the cytoskeleton induces molecular signaling events that lead to contact inhibition, cell cycle arrest, and endothelial cell survival (13-16). In vasculogenesis, a null mutation in the VE-cadherin gene is embryonic lethal because of deficient vascular remodeling in yolk sac and embryo proper. Cells devoid of VE-cadherin demonstrate an inability to organize into vascular-like structures, pointing to the central role played by VE-cadherin in developmental angiogenesis (11).
VE-cadherin has recently become a target for the inhibition of pathological angiogenesis (17,18). Inhibition of VE-cadherin may disrupt the transmission of intracellular signals induced by the angiogenic factor VEGF and expression of VE-cadherin with a truncated cytoplasmic domain results in impaired vascular remodeling and maturation of the vascular network (11). Because VE-cadherin regulates vascular permeability, the use of antibody-based anti-angiogenic treatments risks eliciting widespread systemic vascular permeability increases (19). In the present study we have tested the anti-angiogenic properties of a cyclic peptide inhibitor of VE-cadherin directed against the cell-adhesion recognition sequence present on the VE-cadherin ectodomain. This peptide was found to significantly inhibit angiogenesis in a murine model of oxygen-induced retinal neovascularization. Our observations suggest that selective inhibition of VE-cadherin in newly forming vessels may be a useful target for therapeutic intervention in retinal neovascularization.
METHODS
Animal Model
C57BL/6J mice were bred at the University of New Mexico Animal Research Facility. All experiments were consistent with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Retinal angiogenesis was induced using the oxygen induced retinopathy (OIR) model described by Smith et al (20). Age matched mice, housed only in room air served as controls. Experimental mice were injected intraperitoneally on days P12 to P17 with 0.1- 1mg/kg of the VE-cadherin antagonist ADH100191-A (Ac-CDAEC-NH2) dissolved in sterile PBS, (Adherex Technologies Inc., Durham, NC). Some animals received an equivalent dose of a scrambled control peptide ADH100692-A (Ac-CAEDC-NH2). Eyes were collected at P17 and analyzed histologically for the extent of retinal neovascularization.
Quantitation of Retinal Neovascularization
Eyes from control and experimental animals were fixed overnight in 10% neutral buffered formalin and embedded in paraffin. Serial sections (6 μm) parallel to the optic nerve were mounted on gelatin-coated slides. After deparaffinization, sections were treated with 0.0125% Trypsin at 37°C for 10 minutes. Sections were blocked with 10% normal goat serum (NGS) and stained for Type IV collagen (Rockland Inc., Gilbertsville, PA). The sections were mounted with Vectashield containing diamidinophenylindole (DAPI) to stain the nuclei. Nuclei associated with type IV collagen positive vessels on the vitreous side of the inner limiting membrane of the retina were counted in every third section using a Zeiss inverted fluorescence microscope. Slides were masked prior to counting.
Whole mount Retinal Vessel Staining
Eyes from day 17 animals were removed and fixed with 2% paraformaldehyde in PBS for 2 hours at 4°C. The retina was dissected free and incubated in cold 70% ethanol for 20 minutes followed by cold PBS/1% triton X-100 for an additional 30 minutes. The retinas were incubated overnight at 4°C in PBS containing 5mg/ml Isolectin GS-IB4 conjugated to Alexa fluor 488 (Invitrogen), washed extensively with PBS and mounted on glass slides. Images of the vasculature were obtained at 10X magnification and pseudo-colored.
Cell Culture
Bovine retinal microvascular endothelial cells were obtained from VEC Technologies, Inc., (Rensselaer, NY) and grown on fibronectin-coated cell culture dishes in MCDB-131 media supplemented with 10 % FBS, 10ng/ml EGF, 1 μg/ml Hydrocortisone, 0.2mg/ml EndoGro and 0.09mg/ml heparin. Passages 3–10 were used for all experiments.
Cell Proliferation Assay
Endothelial cells were plated onto fibronectin-coated 24-well plates at a density of 20,000 cells/well. Cells were stimulated with VEGF (40ng/ml) in the presence or absence of the VE-cadherin antagonist (1mg/ml) and a control peptide (1mg/ml). After 48 hours of incubation, cellular proliferation was quantitated using the Vybrant R cell proliferation assay kit (Invitrogen, Carlsbad, California, USA).
Cell Migration Assay
Cell migration assays were performed using Falcon cell culture inserts containing a membrane with 3.0 μm pores. Bovine retinal microvascular endothelial cells (2.5×104 cells) were plated onto membranes coated with fibronectin in serum-free MCDB-131 media. The lower chamber contained serum-free media with or without 40 ng/ml vascular endothelial growth factor (VEGF) added as a chemo-attractant. The upper chamber of the cell culture insert received specified doses of the VE-cadherin antagonist (1mg/ml), Control peptide (1mg/ml) or VE-cadherin neutralizing antibody (100ng/ml). The cells were allowed to migrate for 18 hours and the cells on the upper surface of the membrane were removed with a cotton swab. The cells attached to the lower surface of the membrane were fixed with 100% methanol for 5 minutes and rinsed with PBS. The membranes were excised, mounted on glass slides with Vectashield containing DAPI to stain the cell nucleus. The number of migrated cells was determined at 10X magnification in four peripheral fields and one central field. The number of migrated cells was averaged from 5 fields per membrane.
Tubule Formation Assay
Bovine retinal microvascular endothelial cells (2×105/ml) were seeded into growth factor-reduced Matrigel (BD Biosciences, San Jose, CA) and allowed to gel on glass coverslips. Gels were incubated in complete media containing VEGF (40ng/ml) with either control peptide or VE-cadherin antagonist (1mg/ml) for 48 hours. Gels were fixed in 4% Paraformaldehyde and examined at 20X magnification.
Monolayer Permeability assay
Bovine microvascular retinal endothelial cells were seeded onto fibronectin-coated transwell inserts (0.4um pore size) (BD Biosciences, San Jose, CA) in a modified Boyden chamber. Trans-endothelial resistance (TER) was measured every day until the TER of the monolayer read above 200 mOhms, at which point the cells were regarded as confluent. At the start of the experiment, 5μl of 40K FITC- dextran (40mg/ml) was added to the top chamber. Inserts were treated with either the VE-cadherin antagonist (1mg/ml) or control peptide (1mg/ml). Aliquots of media were removed from the bottom chamber after 24 hours and read at 485nm in a spectrofluorimeter (Perkin Elmer).
Immunofluorescence Microscopy
Cells were seeded at low density onto glass coverslips coated with fibronectin and allowed to attach and spread. The cells were incubated for 3 hours with the VE-cadherin antagonist or control peptide and fixed with an ice-cold solution of methanol/acetic acid (3:1) for 2 min and washed with PBS. Coverslips were blocked with 10% goat serum and incubated with either anti-β catenin antibody (BD Biosciences, San Jose, CA) or anti-VE-cadherin (Alexis) for one hour at room temperature. Coverslips were rinsed three times for 10 min in TBS/0.1% Tween-20 and incubated with a fluorescently-labeled secondary antibody, washed and mounted with Vectashield (Vector Laboratories, Burlingame, CA) and examined by fluorescence microscopy.
Statistical Analysis
All statistical analysis was performed using Graph-pad prism software. Data were analyzed by either Students t-test or by one way ANOVA followed by Bonferroni’s Multiple Comparison test.
RESULTS
A VE-cadherin antagonist suppresses retinal neovascularization
Animals with oxygen induced retinopathy were treated with a VE-cadherin antagonist peptide. The mice received an intraperitoneal injection of either the antagonist (0.1, 0.5 or 1 mg/day) or control peptide (1mg/day) from P12 to P16. Eyes were collected on P17 and the angiogenic response was quantitated as described. A significant 54% reduction in the degree of retinal neovascularization was found in mice treated at the highest dose (1mg) of the antagonist (Figure 1; p< 0.01). Other doses at 0.1 mg or 0.5 mg/kg did not show any significant inhibition of retinal neovascularization. No obvious toxic effects on the normal vasculature were observed in retinas of animals receiving the VE-cadherin antagonist or control peptide (Figure 2).
Figure 1. Quantitation of the neovascular response in the retinas of experimental animals treated with a VE-cadherin antagonist or control peptide.
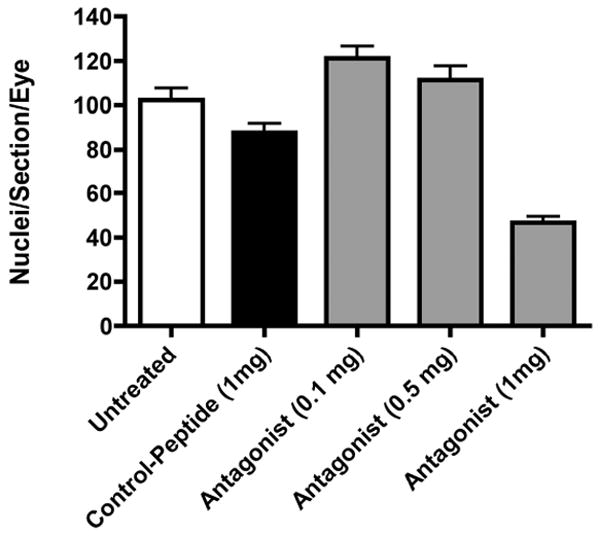
Data are the mean +/- SEM for N= 4 animals per group. Differences among means were tested by ANOVA and corrected using the Bonferroni post test. Values for p<0.05 were considered significant. Untreated vs. Antagonist (1mg), p<0.01; Control Peptide (1mg) vs. Antagonist (1mg), p<0.01; Antagonist (0.1mg) vs. Antagonist (1mg), p<0.01; Antagonist (0.5mg) vs. Antagonist (1mg), p<0.01; all other pairings are not significantly different.
Figure 2. The normal retinal vasculature is not effected by the VE-cadherin antagonist.
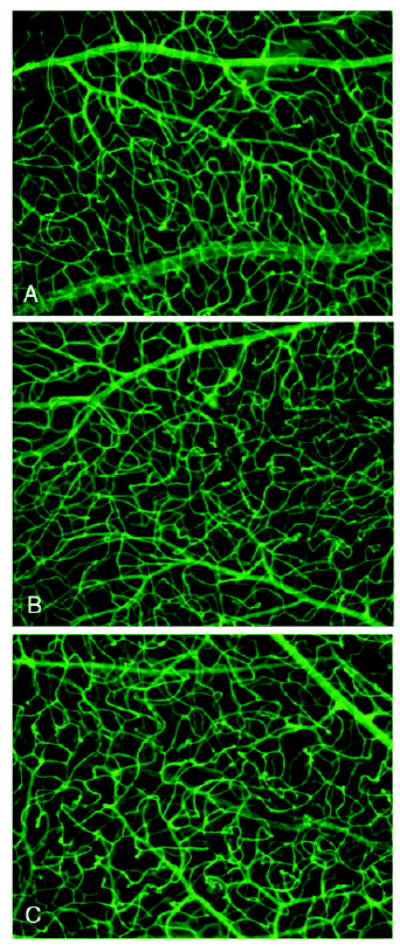
Representative images of vessels from d17 retinal whole mounts stained with FITC-GSA lectin. The vascular morphology and density of vessels within the retinal tissue is similar from animals treated with the VE-cadherin antagonist (A), the control peptide (B) or vehicle only (C).
VE-cadherin Antagonist Inhibits Endothelial Cell Behaviors in vitro
The mechanism by which the VE-cadherin antagonist inhibits retinal angiogenesis was investigated using isolated bovine retinal endothelial cells. Individual cells were suspended in a 3-dimensional extracellular matrix and allowed to form tubes in the presence or absence of the VE-cadherin antagonist and control peptide. In cultures without treatment, the cells organized themselves into extensive branching networks typical for these cells grown in a Matrigel containing matrix (Figure 3). Cells treated with the VE-cadherin antagonist exhibited far fewer interconnected tubules and organized primarily as small groups of elongated cells. Cultures treated with control peptide looked similar to untreated cells.
Figure 3. VE-cadherin antagonist prevents tubule assembly in vitro.
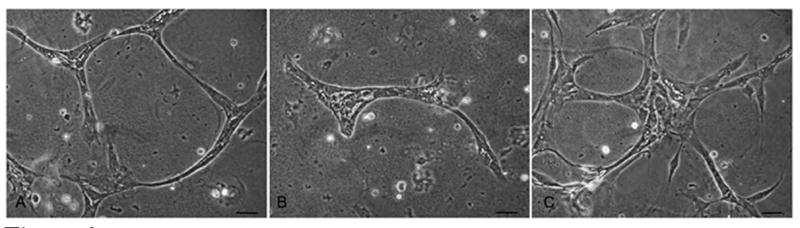
Bovine retinal microvascular endothelial cells were seeded into a Matrigel matrix. Untreated cells (A) organized into branching cordlike structures after 48 hours in culture. In the presence of the VE-cadherin antagonist (B) the formation of the cellular network was inhibited. Cells treated with control peptide (C) showed a normal pattern of tubule morphogenesis comparable to untreated cells. Bar=20um.
The final angiogenic step of tube formation and stabilization is preceded by proliferation and migration of cells derived from either the existing vessels or from bone marrow derived endothelial progenitor cells. We next examined whether the disruption of tubule formation seen in antagonist treated cultures might be due in part to a migration defect in the endothelial cells. The migration of bovine retinal endothelial cells was examined in vitro as described above. Cells plated onto fibronectin-coated migration inserts and stimulated with VEGF showed extensive migration after 18 hours. When the VE-cadherin antagonist was added at the time of plating the cells showed a significant 46% reduction in the extent of migration (Figure 4). No significant effect was seen when cells were incubated in the presence of the control peptide, or when cells were incubated in the presence of an anti-VE-cadherin antibody.
Figure 4. In Vitro cell migration is inhibited by the VE-cadherin antagonist.
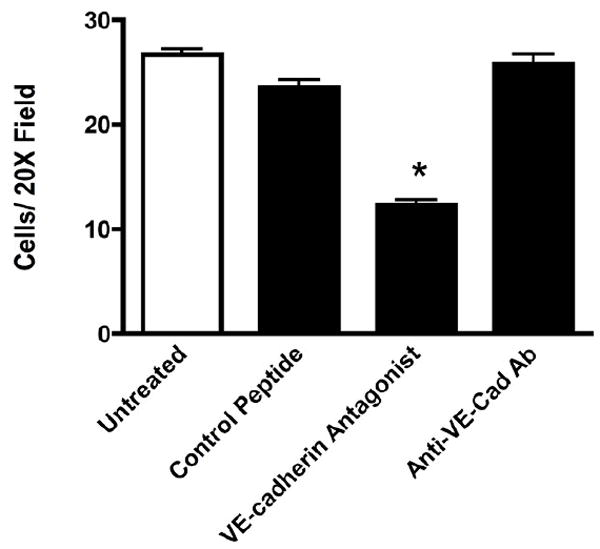
In vitro migration of bovine retinal endothelial cells was quantitated using fibronectin-coated cell culture inserts. As compared to untreated and control peptide treated cells, cells treated with the antagonist showed significantly less migration after 18 hours. The presence of a neutralizing VE-cadherin antibody did not alter the cells capacity to migrate. Data are the mean +/- SEM for N= 3 wells. * Significantly less than untreated (P= 0.0001), control peptide (P= 0.0001) and VE-cadherin antibody treated cells (P = 0.001).
During contact-inhibition, the recognition and engagement of CAR sequences in VE-cadherin ectodomains on adjacent cells can lead to inhibition of cellular proliferation and responsiveness to growth factors (11, 21, 22). We thus investigated whether the proliferation of isolated bovine retinal endothelial cells might be disrupted by the VE-cadherin antogonist. Cells were grown in serum-free conditions and stimulated to proliferate by addition of VEGF in the presence or absence of the VE-cadherin antagonist. After 48 hours of incubation, cellular proliferation was quantitated using the Vybrant R cell proliferation assay kit. In the presence of the VE-cadherin antagonist cells demonstrated a significantly reduced rate of proliferation as compared to untreated cells or cells incubated with the control peptide (Figure 5).
Figure 5. VE-cadherin antagonist inhibits cell proliferation in vitro.
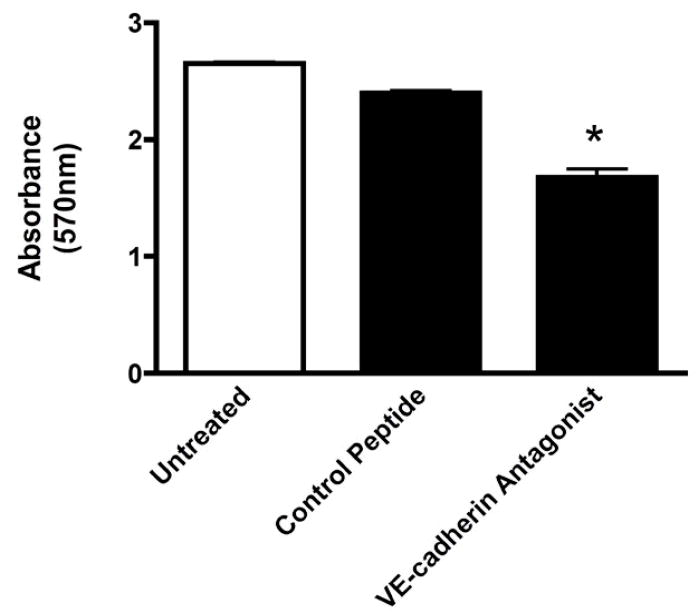
Bovine retinal endothelial cells were stimulated with VEGF and analyzed using the Vybrant R cell proliferation assay. Cells treated with the VE-cadherin antagonist demonstrated a significantly reduced rate of proliferation as compared to untreated cells or cells treated with the control peptide. Values are the mean +/- SEM of N = 3 experiments. * Significantly less than untreated and control peptide-treated cells (P=0.0002)
β-catenin Redistribution Following Antagonist Treatment
To elucidate the molecular effects of a contact inhibition-like signal by the VE-cadherin antagonist, cells were stained for β-catenin. The β-catenin protein is localized to the intracellular aspect of the cell membrane when VE-cadherin proteins are engaged in a confluent monolayer of cells (Figure 6). Immunocytochemical analysis of β-catenin in a sparse culture of cells treated with the control peptide showed a lack of β-catenin in completely isolated cells, and the presence of β-catenin in vesicle-like structures in the cytoplasm and at the membrane where cells are in contact with one another. In cells treated with the VE-cadherin antagonist, β-catenin was dramatically upregulated and appeared to be localized to the peripheral edges and processes of the cell.
Figure 6. The VE-cadherin antagonist induces β-catenin reorganization in cultured endothelial cells.
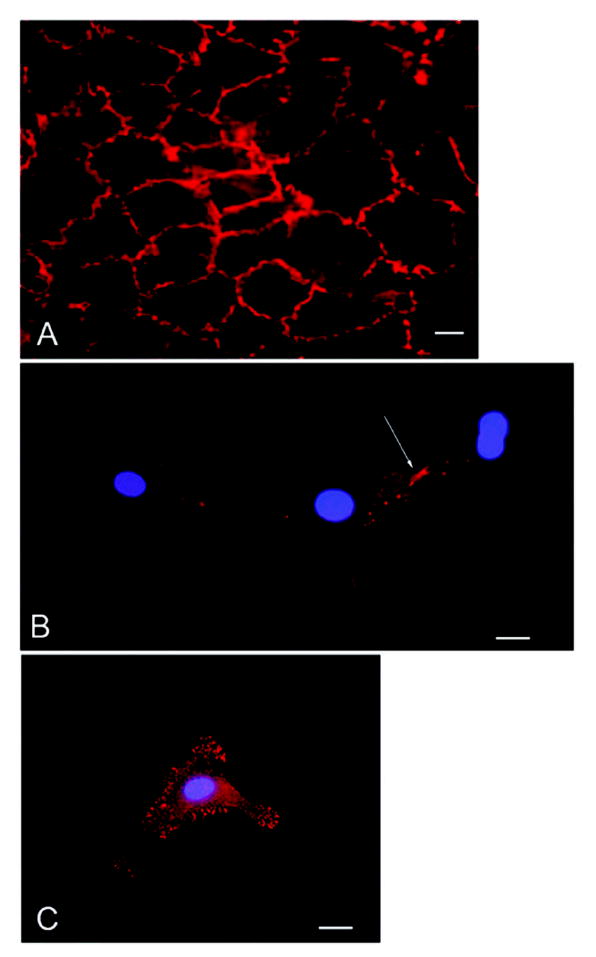
Confluent or subconfluent bovine retinal endothelial cells were stained for β-catenin. β-catenin is present in a continuous distribution along intercellular junctions in a confluent monolayer of cells (A). Isolated cells show no β-catenin staining in the cytoplasm or at the cell membrane except where cell-cell contact is occurring (B arrow). Isolated cells incubated with the VE-cadherin antagonist demonstrate β-catenin staining both in the cytoplasm as well as at discrete locations along the cell membrane (C). Bar=20um.
The VE-cadherin Antagonist Does Not Disrupt Existing Endothelial Cell Monolayers
To test the specificity and selectivity of the antagonist peptide, confluent monolayers of endothelial cells were treated with the antagonist peptide and the staining pattern of VE-cadherin and monolayer permeability was studied. Treatment of confluent monolayers with the antagonist had no effect on the structural integrity of the monolayer as demonstrated by continuous VE-cadherin staining along cell borders (Figure 7). The functional integrity of the monolayer was also unaffected by the antagonist as the permeability of FITC-dextran across the monolayer was comparable between antagonist peptide-treated, control peptide-treated and untreated cells (Figure 7).
Figure 7. The VE-cadherin antagonist does not disrupt existing endothelial cell junctions.
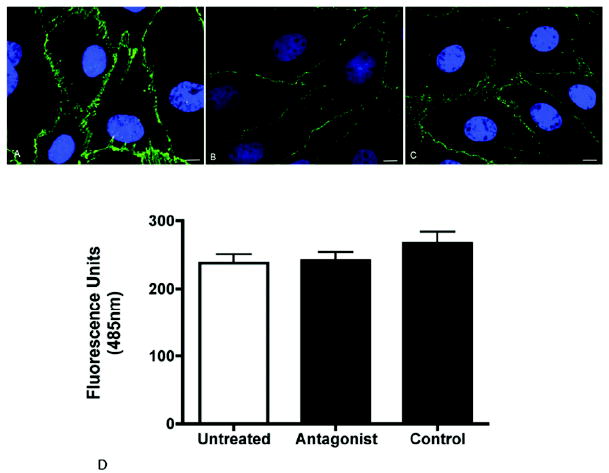
Representative images of confluent bovine retinal endothelial cell monolayers stained for VE-cadherin in untreated (A), control peptide-treated (B) or antagonist-treated cells (C). The antagonist had no effect on the structural integrity of the monolayer as demonstrated by continuous VE-cadherin staining along the cell borders. The function of the cell junctions was assessed by measuring the permeability of the monolayer using FITC-dextran (D). The permeability of the antagonist treated cells was not significantly different from untreated cells or cells treated with control peptide. Values are the mean +/- SEM from N = 4 wells for each treatment. Bar = 10μm
COMMENT
This study addresses the role of the endothelial cell adhesion molecule, VE-cadherin, in the regulation of new vessel formation in a model of oxygen-induced retinal angiogenesis. Animals treated with a peptide antagonist which binds to the extracellular domain of VE-cadherin exhibited a 54% reduction in the neovascular response as compared to animals treated with a control peptide. This degree of inhibition was within the range of inhibition seen with other agents including inhibitors of proteinases (23-25), COX-2 (26), integrins (27) and VEGF (28).
The action of the antagonist was investigated using isolated bovine retinal endothelial cells. The antagonist appeared to be directed toward actively growing endothelial cells as it failed to disrupt an existing monolayer of endothelial cells with homotypically engaged adhesion proteins. Numerous stages of the angiogenic process could be disrupted by the antagonist including cell migration, proliferation and tubule formation. These endothelial cell behaviors were disrupted concomitant with a redistribution of the accessory adhesion protein β-catenin suggesting that the antagonist may act as a decoy receptor. The antagonist likely binds to unengaged VE-cadherin molecules on the cell surface simulating cell-cell interactions. A VE-cadherin antibody was unable to inhibit endothelial cell behavior as the antibody binds to regions of VE-cadherin other than the cell recognition sequence, thus failing to initiate a decoy response.
The structural and functional integrity of the micro-vasculature is partially dependent upon the formation of adhesive junctions between adjacent endothelial cells of the capillary. A major component of the adherens junction in endothelial cells is the VE-cadherin protein. VE-cadherin, like other members of the cadherin family, is a transmembrane protein containing an extracellular domain composed of 5 subunits and a cytoplasmic domain which associates with the actin cytoskeleton. Homophilic interactions of VE-cadherins on adjacent cells, and interactions of VE-cadherin with the cytoskeleton, are facilitated and stabilized by additional proteins including β-catenin, α-catenin, plakoglobin and p120 (21,22). In addition to providing for cell-cell adhesion, VE-cadherin has also been shown to regulate numerous endothelial cell behaviors including intercellular permeability, cell proliferation and cell survival (11, 14,19, 29).
An important role for VE-cadherin in angiogenesis has been previously reported (30-32). Deletion of the VE-cadherin gene is lethal to mice during development due to severe vascular defects (33). Endothelial cells in these animals could adhere to one another but failed to undergo correct vascular morphogenesis (migration, branching and tubule formation). Studies also demonstrate that pathological angiogenesis can be regulated by VE-cadherin function (17, 18). In these studies specific antibodies to VE-cadherin were shown to block tumor growth coincident with a decrease in the extent of tumor vascularization. These effects were seen independent of changes in permeability or changes in cellular adhesion suggesting that the effective antibodies were targeting the developing vasculature and had no effect on the existing normal vessels. Similar findings were reported in this study for the activity of the VE-cadherin antagonist. The antagonist was directed to the cell-adhesion recognition (CAR) sequence that facilitates homotypic recognition, dimerization and cell-cell adhesion. These studies suggest that targeting an epitope on the VE-cadherin protein which is accessible only in loosely organized cells (i.e. endothelial cells during angiogenesis) may be an effective way to target only the angiogenic cells while sparing the normal vasculature. This is important for the antagonist to be considered for therapy as any unintended side effects on the normal existing vasculature would be absent.
The peptide used in our studies was directed against the VE-cadherin CAR sequence, which antagonizes it ability to form stable adherens junctions with another endothelial cell. We speculate that this antagonist activity delivers a decoy adhesion signal to the cell, thereby eliciting a range of cell behaviors aimed at quiescence rather than angiogenesis. This is supported by the finding of decreased proliferation and migration in response to the peptide as well as the redistribution of β-catenin within the cell.
The contact inhibition-like signal induced by treatment with the antagonist might also decrease the responsiveness of endothelial cells to angiogenic cytokines and growth factors such as VEGF. Contact inhibition is mediated largely by the establishment of stable cadherin junctions between adjacent endothelial cells. Caveda et al demonstrated that cells plated onto a substrate containing the VE-cadherin ectodomain displayed decreased cell division and proliferation (14). Several signaling pathways have been identified with VE-cadherin-dependent cell cycle arrest and inhibition of cell proliferation. In actively growing cells, β- catenin is found in the nucleus where it activates the transcription of many proteins important in the regulation of cell proliferation (34,35). The cytoplasmic and membrane localization of β-catenin in response to antagonist stimulation seen in the present study might prevent the transcription of several proteins required to orchestrate cell cycle progression. In angiogenic cells, VE-cadherin is associated with the VEGF receptor and Src leading to phosphorylation of VE-cadherin’s cytoplasmic tail and the induction of cell proliferation facilitated by ERK/MAPK signaling. In addition, the VEGF receptor/ VE-cadherin association induces cell survival through a PI3-kinase/Akt signaling pathway (11). The delivery of a decoy signal might inhibit these molecular events and thus decrease cellular proliferation in endothelial cells, even in the presence of an angiogenic stimulus like VEGF.
Data from this study supports the hypothesis that VE-cadherin plays an important role in the development of pathological ocular angiogenesis and identifies VE-cadherin as a potentially useful therapeutic target for these devastating diseases.
Acknowledgments
The authors would like to acknowledge the funding provided by the National Institutes of Health Grant, RO1 EY12604. The authors would also like to acknowledge Adherex Technologies Inc. for providing the VE-cadherin antagonist and control peptide. The authors would also like to acknowledge the expert technical assistance of Ms. Gina Menicucci and Ms. Dana Wilging.
Footnotes
Financial Disclosures: None.
References
- 1.Das A, McGuire PG. Retinal and Choroidal Angiogenesis. Pathophysiology and strategies for inhibition. Prog Retin Eye Res. 2003;22:721–748. doi: 10.1016/j.preteyeres.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 2.Carmeliet P, Jain RK. Angiogensis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 3.Carmeliet P. Mechanisms of angiogensis and arteriogenesis. Nature Medicine. 2000;6:389–395. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 4.Ritter MR, Banin E, Moreno SK, Aguilar E, Dorrell MI, Friedlander M. Myeloid progenitors differentiate into microglia and promote vascular repair in a model of ischemic retinopathy. J Clin Invest. 2006;116:3266–3276. doi: 10.1172/JCI29683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grant MB, May WS, Caballero S, Brown GAJ, et al. Adult hematopoietic stem cells provide functional hemangioblast activity during retinal neovascularization. Nature Medicine. 2002;8:607–612. doi: 10.1038/nm0602-607. [DOI] [PubMed] [Google Scholar]
- 6.Yang S, Graham J, Kahn JW, Schwartz EA, Gerritsen ME. Functional Roles for PECAM-1 (CD31) and VE-Cadherin (CD144) in Tube Assembly and Lumen Formation in Three-Dimensional Collagen Gels. Am J Pathol. 1999;155:887–895. doi: 10.1016/S0002-9440(10)65188-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dejana E. Endothelial adherens junctions: implications in the control of vascular permeability and angiogenesis. J Clin Invest. 1996;98:1949–1953. doi: 10.1172/JCI118997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dejana E, Bazzoni G, Lampugnani MG. Vascular endothelial (VE)-cadherin: only an intercellular glue? Exp Cell Res. 1999;252:13–19. doi: 10.1006/excr.1999.4601. [DOI] [PubMed] [Google Scholar]
- 9.Bazzoni G, Dejana E, Lampugnani MG. Endothelial adhesion molecules in the development of vascular tree: the garden of forking paths. Curr Opin Cell Biol. 1999;11:573–581. doi: 10.1016/s0955-0674(99)00023-x. [DOI] [PubMed] [Google Scholar]
- 10.Wallez Y, Vilgrain I, Huber P. Angiogenesis. The VE-cadherin switch. Trends Cardiovasc Med. 2006;16:55–59. doi: 10.1016/j.tcm.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 11.Carmeliet P, Lampugnani MG, Moons L, et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 1999;98:147–157. doi: 10.1016/s0092-8674(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 12.Corada M, Mariotti M, Thurston G, et al. Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc Natl Acad Sci U S A. 1999;96:9815–9820. doi: 10.1073/pnas.96.17.9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conacci-Sorrell M, Zhurinsky J, Ben-Ze’ev A. The cadherin-catenin adhesion system in signaling and cancer. J Clin Invest. 2002;109:987–991. doi: 10.1172/JCI15429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caveda L, Martin-Padura I, Navarro P, et al. Inhibition of cultured cell growth by vascular endothelial cadherin (cadherin-5/VE-cadherin) J Clin Invest. 1996;98:886–893. doi: 10.1172/JCI118870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Navarro P, Caveda L, Breviario F, Mandoteanu I, Lampugnani MG, Dejana E. Catenin-dependent and -independent functions of vascular endothelial cadherin. J Biol Chem. 1995;270:30965–30972. doi: 10.1074/jbc.270.52.30965. [DOI] [PubMed] [Google Scholar]
- 16.Aberle H, Schwartz H, Kemler R. Cadherin-catenin complex: protein interactions and their implications for cadherin function. J Cell Biochem. 1996;61:514–523. doi: 10.1002/(SICI)1097-4644(19960616)61:4%3C514::AID-JCB4%3E3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 17.Corada M, Zanetta L, Orsenigo F, Breviario F, Lampugnani MG, Bernasconi S, Liao F, Hicklin DJ, Bohlen P, Dejana E. A monoclonal antibody to vascular endothelial-cadherin inhibits tumor angiogenesis without side effects on endothelial permeability. Blood. 2002;100:905–911. doi: 10.1182/blood.v100.3.905. [DOI] [PubMed] [Google Scholar]
- 18.Liao F, Doody JF, Overholser J, Finnerty B, Bassi R, Wu Y, Dejana E, Kussie P, Bohlen P, Hicklin DJ. Selective targeting of angiogenic tumor vasculature by vascular endothelial-cadherin antibody inhibits tumor growth without affecting vascular permeability. Cancer Res. 2002;62:2567–2575. [PubMed] [Google Scholar]
- 19.Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: Molecular organization and role in vascular homeostasis. Physiol Rev. 2004;84:869–901. doi: 10.1152/physrev.00035.2003. [DOI] [PubMed] [Google Scholar]
- 20.Smith LEH, Wesolowski E, McLellan A, et al. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci. 1994;35:101–111. [PubMed] [Google Scholar]
- 21.Blaschuk OW, Rowlands TM. Plasma membrane components of adherens junctions. Mol Membr Biol. 2002;19:75–80. doi: 10.1080/09687680210132467. [DOI] [PubMed] [Google Scholar]
- 22.Blaschuk OW, Sullivan R, David S, Pouliot Y. Identification of a cadherin cell adhesion recognition sequence. Dev Biol. 1990;139:227–229. doi: 10.1016/0012-1606(90)90290-y. [DOI] [PubMed] [Google Scholar]
- 23.Das A, McLamore A, Song WM, McGuire PG. Retinal neovascularization is suppressed with a matrix metalloproteinase inhibitor. Archives Of Ophthalmology. 1999;117:498–503. doi: 10.1001/archopht.117.4.498. [DOI] [PubMed] [Google Scholar]
- 24.McGuire PG, Jones TR, Talarico N, Warren E, Das A. The Urokinase/Urokinase receptor system in retinal neovascularization: Inhibition by angstrom 6 suggests a new therapeutic target. Investigative Ophthalmology & Visual Science. 2003;44:2736–2742. doi: 10.1167/iovs.02-1160. [DOI] [PubMed] [Google Scholar]
- 25.Wilkinson-Berka JL, Alousis NS, Kelly DJ, Gilbert RE. COX-2 inhibition and retinal angiogenesis in a mouse model of retinopathy of prematurity. Investigative Ophthalmology & Visual Science. 2003;44:974–979. doi: 10.1167/iovs.02-0392. [DOI] [PubMed] [Google Scholar]
- 26.Santulli RJ, Kinney WA, Ghosh S, et al. Studies with an orally bioavailable alpha(v) integrin antagonist in animal models of ocular vasculopathy: Retinal neovascularization in mice and retinal vascular permeability in diabetic rats. Journal Of Pharmacology And Experimental Therapeutics. 2008;324:894–901. doi: 10.1124/jpet.107.131656. [DOI] [PubMed] [Google Scholar]
- 27.Das A, Fanslow W, Cerretti D, Warren E, Talarico N, McGuire PG. Angiopoietin/Tek Interactions Regulate MMP-9 Expression and Retinal Neovascularization. Laboratory Investigation. 2003;83(11):1637. doi: 10.1097/01.lab.0000097189.79233.d8. [DOI] [PubMed] [Google Scholar]
- 28.Aiello LP, Pierce EA, Foley E, et al. Suppression of Retinal Neovascularization In-Vivo by Inhibition of Vascular Endothelial Growth-Factor (VEGF) Using Soluble VEGF-Receptor Chimeric Proteins. Proc Natl Acad Sci. 1995;92:10457–10461. doi: 10.1073/pnas.92.23.10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vincent PA, Xiao K, Buckley KM, Kowalczyk AP. VE-Cadherin: adhesion at arm’s length. Am J Physiol Cell Physiol. 2004;286:C987–C997. doi: 10.1152/ajpcell.00522.2003. [DOI] [PubMed] [Google Scholar]
- 30.Wheelock MJ, Johnson KR. Cadherins as modulators of cellular phenotype. Annu Rev Cell Dev Biol. 2003;19:207–235. doi: 10.1146/annurev.cellbio.19.011102.111135. [DOI] [PubMed] [Google Scholar]
- 31.Wallez Y, Vilgrain I, Huber P. Angiogenesis: The VE-cadherin switch. Trends Cardiovasc Med. 2006;16:55–59. doi: 10.1016/j.tcm.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 32.Dejana E. Endothelial adherens junctions: Implications in the control of vascular permeability and angiogenesis. J Clin Invest. 1996;98:1949–1953. doi: 10.1172/JCI118997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gory-Faure S, Prandini MH, Pointu H, Roullot V, Pignot-Paintrand I, Vernet M, Huber P. Role of vascular endothelial-cadherin in vascular morphogenesis. Development. 1999;126:2093–2102. doi: 10.1242/dev.126.10.2093. [DOI] [PubMed] [Google Scholar]
- 34.Conacci-Sorrell M, Zhurinsky J, Ben-Ze’ev A. The cadherin-catenin adhesion system in signaling and cancer. J clin Invest. 2002;109:987–991. doi: 10.1172/JCI15429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lambeng N, Wallez Y, Rampon C, et al. Vascular endothelial-cadherin tyrosine phosphorylation in angiogenic and quiescent adult tissues. Circ Res. 2005;96:384–391. doi: 10.1161/01.RES.0000156652.99586.9f. [DOI] [PMC free article] [PubMed] [Google Scholar]