

Abstract
The elucidation of the structure of glycosaminoglycan has proven to be challenging for analytical chemists. Molecules of glycosaminoglycan have a high negative charge and are polydisperse and microheterogeneous, thus requiring the application of multiple analytical techniques and methods. Heparin and heparan sulfate are the most structurally complex of the glycosaminoglycans and are widely distributed in nature. They play critical roles in physiological and pathophysiological processes through their interaction with heparin-binding proteins. Moreover, heparin and low-molecular weight heparin are currently used as pharmaceutical drugs to control blood coagulation. In 2008, the health crisis resulting from the contamination of pharmaceutical heparin led to considerable attention regarding their analysis and structural characterization. Modern analytical techniques, including high-performance liquid chromatography, capillary electrophoresis, mass spectrometry, and nuclear magnetic resonance spectroscopy, played critical roles in this effort. A successful combination of separation and spectral techniques will clearly provide a critical advantage in the future analysis of heparin and heparan sulfate. This review focuses on recent efforts to develop hyphenated techniques for the analysis of heparin and heparan sulfate.
Keywords: Heparin/heparan sulfate, High-performance liquid chromatography, Capillary electrophoresis, Tandem mass spectrometry, Nuclear magnetic resonance spectroscopy, Hyphenated techniques
Introduction
Heparin and heparan sulfate (HS) are linear, highly charged, anionic polysaccharides that belong to the glycosaminoglycan (GAG) family. They are widely present on cell surfaces, inside cells and in the extracellular matrix. Heparin and HS play many important roles in physiological and pathophysiological processes. Recent studies have established the specificity of HS interactions with chemokines, cytokines and growth factor receptors [1-3]. These interactions are critical in cell adhesion, proliferation, motility and differentiation, viral and bacterial infection, cancer, and inflammation [4-8]. Therefore, considerable attention has been focused on characterizing the fine structure of heparin/HS and in elucidating their interactions with a wide array of proteins, ligands, receptors and pathogens. Heparin and low-molecular weight heparin (LMWH) inhibit blood coagulation by binding and activating antithrombin III, a coagulation protease inhibitor. Heparin is one of the oldest anticoagulant drugs and is currently in widespread clinical use. In 2008, the health crisis resulting from the contamination of lots of pharmaceutical heparin with chemically modified chondroitin sulfate resulted in the introduction of sophisticated analytical controls to secure the quality and safety of this critical pharmaceutical agent [9].
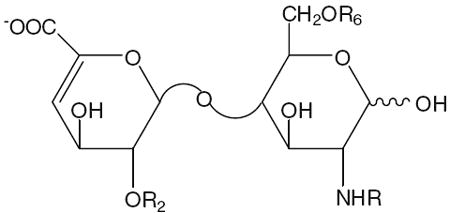
The polysaccharide chains of heparin and HS have closely related structures and consist of a repeating disaccharide structure of 1,4-linked hexuronic acid and d-glucosamine residues with molecular weights ranging from 5 to 70 kDa. The hexuronic acid of heparin/HS can either be d-glucuronic acid (GlcA) or l-iduronic acid (IdoA), both of which can be 2-O-sulfonated. The glucosamine residue in heparin/HS can be N-acetylated (GlcNAc), sulfonated (GlcNS), or unsubstituted (GlcN), and can also be 3- and/or 6-O-sulfonated. HS has a more highly variable structure than heparin, with fewer sulfo groups, and is rich in GlcA and GlcNAc residues [1, 10]. Eight commercially available and enzymatically prepared disaccharide standards are described in Table 1. The structural complexity of heparin/HS is attributed to a mixed combination of different disaccharide units with variable patterns of sulfation and C5 hexuronic acid epimers. This microheterogeneity (sequence variability) depends on species, individual organism, organ, tissue, cell type, environmental conditions and developmental stage. During biosynthesis, the nascent heparin/HS chains on the core protein, which is called heparosan and consists of a simple GlcA–GlcNAc repeat, are acted upon by a series of enzymes, including epimerase, N-deacetylases and N- and O-sulfotransferases, within the Golgi apparatus. The glucosaminyl N-deacetylase/N-sulfotransferase (NDST) converts certain GlcNAc residues into GlcNS. After the N-sulfonation, C5-epimerase converts certain GlcA residues to IdoA. The polysaccharide is then further sequentially modified by 2-O-sulfotransferase (2-OST), 6-O-sulfotransferase (6-OST), and 3-O-sulfotransferase (3-OST), incorporating sulfo groups at the 2-positions of certain IdoA and GlcA and the 6- and 3-positions of certain GlcN residues [3, 11]. The biosynthesis of heparin/HS is not template driven, resulting in variable chain lengths, compositions and sequences.
Table 1.
The structures of the eight disaccharide standards prepared from heparan sulfate/heparin using heparinases
 | ||||||
|---|---|---|---|---|---|---|
| Reference number | Disaccharide | Formulas | R2 | R6 | R | Theoretical molecular mass |
| 1 | 0S | ΔUA-GlcNAc | H | H | Ac | 379.3 |
| 2 | NS | ΔUA-GlcNS | H | H | SO3- | 417.3 |
| 3 | 6S | ΔUA-GlcNAc(6S) | H | SO3- | Ac | 459.4 |
| 4 | 2S | ΔUA(2S)-GlcNAc | SO3- | H | Ac | 459.4 |
| 5 | NS6S | ΔUA-GlcNS(6S) | H | SO3- | SO3- | 497.4 |
| 6 | 2SNS | ΔUA(2S)-GlcNS | SO3- | H | SO3- | 497.4 |
| 7 | 2S6S | ΔUA(2S)-GlcNAc(6S) | SO3- | SO3- | Ac | 539.4 |
| 8 | 2SNS6S | ΔUA(2S)-GlcNS(6S) | SO3- | SO3- | SO3- | 577.5 |
A detailed knowledge of heparin/HS structures is required for an in-depth understanding of their biological roles. Information on heparin/HS structure is also critical for securing the quality and safety of heparin-based drugs. Heparin/HS is extremely difficult to analyze because of its high negative charge, polydispersity, and microheterogeneity. A common strategy for the detailed structural analysis of heparin/HS involves either complete or partial depolymerization by either enzymatic or chemical means to obtain constituent disaccharides for disaccharide analysis, or a range of oligosaccharide fragments for oligosaccharide mapping. One of the most common depolymerization approaches utilizes bacterial enzymes known as heparanases (heparin lyases) that catalyze the β-eliminative cleavage of heparin/HS, affording disaccharide and oligosaccharide products with 4,5-unsaturated uronic acid residues (ΔUA) at their nonreducing ends, which absorb in the UV at 232 nm [12, 13]. Structurally defined heparin/HS oligosaccharides are also important for understanding recognition systems involving specific protein–carbohydrate interactions. Modern separation techniques, including high-performance liquid chromatography (HPLC) [14-16], gel permeation chromatography (GPC) [17-19], polyacrylamide gel electrophoresis (PAGE) [20-22], and capillary electrophoresis (CE) [23-25] have been used to prepare heparin/HS disaccharides and oligosaccharides in order to help solve many complex structures. Nuclear magnetic resonance (NMR) spectroscopy is another important tool for the structural elucidation of heparin/HS, providing valuable information on monosaccharide composition, glycosidic linkage, uronic acid type, and sulfation patterns [21, 26, 27]. Mass spectrometry has become increasingly important for the analysis of heparin/HS oligosaccharides with the development of the soft ionization methods of electrospray ionization (ESI) and matrix-assisted laser desorption/ionization (MALDI). The high sensitivity, high accuracy, and fast sample processing of MS offer both rapid screening and detailed structural analysis [28-30].
The successful combination of separation and spectral techniques clearly provides a critical advantage in understanding heparin structure. Recent efforts to develop methodologies for heparin/HS analysis have coupled electrophoresis and chromatographic separation to fluorescence, MS and NMR analyses to enhance structural characterization. This review discusses developments in hyphenated techniques for the separation and structural characterization of heparin/HS.
High-performance liquid chromatography
Strong anion exchange high-performance liquid chromatography
Strong anion exchange (SAX)-HPLC is a traditional preparative and analytical method for the separation of heparin/HS (intact GAG), oligosaccharides, and disaccharide mixtures [19, 31]. Oligosaccharide and disaccharide mixtures prepared from heparin/HS isolated from small quantities of tissues or from cultured cells using heparinases are often difficult to detect and identify based on their UV absorbance. SAX-HPLC coupled with in-line fluorescence detection can be more useful for such heparin/HS micro-analysis after fluorescent labeling. The fluorophore 2-aminobenzamide (2-AB) can be conveniently linked at the reducing ends of heparin/HS-derived disaccharides and oligosaccharides through reductive amination. The resulting labeled compounds can be well separated and quantified with a low picomole level detection limit by SAX-HPLC with fluorescence detection [32, 33], and this approach has been used to determine the disaccharide composition of heparin/HS obtained from small biological samples [34, 35]. BODIPY (4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionic acid) hydrazide is a promising fluorophore that offers the prospect of improved labeling yields due to its more reactive hydrazide group and a high extinction coefficient (ε) of 71,000 M−1 cm−1 at 503 nm, closer to the wavelengths of widely available lasers (e.g., 488 nm). Since BODIPY hydrazide is uncharged or positively charged, excess tag can be easily removed from labeled disaccharides and oligosaccharides. Eight heparin/HS disaccharide standards (Table 1) derivatized with the BODIPY hydrazide through the formation of a Schiff base can easily be separated by SAX-HPLC on a ProPac PA1 column (Fig. 1). The retention times of the eight disaccharides were consistent, with standard deviations (σn−1, n=9) ranging from 0.7 to 2.0%. The unreacted BODIPY hydrazide tag passed through the column well before the disaccharide peaks [36, 37].
Fig. 1.
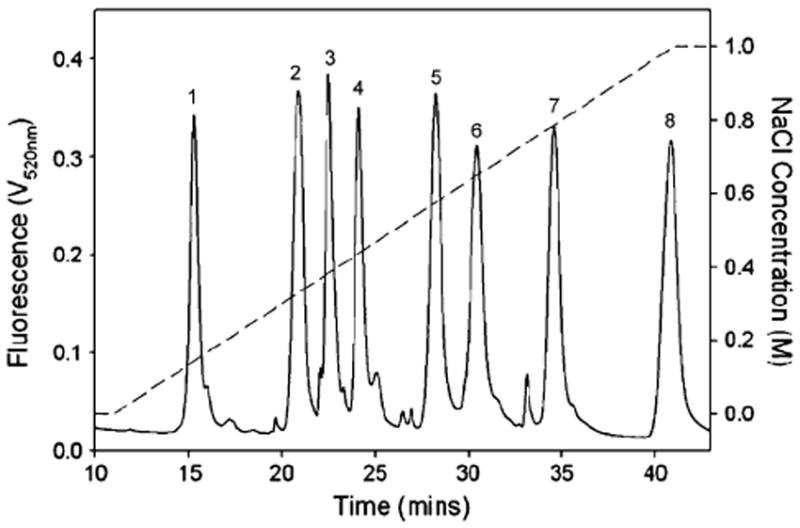
Separation of BODIPY-labeled disaccharide standards (solid line, see Table 1 for structures of compounds 1–8) on HPAEC using a linear gradient of NaCl (dotted line) over 30 min in the presence of isocratic sodium hydroxide (150 mM). A ProPac PA1 column was used with fluorescence detection (λem=488 nm/λex=520 nm). Adapted from [36] with permission
An alternative method for the detection of underivatized carbohydrates with low-pmol sensitivity relies on electrochemical detection by pulsed amperometric detection (PAD). High-performance anion exchange chromatography, (HPAEC)-PAD, has been applied widely in the monosaccharide composition analysis of N- and O-linked glycoproteins [38-40]. It has also been used with good sensitivity and selectivity for the acid hydrolysates of heparin. All of the monosaccharides of heparin can be separated in a single chromatographic step and the content of l-iduronic acid was determined in low-microgram samples [41]. This method has also been applied to the analysis of heparin immobilized on the surfaces of intraocular lenses [42] and to the determination of heparin/HS in plasma and serum [43]. Recently, a new approach was reported that combines nitrous acid chemical degradation of heparin/HS followed by HPAEC separation on a ProPac PA1 column (Fig. 2A) and UV–MALDI–time of flight (TOF)–MS direct, off-line, analysis. Heparin/HS can be selectively cleaved at GlcNS residues by treating with nitrous acid at pH 1.5 under reducing conditions, leading to 2,5-anhydromannitol terminal units (Fig. 2B). The UV-MALDI-TOF spectra in the negative-ion mode of one peak are shown in Fig. 2C–E. The molecular ion for this hexasulfated hexasaccharide showed an m/z of 1536.9 (disodium salt) and an m/z of 1513.8 (monosodium salt). Mass calculations showed no GlcNAc; two GlcNS residues were present instead. Fragmentation leads to a peak at m/z 1351.2 (C5 + Na-H2O), indicating that the reducing terminal anhydromannitol residue was not sulfated (Fig. 2B and C). A C2 fragment observed at m/z 593.0 and a 1,5A2 fragment at m/z 548.0 suggested the presence of two additional sulfo groups at the nonreducing end of the disaccharide (Fig. 2D). A 3,5X2–H2O fragment at m/z 568.1 confirmed the presence of GlcNS6S, and the remaining sulfo group could be located at either the 2-position of IdoA or (less likely) at the 3-position of GlcNS. A Y3 fragment observed at m/z 837.8 corresponds to an anhydromannitol containing a trisaccharide with three sulfo groups (Fig. 2E). In a one-day analysis, six oligosaccharides, eluting as a function of total sulfate content, were collected and characterized by MALDI-MS [44].
Fig. 2.
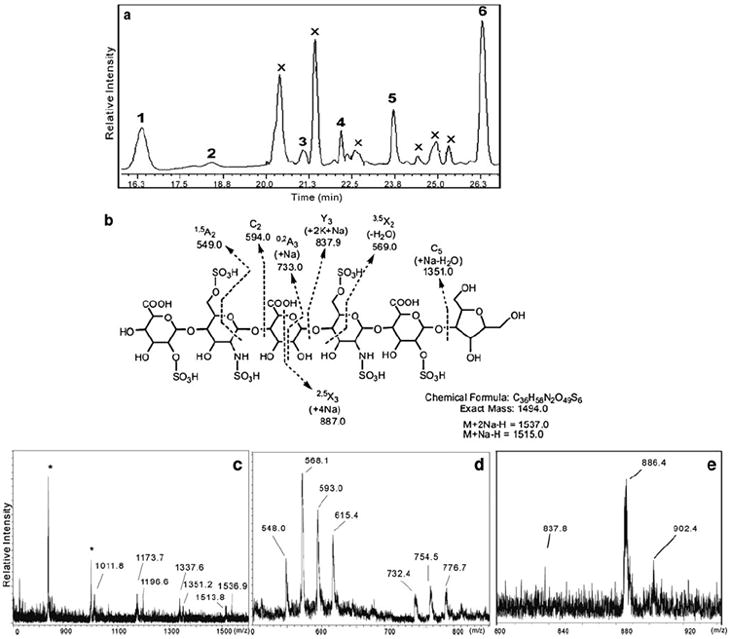
a–e Analysis of heparin fragments prepared through nitrous acid degradation. a HPAEC-PAD separation with peaks numbered from 1 to 6 and with X corresponding to unanalyzed peaks; b The structure and fragmentation assignments for peak 5. c–e UV-MALDI-TOF spectra obtained in the negative-ion mode of peak 5 using DHB (c) or norharmane (d, e) as matrix. Peaks labeled with an asterisk correspond to adducts of matrix molecules. Adapted from [44] with permission
Reversed-phase high-performance liquid chromatography
Reversed-phase high-performance liquid chromatography (RP-HPLC) can provide improved resolution using readily available, long-lifetime C18 columns. While carbohydrates are not retained on reversed-phase stationary phases, labeling with a hydrophobic fluorophore improves both chromatographic properties and sensitivity. Eight 2-aminoacridone (AMAC)-labeled heparin/HS disaccharides were fully resolved by C18 RP-HPLC (Fig. 3A). Excess hydrophobic 2-AMAC fluorophore is strongly retained on the RP column and thus does not require removal from the samples prior to analysis. AMAC labeling and RP-HPLC was then used to analyze 10 ng of cell-derived or tissue-derived heparin/HS. Disaccharide compositional analysis of heparin/HS can be achieved using a partially purified GAG mixture prepared in a 10-cm dish of ~50% confluent MDCK cells (Fig. 3B). Rat liver HS has been similarly analyzed (Fig. 3C) [45]. Disaccharide standards from heparin/HS have also been labeled with BODIPY fluorophore and separated using the RP-HPLC technique [36]. Acid hydrolysis of GAG to GlcN and galactosamine followed by reductive coupling to o-phthaldialdehyde and 3-mercaptopropionic acid affords fluorescent isoindole derivatives that can be separated and quantified by reverse-phase HPLC with a C12 column and detected with an excellent sensitivity of 0.04 pmol (linearity was from 2.5 to 1280 pmol) [46]. A 1-phenyl-3-methyl pyrazolone (PMP) derivatization technique has been developed for the quantification of GAG metabolites from urine or plasma and to monitor the dose response of enzyme replacement therapy in feline mucopolysaccharidosis using on-line RP-HPLC/MS [47-49].
Fig. 3.
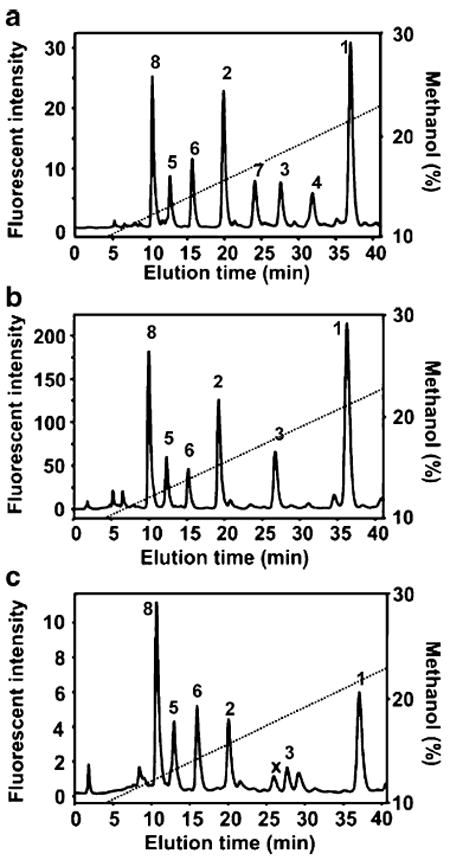
a–c Analysis of AMAC-labeled heparin/HS disaccharides. a RP-HPLC separation of a mixture of eight AMAC-labeled heparin/HS disaccharide standards. b Heparin/HS disaccharide components of GAG isolated from cultured MDCK cells. c Heparin/HS disaccharides of GAG isolated from rat livers. AMAC-labeled disaccharides separated by C18 RP-HPLC were detected using fluorescence. The numbers over the disaccharide peaks correspond to the AMAC-labeled disaccharides shown in Table 1. Adapted from [45] with permission
Reversed-phase ion-pair high-performance liquid chromatography
Reversed-phase ion-pair (RPIP)-HPLC is a promising and increasingly popular method for the analysis of heparin/HS disaccharides and oligosaccharides. Lipophilic ion-pairing reagents acting as mobile phase modifiers aid in the retention and resolution of charged species on hydrophobic stationary phases [50, 51]. RPIP-HPLC with in-line fluorescence detection or laser-induced fluorescence (LIF) detection has been used to determine the composition of fluorescently labeled disaccharides derived from heparin and HS [52-62]. The RPIP-HPLC technique is compatible with both evaporative light scattering detection (ELSD) [63] and on-line MS detection [64-67].
Fluorescence detection greatly improves sensitivity in carbohydrate analysis. A method of heparin/HS unsaturated disaccharide analysis has been developed that relies on RPIP-HPLC with postcolumn fluorescent labeling using 2-cyanoacetamide, and is monitored by an in-line high-sensitivity fluorescence detector. GAGs obtained from mosquito organs were depolymerized to disaccharides using heparinase and analyzed by this method [56]. The method was applied to the disaccharide composition analysis of GAG samples from animal and human tissues [59, 60, 62]. Toyoda et al. developed a rapid and sensitive postcolumn fluorometric method for the analysis of heparin/HS disaccharide composition using RPIP-HPLC on a 2 μm porous silica gel column that allowed disaccharide separation within 15 min (linearity was between 1 ng and 1 μg), and applied it to determine heparin/HS in human urine and very small tissue samples [54, 55, 57, 58]. RPIP-HPLC has also been employed to analyze LMWHs using on-line ELSD [63]. In his work, several parameters were investigated, including the concentration of organic modifier, different ion-pairing reagents, the concentration of ion-pairing reagent, and the pH of the mobile phase. This methodology clearly differentiated three LMWHs, with each giving chromatograms with sharp peaks at consistent retention times, making this method potentially useful for pharmaceutical analysis and stability determination of LMWH.
RPIP-HPLC provides excellent chromatographic resolution, but generally relies on high concentrations of nonvolatile quaternary ammonium salts, making it incompatible for use with ESI-MS [43]. Volatile ion-pairing reagents, including primary, secondary and tertiary amines [68], can allow efficient postcolumn removal of ion-pairing reagents with an in-line membrane [69], with the addition of a postcolumn sheath liquid, and by splitting the eluent flow [70]. These approaches have allowed the development of the on-line separation and structural identification of heparin/HS-derived oligosaccharides. Kuberan et al. systematically studied the ionization efficiencies of different volatile amine ion-pairing reagents, such as triethylamine, dibutylamine, tributylamine, tripentylamine, tetrapropylamine and tetrabutylamine. They demonstrated that RPIP-HPLC-ESI-TOF-MS, using C18 capillary columns with methanol gradients and an ion-pairing reagent of 5 mM dibutylammonium acetate, separated heparosan oligosaccharides (ΔUA[GlcNAc-GlcA]nGlcNAc, n=2−19) [66]. Thanawiroon et al. developed RPIP-HPLC/ESI-MS for the analysis of highly sulfated heparin oligosaccharides (ΔUA2S[GlcNS6S-IdoA2S]nGlcNS6S, n=0−13) using 15 mM tributylamine/50 mM ammonium acetate as the ion-pairing agent. High-resolution mass spectrometry in combination with UV detection afforded the identification of a series of oligosaccharide compositions that contain either the reducing or the nonreducing end of the parent heparin chain. The structural identification of these oligosaccharides provided sequence information from a reading frame beginning at the nonreducing terminus of the heparin chain. This method has been improved and applied to heparin/HS disaccharide compositional analysis of GAGs from animal tissues [71] and to the compositional analysis of heparin/HS interacting with fibroblast growth factor receptor complexes [20]. Mass tagging in combination with RPIP-HPLC/MS on a C18 column with dibutylamine (DBA) as an ion-pairing agent has been used for the quantitative analysis of disaccharide composition and to make ratiometric comparisons between samples. The reducing ends of the heparinase-generated disaccharides were tagged with aniline-containing stable isotopes (12C6 and 13C6). The differentially isotope-tagged samples can be compared simultaneously by combination with and quantified by admixture with known amounts of standards. The different isotope tags have no effect on chromatography retention times but can be easily discriminated by a mass detector. Chemoenzymatic synthesis has been used to prepare isotopically enriched heparin/HS disaccharides from a uniformly 13C, 15N-labeled N-acetylheparosan [−GlcA(1−4)GlcNAc−] obtained by the fermentation of E. coli K5. Quantification of heparin/HS disaccharides was achieved by reversed-phase ion-pairing microflow high-performance liquid chromatography (RPIP-Mf-HPLC) with on-line ESI-MS using these isotopically enriched disaccharides as internal standards. Eight HP/HS disaccharides were separated by RPIP-Mf-HPLC and detected by extracted ion chromatography (EIC, Fig. 4A). The separation observed in EIC resolved the α- and β-anomeric forms present in the 6S, 2S, and 2S6S disaccharides. In addition to excellent separation, ESI-MS affords the mass of each disaccharide. A peak was observed for each disaccharide at m/z 378.1, 416.1, 458.1, 496.0, 538.0, and 575.9, respectively (Fig. 4B–I). The anomers 6S/2S and NS6S/NS2S have the same m/z values (458.1 and 496.0, respectively; Table 1). Thus, each HS/heparin disaccharide could be unambiguously identified by both retention time and mass using RPIP-Mf-HPLC-ESI-MS. Furthermore, no multiply charged ions were observed, even for highly charged disaccharides. This may be due to the relatively high concentration of the TrBA ion-pairing reagent, which helped to avoid multiple ionization. The ratio of intensities between each pair of enriched and nonenriched disaccharides showed a linear relationship with concentration. Using these calibration curves, the amount of each disaccharide (2 ng/disaccharide) could be quantified in four heparin sulfate samples analyzed by this method [65]. Thus, isotope mass tagging in combination with RPIP-Mf-HPLC-ESI-MS provides a more robust, reliable, and sensitive means of quantitatively evaluating heparin/HS disaccharide composition.
Fig. 4.

a–i LC-MS analysis of the disaccharides most commonly found in heparin/HS. a Extracted ion chromatography (EIC) of disaccharides. b–i Mass spectra of the 0S, NS, 6S, 2S, NS6S, NS2S, 2S6S, and TriS disaccharides, respectively. Taken from [65] with permission
Ultraperformance liquid chromatography
Ultraperformance liquid chromatography (UPLC) separations are performed at high pressures (up to 108 Pa) on columns packed with 1.7 μm particles. This new category of analytical separation science retains the practicality and principles of HPLC while yielding a major improvement in chromatographic performance. Compared to traditional HPLC analysis, UPLC takes advantage of technological strides made in resolution, peak capacity, sensitivity, efficiency, and speed of analysis [72, 73]. An RPIP-HPLC separation method using tributylamine (TrBA) as ion-pairing reagent has recently been adapted for RPIP-UPLC-ESI-TOF-MS and used for compositional profiling and quantification of heparin/HS [74]. Separations of heparin/HS disaccharides were performed on a 2.1×100 mm UPLC BEH C18 column packed with 1.7 μm particles at 40 °C using 5 mM TrBA and 50 mM aqueous ammonium acetate, pH 7, and acetonitrile gradient elution at 0.5 mL/min. This fast and highly sensitive method was used for the disaccharide compositional analysis of porcine and bovine heparin and bovine heparan sulfate. Highly sulfated heparin tetrasaccharides were also resolved by a similar method and detected by MS [75].
The structural characterization of heparin and HS is a challenging analytical problem due to their high negative charges and microheterogeneity. A rapid, robust, and simple method for heparin oligosaccharide analysis relies on RPIP-UPLC-ESI-QTOF-MS. This method utilizes an optimized buffer system containing a linear pentylamine (PTA) and a unique additive, 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP), to resolve heparin oligosaccharides and give an enhanced MS response (Fig. 5A). The accurate molecular weights of hexasaccharide to octadecasaccharide were assigned in the negative ion ESI spectra. Chromatographic conditions also enabled the baseline resolution of isomeric heparin hexasaccharides (Fig. 5B) and produced intact molecular ions with no SO3 loss in positive-ion ESI-MS. The chromatographic separation and the positive-ion ESI-MS spectra of the hexasaccharides are shown in Fig. 5C and D. These conditions also allow detection in the positive-ion mode and the identification of structural isomers by MS/MS. Potential applications of this new method include the analysis of LMWHs [76].
Fig. 5.
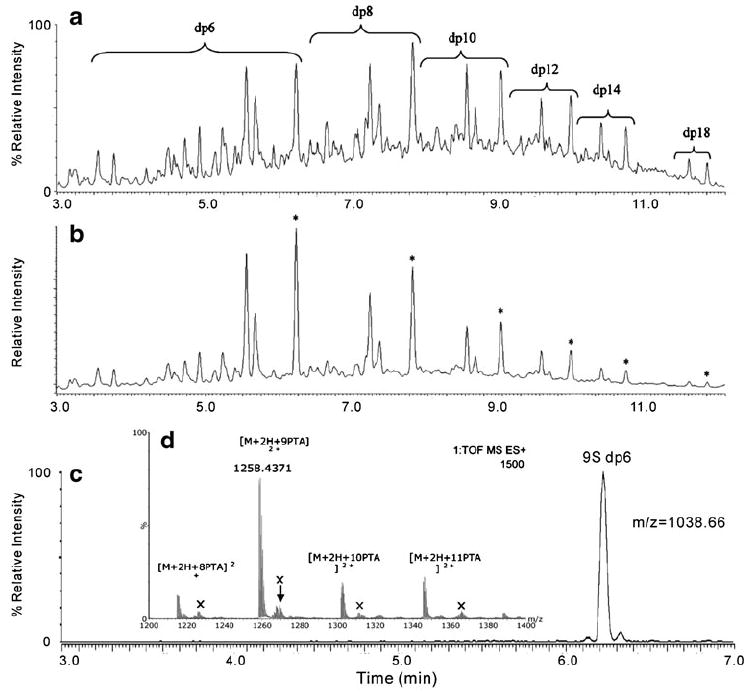
a–c Analysis of heparin oligosaccharides. a Total ion chromatogram. b UV trace at 232 nm observed for the IPRP-UPLC separation of a test mixture containing 100 μg/mL of each of the hexa- (dp6) to tetradecasaccharides (dp14) and octadecasaccharides (dp18). Peaks labeled with an asterisk in the UV trace correspond to the fully sulfated (three sulfo groups/disaccharide unit) heparin oligosaccharides. Separation was performed on an Acquity UPLC BEH C18 column (2.1 mm×150 mm, 1.7 μm) at a flow rate of 0.4 mL/min and a column temperature of 45 °C. Gradient conditions: 10-40% B in 10 min; eluent A=15 mM pentylamine (PTA), 50 mM HFIP, pH 8.8. Eluent B=75% acetonitrile, 15 mM PTA, 50 mM hexafluoro-2-propanol (HFIP). c Chromatographic separation of a nonasulfated heparin hexasaccharide. d Positive-ion ESI-MS of a nanosulfated hexasaccharide. Adapted from [76] with permission
The development of RPIP-HPLC and RPIP-UPLC separations of heparin/HS oligosaccharides with on-line MS detection is a promising approach to analysis with minimal sample preparation. Factors including type and concentration of the ion-pairing reagent, mobile-phase pH, organic modifier, ionic strength, and stationary phase all play roles in the overall efficiency of these separations. The role that competition plays between ion-pairing reagents with different steric bulks and hydrophobicities in the separation of isomeric heparin/HS disaccharides has also been investigated using UPLC. Ion-pair competition could lead to new methods for the separation of complex mixtures of larger heparin/HS oligosaccharides [77].
Hydrophilic interaction chromatography
Hydrophilic interaction chromatography (HILIC)-MS is a promising new on-line platform developed for the analysis of glycoprotein [78, 79] and GAG oligosaccharides [80-83]. HILIC separates oligosaccharides on the basis of their overall polarity [84]. Solvent modifiers are required and MS-compatible ammonium salts are often used for on-line HILIC-MS [85]. Unsaturated disaccharides produced by the heparinase digestion of HS from mouse brain and liver and tumor tissues can be determined using a short (35 mm×2 mm) Capcell Pak NH2 UG80 column [86]. A hydrophobic antithrombin III trapping method has been developed for screening and quantitatively analyzing a library of heparin/HS oligosaccharides using on-line Amide-80 HILIC-MS. Although this system does not separate isomeric compositions efficiently, it has the advantage of resolving heparin/HS-derived oligosaccharides based on properties that dictate the overall polarity of an oligosaccharide, such as size, sulfation and acetyl content [80]. This novel chip-based amide-HILIC system for negative ion LC-MS of heparin/HS removes much of the variability with permission associated with the spray interface, giving robust performance in the negative-ion mode, and the built-in trapping cartridge reduces background from other contaminants in the biological sample [82]. In more recent work, the addition of postcolumn makeup flow to the amide-HPLC-chip configuration has permitted even more robust and reproducible analyses of heparin/HS oligosaccharides [83].
Capillary electrophoresis
Capillary electrophoresis (CE) is one of the most powerful techniques for GAG analysis. CE provides many advantages in comparison to a variety of other analytical methods, including high separation efficiency, high sensitivity, short analysis time, straightforward operation, on-line detection, and flexibility of separation mode (i.e., either normal-polarity or reversed-polarity modes can be used). Furthermore, CE requires small amounts of sample and buffer, and it exhibits compatibility with a variety of detection methods, including MS, LIF, and most recently NMR spectroscopy.
The separation principle for resolving heparin/HS disaccharides and oligosaccharides by CE has already been reviewed [24, 87]. Reversed-polarity CE in phosphate pH 3 buffer was used to address the contamination of heparin with oversulfated chondroitin sulfate (OSCS) and dermatan sulfate (DS) impurities. CE with UV detection is a robust, fast and reproducible method for the detection of the toxic contaminant OSCS in heparin [88]. A simple CE method for the determination and separation of LMWHs and heparin utilizes a 70 cm×50 μm silica capillary run with 50 mM phosphate buffer at pH 3.5, performed at 30 kV [89]. Heparin and LMWH oligosaccharides prepared by controlled heparinase catalyzed depolymerization are well resolved using normal-polarity CE performed at 20 kV with 10 mM sodium borate and 50 mM sodium dodecyl sulfate at pH 8.8 [90]. Heparin/HS disaccharides were separated by reversed-polarity CE performed at 12–20 kV in 15–20 mM phosphate pH 3.5. This technique gave better separation efficiency, resolution, and sensitivity than a previously reported normal-polarity CE method [91].
CE with laser-induced fluorescence detection
Heparin/HS disaccharide and oligosaccharide samples prepared using heparinase that had been separated using CE can be easily detected by UV absorbance at 232 nm. Higher sensitivity is often required for the determination of heparin/HS saccharides in complex biological samples. In addition, the heparin/HS saccharides produced by most chemical methods do not have a terminal ΔUA residue, so they cannot be sensitively detected by UV absorbance at 232 nm. LIF detection increases the sensitivity and lowers the detection limit for heparin/HS saccharides after CE separation. Fluorescence labeling can also improve CE resolution. A number of labeling reagents have been exploited for heparin/HS analyses, including 2-aminopyridine, p-aminobenzoic acid, 8-aminopyrene-1,3,6-trisulfonic acid (APTS), AMAC and BODIPY. A reductive amination reaction relying on the initial formation of a Schiff base between the reducing-end aldehyde of the saccharide and the amino group of the labeling reagent followed by its reduction is the method most frequently used for the fluorescence derivatization of glycans. Monosaccharides formed through the acid-catalyzed hydrolysis of GAGs have been reductively aminated with APTS (λex=455 nm, λem=512 nm) and analyzed in 100 mM acetate buffer at pH 4.5 by CE-LIF [92]. Heparin/HS disaccharides derivatized with AMAC (λex=425 nm, λem=520 nm) were separated by reversed-polarity CE in 50 mM pH 3.5 phosphate buffer at 30 kV and detected at the attomole level (100-fold more sensitive than UV detection at 232 nm) by LIF using an Ar-ion laser [93]. CE-LIF has also been performed using carbodiimide coupling of the carboxylate group of the heparin/HS disaccharide and the amino group of 7-aminonaphthalene-1,3-disulfonic acid (ANDSA) (λex=315 nm, λem=420 nm), and this provided 100-fold and 1000-fold improvements in detection sensitivity over standard fluorescence and UV detection, respectively [94]. Excess amounts of the labeling reagent can ensure the complete reductive amination of heparin/HS disaccharides, but additional purification steps are needed to remove excess tag prior to analysis. Hydrophilic interaction chromatography on microspin columns containing cellulose, diol, amino, silica, and cyano resins has been used to increase the recoveries of the AMAC-labeled disaccharides from excess tag. The highest recovery was obtained when the cellulose microspin column was used; this increased analytical sensitivity 100-fold. CE-LIF performed in reversed-polarity mode following exhaustive heparinase digestion of 1 μg bovine kidney HS and followed by AMAC derivatization and cellulose column cleanup showed that seven heparin/HS disaccharides were present in the electropherogram (Fig. 6A). The percentage composition of each disaccharide obtained from bovine kidney HS is shown in Fig. 6B [95].
Fig. 6.
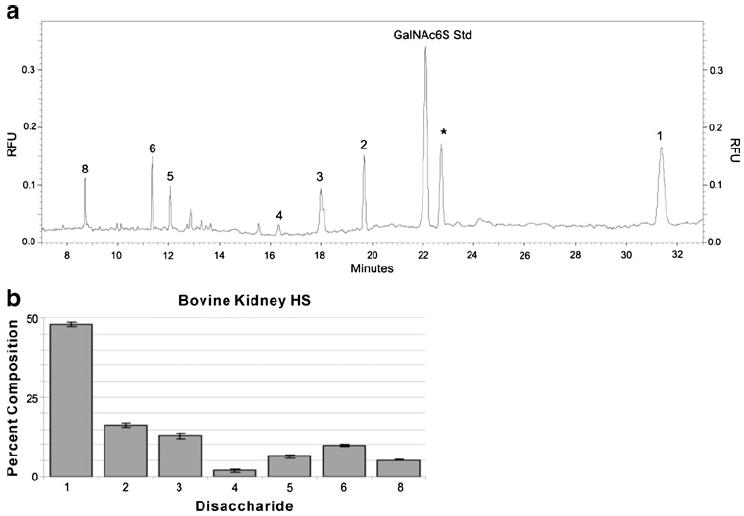
a–b Analysis of bovine kidney HS. a Electropherogram (obtained by performing CE-LIF in reversed-polarity mode) of bovine kidney HS (0.05 μg) subjected to exhaustive heparinase-catalyzed depolymerization, optimized AMAC derivatization, and cellulose cleanup. The disaccharide peaks are labeled with numbers that correspond to the structures shown in Table 1. The asterisk represents an unidentified peak deriving from the bovine HS sample. b Bar graph representation of the CE-LIF disaccharide analysis of bovine kidney HS. Adapted from [95] with permission
Capillary electrophoresis–mass spectrometry
CE-MS is a promising and increasingly popular approach for the analysis of carbohydrates. An appealing advantage of this hyphenated technique is that both methods offer excellent resolution, sensitivity and structural information in a single experiment. The widespread application of CE-MS for heparin/HS oligosaccharide analysis still faces numerous significant challenges. The most well-developed CE buffers rely on high concentrations of nonvolatile salts that are usually incompatible with the electrospray process. Direct coupling between CE and ionspray mass spectrometry initially explored volatile ammonium acetate buffer. The CE/MS interface required optimization of the buffer flow rate, the composition of the sheath liquid, the sheath gas flow, and the positioning of the CE capillary in the electrospray needle. Aqueous ammonium acetate electrolytes adjusted to either pH 3.5 or 9.2 were compatible with CE-MS coupling through a sheath liquid/sheath gas ionspray interface. Both normal- and reversed-polarity CE can be coupled to either positive-ion or negative-ion ESI-MS modes. This allows four combinations of CE-MS and provides the opportunity to obtain complementary structural information. Optimized CE-MS was first used to analyze eight heparin/HS disaccharides (Table 1), and was further applied to the characterization of the disaccharide composition of heparin depolymerized with heparinase [96]. Subsequently, a pressure-assisted CE-ion trap MS was used in the analysis of heparin/HS disaccharides. Pressure assistance improves the stability of the electrospray conditions. Reversed-polarity CE-MS has also relied on formic acid, with a sheath flow of 2-propanol in formic acid at pH 3.2 [97]. Volatile solvent systems for the CE separation of high-molecular-mass heparin oligosaccharides compatible with mass spectrometric detection have also been explored. Structurally defined heparin oligosaccharides ranging in size from tetrasaccharides to tetradecasaccharides, prepared through controlled heparinase treatment, showed good separation efficiency and resolution in normal-polarity mode using ammonium bicarbonate and triethylamine at pH 8.50 [98]. CE-MS analysis of GAG-derived oligosaccharides has been a source of intense research interest [99, 100], and has recently been applied to the detection of the binding of heparin/HS to chemokine stromal cell-derived factor-1 and antithrombin III [101, 102].
Tandem mass spectrometry
Mass spectrometry is an important tool for the structural analysis of heparin/HS, offering accurate molecular weight measurements for intact oligosaccharides and an approach for deducing oligosaccharide sequences through fragmentation. Fast atom bombardment (FAB), one of the first techniques to provide good sensitivity in heparin oligosaccharide analysis [103, 104], has been largely replaced with the modern soft ionization methods of ESI and MALDI. MS offers the advantages of high sensitivity, precise results, rich data and analytical versatility. MALDI-MS [28, 76], while very sensitive and offering a high mass range, is most useful for the analysis of protein/peptide–heparin/HS oligosaccharide complexes [105, 106]. MALDI only poorly ionizes uncomplexed heparin/HS oligosaccharides from conventional matrices [107]. While some progress has been made in developing novel matrices [39, 44, 107-110], MALDI-MS uses a solid target, so it may not be ideally suited for hyphenated analytical techniques. Heparin/HS oligosaccharides readily ionize under ESI-MS, and ions are often detected as their alkali or ammonium salts in the negative ion mode [15, 28-30], making ESI-MS applicable to hyphenated techniques involving HPLC and CE. The development of ESI-MS of heparin/HS oligosaccharides has also posed problems, including in-source fragmentation with the loss of SO3.
Collisionally induced dissociation tandem mass spectrometry
The application of tandem MS to the sequencing of heparin/HS oligosaccharides is extremely challenging because the S–O bonds of their sulfo groups are more labile than the glycosidic bonds that provide sequence-informative fragmentation. Despite the major research effort devoted to using tandem MS for heparin/HS oligosaccharide analysis [111, 112], there have been few successful applications. Negative-ion FAB-CID-MS/MS and ESI-CID-MS/MS have been successfully used in the characterization of heparin/HS disaccharides [113, 114]. The mechanisms of dissociation for isomeric heparin/HS disaccharides (Table 1) have been established using isotope labeling and ion-trap tandem mass spectrometry. No SO3 loss was observed in the MS1 spectra of these disaccharides, and the addition of ammonium hydroxide reduced the number of sodiated adducts, simplifying the spectra. The most abundant ion in each case was the molecular ion: [M-H]− for the non- and monosulfated disaccharides, [M-2H]2− for the disulfated disaccharides, and [M-3H]3− for the trisulfated disaccharide. Some of these disaccharides can be distinguished directly by MS1 based their different molecular masses, while others were differentiated using product-ion spectra generated by MSn. Through-glycosidic fragmentation observed in the MSn spectra as a diagnostic ion as well as 0,2 A2 and 0,4A2 are also critical for assigning structure. The linearity of an equimolar mixture of the heparin/HS disaccharides ranged from 1 to 100 pmol. Disaccharide mixtures with different concentrations of each disaccharide could be quantitatively analyzed by MS with the internal standard, and tandem MS was used to distinguish the isomers [115-117].
The structural characterization of heparin/HS oligosaccharides has also been performed by multistage ESI tandem mass spectrometric analysis. CID-generated product ions arise from the multiply charged molecular ion peaks. Those product ions bound to sodium cations and those bound to calcium cations in the tetrasulfated trisaccharide, pentasulfated tetrasaccharide, and octasulfated pentasaccharide result in abundant fragment ions corresponding to glycosidic bond and cross-ring cleavage. The results demonstrated that abundant glycosidic bond cleavages could be obtained provided that most of the sulfo groups are charged. Repulsions from such charges are likely to destabilize glycosidic bonds, making their rupture more energetically favorable than the loss of SO3 from the precursor ion. The nomenclature used assumes that the protons are displaced by metal cations, and only the loss of protons confers charge on the ion. More sequence information is obtained from the CID mass spectra of precursor ions with metal cations than from ions without metal cations as a result of additional glycosidic-bond and cross-ring cleavage. Furthermore, it was observed that sulfo group lability follows the order SO3− > SO3Na > SO3(1/2) Ca+. Pairing the sulfated oligosaccharide anions with metal cations serves to increase sulfo group stability so that abundant backbone cleavage fragments are observed [30, 117, 118]. In conjunction with the ESI tandem MS analysis of heparin/HS oligosaccharides, an effective strategy can be designed for sequencing. An MS software package known as HOST (heparin oligosaccharide sequencing tool) has also been developed to help analyze sequences [119]. The basic function of HOST is to aid the user by accepting and merging the data acquired from two sets of experiments: the disaccharide composition obtained upon enzymatic digestion and topological information obtained from a set of MSn experiments applied to the intact heparin oligosaccharide. After the information has been obtained, HOST lists all possible structures and generates a scoring system that puts the most likely structures at the top of the list and relates subsequent structural sequences to the best match of the series. It automates the interpretation of the MSn data generated from glycosaminoglycans, providing a practical methodology for the future analysis of heparin/HS oligosaccharides of unknown structure.
Electron detachment dissociation tandem mass spectrometry
The recombination of low-energy electrons (≤1 eV) with multiply charged precursor ions, known as electron capture dissociation (ECD), has found widespread use in biomolecular analysis. Electron detachment dissociation (EDD), the negative-ion complement of ECD, has been used for the characterization of acidic molecules that do not easily form positive ions. In EDD, multiply charged precursor anions are irradiated with moderate-energy (19 eV) electrons, resulting in electron ejection from the precursor ion. Similar to ECD, a radical species is generated that fragments differently compared with the dissociation pathways of even-electron ions (which undergo low-energy or threshold dissociation). Compared to collisionally activated dissociation (CAD) and infrared multiphoton dissociation (IRMPD) MS, EDD provides improved cross-ring fragmentation, which is important for determining the patterns of sulfation and acetylation and the hexuronic acid stereochemistry of GAG oligosaccharides (Fig. 7A–C). EDD has demonstrated its utility for GAG-derived oligosaccharides ranging in size from tetrasaccharides to decasaccharides. Recently, EDD has been developed to distinguish between the IdoA and GlcA epimers in four HS-derived tetrasaccharides: ΔUA-GlcN-GlcA-GlcNAc, ΔUA-GlcNAc-IdoA-GlcNAc, ΔUA-GlcNS-GlcA-GlcNAc, and ΔUA-GlcNS-IdoA-GlcNAc [120-123]. The 0,2A3, [B3-H] and [B3′-CO2] product ions are detected in the MS/MS spectrum of ΔUA-GlcN-GlcA-GlcNAc (Fig. 7A) but they are absent in the spectrum of ΔUA-GlcNAc-IdoA-GlcNAc (Fig. 7E). Therefore, the 0,2A3, [B3-H] and [B3′-CO2] product ions are diagnostic ions for distinguishing GlcA from IdoA. The same results were obtained for ΔUA-GlcNS-GlcA-GlcNAc and ΔUA-GlcNS-IdoA-GlcNAc. These diagnostic product ions can be rationalized as arising from a proposed radical species whose subsequent fragmentation is influenced by C5 stereochemistry.
Fig. 7.
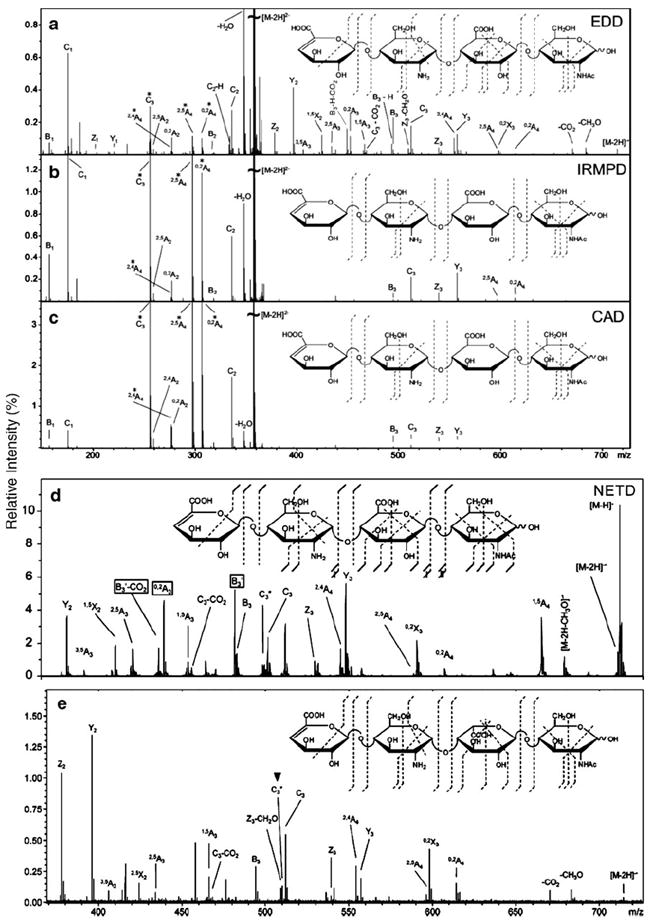
a–e Tandem mass spectra of the [M-2H]2− precursor ion of an HS tetrasaccharide (ΔUA-GlcN-GlcA-GlcNAc) obtained using a EDD, b IRMPD, c CAD, and d NETD. e Tandem mass spectrum of ΔUA-GlcN-IdoA-GlcNAc obtained using EDD. Doubly charged product ions are indicated with an asterisk. Adapted from [120, 121, 124] with permission
Most recently, negative electron transfer dissociation (NETD) has been applied to the analysis of HS oligosaccharides using fluoranthene or xenon as the reagent gas. NETD produces fragmentation that is very similar to the previously observed EDD fragmentation (Fig. 7A and D). Using fluoranthene or xenon, both glycosidic and cross-ring cleavages are observed, as well as even and odd electron products. The loss of SO3 can be minimized, and an increase in cross-ring cleavages is observed if a negatively charged carboxylate is present during NETD. This can be controlled by altering the charge state or through the addition of sodium. Both EDD and NETD are useful for distinguishing GlcA from IdoA in HS tetrasaccharides. NETD effectively dissociates larger GAG oligosaccharides, although the low resolution of the ion trap makes assigning product ions more difficult. These results demonstrate that NETD is effective at characterizing GAG oligosaccharides in a single tandem mass spectrometry experiment on a widely available mass spectrometry platform [124].
Nuclear magnetic resonance spectroscopy
NMR spectroscopy is a nondestructive technique that provides critical qualitative and quantitative structural information in heparin/HS analysis. Although NMR requires far more analyte, this information-rich technique has helped to elucidate the structures of complex heterogeneous mixtures such as heparin/HS GAGs. NMR is currently also the most reliable method for distinguishing GlcA from IdoA and positioning sulfo groups in oligosaccharide sequences. One-dimensional (1D) 1H- and 13C-NMR experiments are routinely performed on a wide variety of heparin/HS samples ranging from intact polysaccharides to oligosaccharides and disaccharides. Two-dimensional (2D) and multidimensional NMR techniques often require more analyte, skill and time, but are particularly beneficial for making full assignments and studying dynamic processes in conformational analysis and protein-binding studies. Correlation spectroscopy (H–H COSY), heteronuclear multiple quantum coherence spectroscopy (C–H HMQC), total correlation spectroscopy (TOCSY) and nuclear Overhauser effect spectroscopy (NOESY and ROESY) have been widely used for the structural elucidation of heparin/HS. While COSY, TOCSY and HMQC can be used to assign the individual saccharide resonances, NOESY and ROESY have been employed for sequencing sugar units and for the conformation analysis of GAGs [125]. A combination of 1D and 2D NMR experiments is often essential for determining the detailed structure of heparin/HS [125-128].
NMR sensitivity can be defined in terms of mass and/or concentration sensitivity. A mass-limited sample will yield the highest S/N in a microcoil NMR probe, while a concentration-limited sample is most effectively analyzed in a larger traditional NMR probe. The S/N ratio increases as the size of the NMR coil decreases for mass-limited samples. For example, while the mass sensitivity of a 1.5 μL CapNMR probe is ~10× higher than a conventional 5 mm NMR probe, the concentration sensitivity of the 1.5 μL CapNMR probe is approximately ~13× lower than that of the 5 mm NMR probe [129]. Since heparin/HS samples are generally highly water soluble, NMR analyses of them are typically mass limited. While the major drawback of NMR spectroscopy is its relatively poor sensitivity compared with other conventional analytical techniques, recent technological advances in cyroprobe and microprobe technologies and newly improved higher magnetic field strength instruments have dramatically increased their sensitivity, facilitating the analysis of mass-limited samples [130-132].
Capillary electrophoresis with nuclear magnetic resonance spectroscopy detection
CE-NMR offers the high resolution of CE and the information-rich analysis of NMR, and can thus facilitate the on-line or off-line identification and structure elucidation of components in complex mixtures. Although there are several published applications of CE coupled to NMR for other types of compounds [133, 134], there are few reports on GAG structural analysis by CE-NMR. A novel computational approach that integrates NMR spectroscopy and CE data off-line has been used in the unbiased structural assignment of HS oligosaccharides. This approach relies on a numerical property encoding nomenclature (PEN) framework for the unbiased and systematic processing of data sets [135]. In a second off-line study, NMR spectroscopy was used as pH detector to determine the acidity of the carboxylic acid and ammonium moieties of commercially available heparin disaccharide standards. These pKa values were used to calculate the net charge of each disaccharide in order to provide a better understanding of the migration order obtained by CE [25].
Capillary isotachophoresis with NMR detection
Capillary isotachophoresis (cITP) NMR is a relatively new, robust and efficient method for the identification of components in complex mixtures such as heparin/HS oligosaccharides. cITP is coupled to NMR in order to overcome the lack of sensitivity for detecting concentration-limited samples that is typical of microcoil NMR. Approximately one microliter of the sample is injected into the cITP system, separated, and concentrated prior to NMR analysis. This allows the acquisition of NMR spectra for separated components of complex mixtures like heparin/HS disaccharides or oligosaccharides.
In cITP, a fused silica capillary is used to reduce electroosmotic flow (EOF). The leading electrolyte (LE) and the tailing electrolyte (TE) have high and low electrophoretic mobilities, respectively. The sample is introduced between the LE and TE, and by adjusting the concentration of the LE it is possible to separate the components of the sample and to concentrate the analytes 10- to 100-fold. cITP can be directly coupled with on-line microcoil NMR detection for the separation and analysis of nmol quantities of heparin/HS oligosaccharides. In cITP-NMR, 1H NMR spectra were recorded using a 45° pulse, a spectral width of 7.8 kHz, and an acquisition time of 1.4 s at 15 kV. Eight FIDs, giving a time resolution of 11.2 s, were collected for each spectrum. Heparin/HS disaccharides were analyzed using on-line cITP-NMR, and this approach was found to give an ~2× increase in sensitivity compared to conventional NMR probes [136].
Using a combination of cITP-NMR at 12 kV and a CapNMR microcoil probe, the S/N was enhanced 2.4-fold and spectral resolution was increased. The cITP-NMR spectrum obtained by the postacquisition coaddition of the spectra of 5.8 μg of an unknown crude heparin oligosaccharide sample (including buffer salts) is shown in Fig. 8A. The four anomeric protons marked with asterisks in this figure indicate that the unknown oligosaccharide is a tetrasaccharide. The tetrasaccharide contained a significant amount of salt, so 1H NMR spectra of the heparin tetrasaccharide were acquired using the CapNMR probe (Fig. 8B). The CapNMR probe is very useful for mass- and concentration-limited samples, and its performance is unaffected by the presence of buffer salts. The heparin tetrasaccharide spectrum obtained using CapNMR has a high resolution and a good signal-to-noise ratio. The 2D COSY, TOCSY, and ROESY NMR techniques were used to assign all of the resonances and establish the sequence of the heparin tetrasaccharide. In conclusion, a combined cITP-NMR and CapNMR strategy can be used to define impurities and completely characterize the heparin oligosaccharides in microgram samples with improved sensitivity and spectral resolution [137].
Fig. 8.
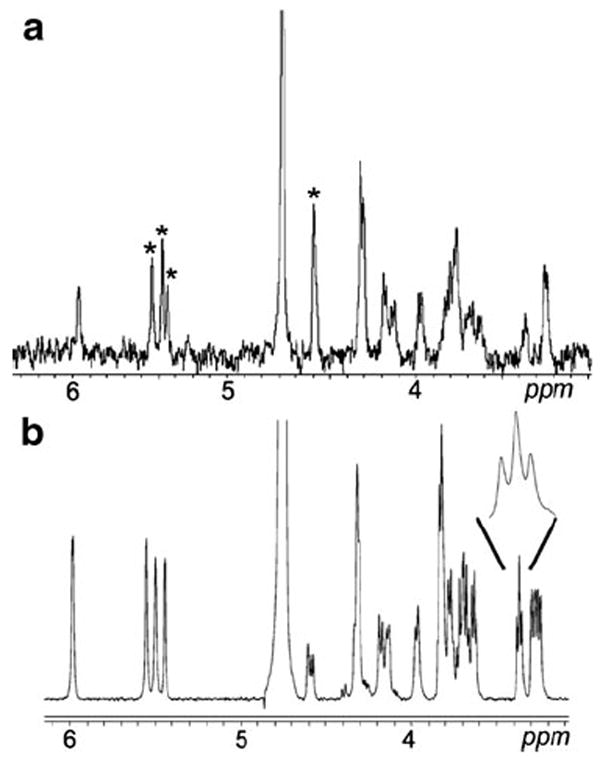
a–b 1H-NMR spectrum obtained by postacquisition coaddition of the cITP-NMR spectra of 2.5 μg of the pure heparin oligosaccharide. a. The four anomeric protons marked with asterisks indicate that the unknown oligosaccharide is a tetrasaccharide. 1H NMR spectrum of the heparin tetrasaccharide acquired using the CapNMR probe with 30 μg of the oligosaccharide in the probe flowcell. b. Enlarged portion of the spectrum shows the characteristic d-glucuronate H-4 triplet at 3.38 ppm. Adapted from [137] with permission
High-performance liquid chromatography with nuclear magnetic resonance spectroscopy detection
Intact GAG heparin and the contaminant oversulfated chondroitin sulfate (OSCS) have been analyzed using weak anion exchange chromatography (WAX) combined with both UV and on-line/off-line 1H NMR detection. Separation was achieved using a Shodex Asahipak NH2P-50E amino-bonded column with a 0.1 M/min sodium chloride gradient at pH 9.65. Although UV detection is more sensitive than NMR, it requires additional standards to detect unknown contaminants. Additionally, MS detection is incompatible with a WAX separation requiring high salt concentrations. The on-flow and stop-flow WAX-NMR spectra of heparin and OSCS can be used to identify the characteristic 1H NMR signals of these polysaccharides (Fig. 9). Characteristic 1H NMR peaks of heparin and OSCS can be easily detected in on-flow WAX-NMR spectra (Fig. 9A). However, the signal-to-noise ratio of the NMR spectrum is relatively low, and if there are small amounts of impurities present in the heparin sample this technique may not be capable of detecting them. Therefore, HPLC-NMR experiments have usually been performed in the stop-flow mode, where peaks are first detected in the UV chromatogram and then collected in the NMR flow cell. The peak intensity in stop-flow mode is much higher than the peak intensity obtained using on-flow WAX-NMR analysis. In initial experiments, the same amount (40 mg/mL) of heparin and OSCS was used, although in an actual contaminated heparin sample the amount of impurity is generally much lower. As a result, 40 mg/mL of heparin containing 2 mg/ml OSCS were used to establish the utility of the method. The peaks for OSCS were very weak, even when a high number of scans were performed. OSCS was trapped in the WAX column for ten replicate injections to solve this sensitivity problem [138].
Fig. 9.
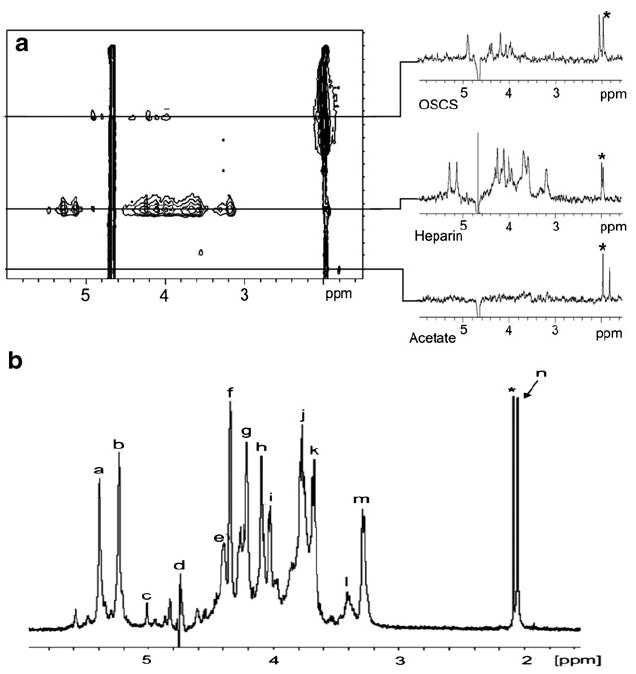
a On-flow 1H NMR-detected WAX chromatogram of heparin and OSCS. The slices taken through the chromatographic peaks show the 1H NMR spectra of OSCS, heparin, and acetate, which was present as an unexpected impurity. The asterisk represents residual acetonitrile as a column contaminant. b Stop-flow 1H NMR spectrum of heparin. The spectrum was acquired from a single 25 μL injection of a sample containing 40 mg/mL of heparin. Signals: a, H-1 of GlcNS; b, H-1 of Ido2S; c, H-1 of IdoA; d, HDO; e, H-6 of GlcNS6S; f, H-2 of IdoA2S; g, H-3 of IdoA2S; h, H-4 of IdoA2S; i, H-5 of GlcNS6S; j, H-4 of GlcNS6S; k, H-3 of GlcNS; l, H-2 of GlcA; m, H-2 of GlcNS6S; n, N-acetyl; asterisk, residual acetonitrile. Adapted from [138] with permission
Conclusions and perspectives
In summary, HPLC-based hyphenated techniques are robust and represent important tools for the structural elucidation of heparin/HS. Coupling traditional separation techniques, including HPAEC, HPSEC, RP-HPLC and RPIP-HPLC, to high-sensitivity spectroscopic detectors can often provide excellent resolution, sensitivity and structural information. New developments in UPLC and graphitized carbon chromatography (GCC) have provided the highest resolution yet of any chromatography system for GAG-oligosaccharide separations. Moreover, HILIC and chip-based HILIC technology are leading to automation and miniaturization, enabling rapid and sensitive analysis. Hyphenated CE-based techniques have emerged as a highly promising and increasingly popular approach for GAG oligosaccharide analysis due to their high separation efficiencies, short analytical times and low sample requirements. The next developmental step appears to be the expanded use of CE-MS and CE-MS/MS for heparin/HS oligosaccharide separation and sequencing, and for the identification of single components in complex biological mixtures. Although the application of tandem MS to the sequencing of heparin/HS oligosaccharides is complicated by their highly charged nature, the modern approaches of EDD and NETD should provide significantly more sequence-informative fragmentation. NMR spectroscopy, a powerful technique for the structure elucidation of heparin/HS, provides complementary information to that obtained using HPLC, CE, and MS. cITP-NMR is a relatively new, robust and efficient method for the identification of heparin/HS oligosaccharides that can overcome the lack of sensitivity typically associated with NMR spectroscopy. Although the analysis of heparin/HS remains a challenge, new developments in hyphenated techniques will undoubtedly yield improvements that will lead to a better understanding of the structure–activity relationships of these biologically important molecules.
Acknowledgments
The authors thank the NIH for supporting this work with grants GM38060, GM090257, HL096972, and HL101721.
References
- 1.Capila I, Linhardt RJ. Angew Chem Int Ed Engl. 2002;41(3):391–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 2.Olsen SK, Li JY, Bromleigh C, Eliseenkova AV, Ibrahimi OA, Lao Z, Zhang F, Linhardt RJ, Joyner AL, Mohammadi M. Genes Dev. 2006;20(2):185–198. doi: 10.1101/gad.1365406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laremore TN, Zhang F, Dordick JS, Liu J, Linhardt RJ. Curr Opin Chem Biol. 2009;13:633–640. doi: 10.1016/j.cbpa.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hacker U, Nybakken K, Perrimon N. Nat Rev Mol Cell Biol. 2005;6(7):530–541. doi: 10.1038/nrm1681. [DOI] [PubMed] [Google Scholar]
- 5.Tumova S, Woods A, Couchman JR. Int J Biochem Cell Biol. 2000;32(3):269–288. doi: 10.1016/s1357-2725(99)00116-8. [DOI] [PubMed] [Google Scholar]
- 6.Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Annu Rev Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 7.Petitou M, Herault JP, Bernat A, Driguez PA, Duchaussoy P, Lormeau JC, Herbert JM. Nature. 1999;398(6726):417–422. doi: 10.1038/18877. [DOI] [PubMed] [Google Scholar]
- 8.Wang L, Fuster M, Sriramarao P, Esko JD. Nat Immunol. 2005;6(9):902–910. doi: 10.1038/ni1233. [DOI] [PubMed] [Google Scholar]
- 9.Guerrini M, Beccati D, Shriver Z, Naggi A, Viswanathan K, Bisio A, Capila I, Lansing JC, Guglieri S, Fraser B, Al-Hakim A, Gunay NS, Zhang Z, Robinson L, Buhse L, Nasr M, Woodcock J, Langer R, Venkataraman G, Linhardt RJ, Casu B, Torri G, Sasisekharan R. Nat Biotechnol. 2008;26(6):669–675. doi: 10.1038/nbt1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Linhardt RJ. J Med Chem. 2003;46(13):2551–2564. doi: 10.1021/jm030176m. [DOI] [PubMed] [Google Scholar]
- 11.Martin JG, Gupta M, Xu Y, Akella S, Liu J, Dordick JS, Linhardt RJ. J Am Chem Soc. 2009;131(31):11041–11048. doi: 10.1021/ja903038d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jandik KA, Kruep D, Cartier M, Linhardt RJ. J Pharm Sci. 1996;85(1):45–51. doi: 10.1021/js9502736. [DOI] [PubMed] [Google Scholar]
- 13.Cohen DM, Linhardt RJ. Biopolymers. 1990;30(7–8):733–741. doi: 10.1002/bip.360300708. [DOI] [PubMed] [Google Scholar]
- 14.Rice KG, Kim YS, Grant AC, Merchant ZM, Linhardt RJ. Anal Biochem. 1985;150(2):325–331. doi: 10.1016/0003-2697(85)90518-4. [DOI] [PubMed] [Google Scholar]
- 15.Vives RR, Pye DA, Salmivirta M, Hopwood JJ, Lindahl U, Gallagher JT. Biochem J. 1999;339(Pt 3):767–773. [PMC free article] [PubMed] [Google Scholar]
- 16.Yamada S, Sakamoto K, Tsuda H, Yoshida K, Sugiura M, Sugahara K. Biochem. 1999;38(2):838–847. doi: 10.1021/bi981889n. [DOI] [PubMed] [Google Scholar]
- 17.Chuang WL, McAllister H, Rabenstein L. J Chromatogr A. 2001;932(1–2):65–74. doi: 10.1016/s0021-9673(01)01241-9. [DOI] [PubMed] [Google Scholar]
- 18.Hileman RE, Smith AE, Toida T, Linhardt RJ. Glycobiology. 1997;7(2):231–239. doi: 10.1093/glycob/7.2.231. [DOI] [PubMed] [Google Scholar]
- 19.Pervin A, Gallo C, Jandik KA, Han XJ, Linhardt RJ. Glycobiology. 1995;5(1):83–95. doi: 10.1093/glycob/5.1.83. [DOI] [PubMed] [Google Scholar]
- 20.Zhang F, Zhang Z, Lin X, Beenken A, Eliseenkova AV, Mohammadi M, Linhardt RJ. Biochem. 2009;48(35):8379–8386. doi: 10.1021/bi9006379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Z, Li B, Suwan J, Zhang F, Wang Z, Liu H, Mulloy B, Linhardt RJ. J Pharm Sci. 2009;98(11):4017–4026. doi: 10.1002/jps.21729. [DOI] [PubMed] [Google Scholar]
- 22.Rice KG, Rottink MK, Linhardt RJ. Biochem J. 1987;244(3):515–522. doi: 10.1042/bj2440515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Volpi N, Maccari F, Linhardt RJ. Anal Biochem. 2009;388(1):140–145. doi: 10.1016/j.ab.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Volpi N, Maccari F, Linhardt RJ. Electrophoresis. 2008;29(15):3095–3106. doi: 10.1002/elps.200800109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eldridge SL, Higgins LA, Dickey BJ, Larive CK. Anal Chem. 2009;81(17):7406–7415. doi: 10.1021/ac901218q. [DOI] [PubMed] [Google Scholar]
- 26.Bigler P, Brenneisen R. J Pharm Biomed Anal. 2009;49(4):1060–1064. doi: 10.1016/j.jpba.2009.01.017. [DOI] [PubMed] [Google Scholar]
- 27.Chai W, Hounsell EF, Bauer CJ, Lawson AM. Carbohydr Res. 1995;269(1):139–156. doi: 10.1016/0008-6215(94)00349-k. [DOI] [PubMed] [Google Scholar]
- 28.Chai W, Luo J, Lim CK, Lawson AM. Anal Chem. 1998;70(10):2060–2066. doi: 10.1021/ac9712761. [DOI] [PubMed] [Google Scholar]
- 29.Zaia J, Costello CE. Anal Chem. 2001;73(2):233–239. doi: 10.1021/ac000777a. [DOI] [PubMed] [Google Scholar]
- 30.Zaia J, Costello CE. Anal Chem. 2003;75(10):2445–2455. doi: 10.1021/ac0263418. [DOI] [PubMed] [Google Scholar]
- 31.Linhardt RJ, Rice KG, Kim YS, Engelken JD, Weiler JM. J Biol Chem. 1988;263(26):13090–13096. [PubMed] [Google Scholar]
- 32.Kitagawa H, Kinoshita A, Sugahara K. Anal Biochem. 1995;232(1):114–121. doi: 10.1006/abio.1995.9952. [DOI] [PubMed] [Google Scholar]
- 33.Kinoshita A, Sugahara K. Anal Biochem. 1999;269(2):367–378. doi: 10.1006/abio.1999.4027. [DOI] [PubMed] [Google Scholar]
- 34.Yamada S, Morimoto H, Fujisawa T, Sugahara K. Glycobiology. 2007;17(8):886–894. doi: 10.1093/glycob/cwm051. [DOI] [PubMed] [Google Scholar]
- 35.Lv H, Yu G, Sun L, Zhang Z, Zhao X, Chai W. Oncology. 2007;72(5–6):347–356. doi: 10.1159/000113145. [DOI] [PubMed] [Google Scholar]
- 36.Skidmore MA, Guimond SE, Dumax-Vorzet AF, Atrih A, Yates EA, Turnbull JE. J Chromatogr A. 2006;1135(1):52–56. doi: 10.1016/j.chroma.2006.09.064. [DOI] [PubMed] [Google Scholar]
- 37.Skidmore M, Atrih A, Yates E, Turnbull JE. Meth Mol Biol. 2009;534:157–169. doi: 10.1007/978-1-59745-022-5_12. [DOI] [PubMed] [Google Scholar]
- 38.Adamo M, Qiu D, Dick LW, Jr, Zeng M, Lee AH, Cheng KC. J Pharm Biomed Anal. 2009;49(2):181–192. doi: 10.1016/j.jpba.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 39.Finke B, Stahl B, Pfenninger A, Karas M, Daniel H, Sawatzki G. Anal Chem. 1999;71(17):3755–3762. doi: 10.1021/ac990094z. [DOI] [PubMed] [Google Scholar]
- 40.Stadheim TA, Li H, Kett W, Burnina IN, Gerngross TU. Nat Protoc. 2008;3(6):1026–1031. doi: 10.1038/nprot.2008.76. [DOI] [PubMed] [Google Scholar]
- 41.Whitfield DM, Stojkovski S, Pang H, Baptista J, Sarkar B. Anal Biochem. 1991;194(2):259–267. doi: 10.1016/0003-2697(91)90228-l. [DOI] [PubMed] [Google Scholar]
- 42.Ander B, Karlsson A, Ohrlund A. J Chromatogr A. 2001;917(1–2):105–110. doi: 10.1016/s0021-9673(01)00661-6. [DOI] [PubMed] [Google Scholar]
- 43.Campo GM, Campo S, Ferlazzo AM, Vinci R, Calatroni A. J Chromatogr B. 2001;765(2):151–160. doi: 10.1016/s0378-4347(01)00427-3. [DOI] [PubMed] [Google Scholar]
- 44.Bultel L, Landoni M, Grand E, Couto AS, Kovensky J. J Am Soc Mass Spectrom. 2010;21(1):178–190. doi: 10.1016/j.jasms.2009.09.025. [DOI] [PubMed] [Google Scholar]
- 45.Deakin JA, Lyon M. Glycobiology. 2008;18(6):483–491. doi: 10.1093/glycob/cwn028. [DOI] [PubMed] [Google Scholar]
- 46.Studelska DR, Giljum K, McDowell LM, Zhang L. Glycobiology. 2006;16(1):65–72. doi: 10.1093/glycob/cwj037. [DOI] [PubMed] [Google Scholar]
- 47.Mason KE, Meikle PJ, Hopwood JJ, Fuller M. Anal Chem. 2006;78(13):4534–4542. doi: 10.1021/ac052083d. [DOI] [PubMed] [Google Scholar]
- 48.Ramsay SL, Meikle PJ, Hopwood JJ. Mol Genet Metab. 2003;78(3):193–204. doi: 10.1016/s1096-7192(03)00018-0. [DOI] [PubMed] [Google Scholar]
- 49.Crawley A, Ramsay SL, Byers S, Hopwood J, Meikle PJ. Pediatr Res. 2004;55(4):585–591. doi: 10.1203/01.PDR.0000113789.30640.5C. [DOI] [PubMed] [Google Scholar]
- 50.Cecchi T, Passamonti P. J Chromatogr A. 2009;1216(10):1789–1797. doi: 10.1016/j.chroma.2008.10.031. [DOI] [PubMed] [Google Scholar]
- 51.Cecchi T, Pucciarelli F, Passamonti P. Anal Chem. 2001;73(11):2632–2639. doi: 10.1021/ac001341y. [DOI] [PubMed] [Google Scholar]
- 52.Karamanos NK, Vanky P, Tzanakakis GN, Tsegenidis T, Hjerpe A. J Chromatogr A. 1997;765(2):169–179. doi: 10.1016/s0021-9673(96)00930-2. [DOI] [PubMed] [Google Scholar]
- 53.Thanawiroon C, Linhardt RJ. J Chromatogr A. 2003;1014(1–2):215–223. doi: 10.1016/s0021-9673(03)00779-9. [DOI] [PubMed] [Google Scholar]
- 54.Toyoda H, Yamamoto H, Ogino N, Toida T, Imanari T. J Chromatogr A. 1999;830(1):197–201. [Google Scholar]
- 55.Toyoda H, Nagashima T, Hirata R, Toida T, Imanari T. J Chromatogr B. 1997;704(1–2):19–24. doi: 10.1016/s0378-4347(97)00478-7. [DOI] [PubMed] [Google Scholar]
- 56.Sinnis P, Coppi A, Toida T, Toyoda H, Kinoshita-Toyoda A, Xie J, Kemp MM, Linhardt RJ. J Biol Chem. 2007;282(35):25376–25384. doi: 10.1074/jbc.M704698200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Toyoda H, Kinoshita-Toyoda A, Fox B, Selleck SB. J Biol Chem. 2000;275(29):21856–21861. doi: 10.1074/jbc.M003540200. [DOI] [PubMed] [Google Scholar]
- 58.Toyoda H, Kinoshita-Toyoda A, Selleck SB. J Biol Chem. 2000;275(4):2269–2275. doi: 10.1074/jbc.275.4.2269. [DOI] [PubMed] [Google Scholar]
- 59.Ha YW, Jeon BT, Moon SH, Toyoda H, Toida T, Linhardt RJ, Kim YS. Carbohydr Res. 2005;340(3):411–416. doi: 10.1016/j.carres.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 60.Vongchan P, Warda M, Toyoda H, Toida T, Marks RM, Linhardt RJ. Biochim Biophys Acta. 2005;1721(1–3):1–8. doi: 10.1016/j.bbagen.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 61.Linhardt RJ, Gu KN, Loganathan D, Carter SR. Anal Biochem. 1989;181(2):288–296. doi: 10.1016/0003-2697(89)90245-5. [DOI] [PubMed] [Google Scholar]
- 62.Warda M, Toida T, Zhang F, Sun P, Munoz E, Xie J, Linhardt RJ. Glycoconj J. 2006;23(7–8):555–563. doi: 10.1007/s10719-006-7668-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Patel RP, Narkowicz C, Jacobson GA. Anal Biochem. 2009;387(1):113–121. doi: 10.1016/j.ab.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 64.Thanawiroon C, Rice KG, Toida T, Linhardt RJ. J Biol Chem. 2004;279(4):2608–2615. doi: 10.1074/jbc.M304772200. [DOI] [PubMed] [Google Scholar]
- 65.Zhang Z, Xie J, Liu H, Liu J, Linhardt RJ. Anal Chem. 2009;81(11):4349–4355. doi: 10.1021/ac9001707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuberan B, Lech M, Zhang L, Wu ZL, Beeler DL, Rosenberg RD. J Am Chem Soc. 2002;124(29):8707–8718. doi: 10.1021/ja0178867. [DOI] [PubMed] [Google Scholar]
- 67.Volpi N, Linhardt RJ. Nat Protoc. 2010;5(6):993–1004. doi: 10.1038/nprot.2010.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Storm T, Reemtsma T, Jekel M. J Chromatogr A. 1999;854(1–2):175–185. doi: 10.1016/s0021-9673(99)00525-7. [DOI] [PubMed] [Google Scholar]
- 69.Conboy JJ, Henion JD, Martin MW, Zweigenbaum JA. Anal Chem. 1990;62(8):800–807. [Google Scholar]
- 70.Witters E, Van Dongen W, Esmans EL, Van Onckelen HA. J Chromatogr B. 1997;694(1):55–63. doi: 10.1016/s0378-4347(96)00538-5. [DOI] [PubMed] [Google Scholar]
- 71.Zhang F, Zhang Z, Thistle R, McKeen L, Hosoyama S, Toida T, Linhardt RJ, Page-McCaw P. Glycoconj J. 2009;26(2):211–218. doi: 10.1007/s10719-008-9177-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Swartz ME. J Liq Chromatogr R T. 2005;28(7):1253–1263. [Google Scholar]
- 73.MacNair JE, Lewis KC, Jorgenson JW. Anal Chem. 1997;69(6):983–989. doi: 10.1021/ac961094r. [DOI] [PubMed] [Google Scholar]
- 74.Korir AK, Limtiaco JF, Gutierrez SM, Larive CK. Anal Chem. 2008;80(4):1297–1306. doi: 10.1021/ac702235u. [DOI] [PubMed] [Google Scholar]
- 75.Eldridge SL, Korir AK, Gutierrez SM, Campos F, Limtiaco JF, Larive CK. Carbohydr Res. 2008;343(17):2963–2970. doi: 10.1016/j.carres.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 76.Doneanu CE, Chen W, Gebler JC. Anal Chem. 2009;81(9):3485–3499. doi: 10.1021/ac802770r. [DOI] [PubMed] [Google Scholar]
- 77.Jones CJ, Membreno N, Larive CK. J Chromatogr A. 2010;1217(4):479–488. doi: 10.1016/j.chroma.2009.11.064. [DOI] [PubMed] [Google Scholar]
- 78.Royle L, Roos A, Harvey DJ, Wormald MR, van Gijlswijk-Janssen D, Redwan E-RM, Wilson IA, Daha MR, Dwek RA, Rudd PM. J Biol Chem. 2003;278(22):20140–20153. doi: 10.1074/jbc.M301436200. [DOI] [PubMed] [Google Scholar]
- 79.Thomsson KA, Karlsson NG, Hansson GC. J Chromatogr A. 1999;854(1–2):131–139. doi: 10.1016/s0021-9673(99)00625-1. [DOI] [PubMed] [Google Scholar]
- 80.Naimy H, Leymarie N, Bowman MJ, Zaia J. Biochem. 2008;47(10):3155–3161. doi: 10.1021/bi702043e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hitchcock AM, Yates KE, Costello CE, Zaia J. Proteomics. 2008;8(7):1384–1397. doi: 10.1002/pmic.200700787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Staples GO, Bowman MJ, Costello CE, Hitchcock AM, Lau JM, Leymarie N, Miller C, Naimy H, Shi X, Zaia J. Proteomics. 2009;9(3):686–695. doi: 10.1002/pmic.200701008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Staples GO, Naimy H, Yin H, Kileen K, Kraiczek K, Costello CE, Zaia J. Anal Chem. 2010;82(2):516–522. doi: 10.1021/ac901706f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hemstrom P, Irgum K. J Sep Sci. 2006;29(12):1784–1821. doi: 10.1002/jssc.200600199. [DOI] [PubMed] [Google Scholar]
- 85.Zaia J. Mass Spectrom Rev. 2009;28(2):254–272. doi: 10.1002/mas.20200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Oguma T, Toyoda H, Toida T, Imanari T. J Chromatogr B. 2001;754(1):153–159. doi: 10.1016/s0378-4347(00)00601-0. [DOI] [PubMed] [Google Scholar]
- 87.Mao W, Thanawiroon C, Linhardt RJ. Biomed Chromatogr. 2002;16(2):77–94. doi: 10.1002/bmc.153. [DOI] [PubMed] [Google Scholar]
- 88.Somsen GW, Tak YH, Torano JS, Jongen PM, de Jong GJ. J Chromatogr A. 2009;1216(18):4107–4112. doi: 10.1016/j.chroma.2009.02.063. [DOI] [PubMed] [Google Scholar]
- 89.Patel RP, Narkowicz C, Hutchinson JP, Hilder EF, Jacobson GA. J Pharm Biomed Anal. 2008;46(1):30–35. doi: 10.1016/j.jpba.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 90.Desai UR, Wang H, Ampofo SA, Linhardt RJ. Anal Biochem. 1993;213(1):120–127. doi: 10.1006/abio.1993.1394. [DOI] [PubMed] [Google Scholar]
- 91.Pervin A, Al-Hakim A, Linhardt RJ. Anal Biochem. 1994;221(1):182–188. doi: 10.1006/abio.1994.1395. [DOI] [PubMed] [Google Scholar]
- 92.Ruiz-Calero V, Puignou L, Galceran MT. J Chromatogr B. 2003;791(1–2):193–202. doi: 10.1016/s1570-0232(03)00214-9. [DOI] [PubMed] [Google Scholar]
- 93.Militsopoulou M, Lamari FN, Hjerpe A, Karamanos NK. Electrophoresis. 2002;23(7–8):1104–1109. doi: 10.1002/1522-2683(200204)23:7/8<1104::AID-ELPS1104>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 94.el Rassi Z, Postlewait J, Mechref Y, Ostrander GK. Anal Biochem. 1997;244(2):283–290. doi: 10.1006/abio.1996.9905. [DOI] [PubMed] [Google Scholar]
- 95.Hitchcock AM, Bowman MJ, Staples GO, Zaia J. Electrophoresis. 2008;29(22):4538–4548. doi: 10.1002/elps.200800335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Duteil S, Gareil P, Girault S, Mallet A, Feve C, Siret L. Rapid Commun Mass Spectrom. 1999;13(19):1889–1898. doi: 10.1002/(SICI)1097-0231(19991015)13:19<1889::AID-RCM719>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 97.Ruiz-Calero V, Moyano E, Puignou L, Galceran MT. J Chromatogr A. 2001;914(1–2):277–291. doi: 10.1016/s0021-9673(00)01181-x. [DOI] [PubMed] [Google Scholar]
- 98.Gunay NS, Linhardt RJ. J Chromatogr A. 2003;1014(1–2):225–233. doi: 10.1016/s0021-9673(03)01288-3. [DOI] [PubMed] [Google Scholar]
- 99.Zamfir A, Seidler DG, Kresse H, Peter-Katalinic J. Rapid Commun Mass Spectrom. 2002;16(21):2015–2024. doi: 10.1002/rcm.820. [DOI] [PubMed] [Google Scholar]
- 100.Kuhn AV, Ruttinger HH, Neubert RH, Raith K. Rapid Commun Mass Spectrom. 2003;17(6):576–582. doi: 10.1002/rcm.950. [DOI] [PubMed] [Google Scholar]
- 101.Fermas S, Gonnet F, Varenne A, Gareil P, Daniel R. Anal Chem. 2007;79(13):4987–4993. doi: 10.1021/ac070146h. [DOI] [PubMed] [Google Scholar]
- 102.Fermas S, Gonnet F, Sutton A, Charnaux N, Mulloy B, Du Y, Baleux F, Daniel R. Glycobiology. 2008;18(12):1054–1064. doi: 10.1093/glycob/cwn088. [DOI] [PubMed] [Google Scholar]
- 103.Linhardt RJ, Wang HM, Loganathan D, Lamb DJ, Mallis LM. Carbohydr Res. 1992;225(1):137–145. doi: 10.1016/0008-6215(92)80045-3. [DOI] [PubMed] [Google Scholar]
- 104.Mallis LM, Wang HM, Loganathan D, Linhardt RJ. Anal Chem. 1989;61(13):1453–1458. doi: 10.1021/ac00188a030. [DOI] [PubMed] [Google Scholar]
- 105.Kumar V, Hassan MI, Kashav T, Singh TP, Yadav S. Mol Reprod Dev. 2008;75(12):1767–1774. doi: 10.1002/mrd.20910. [DOI] [PubMed] [Google Scholar]
- 106.Ori A, Free P, Courty J, Wilkinson MC, Fernig DG. Mol Cell Proteomics. 2009;8(10):2256–2265. doi: 10.1074/mcp.M900031-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Laremore TN, Murugesan S, Park TJ, Avci FY, Zagorevski DV, Linhardt RJ. Anal Chem. 2006;78(6):1774–1779. doi: 10.1021/ac051121q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Laremore TN, Linhardt RJ. Rapid Commun Mass Spectrom. 2007;21(7):1315–1320. doi: 10.1002/rcm.2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Laremore TN, Zhang F, Linhardt RJ. Anal Chem. 2007;79(4):1604–1610. doi: 10.1021/ac061688m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tissot B, Gasiunas N, Powell AK, Ahmed Y, Zhi ZL, Haslam SM, Morris HR, Turnbull JE, Gallagher JT, Dell A. Glycobiology. 2007;17(9):972–982. doi: 10.1093/glycob/cwm072. [DOI] [PubMed] [Google Scholar]
- 111.Yu G, Zhao X, Yang B, Ren S, Guan H, Zhang Y, Lawson AM, Chai W. Anal Chem. 2006;78(24):8499–8505. doi: 10.1021/ac061416j. [DOI] [PubMed] [Google Scholar]
- 112.Yang B, Yu G, Zhao X, Jiao G, Ren S, Chai W. FEBS J. 2009;276(7):2125–2137. doi: 10.1111/j.1742-4658.2009.06947.x. [DOI] [PubMed] [Google Scholar]
- 113.Ii T, Kubota M, Okuda S, Hirano T, Ohashi M. Glycoconj J. 1995;12(2):162–172. doi: 10.1007/BF00731361. [DOI] [PubMed] [Google Scholar]
- 114.Behr JR, Matsumoto Y, White FM, Sasisekharan R. Rapid Commun Mass Spectrom. 2005;19(18):2553–2562. doi: 10.1002/rcm.2079. [DOI] [PubMed] [Google Scholar]
- 115.Saad OM, Leary JA. Anal Chem. 2003;75(13):2985–2995. doi: 10.1021/ac0340455. [DOI] [PubMed] [Google Scholar]
- 116.Chi L, Amster J, Linhardt RJ. Curr Anal Chem. 2005;1(1):223–240. doi: 10.2174/157341105774573929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang Z, Linhardt RJ. Curr Anal Chem. 2009;5(3):225–237. doi: 10.2174/157341109788680291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zaia J. Mass Spectrom Rev. 2004;23(3):161–227. doi: 10.1002/mas.10073. [DOI] [PubMed] [Google Scholar]
- 119.Saad OM, Leary JA. Anal Chem. 2005;77(18):5902–5911. doi: 10.1021/ac050793d. [DOI] [PubMed] [Google Scholar]
- 120.Wolff JJ, Amster IJ, Chi L, Linhardt RJ. J Am Soc Mass Spectrom. 2007;18(2):234–244. doi: 10.1016/j.jasms.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wolff JJ, Laremore TN, Busch AM, Linhardt RJ, Amster IJ. J Am Soc Mass Spectrom. 2008;19(6):790–798. doi: 10.1016/j.jasms.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wolff JJ, Laremore TN, Aslam H, Linhardt RJ, Amster IJ. J Am Soc Mass Spectrom. 2008;19(10):1449–1458. doi: 10.1016/j.jasms.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wolff JJ, Chi L, Linhardt RJ, Amster IJ. Anal Chem. 2007;79(5):2015–2022. doi: 10.1021/ac061636x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wolff JJ, Leach FE, 3rd, Laremore TN, Kaplan DA, Easterling ML, Linhardt RJ, Amster IJ. Anal Chem. 2010;82(9):3460–3466. doi: 10.1021/ac100554a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Iacomini M, Casu B, Guerrini M, Naggi A, Pirola A, Torri G. Anal Biochem. 1999;274(1):50–58. doi: 10.1006/abio.1999.4230. [DOI] [PubMed] [Google Scholar]
- 126.Yates EA, Santini F, Guerrini M, Naggi A, Torri G, Casu B. Carbohydr Res. 1996;294:15–27. doi: 10.1016/s0008-6215(96)90611-4. [DOI] [PubMed] [Google Scholar]
- 127.Mulloy B, Mourao PA, Gray E. J Biotechnol. 2000;77(1):123–135. doi: 10.1016/s0168-1656(99)00211-4. [DOI] [PubMed] [Google Scholar]
- 128.Hricovini M, Guerrini M, Bisio A, Torri G, Petitou M, Casu B. Biochem J. 2001;359(Pt 2):265–272. doi: 10.1042/0264-6021:3590265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Olson DL, Norcross JA, O’Neil-Johnson M, Molitor PF, Detlefsen DJ, Wilson AG, Peck TL. Anal Chem. 2004;76(10):2966–2974. doi: 10.1021/ac035426l. [DOI] [PubMed] [Google Scholar]
- 130.Webb AG. Ann Rep NMR Spectrosc. 2006;58:1–50. [Google Scholar]
- 131.Spraul M, Freund AS, Nast RE, Withers RS, Maas WE, Corcoran O. Anal Chem. 2003;75(6):1536–1541. doi: 10.1021/ac026203i. [DOI] [PubMed] [Google Scholar]
- 132.Lacey ME, Subramanian R, Olson DL, Webb AG, Sweedler JV. Chem Rev. 1999;99(10):3133–3152. doi: 10.1021/cr980140f. [DOI] [PubMed] [Google Scholar]
- 133.Behnke B, Schlotterbeck G, Tallarek U, Strohschein S, Tseng L-H, Keller T, Albert K, Bayer E. Anal Chem. 1996;68(7):1110–1115. doi: 10.1021/ac950925a. [DOI] [PubMed] [Google Scholar]
- 134.Wu N, Peck TL, Webb AG, Magin RL, Sweedler JV. J Am Chem Soc. 1994;116(17):7929–7930. [Google Scholar]
- 135.Guerrini M, Raman R, Venkataraman G, Torri G, Sasisekharan R, Casu B. Glycobiology. 2002;12(11):713–719. doi: 10.1093/glycob/cwf084. [DOI] [PubMed] [Google Scholar]
- 136.Korir AK, Almeida VK, Malkin DS, Larive CK. Anal Chem. 2005;77(18):5998–6003. doi: 10.1021/ac050669u. [DOI] [PubMed] [Google Scholar]
- 137.Korir AK, Larive CK. Anal Bioanal Chem. 2007;388(8):1707–1716. doi: 10.1007/s00216-007-1400-2. [DOI] [PubMed] [Google Scholar]
- 138.Limtiaco JF, Jones CJ, Larive CK. Anal Chem. 2009;81(24):10116–10123. doi: 10.1021/ac901812y. [DOI] [PubMed] [Google Scholar]