

Abstract
Objective:
Some pathologic features of the FHL1 myopathies and the myofibrillar myopathies (MFMs) overlap; we therefore searched for mutations in FHL1 in our cohort of 50 patients with genetically undiagnosed MFM.
Methods:
Mutations in FHL1 were identified by direct sequencing. Polymorphisms were excluded by using allele-specific PCR in 200 control subjects. Structural changes in muscle were analyzed by histochemistry, immunocytochemistry, and electron microscopy.
Results:
We detected 2 novel and 1 previously identified missense mutation in 5 patients. Patients 1–4 presented before age 30, display menadione–nitro blue tetrazolium–positive reducing bodies, and harbor mutations in the FHL1 LIM2 domain. Patient 5 presented at age 75 and has no reducing bodies, and his mutation is not in a LIM domain. The clinical features include progressive muscle weakness, hypertrophied muscles, rigid spine, and joint contractures, and 1 patient also has peripheral neuropathy. High-resolution electron microscopy reveals the reducing bodies composed of 13-nm tubulofilaments initially emanating from Z-disks. At a more advanced stage, abundant reducing bodies appear in the cytoplasm and nuclei with concomitant myofibrillar disintegration, accumulation of cytoplasmic degradation products, and aggregation of endoplasmic reticulum and sarcotubular profiles.
Conclusions:
FHL1 dystrophies can be associated with MFM pathology. Mutations in the LIM2 domain are associated with reducing bodies composed of distinct tubulofilaments. A mutation extraneous to LIM domains resulted in a mild late-onset phenotype with MFM pathology but no reducing bodies.
The four-and-a-half LIM domain protein 1 (FHL1), encoded by FHL1, is located on Xq26.3. It is highly expressed in skeletal and cardiac muscle and less abundantly in brain, placenta, lung, liver, kidney, pancreas, and testis.1–3 Mutations in FHL1 have been associated with diverse chronic myopathies.4 These include late-onset X-linked scapulo-axio-peroneal myopathy with bent spine syndrome,5 reducing body myopathy,6,7 X-linked dominant scapuloperoneal muscular dystrophy,8 rigid spine syndrome,9 and contractures and cardiomyopathy mimicking Emery-Dreifuss muscular dystrophy.10,11 Most FHL1 mutations appear in the second or fourth LIM domains with a single mutation in the third LIM domain.
Myofibrillar myopathies (MFMs) arise from primary degeneration of the muscle fibers and represent a morphologically distinct but genetically heterogeneous subset of muscular dystrophies. The unifying histologic findings are early disintegration of the Z-disk followed by myofibrillar disintegration, aggregation of degraded filaments into pleomorphic granular or hyaline inclusions, and ectopic expression of multiple Z-disk–related and other proteins in the affected muscle fibers.12–14 Although some pathologic features of the FHL1 muscular dystrophies and the MFMs overlap, reducing bodies have been detected only in FHL1 dystrophies.7,15 We here report 2 novel and a previously published missense mutation in FHL1 in 4 kinships. One mutation, which is novel, is located outside the LIM domains, and 2 others are in the second LIM domain. We examine the structural consequences of the mutations, analyze the ultrastructure of reducing bodies at high resolution, and expand the structural and clinical features of the FHL1 dystrophies.
METHODS
Patients.
Five patients were investigated. Clinically affected limb muscles, grade 3–4 on the Medical Research Council scale, were biopsied in patients 2–5. Genomic DNA was isolated from peripheral blood or frozen muscle.
Standard protocol approvals, registrations, and patient consents.
All studies were in accord with the guidelines of the institutional review board of the Mayo Clinic.
Mutation analysis.
PCR primers were designed to analyze all coding exons and their flanking noncoding regions of FHL1 (GenBank reference number NG_015895.1). PCR-amplified fragments were sequenced using fluorescently labeled dideoxy terminators. FHL1 nucleotides were numbered according to the mRNA sequence (GenBank reference number NM_001449). Allele-specific PCR was used to screen for the novel mutations in the patients and unrelated normal control subjects.
Histochemistry and immunostains.
Conventional histochemical studies were performed, and congophilic deposits were visualized as described previously.16 Reducing bodies were identified by menadione–nitro blue tetrazolium (NBT) staining without α-glycerophosphate substrate.17,18 Six to 10-μm-thick cryostat sections were treated with monoclonal antibodies against desmin (Dako, Carpinteria, CA), αB-crystallin (Stressgen, Ann Arbor, MI), myotilin (Novocastra, Bannockburn, IL), dystrophin (Novocastra), neural cell adhesion molecule (Cell Sciences, Canton, MA), ubiquitin (Chemicon, Temecula, CA), CD4 and CD8 (BD Biosciences, San Jose, CA), and CD22 (Dako). FHL1 (directed against the C-terminal region of FHL1A isoform; Abcam, Cambridge, MA) and CD3 (Dako) were immunolocalized with polyclonal antibodies. The immunoreactive sites were then visualized with appropriate second antibodies using immunoperoxidase and immunofluorescence methods, as described previously.14 Adjacent sections in the series were stained with trichrome. Controls consisted of replacement of antibodies with nonimmune immunoglobulin G of the same subclass and concentration as the primary antibody.
Electron microscopy.
Electron microscopy was performed on muscle specimens fixed at rest length and processed by standard methods.16
RESULTS
Clinical features.
Three patients were female and 2 were male. The age at onset of symptoms ranged from 6 to 75 years and the age at diagnosis from 10 to 77 years (table). All patients but patient 4 presented with progressive muscle weakness; he had hypertrophied muscles, limited neck movements, a rigid spine, and progressive joint contractures (figure 1, A and B). Patients 1 and 3 had conspicuous asymmetric muscle atrophy. Patient 5 also had peripheral neuropathy with distal pain, temperature, and vibration sense loss in the legs. None of the patients had cardiac symptoms, but multiple nuclear family members of patient 1 had died prematurely of coronary artery disease and one of her sisters had a cardiac pacemaker, but none were aware of muscle weakness. No DNA was available from deceased family members. The son of the brother of patient 1 also had the cardiac disease; therefore, cardiac disease in this family is not related to the FHL1 mutation. All patients had a normal EKG and echocardiogram. Holter monitoring of patient 4 revealed no arrhythmia.
Table.
Clinical data and mutation analysis
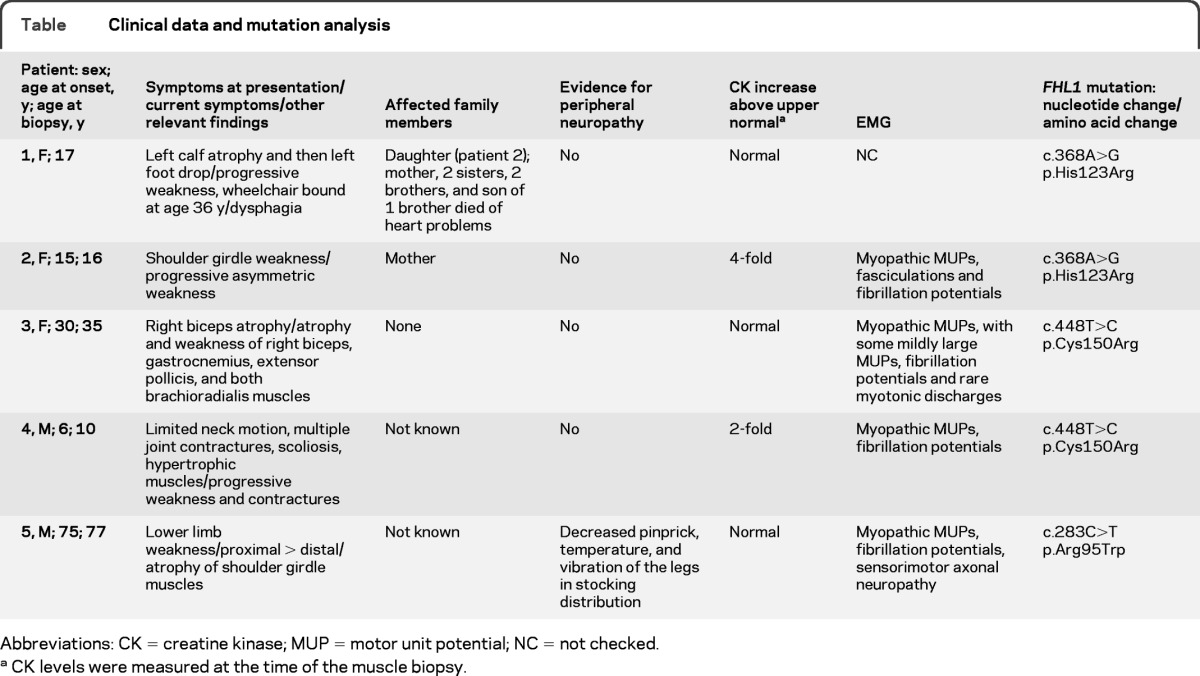
Abbreviations: CK = creatine kinase; MUP = motor unit potential; NC = not checked
CK levels were measured at the time of the muscle biopsy.
Figure 1. Patient 4 at age 10 (A) and 11 (B) years, and FHL1 gene structure, splice variants, identified mutations (C), and alignment of amino acid sequences (D).

(A, B) Note quadriceps muscle hypertrophy, ankle and elbow contractures, and worsening of elbow contracture over 1 year. The right leg is in a cast owing to heel cord lengthening surgery. (C) Exons 1 and 2 are noncoding. The last exon of FHL1A has a different amino acid (aa) sequence than the last exon of FHL1B and FHL1C. Three nuclear localization signals (NLS) and a nuclear export sequences (NES) are present only in FHL1B. (D) The mutated residues are conserved across vertebrate species.
EMG studies of patients 2–5 showed myopathic motor unit potentials with abnormal electrical irritability including myotonic discharges, and patient 5 also showed axonal sensorimotor neuropathy.
Mutation analysis.
The MFM features of our patients' muscle specimens prompted us to sequence the genes encoding desmin, αB-crystallin, myotilin, Zasp, filamin C, and Bag3, but we detected no mutations. Because previously reported that patients with FHL1 mutations also had pathologic features of MFM,7,15 we also sequenced this gene in our cohort of patients with MFM. This revealed a novel heterozygous c.368A>G transition that predicts p.His123Arg substitution in patient 1 and her daughter (patient 2) (table; figure 1, C and D). None of the unaffected family members (3 brothers, 2 sisters, and the son of patient 1) carries this mutation. Direct sequencing of FHL1 in patients 3 and 4 revealed a previously reported heterozygous and hemizygous c.448T>C transition that predicts p.Cys150Arg substitution. Direct sequencing of FHL1 in patient 5 revealed a novel hemizygous c.283C>T transition that predicts p.Arg95Trp substitution. The novel mutations were not detected by allele-specific PCR in 200 unrelated individuals. All mutated residues are evolutionarily conserved across vertebrate species (figure 1D).
Structural observations.
Light microscopy.
In each patient, the light microscopic findings were typical of MFM (figure 2). The muscle fibers varied from 9 to more than 120 μm in diameter. Trichrome-stained sections revealed pleomorphic granular or hyaline material and few to many cytoplasmic bodies in the abnormal fibers (figure 2, A, D, and J). In patients 2–4, some nuclei were surrounded by granular material (asterisk on figure 2A). Except in patient 5, some abnormal fiber regions reacted for menadione-NBT without substrate, signaling the presence of reducing bodies (figure 2C). The hyaline masses were devoid of oxidative enzyme activity (figure 2E). Increases in acid phosphatase and periodic acid-Schiff–positive material (figure 2K) appeared near many inclusions. Numerous fibers harbored intensely congophilic inclusions (figure 2L). Some fibers were necrotic, and a comparable number were regenerating. A fair number of fibers displayed internal nuclei. Scattered fibers contained multiple vacuoles filled with granular material (figure 2A). In adenosine triphosphatase-reacted sections, the atrophic fibers were of either histochemical type. There was a mild to moderate increase of endomysial and a moderate increase of perimysial fibrous and fatty connective tissue. In patient 2, small collections of mononuclear cells appeared at few perivascular sites in perimysium and at multiple sites in endomysium where they surrounded, invaded, and partially replaced highly degenerate muscle fibers (figure 2J).
Figure 2. Histologic and immunocytochemical findings.

(A, D, J) Pleomorphic granular or hyaline material and cytoplasmic bodies in the abnormal fibers in trichrome-stained sections. Note a nucleus surrounded by dark blue granular material (asterisk, A) and inflammatory exudate surrounding and replacing abnormal fibers (J). (E) The hyaline masses are devoid of NADH dehydrogenase activity. (K) Periodic acid-Schiff–positive material appears near inclusions. (L) Fibers harbor intensely congophilic inclusions. Nonconsecutive sections in the same series show abnormal fiber regions that immunoreact strongly for FHL1 (B) and part of the same regions react for menadione–nitro blue tetrazolium without substrate (C). Note abnormal accumulation of αB-crystallin (F), myotilin (G), dystrophin (H), and ubiquitin (I) in the structurally abnormal fibers. Samples were from the right soleus muscle of patient 4 (A) and from the right triceps muscle of patient 2 (B–L). (A, D–L) Scale bar = 50 μm. (B, C) Scale bar = 20 μm.
Immunohistochemistry.
In all specimens, more than 75% of the abnormal fibers reacted strongly for FHL1 (figure 2B), desmin, αB-crystallin (figure 2F), and myotilin (figure 2G). Approximately 25% of the abnormal fibers reacted for dystrophin (figure 2H) and ubiquitin (figure 2I), and a few reacted for neural cell adhesion molecule. All menadione-NBT–positive areas reacted for FHL1, but the FHL1 immunoreactivity was more widespread than the menadione-NBT positivity (compare figure 2, B and C).
The autoaggressive inflammatory cells in patient 2 were CD3+CD4+ T cells with a lesser admixture of CD3+CD8+ T cells. Only rare CD22+ B cells were present (figure e-1 on the Neurology® Web site at www.neurology.org).
Electron microscopy.
This was performed in patients 2, 3, and 4 at the ages of 16, 35, and 10 years. The earliest pathologic alterations involved the Z-disk. Some Z-disks showed streaming (figure 3A) and others gave rise to expanses of tubulofilamentous material (figure 3B). The same material occupied vast expanses of the interior of many abnormal fibers, sometimes encasing nuclei or accumulating in other fiber regions (figures 3, C and D, and 4, D and E), probably corresponding to the reducing bodies seen by light microscopy. The larger tubulofilamentous deposits also entrained clusters of glycogen granules. At high resolution, the tubulofilaments were nonbranching and linear or slightly curving with an apparent diameter of 13 ± 1.2 nm (mean ± SD, n = 28) (figure 3E). In favorable sections, the tubulofilaments were decorated with ∼1.2 nm spicules orthogonal to their long axis. Other inclusions, sometimes abutting on the tubulofilamentous inclusions, consisted of aggregates of rough endoplasmic reticulum (figure 4A), clusters of dilated sarcotubular profiles (figure 4B), cytoplasmic bodies (figure 3C), and fine irregularly oriented filament fragments (figure 3C). At a still more advanced stage, the muscle fibers were occupied by remnants of myofibrils surrounded by degenerating organelles (figure 4G), or multiple small vacuoles sometimes intermingled with the tubular profiles. Occasionally, some nuclei were surrounded by a matrix of irregularly oriented fine filaments intermingled with ribosomes (figure 4C).
Figure 3. Electron microscopy findings.

(A) Z-disk streaming and (B) Z-disk giving rise to expanses of tubulofilamentous material. (C, D) The tubulofilamentous material occupies vast expanses of the sarcoplasm. The imaged field (C) also displays a cytoplasmic body (CB) and fine irregularly oriented fragments of fine filaments (asterisk). (E) High-resolution image of the tubulofilaments. (A–D) Scale bar = 1 μm. (E) Scale bar = 0.2 μm.
Figure 4. Electron microscopy findings.

(A) Aggregate of rough endoplasmic reticulum profiles. (B) Clusters of dilated sarcotubular profiles intermingled with tubulofilamentous material. (C–F) Nuclear alterations. (C, E) The nuclei are filled with the tubulofilamentous material. (D) This material encases the nucleus. A matrix of irregularly oriented fine filaments intermingled with ribosomes is also seen (C). (F) A highly degenerate nucleus is filled with multiple small myeloid structures and debris. (G) A highly degenerate fiber containing remnants of myofibrils surrounded by degenerating organelles. (H) An abnormal fiber is invaded by a mononuclear cell. (A) Scale bar = 0.5 μm. (B–H) Scale bar = 1 μm in (B–H).
Of the nuclei, 13% (n = 146) were completely filled with the densely packed tubulofilamentous deposits (figure 4, C and E). Few other highly degenerate nuclei contained multiple small myeloid structures and debris (figure 4F). Rare nuclei harbored nemaline rods.
In patient 2, consistent with the autoaggressive inflammatory exudate noted by light microscopy (figure 2J), some abnormal fibers were focally invaded by mononuclear cells (figure 4H).
DISCUSSION
We identified 3 FHL1 mutations, 2 of which are novel in 4 kinships whose muscle specimens displayed MFM pathology. FHL1 has 3 recognized alternatively spiced isoforms with different tissue localizations and protein interactions; the mutations identified are present in each isoform (figure 1, C and D). Patient 1 and her daughter patient 2 carry His123Arg in the LIM2 domain. His123 is 1 of the 4 zinc atom ligands in this domain.19–22 Mutations of this residue to glutamine, tyrosine, or leucine in other patients have caused a severe early-onset myopathy. The loss of a zinc binding of His123 was proposed to be important for the pathogenesis,7 but more recent studies suggest that mutations of residue 123 expose a nonpolar surface causing protein aggregation.23 Patients 1 and 2 harboring His123Arg presented in their teens and were less severely affected than other patients harboring His123 mutations. A possible reason for this that is arginine has a basic side chain and is hydrophilic like histidine, whereas glutamine is acidic, and leucine and tyrosine are hydrophobic.
Patients 3 and 4 carry a previously reported Cys150Arg mutation. Cys150 is a crucial coordinating residue in the second LIM domain.24 Patient 3, a 35-year-old woman, has mild asymmetric atrophy and weakness of selected muscles that presented at approximately age 30. Patient 4, a 10-year-old boy presented at age 6 years with a rigid spine and rapidly progressive joint contractures. The rapid evolution of the joint contractures pointed away from the diagnosis of Emery-Dreifuss dystrophy. His clinical features are similar to those noted in a recently reported German family with the same mutation25; however, he also had a pseudoathletic appearance, a feature thought to be specific for late-onset X-linked scapulo-axio-peroneal myopathy with bent spine caused by mutations in the LIM4 domain.15 X-linked inactivation studies were not performed in our patients. Because of X chromosome inactivation, heterozygous women are mosaic for X-linked gene expression. This may explain the much milder phenotype in patient 3 compared with that in patient 4.
Patient 5 presented with mild muscle weakness in the seventh decade. He carries an Arg95Trp mutation, which is the first identified mutation external to an FHL1 LIM domain. The SIFT (Sorting Intolerant From Tolerant) database predicts that the mutation affects protein function, the PolyPhen program deems it probably damaging, and the dbSNP Build 133 database does not list it as a polymorphism. Because no family members of patient 5 were available, no segregation studies could be performed and its pathogenic role was not further confirmed. The pathology is typical of MFM, but this mutation is not associated with reducing bodies. Patient 5 also has peripheral sensorimotor neuropathy. FHL1 is expressed in different organs including the brain, but its expression in peripheral nerve has not been documented. The peripheral neuropathy was not reported previously in association with FHL1 mutations and whether it is caused by a defect in FHL1 remains an unresolved question.
The 3 patients harboring mutations in the second LIM domain have typical MFM pathology plus reducing bodies identified by the menadione-NBT reaction without substrate. Of special interest, the initial alteration involves not only streaming of the Z-disk but also reducing body material emanating from it that has not been observed in other types of MFM.26,27 High-resolution electron microscopy shows the reducing bodies composed of 13 ± 1.2-nm tubulofilaments. In fibers with more advanced changes, the tubulofilamentous bodies inundate the sarcoplasm and appear around and within numerous nuclei. Intranuclear tubulofilamentous inclusions are also characteristic features of oculopharyngeal muscular dystrophy, and cytoplasmic and intranuclear inclusions are features of sporadic and hereditary forms of inclusion body myositis. The tubulofilaments in oculopharyngeal dystrophy are 8.5 nm in diameter, are unbranched, course rectilinearly,28 and immunoreact for the mutant PABPN1 protein.29 The tubulofilaments in inclusion body myositis measure 18 ± 2 nm in diameter in the cytoplasm and 10–16 nm in the nuclei; they course rectilinearly, are arranged in parallel or at random, and, in some cases, are decorated with 5 ± 1-nm striations.30–33 In our patients, the tubulofilaments are nonbranching, are linear or slightly curving with a diameter of 13 ± 1.2 nm, and are decorated with ∼1.2-nm spicules. The inclusions of FHL1 dystrophy are at least partly composed of mutant FHL1 protein.6 The large aggregates of rough endoplasmic reticulum and clusters of dilated sarcotubular profiles are not observed in other types of MFM.26,27
The autoaggressive inflammatory exudate composed mostly of CD3+CD4+ T cells in patient 2 was also of interest. A plausible explanation for this finding is that degraded fiber organelles instigate a low-grade cell-mediated immune response. We have detected similar cell-mediated myocytotoxicity in a patient with myotilinopathy.34 Immunotherapy failed to mitigate the clinical course of either patient.
Are the MFMs, including FHL1-related myopathies, muscular dystrophies? We feel they are on the basis of the classic definition of Erb35 that muscular dystrophies are diseases caused by the primary degeneration of the muscle fiber. Muscle biopsies in MFM indeed show primary degeneration of the muscle fibers, with necrotic and regenerating fibers as well as replacement by fibrous connective tissue in advanced stages. The phenotypic features of different MFMs are like those of muscular dystrophies, and some were first reported as such. For example, myotilinopathy was reported as limb girdle muscular dystrophy 1A by the clinical criteria,36,37 and the αB-crystallinopathy of Canadian aboriginals was reported under the rubric of a fatal infantile muscular dystrophy.38,39
Finally, the clinical and pathologic features of the FHL1 muscular dystrophies can be typical of MFMs, but MFM pathology has not been described or may have been overlooked in some cases of FHL1-related muscular dystrophies. The presence of reducing bodies points to a mutation in the LIM2 domain of FHL1. Menadione stain is a useful, but not foolproof, marker for an FHL1 mutation in patients with MFM pathology.
Supplementary Material
GLOSSARY
- MFM
myofibrillar myopathy
- NBT
nitro blue tetrazolium.
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Dr. Selcen contributed to study concept, acquisition, analysis and interpretation of data and drafted the manuscript for content, including medical writing for content. Dr. Bromberg contributed to the acquisition and interpretation of data. Dr. Chen contributed to the acquisition and interpretation of data. Dr. Engel contributed to study concept, acquisition, analysis, and interpretation of data, and medical writing for content.
DISCLOSURE
Dr. Selcen has received research support from the NIH. Dr. Bromberg is a medical advisor for Accordant Health Care; has received funding from Rx Solutions and Talecris; is an Assistant Editor for Muscle & Nerve, Clinical Neurophysiology, and the Journal of Neuromuscular Diseases; receives royalties from Handbook of Peripheral Neuropathy and UpToDate; and is on the speaker's bureau of Talecris Biotherapeutics. Dr. Chin holds stock in Myriad Genetics, Inc. Dr. Engel serves as an Associate Editor of Neurology®; receives publishing royalties for Myology, 3rd ed. (McGraw-Hill, 2004); and has received research support from the NIH and the Muscular Dystrophy Association. Dr. Bromberg serves on a scientific advisory board for Accordant Health Services, Inc.; has received funding for travel and speaker honoraria from Rx Solutions and Talecris Biotherapeutics; serves as an Assistant Editor for Muscle & Nerve and the Journal of Neuromuscular Diseases and as Editor of Clinical Neurophysiology; receives publishing royalties for Handbook of Peripheral Neuropathy (Taylor & Francis, 2005) and Quality of Life Measurement in Neurodegenerative and Related Conditions (Cambridge, 2010); serves on the speakers' bureau for Talecris Biotherapeutics; and receives research support from Knopp Neurosciences Inc. and the NIH.
REFERENCES
- 1. Morgan MJ, Madgwick AJ. The LIM proteins FHL1 and FHL3 are expressed differently in skeletal muscle. Biochem Biophys Res Commun 1999; 255: 245– 250 [DOI] [PubMed] [Google Scholar]
- 2. Chu PH, Ruiz-Lozano P, Zhou Q, Cai C, Chen J. Expression patterns of FHL/SLIM family members suggest important functional roles in skeletal muscle and cardiovascular system. Mech Dev 2000; 95: 259– 265 [DOI] [PubMed] [Google Scholar]
- 3. Ng EK, Lee SM, Li HY, et al. Characterization of tissue-specific LIM domain protein (FHL1C) which is an alternatively spliced isoform of a human LIM-only protein (FHL1). J Cell Biochem 2001; 82: 1– 10 [DOI] [PubMed] [Google Scholar]
- 4. Cowling BS, Cottle DL, Wilding BR, D'Arcy CE, Mitchell CA, McGrath MJ. Four and a half LIM protein 1 gene mutations cause four distinct human myopathies: a comprehensive review of the clinical, histological and pathological features. Neuromuscul Disord 2011; 21: 237– 251 [DOI] [PubMed] [Google Scholar]
- 5. Windpassinger C, Schoser B, Straub V, et al. An X-linked myopathy with postural muscle atrophy and generalized hypertrophy, termed XMPMA, is caused by mutations in FHL1. Am J Hum Genet 2008; 82: 88– 99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schessl J, Zou Y, McGrath MJ, et al. Proteomic identification of FHL1 as the protein mutated in human reducing body myopathy. J Clin Invest 2008; 118: 904– 912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schessl J, Taratuto AL, Sewry C, et al. Clinical, histological and genetic characterization of reducing body myopathy caused by mutations in FHL1. Brain 2009; 132: 452– 464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Quinzii CM, Vu TH, Min KC, et al. X-linked dominant scapuloperoneal myopathy is due to a mutation in the gene encoding four-and-a-half-LIM protein 1. Am J Hum Genet 2008; 82: 208– 213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shalaby S, Hayashi YK, Goto K, et al. Rigid spine syndrome caused by a novel mutation in four-and-a-half LIM domain 1 gene (FHL1). Neuromuscul Disord 2008; 18: 959– 961 [DOI] [PubMed] [Google Scholar]
- 10. Gueneau L, Bertrand AT, Jais JP, et al. Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. Am J Hum Genet 2009; 85: 338– 353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Knoblauch H, Geier C, Adams S, et al. Contractures and hypertrophic cardiomyopathy in a novel FHL1 mutation. Ann Neurol 2010; 67: 136– 140 [DOI] [PubMed] [Google Scholar]
- 12. De Bleecker JL, Engel AG, Ertl BB. Myofibrillar myopathy with abnormal foci of desmin positivity. II. Immunocytochemical analysis reveals accumulation of multiple other proteins. J Neuropathol Exp Neurol 1996; 55: 563– 577 [DOI] [PubMed] [Google Scholar]
- 13. Nakano S, Engel AG, Waclawik AJ, Emslie-Smith AM, Busis NA. Myofibrillar myopathy with abnormal foci of desmin positivity. I. Light and electron microscopy analysis of 10 cases. J Neuropathol Exp Neurol 1996; 55: 549– 562 [DOI] [PubMed] [Google Scholar]
- 14. Selcen D, Ohno K, Engel AG. Myofibrillar myopathy: clinical, morphological and genetic studies in 63 patients. Brain 2004; 127: 439– 451 [DOI] [PubMed] [Google Scholar]
- 15. Schoser B, Goebel HH, Janisch I, et al. Consequences of mutations within the C terminus of the FHL1 gene. Neurology 2009; 73: 543– 551 [DOI] [PubMed] [Google Scholar]
- 16. Engel AG. The muscle biopsy. In: Engel AG, Franzini-Armstrong C, eds. Myology, 3rd ed New York: McGraw-Hill; 2004: 681– 690 [Google Scholar]
- 17. Wattenberg LW, Leong JL. Effects of coenzyme Q10 and menadione on succinic dehydrogenase activity as measured by tetrazolium salt reduction. J Histochem Cytochem 1960; 8: 296– 303 [DOI] [PubMed] [Google Scholar]
- 18. Brooke MH, Neville HE. Reducing body myopathy. Neurology 1972; 22: 829– 840 [DOI] [PubMed] [Google Scholar]
- 19. Way JC, Chalfie M. mec-3, a homeobox-containing gene that specifies differentiation of the touch receptor neurons in C. elegans. Cell 1988; 54: 5– 16 [DOI] [PubMed] [Google Scholar]
- 20. Freyd G, Kim SK, Horvitz HR. Novel cysteine-rich motif and homeodomain in the product of the Caenorhabditis elegans cell lineage gene lin-11. Nature 1990; 344: 876– 879 [DOI] [PubMed] [Google Scholar]
- 21. Karlsson O, Thor S, Norberg T, Ohlsson H, Edlund T. Insulin gene enhancer binding protein Isl-1 is a member of a novel class of proteins containing both a homeo- and a Cys-His domain. Nature 1990; 344: 879– 882 [DOI] [PubMed] [Google Scholar]
- 22. Schmeichel KL, Beckerle MC. Molecular dissection of a LIM domain. Mol Biol Cell 1997; 8: 219– 230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen DH, Raskind WH, Parson WW, et al. A novel mutation in FHL1 in a family with X-linked scapuloperoneal myopathy: phenotypic spectrum and structural study of FHL1 mutations. J Neurol Sci 2010; 296: 22– 29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Michelsen JW, Sewell AK, Louis HA, et al. Mutational analysis of the metal sites in an LIM domain. J Biol Chem 1994; 269: 11108– 11113 [PubMed] [Google Scholar]
- 25. Schessl J, Columbus A, Hu Y, et al. Familial reducing body myopathy with cytoplasmic bodies and rigid spine revisited: identification of a second LIM domain mutation in FHL1. Neuropediatrics 2010; 41: 43– 46 [DOI] [PubMed] [Google Scholar]
- 26. Claeys KG, Fardeau M, Schroder R, et al. Electron microscopy in myofibrillar myopathies reveals clues to the mutated gene. Neuromuscul Disord 2008; 18: 656– 666 [DOI] [PubMed] [Google Scholar]
- 27. Selcen D. Myofibrillar myopathies. Curr Opin Neurol 2010; 23: 477– 481 [DOI] [PubMed] [Google Scholar]
- 28. Tome FM, Chateau D, Helbling-Leclerc A, Fardeau M. Morphological changes in muscle fibers in oculopharyngeal muscular dystrophy. Neuromuscul Disord 1997; 7 (suppl 1): S63– S69 [DOI] [PubMed] [Google Scholar]
- 29. Calado A, Tome FM, Brais B, et al. Nuclear inclusions in oculopharyngeal muscular dystrophy consist of poly(A) binding protein 2 aggregates which sequester poly(A) RNA. Hum Mol Genet 2000; 9: 2321– 2328 [DOI] [PubMed] [Google Scholar]
- 30. Chou SM. Myxovirus-like structures and accompanying nuclear changes in chronic polymyositis. Arch Pathol 1968; 86: 649– 658 [PubMed] [Google Scholar]
- 31. Yunis EJ, Samaha FJ. Inclusion body myositis. Lab Invest 1971; 25: 240– 248 [PubMed] [Google Scholar]
- 32. Sato T, Walker DL, Peters HA, Resse HH, Chou SM. Chronic polymyositis and myxovirus-like inclusions: electron microscopic and viral studies. Arch Neurol 1971; 24: 409– 418 [DOI] [PubMed] [Google Scholar]
- 33. Lotz BP, Engel AG, Nishino H, Stevens JC, Litchy WJ. Inclusion body myositis: observations in 40 patients. Brain 1989; 112: 727– 747 [DOI] [PubMed] [Google Scholar]
- 34. Selcen D, Engel AG. Mutations in myotilin cause myofibrillar myopathy. Neurology 2004; 62: 1363– 1371 [DOI] [PubMed] [Google Scholar]
- 35. Erb W. Dystrophia muscularis progressiva: klinische und pathologisch-anatomische studien. Dtsch Z Nervenheilkd 1891; 1: 173– 261 [Google Scholar]
- 36. Gilchrist JM, Pericak-Vance M, Silverman L, Roses AD. Clinical and genetic investigation in autosomal dominant limb-girdle muscular dystrophy. Neurology 1988; 38: 5– 9 [DOI] [PubMed] [Google Scholar]
- 37. Hauser MA, Horrigan SK, Salmikangas P, et al. Myotilin is mutated in limb girdle muscular dystrophy 1A. Hum Mol Genet 2000; 9: 2141– 2147 [DOI] [PubMed] [Google Scholar]
- 38. Lacson AG, Seshia SS, Sarnat HB, et al. Autosomal recessive, fatal infantile hypertonic muscular dystrophy among Canadian Natives. Can J Neurol Sci 1994; 21: 203– 212 [DOI] [PubMed] [Google Scholar]
- 39. Del Bigio MR, Chudley AE, Sarnat HB, et al. Infantile muscular dystrophy in Canadian aboriginals is an αB-crystallinopathy. Ann Neurol 2011; 69: 866– 871 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.