

Abstract
A novel series of 5,7-dibromoisatin analogs were synthesized and evaluated for their cytotoxicities against four human cancer cell lines including colon HT29, breast MCF-7, lung A549 and melanoma UACC903. Analogs 6, 11 and 13 displayed good in vitro anticancer activity on the HT29 human colon cancer cell line in the 1 µM range. Analogs 5, 9 and 12, containing a selenocyanate group in the alkyl chain were the most promising compounds on the breast cancer MCF-7 cell line. Biological assays relating to apoptosis were performed to understand the mechanism of action of these analogs. Compounds 5 and 6 were found to inhibit tubulin polymerization to the same extent as the anticancer drug vinblastine sulfate, but compounds 11 and 13 inhibited significantly better than vinblastine. Further western blot analysis suggested that compound 6 at 2 µM reduced both levels and phosphorylation state of Akt. Compounds 11 and 13 at 1 µM caused reduced Akt protein levels and strongly suppressed the phosphorylation of Akt. Therefore, 11 and 13 were demonstrated as efficient dual inhibitors of both tubulin polymerization and the Akt pathway and good candidates for further study. More importantly, the strategy of microtubule and Akt dual inhibitors might be a promising direction for developing novel drugs for cancer.
Keywords: 5, 7-Dibromoisatin; Synthesis; cytotoxicity; Apoptosis; Tubulin polymerization
1. Introduction
The lack of selectivity of many anti-cancer agents and the occurrence of intrinsic or acquired resistance of tumors to chemotherapy has been major obstacles in the treatment of cancer. Microtubules have an important role in a variety of cellular process, including mitosis and cell division1, 2. Various anti-mitotic agents interfering with the natural dynamics of tubulin, the major protein component of microtubules, inhibit cancer cell growth3. Anti-mitotic agents such as paclitaxel stabilize microtubules by preventing the depolymerization of tubulin. The vinca alkaloids and colchicines inhibit the polymerization of tubulin. Anti-mitotic compounds have been used clinically in the treatment of different cancers. Although several antimitotic agents are available, due to the development of drug resistance, side effects and the structural complexity of vinca alkaloids and taxoids, there is still a need to identify novel anticancer drugs that effectively target microtubules4, 5.
Protein kinase B, also known as Akt, is a 57-kDa serine/threonine kinase plays a critical role in anti-apoptotic processes6. Overexpression of Akt can result from inactivation of the tumor suppressor PTEN and has been correlated with an increasing number of human cancers7. Akt is also responsible for promoting survival signals that down regulate apoptotic pathways and contribute to cancer progression. Correlation between resistance to chemotherapy and Akt activation has also been observed in prostate cancer cell lines and in human tumor tissue8. Inhibition of Akt alone or in combination with other standard cancer chemotherapeutics results in increased programmed death of cancer cells leading to decreased tumor growth and tumor resistance to chemotherapy.
The isatin (1H-indole-2,3-dione) 1 is found as an endogenous molecule in humans and other mammals and its analogs display diverse types of biological activities including anticancer activities9–11. It is an oxidized derivative of an indole moiety; many of the indole heterocycles are tubulin polymerization inhibitors12–14. In addition, many indole-based compounds appear to act as inhibitors of various protein kinase families, particularly receptor tyrosine kinases (RTKs) and serine/threonine-specific protein kinases such as the cyclic-dependent kinases (CDKs). SU11248 (Sutent), a 5-fluoro-3-substituted-2-oxoindole is approved by the US FDA for the treatment of advanced renal carcinoma and gastrointestinal stromal tumors15, 16. Recently, it has been reported that 5,7-dibromoisatin 2 is significantly more potent in vitro as a cytotoxic agent than the parent molecule 1 (Scheme 1) against U937 (human monocyte-like histiocytic lymphoma) cells11. Moreover, N-benzylation of 5,7-dibromoisatin 2 further increased the cytotoxicity and targeting of microtubules in these lymphoma cells and was potent against a range of human cancer cell lines including a metastatic breast adenocarcinoma cell line (MDA-MB-231)17. In this context, it was of interest to investigate further the cytotoxicity of N-alkylated 5,7-dibromoisatin analogs by altering the chain length at N-1 to increase the lipophilicity and substitution of the functional groups containing isothiocyanate (ITCs), thiocyanate and selenocyanate in the alkyl chain. These functionalities were chosen because of the well-known anti-cancer properties shown by the agents having these moieties. For example, ITCs, popular chemopreventive agents present in cruciferous vegetables in the form of glucosinolates, provide growth-inhibiting and apoptosis-inducing activities in cancer cell lines in vitro18–20. Isothiocyanates are among the most effective naturally occurring cancer chemopreventive agents in animal models21, 22. In addition, epidemiological studies have demonstrated that the human consumption of isothiocyanates in vegetables decreases cancer risk23, 24. ITCs have been shown to exhibit the anticarcinogenic effects through dual mechanisms occurring at the level of initiation of carcinogenesis by blocking phase I enzymes (cytochrome P-450) that activate procarcinogens and by inducing phase II enzymes that detoxify electrophilic metabolites generated by phase I enzymes25–29. Certain studies suggest the mechanism of action of ITCs is inhibition of the PI3 kinase pathway21, 30. Our recent studies have also shown that isothiocyanate/isoselenocyanate compounds to be effective in inhibiting PI3K/Akt pathway31–35. Therefore, the use of this functional group was hoped to impart Akt inhibition to the isatin compounds. Selenium is also an effective chemopreventive agent and is known to modulate Akt activity36–38. The rationale for adding the selenocyanate group was that the selenium compounds have been found to inhibit and/or retard tumorigenesis in a variety of experimental animal models39–41. Epidemiologic studies have reported an inverse association between the nutritional selenium status and cancer risk42, suggesting that a relatively low Se status may be among the determinants of cancer risk. We have also shown selenium compounds to be effective in inhibiting tumor growth in melanoma31–34 and colon43 xenograft models. Specifically, several synthetic alkyl and aryl selenocyanates have been evaluated for anticarcinogenicity in animal models. The more effective of these are benzylselenocyanate (BSC) and 1,4-phenylenebis-(methylene)selenocyanate (p-XSC)44–47.
Scheme 1.

Synthesis of compounds 2–13 derivatives of 5, 7-dibromoisatin. Reagents and conditions; (a) Br2/EtOH; (b) K2CO3/DMF, RT − 80 °C, Cl(CH2)nBr, 4–18 h; (c) K2CO3/DMF, RT, BrCH2C6H4CH2Br, 8 h; (d) K2CO3/DMF, RT − 80 °C, Br(CH2)3NHBoc,12 h; (e) K2CO3/DMF, RT − 80 °C, BrCH2C6H4CH2NHBoc, 16 h; (f) KI/CH3CN, KSCN or KSeCN; (g) CF3COOH, CH2Cl2, K2CO3, CH2Cl2, CSCl2.
From the combined literature survey of isatin derivatives, ITCs and selenocyanates, we hypothesized that combination of indole heterocycle with thiocyanate, isothiocyanate and selenocyanate moieties would yield novel dual targeted inhibitors for cancer therapy. Here, we provide a first report of the activity of such novel agents, which are significantly toxic to cancer cells in culture by inhibition of both tubulin polymerization and Akt phosphorylation and expression. For these studies, we synthesized 12 derivatives of 5,7-dibromoisatin containing thiocyanate, isothiocyanate, and selenocyanate groups in the alkyl chain. In vitro screening against various cancer cell lines was carried out in order to establish a more comprehensive structure–activity relationship (SAR).
2. Results and discussion
2.1. Synthesis
The general synthesis of N-propyl, N-butyl, and N-benzyl series of 5,7-dibromoisatin listed in Table 1 is shown in Scheme 1. Compounds 2–13 (Table 1) were prepared in good yield in two or three steps. The first step consisted of alkylating 2,7-dibromoisatin with 1-bromo-3-chloropropane, l-bromo-4-chlorobutane and 1,4-bis(bromomethyl)benzene to prepare 5,7-dibromo-N-(3’-chloropropyl)isatin (3), 5, 7-dibromo-N-(4’-chloropropyl)isatin (7) a n d 5, 7-dibromo-N-(p-bromomethylbenzyl)isatin (10). Alkylation was accomplished by first converting 5,7-dibromoisatin to the anionic species using the base, K2CO3 in DMF17. Iodide-catalyzed nucleophilic substitution of the N-propyl or -butyl chloride and N-(p-methylbenzyl) bromide of 2,7-dibromoisatin with KSCN and KSeCN by stirring in anhydrous acetonitrile at RT, afforded the thiocyanates 4, 8, 11 and selenocyanates 5, 9, 12, respectively in good yield (Scheme 1).
Table 1.
Cytotoxicity of 5,7-dibromoisatin derivatives 2–13, on HT29a, MCF-7b, A549c and UACC903d as calculated from dose response curvese.
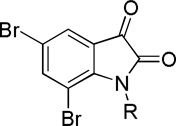 | |||||
|---|---|---|---|---|---|
| Compound | R | IC50 (µM)f ± SD | |||
| HT29 | MCF-7 | A549 | UACC903 | ||
| 48 h | 48 h | 48 h | 48 h | ||
| 2 | H | >50 | 22.45 ± 1.12 | >50 | 26.5 ± 1.8 |
| 3 | —(CH2)3—Cl | 2.57 ± 0.10 | 21.51 ± 1.31 | 5.27 ± 0.32 | 8.57 ± 0.94 |
| 4 | —(CH2)3—SCN | 3.24 ± 0.25 | 9.78 ± 0.53 | 3.53 ± 0.18 | 2.89 ± 0.18 |
| 5 | —(CH2)3—SeCN | 3.18 ± 0.15 | 1.65 ± 0.24 | 3.46 ± 0.27 | 21.64 ± 1.09 |
| 6 | —(CH2)3—N=C=S | 1.56 ± 0.11 | 8.96 ± 0.38 | 2.13 ± 0.19 | 4.37 ± 0.30 |
| 7 | —(CH2)4—Cl | 2.51± 0.21 | 14.86 ± 0.43 | 3.81 ± 0.22 | 3.74 ± 0.26 |
| 8 | —(CH2)4—SCN | 2.42 ± 0.19 | 22.13 ± 1.10 | 5.53 ± 0.18 | 2.06 ± 0.15 |
| 9 | —(CH2)4—SeCN | 2.39 ± 0.30 | 1.53 ± 0.17 | 3.15 ± 0.26 | 9.03 ± 0.34 |
| 10 | 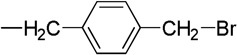 |
5.10 ± 0.22 | 27.74 ± 1.52 | 7.86 ± 0.31 | 2.64 ± 0.16 |
| 11 | 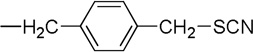 |
1.14 ± 0.16 | 24.70 ± 1.34 | 2.53 ± 0.15 | 2.23 ± 0.11 |
| 12 | 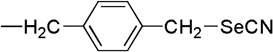 |
2.59 ± 0.33 | 1.45 ± 0.11 | 2.41 ± 0.19 | 12.12 ± 0.78 |
| 13 | 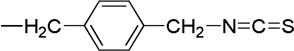 |
1.09 ± 0.09 | 12.01 ± 0.89 | 3.82 ± 0.27 | 3.11 ± 0.29 |
| Vinblastine | 0.55 ± 0.08 | NTg | NT | 1.65 ± 0.16 | |
HT29: human colorectal cancer cells,
MCF-7: human breast cancer (non-metastatic) cell line.
A549: human lung epithelial carcinoma cells,
UACC903: human melanoma cancer cells,
Sigmoidal dose response curves (variable slope) were generated using GraphPad Prism V. 4.02 (GraphPad Software Inc.).
Values are the mean of triplicates of at least two independent experiments.
NT = not tested.
The isothiocyanate derivatives 6 and 13 (Scheme 1) were synthesized by the treatment of 5,7-dibromoisatin with tert-butyl 3-bromopropylcarbamate or tert-butyl(4-bromomethyl benzyl)carbamate in the presence of K2CO3 in DMF, to afford Boc-protected intermediates 14 and 15, respectively. The Boc group in 14 and 15 was removed by trifluroacetic acid, followed by a reaction with thiophosgene with K2CO3 in anhydrous methylene chloride to give 6 and 13 in good yield. All of these compounds were purified by column chromatography or recrystallization and dried under high vacuum. The purity of the compounds was tested by HPLC, ≥ 99% pure compounds were used for biological assays (data not shown).
2.2. Biological Characterization
2.2.1. Cytotoxicity studies
The cytotoxicity of a series of new N-alkyl derivatives of 5,7-dibromoisatin was evaluated against a panel of four different human cancer cell lines including a colon (HT29), breast (MCF-7), lung (A549) and melanoma (UACC903), after a continuous exposure of 48h. The results are summarized in Table 1. All the compounds exhibited significant cytotoxicity with an IC50 values of <5 µM in HT29 cell line; compounds 6, 11 and 13 showed relatively higher potency with IC50 values of 1.56, 1.14 and 1.09 µM, respectively. The results showed that the cytotoxicity of compound 2 significantly increased through N-alkylation, as reported previously for the 5,7-dibromoisatin derivatives17. Compounds 3–6, contain a three-carbon linker, compounds 7–9 contain a four-carbon linker, and compounds 10–13 contain an aromatic ring with a one-carbon linker at the nitrogen N1. The findings showed that, by slightly increasing the hydrophobicity, there is no significant change in the inhibiting activity between N-butyl series (compounds 7–9) and N-propyl series (compounds 3–6). Introduction of N-benzyl with isothiocyanate/thiocyanate groups to the alkyl chain yielded relatively more active compounds (11 and 13) in this series against the growth of HT29 colon cancer cells.
The MCF-7 cell line was susceptible to compounds 5, 9 and 12 with IC50 values of 1.65, 1.53 and 1.45 µM, respectively. All other compounds were less potent against MCF-7 cells. This effect may be due in part to the expression of the anti-apoptotic Bcl-2 group of proteins that have been identified in MCF-7 cells48–51. Substitution of SeCN group in the alkyl chain (5, 9 and 12), showing high inhibitory activity compared to the other compounds against MCF-7 cells, indicates that selenium might be playing a role in the antiproliferative activity presumably through the inhibition of anti-apoptotic Bcl-2 group of proteins. These selenium substituted compounds also showed good inhibitory activity against HT29 and A549 cell lines, but poor inhibitory activity in UACC903 cells. ITCs and thiocyanates did not diminish the cytotoxic activity against the MCF-7 cell lines, but were more potent in colon and lung cancer cell lines. Selenium compounds were more potent than corresponding sulfur containing compounds in killing MCF-7 cells. All the isatin compounds were 10–25 fold more potent than 2 against A549 cell lines. In the presence of 6, 11, and 12 the growth of A549 cells was inhibited, with IC50 values of 2.13, 2.53 and 2.41 µM, respectively. The thiocyanate substituted derivatives 4, 8, 11 showing significant growth inhibitory activity against melanoma UACC903 cells with IC50 values between 2.06 and 2.89 µM. To examine whether isatin compounds are toxic to normal cells, mouse embryonic fibroblasts (MEF) cells were treated with compounds 6 and 11 for 48 h, later viability was evaluated by MTS assay. Both compounds 6 and 11 exhibited IC50 > 20 µM (data not shown).
2.2.2. Apoptosis in cultured colon cancer cells
To assess the effect of isatins on cell growth inhibition and apoptosis, we selected compounds 5, 6, 11 and 13 for an apoptosis study using HT29 cells. HT29 cells were sensitive to these compounds with IC50 values 3.18, 1.56, 1.14 and 1.09 µM respectively (Table 1). We exposed HT29 cells grown in complete medium with 0.1% Me2SO4, to 5, 6, 11 and 13 at 1.25, 2.5 and 5 µM concentrations for 24h. Cells were harvested, and the extent of apoptosis was evaluated by the annexin V/7-Aminoactinomycin D (7-AAD) method. Apoptosis was measured by quantifying the PE annexin V and 7-AAD positive cells using flow cytometry (BD FACScan). Within the apoptotic population, cells in the early stages of apoptosis were annexin V-positive and 7-AAD-negative, whereas those in the late stages were annexin V-positive and 7-AAD–positive (Figure 1). Approximately 6–7% of cells in the control population were undergoing spontaneous apoptosis. Treatment with 5, 6, 11 and 13 induced apoptosis dose-dependently and at 5 µM, 56, 61, 67 and 61% of cells underwent apoptosis, respectively. These populations are combined and presented in (Table 2). Thus growth inhibition appears to correlate well with the concentration of isatins that induce apoptosis in HT29 cells. As an additional indication of apoptosis occurring in those cells, caspases-3/7 activity, which plays a key role in apoptosis, was measured. Caspases-3/7 activity was increased in a dose-dependent manner to a maximum of 16 fold in response to isatins compared to control (Figure 2). To confirm that the observed reduction in viability of HT29 cells occurred via induction of apoptosis, we used TUNEL staining to measure DNA fragmentation as an early hallmark of apoptotic cell death in treated cells. HT29 cells were treated with compounds 6 and 11, at 5 µM concentration for 48 h. Fragmented DNA of apoptotic cells were stained using an Apop Tag Red In Situ Apoptosis Detection Kit and visualized by fluorescence microscopy using appropriate filters. As evidenced in Panels c and d of (Figure 3), compound 6 and 11 resulted in a significant number of TUNEL-positive colon cancer cells.
Figure 1.

Quantitative analysis of cell apoptosis induced by 5, 7- dibromoisatin derivatives. Apoptosis was measured by using the PE Annexin V and 7-AAD staining. Cells were analyzed and quantified by using flow cytometry (BD FACScan). Human colon cancer cells HT29 treated with compounds 5, 6, 11 and 13 (1.25 – 5.0 µM) or DMSO as a control for 24 h at 37 °C. At the end of incubation, cells were harvested and washed twice with cold PBS. Cells were resuspended in binding buffer at a concentration of 1 × 106 cells/ml and stained with 7-AAD and PE Annexin V and incubated for 15 min at RT in the dark.
Table 2.
Summary of % of HT29 cells undergoing apoptosis after a 24 h treatment with compounds 5, 6, 11 and 13 at 1.25, 2.5 and 5 µM concentrations.
| Compound No. | 1.25 µM | 2.5 µM | 5 µM |
|---|---|---|---|
| Control (DMSO) | - | - | 6.8 |
| 5 | 7.92 | 10.44 | 56.28 |
| 6 | 8.54 | 21.23 | 61.5 |
| 11 | 11.47 | 39.46 | 66.98 |
| 13 | 16.38 | 36.64 | 61.23 |
Figure 2.

Isatin analogs induce apoptosis in cultured colon cancer cell lines by the activation of caspase 3/7. Levels of caspase-3/7activity were measured in HT29 cells exposed to compounds 5, 6, 11, and 13 using the Apo-ONE homogeneous caspase-3/7 assay kit. Results show significant dose dependent increases in caspase-3/7 activity relative to DMSO vehicle treated cells. Data are means ± SD of one representative experiment performed in triplicate.
Figure 3.

Detection of in situ DNA fragmentation was analyzed by TUNEL staining after 48 h treatment with the compounds. HT29 cells were grown for 24 hours and treated with 5 µM compound 6, and 11. The blue stain (a, b) represents the nuclear Hoechst stain. The red stain (c, d) determines the extent of DNA fragmentation or damage. The overlay figures (e, f) showing the number of DNA fragmented cells (red) to the total number of cells (blue).
2.2.3. Effects on Tubulin Polymerization and Microtubule Formation
Several tubulin polymerization inhibitors characterized by the presence of an indole nucleus have been obtained from natural sources or have been prepared by semi-synthesis. The indole heterocyclic nucleus is central to a large number of tubulin polymerization inhibitors12, 14, 52. Isatins are oxidized derivatives of an indole moiety, and 5,7-dibromo-N-benzylisatin derivatives interfere with microtubule dynamics. Compounds 5, 6, 11, and 13 were chosen as representative molecules to further investigate their ability to alter tubulin polymerization in vitro. To investigate whether the antiproliferative activities of compounds 5, 6, 11 and 13 derived from an interaction with tubulin, they were evaluated for their inhibition of tubulin polymerization in a cell-free in vitro assay. Paclitaxel and vinblastine sulfate were used as a known microtubule stabilizer and destabilizer, respectively. The results of both paclitaxel and vinblastine were consistent with the literature reports53, 54. At 10 µM, paclitaxel stabilized microtubules, in comparison to the vehicle control, while vinblastine strongly inhibited microtubule formation at the same concentration (Figure 4). The test compounds 11 and 13 more strongly inhibited about 71% and 77% respectively the rate of microtubule polymerization at 10 µM, than vinblastine (66%) (Figure 4). Analogs 5 (63%) and 6 (62%) were almost similar to vinblastine as inhibitors of tubulin assembly. This suggests that both benzyl and thiocyanate/isothiocyanate groups in 11 and 13 are playing a role in the maximum inhibition of tubulin assembly. By comparison, both compounds 6 and 13 have isothiocyanate functional group, compound 6 was slightly less active than 13 as a microtubule destabilizer, suggesting that, N-benzyl substitution is more important than N-propyl for microtubule destabilization.
Figure 4.

The effect of compounds 5, 6, 11, 13, paclitaxel, and vinblastine sulfate at 10 µM on the assembly of purified bovine neuronal tubulin. DMSO was used as a control. A shift of the curve to the left or right of the control is indicative of either an increase or a decrease, respectively, in the rate of tubulin polymerization. Changes in fluorescence were measured at an excitation wavelength of 360±10 nm, and the fluorescence was collected at 440±10 nm. Data points are the means of duplicate experiments.
2.2.4. Inhibition of Akt phosphorylation
To determine the effects of the compounds on Akt, Western blot analysis was performed. Cells were treated for 24 hours, and Western blots were performed on the lysates (Figure 5). The blots were probed for phospho-Akt (top panel) and for total Akt (middle panel). Results show that compound 6 at 1 µM had very little effect on either expression or phosphorylation of Akt, while at 2 µM both levels and phosphorylation state of Akt were reduced. For compounds 11 and 13, Akt levels were reduced but the phosphorylation was virtually eliminated, indicating that the Akt present was not active. These results indicate that both compounds 11 and 13 are more potent Akt inhibitors than compound 6 and that in addition to inhibition of activity, the drugs down regulate the expression of the proteins. Comparison with N-propyl isothiocyanate (6) and N-benzyl thiocyanate/isothiocyanate (11 and 13), benzyl group gave more potency to the isothiocyanate/thiocyanate for the Akt inhibition.
Figure 5.

Inhibition of Akt phosphorylation by compounds 6, 11, and 13. HT29 cells were treated for 24 hours with drugs at either 1 or 2 µM, or with no drug. Cell lysates were assayed by Western blot analysis for phospho Akt or total Akt. Lower panel shows actin loading control.
3. Conclusions
From Table 1, it is interesting to note that the N-alkylation with thiocyanate, isothiocyanate and selenocyanate moieties apparently play an important role in the activity of these compounds. Compounds with N-propyl or N-benzyl are essential for activity, but N-butyl does not improve the potency of the compounds. Compounds 5 and 6 inhibited tubulin polymerization to the same extent as anticancer drug vinblastine sulfate. Compounds 11 and 13 were found to inhibit tubulin polymerization greater than the vinblastine. Further western blot analysis suggested that compound 6, at 1 µM had very little effect on either expression or phosphorylation of Akt, while at 2 µM both levels and phosphorylation state were reduced. Compounds 11 and 13 reduced Akt levels and strongly suppressed the phosphorylation of Akt. Introduction of a selenocyanate moiety in the alkyl chain (compounds 5, 9, and 12) showed higher cytotoxicity than thiocyanate and isothiocyanate against MCF-7 breast cancer cell lines. Overall compounds 11 and 13 emerged as dual inhibitors of tubulin polymerization and the Akt signaling pathway and the most promising candidates for further investigation in vivo as antitumor agents for colon cancer.
4. Experimental Section
4.1. Chemistry
Isatin was purchased from Sigma-Aldrich, 5,7-dibromoisatin was synthesized using previously reported methods11. All other chemicals and solvents were purchased from the major vendors. Anhydrous solvents were used as received. Reactions were carried out using dried glassware and under an atmosphere of nitrogen. Reaction progress was monitored with analytical thin-layer chromatography (TLC) on aluminum backed precoated silica gel 60 F254 plates (E. Merck). The N-alkylisatins were highly colored and would usually be clearly seen on a TLC plate; colorless compounds were detected using UV light and/or iodine vapor. Column chromatography was carried out using silica gel 60 (230–400 mesh, E. Merck) with the solvent system indicated in the individual procedures. All solvent ratios are quoted as vol/vol. NMR spectra were recorded using a Bruker Avance 500 MHz spectrometer. Chemical shifts (δ) were reported in parts per million downfield from the internal standard. The signals are quoted as s (singlet), d (doublet), t (triplet), m (multiplet), dd (doublet of doublet). Spectra are referenced to the residual solvent peak of the solvent stated in the individual procedure. High-resolution mass spectra (HRMS) were determined on Thermo Electron MAT 95XL magnetic sector mass spectrometer operating at 70eV for EI with a source temperature at 180 °C and were referenced with PFK and at 5kV for ESI operating with a source temperature at 250 ° C and were referenced with polyethylene amine. Melting points were determined on a Fischer-Johns melting point apparatus and are uncorrected.
4.1.2. 5, 7-Dibromo-N-(3’-chloropropyl)isatin (3)
5, 7-dibromoisatin 2 (1 g, 3.28 mmol) was taken up in anhydrous DMF (30 mL) and cooled on ice with stirring. Solid K2CO3 (544 mg, 3.94 mmol) was added in one portion, and the dark colored suspension was brought to room temperature and stirred for a further 1h. 1-Bromo-3-chloropropane (620 mg, 3.94 mmol, 0.387 mL) was added slowly with constant stirring and the reaction mixture was stirred at 80 °C for 4–8 h, until the 5,7-dibromoisatin starting material had been consumed (TLC). The reaction mixture was poured into HCl (0.5 M, 50 mL) and extracted with ethyl acetate (3×50 mL). The ethyl acetate layer was washed with brine and dried over MgSO4. The solvent was removed, and the crude product was purified by silica gel column chromatography (CH2Cl2 as eluent) to give pure 3 (0.93 g, 74%) as orange red crystals, mp 130–132 °C. 1H NMR (CDCl3, 500 MHz) δ 2.24–2.39 (m, 2H, H2’), 3.50(t, 1H, J = 6.5 Hz, H3’), 3.67 (t, 1H, J = 6.5 Hz, H3’), 4.31–4.36 (m, 2H, H1’), 7.72 (d, J = 2 Hz, 1H, isatin ArH), 7.91 (d, J = 2.5 Hz, 1H, isatin ArH). HRMS (EI) M+ calcd for C11H8O2N179Br235Cl1, 378.8605; found, 378.8609.
4.1.3. 5, 7-Dibromo-N-(3’-thiocyanatopropyl)isatin (4)
Compound 3 (200 mg, 0.52 mmol) was dissolved in anhydrous acetonitrile (15 mL), and KI (300 mg, 1.81 mmol) and KSCN (200 mg, 2.05 mmol) were added. The mixture was stirred overnight until a substantial amount of product was formed. The reaction mixture was diluted with water and extracted with ethyl acetate (3 × 50 mL). The organic layer was washed with water, dried over anhydrous MgSO4, and evaporated to dryness on the rotary evaporator. The crude product was purified by silica gel column chromatography (CH2Cl2-hexane, 7:3) eluted the product 4 as a yellow-orange solid (140 mg, 65%), mp 118–120 °C. 1H NMR (CDCl3, 500 MHz) δ 2.36 (m, 2H, H2’), 3.06(t, 1H, J = 7Hz, H3’), 4.35 (t, 2H, J = 6.5Hz, H1’), 7.74 (d, J = 2 Hz, 1H, isatin ArH), 7.93 (d, J = 2 Hz, 1H, isatin ArH). 13C NMR (CDCl3, 126 MHz) δ 30.1, 31.2, 39.8, 104.7, 111.7, 117.4, 121.4, 127.8, 145.3, 146.1, 158.3, 180.9. HRMS (EI) M+ calcd for C12H8O2N279Br2S1, 401.8668; found, 401.8675.
4.1.4. 5, 7-Dibromo-N-(3’-selenocyanatopropyl)isatin (5)
The procedure used for 4 was repeated with (200 mg, 0.52 mmol) of 3, KI (300 mg, 1.81 mmol) and KSeCN (200 mg, 1.39 mmol). The product was a bright orange solid (158 mg, 67%), mp 120–122 °C. 1H NMR (CDCl3, 500 MHz) δ 2.45 (m, 2H, H2’), 3.11(t, 1H, J = 7 Hz, H3’), 4.35 (t, 2H, J = 6.5 Hz, H1’), 7.73 (d, J = 2 Hz, 1H, isatin ArH), 7.92 (d, J = 2 Hz, 1H, isatin ArH). 13C NMR (CDCl3, 126 MHz) δ 26.1, 31.0, 40.7, 101.1, 104.7, 117.4, 121.4, 127.8, 145.3, 146.2, 158.5, 180.8. HRMS (EI) M+ calcd for C12H8O2N279Br280Se, 449.8112; found, 449.8113.
4.1.5. 5, 7-Dibromo-N-(3’-isothiocyanatopropyl)isatin (6)
The procedure used for 3 was repeated with (500 mg, 1.64 mmol) of 2, 15 ml of anhydrous DMF, KI (300 mg, 1.81 mmol), K2C03 (272 mg, 1.97 mmol) and (470 mg, 1.97 mmol) of tert-butyl 3-bromopropyl carbamate. The crude product was purified by silica gel column chromatography (hexanes/EtOAc, 80/20) to obtain 5,7-dibromo-N-[3’-(tert-butylcarbamate) propyl]isatin 14 as an orange-yellow solid (470 mg, 62%), mp 94–96 °C. 1H NMR (CDCl3, 500 MHz) δ 1.46 (s, 9H, H t-butyl), 1.98 (m, 2H, H2’), 3.22 (q, 2H, J = 6Hz, H3’), 4.23 (t, 2H, J = 7Hz, H1’), 7.70 (d, J = 2 Hz, 1H, isatin ArH), 7.89 (d, J = 2 Hz, 1H, isatin ArH). HRMS (EI) M+ calcd for C16H18O4N279Br2, 459.9633; found, 459.9613.
To a solution of 5, 7-dibromo-N-[3’-(tert-butylcarbamate)propyl]isatin 14 (400 mg, 0.89 mmol) in methylene chloride (20 mL) was added trifluroacetic acid (1.5 mL) at 0 °C, and the reaction was stirred at room temperature for 12 h. The mixture was then concentrated to give a solid which was washed with diethyl ether, affording trifluroacetate salt of 5,7-dibromo-N-(3’-aminopropyl)isatin as an orange red solid (460 mg, 96%). The whole salt was carried to synthesis of isothiocyanate, suspended in CH2Cl2 and (1.3 g, 9.4 mmol) of K2CO3. Thiophosgene (112 mg, 74 µl, 0.96 mmol) was added slowly and stirred 12 h at RT. The product was extracted with more CH2Cl2 and washed with water, dried with MgSO4 and chromatographed on silica gel column, (hexanes/CH2Cl2, 20/80) obtained 6 as a bright orange red solid (210 mg, 54%), mp 131–133 °C. 1H NMR (CDCl3, 500 MHz) δ2.18 (m, 2H, H2’), 3.22 (t, 2H, J = 6Hz, H3’), 4.34 (t, 2H, J = 7Hz, H1’), 7.72 (d, J = 2 Hz, 1H, isatin ArH), 7.91 (d, J = 2 Hz, 1H, isatin ArH). 13C NMR (CDCl3, 126 MHz) δ 29.7, 39.2, 42.7, 104.7, 117.2, 121.4, 127.7, 131.5, 145.2, 146.2, 158.1, 180.9. HRMS (EI) M+ calcd for C12H8O2N279Br2S, 401.8668; found, 401.8675.
4.1.6. 5, 7-Dibromo-N-(4’-chlorobutyl)isatin (7)
The procedure used for 3 was repeated with (1 g, 3.28 mmol) of 2, (544 mg, 3.94 mmol) of K2CO3, anhydrous DMF (30 mL) and of 1 bromo-4-chlorobutane (452 µL, 3.94 mmol). The product was chromatographed on the silica gel with CH2Cl2 as eluent yielded 7as an orange-red solid (0.98 g, 75%), mp 80–82 °C. 1H NMR (CDCl3, 500 MHz) δ 1.89–1.99 (m, 4H, H2’ and H3’), 3.48(t, 1H, J = 6 Hz, H4’), 3.61 (t, 1H, J = 6.0 Hz, H4’), 4.19 (t, J = 7 Hz, 2H, H1’), 7.71 (d, J = 2 Hz, 1H, isatin ArH), 7.90 (d, J = 2.5 Hz, 1H, isatin ArH). HRMS (EI) M+ calcd for C12H10O2N79Br235Cl, 392.8761; found, 392.8770.
4.1.7. 5, 7-Dibromo-N-(4’-thiocyanatobutyl)isatin (8)
The procedure used for 4 was followed with (200 mg, 3.66 mmol) of 7, KI (300 mg, 1.81 mmol) and KSCN (200 mg, 2.06 mmol) to obtain 8. The product was chromatographed on the silica gel with CH2Cl2-hexane (7:3), yielded 8 as an orange-yellow solid (144 mg, 68%), mp 120–122°C). 1H NMR (CDCl3, 500 MHz) δ 1.96 (m, 4H, H2’ and H3’), 3.05(t, 2H, J = 7Hz, H4’), 4.21 (t, 2H, J = 6.5Hz, H1’), 7.72 (d, J = 2 Hz, 1H, isatin ArH), 7.91 (d, J = 2 Hz, 1H, isatin ArH). HRMS (EI) M+ calcd for C13H10O2N279Br2S, 415.8824; found, 415.8824.
4.1.8. 5,7-Dibromo-N-(4’-selenocyanatobutyl)isatin (9)
The procedure used for 5 was repeated with (200 mg, 3.66 mmol) of 7, (300 mg, 1.81 mmol) of KI, (200 mg, 1.39 mmol) of KSeCN. The product was an orange red solid (158 mg, 67%), mp 123–125 °C. 1H NMR (CDCl3, 500 MHz) δ 1.95–2.06 (m, 4H, H2’and H3’), 3.12(t, 1H, J = 7Hz, H4’), 4.21 (t, 2H, J = 7.5Hz, H1’), 7.71 (d, J = 1.5 Hz, 1H, isatin ArH), 7.90 (d, J = 2 Hz, 1H, isatin ArH). HRMS (EI) M+ calcd for C13H10O2N279Br280Se, 463.8269; found, 463.8273.
4.1.9. 5, 7-Dibromo-N-(p-bromomethylbenzyl)isatin (10)
The procedure used for 3 was repeated with 5,7-dibromoisatin (1 g, 3.28 mmol), (544 mg, 3.94 mmol) of K2CO3, anhydrous DMF (30 mL) and of 1,4-bis(bromomethyl)benzene (1.04 g, 3.94 mmol). Compound 10 was obtained as an orange red solid (1.11 g, 75%), mp 140–142 °C). 1H NMR (CDCl3, 500 MHz) δ 4.49 (s, 2H, H1’), 5.42 (s, 2H, CH2Br), 7.24 (d, J = 8 Hz, 2H, phenyl ArH), 7.39 (d, J = 8.5Hz, 2H, phenyl ArH), 7.75 (d, J = 2 Hz, 1H, isatin ArH) 7.85 (d, J = 2 Hz, 1H, isatin ArH). HRMS (EI) M+ calcd for C16H10O2N79Br3, 484.8256; found, 484.8256.
4.1.10. 5, 7-Dibromo-N-(p-thiocyanomethylbenzyl)isatin (11)
Compound 11 in the pure form was prepared following the procedure used for 4 with (200 mg, 0.44 mmol) of 10, (300 mg, 1.81 mmol) of KI, and (200 mg, 2.06 mmol) of KSCN. The product was a yellow-orange solid (144 mg, 74%), mp 162–164 °C. 1H NMR (CDCl3, 500 MHz): δ 4.16 (s, 2H, H1’), 5.44 (s, 2H, CH2SCN), 7.31 (d, J = 8 Hz, 2H, phenyl ArH), 7.38 (d, J = 8.5Hz, 2H, phenyl ArH), 7.76 (d, J = 2 Hz, 1H, isatin ArH) 7.86 (d, J = 2 Hz, 1H, isatin ArH). 13C NMR (CDCl3, 126 MHz): δ 37.8, 44.4, 105.1, 111.7, 117.4, 121.4, 127.6, 129.5, 134.0, 136.6, 145.3, 146.5, 158.2, 181.1. HRMS (EI) M+ calcd for C17H10O2N279Br2S, 463.8824; found, 463.8831.
4.1.11. 5, 7-Dibromo-N-(p-selenocyanomethylbenzyl)isatin (12)
(200 mg 0.44 mmol) of 10, (300 mg, 1.81 mmol) of KI, (200 mg, 1.39 mmol) of KSeCN, 15 mL of acetonitrile were stirred and processed according to the procedure used for 5. Purification by column chromatography using CH2Cl2-hexane (7:3) as an eluent afforded 12 as a bright red solid (162 mg, 76%), mp 173–175 °C). 1H NMR (CDCl3, 500 MHz): δ 4.30 (s, 2H, H1’), 5.43 (s, 2H, CH2SeCN), 7.29 (d, J = 8 Hz, 2H, phenyl ArH), 7.37 (d, J = 8.5Hz, 2H, phenyl ArH), 7.76 (d, J = 2 Hz, 1H, isatin ArH) 7.85 (d, J = 2 Hz, 1H, isatin ArH). 13C NMR (CDCl3, 126 MHz): δ 32.2, 44.4, 101.6, 105.1, 117.3, 121.4, 127.3, 127.6, 129.5, 135.1, 136.4, 145.3, 146.5, 158.2, 181.1. HRMS (ESI) (M+Na)+ calcd for C17H10O2N279Br280SeNa, 534.8166; found 534.8167.
4.1.12. 5, 7-Dibromo-N-(p-isothiocyanatomethylbenzyl)isatin (13)
The experimental procedure used for 3 was employed with (500 mg, 1.64 mmol) of 2, 15 ml of anhydrous DMF, (100 mg, 0.60 mmol) of KI, (272 mg, 1.97 mmol) of K2CO3, and (590 mg, 1.97 mmol) of tert-butyl(4-bromomethyl-benzyl)carbamate. The product 5, 7-Dibromo-N-(tert-butyl-carbamate-methylbenzyl)isatin 15 was obtained as an orange red solid (620 mg, 72%), mp 125–127 °C. 1H NMR (CDCl3, 500 MHz): δ 1.47 (s, 9H, H t-butyl), 5.42 (s, 2H, H1’), 4.32 (d, J = 5 Hz, 2H, CH2NHBOC), 7.22–7.28 (m, 4H, phenyl ArH), 7.74 (d, J = 2 Hz, 1H, isatin ArH) 7.84 (d, J = 2 Hz, 1H, isatin ArH). HRMS (ESI) (M+Na)+ calcd for C21H20O4N279Br2Na, 546.9662; found, 546.9692. The procedure used for 6 was repeated to convert trifluroacetate salt into isothiocyanate product 13 (277 mg, 52%), mp 167–69 °C. 1H NMR (CDCl3, 500 MHz): δ 5.44 (s, 2H, H1’), 4.73 (s, 2H, CH2NCS), 7.31 (d, J = 3 Hz, 4H, phenyl ArH), 7.76 (d, J = 2 Hz, 1H, isatin ArH) 7.86 (d, J = 2 Hz, 1H, isatin ArH). 13C NMR (CDCl3, 126 MHz): δ 44.4, 48.3, 105.3, 117.5, 121.4, 127.1, 127.5, 127.8, 133.8, 136.0, 145.5, 146.5, 158.3, 181.3. HRMS (EI) M+ calcd for C17H10O2N279Br2S, 463.8824; found, 463.8831.
4.2. Cell Lines and culture conditions
The cell lines used in this study were as follows: HT29 (human colorectal cancer), MCF-7 (human breast cancer), A549 (human lung epithelial carcinoma), and UACC903 (human melanoma cancer) were obtained from American Type Culture Collection (Manassas, VA). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% fetal bovine serum (FBS) and maintained at 37°C in 5% CO2. HT29 cells were cultured in RPMI medium with 10% FBS.
4.3. Cellular Viability assay
A colorimetric cell proliferation assay was used in each of the HT29, MCF-7, A549, and UACC903 to assess the effect on cell proliferation of a series of 5, 7-dibromoisatin derivatives. Cells (5×103) were seeded into each well of a 96-well plate in 100 µl of tissue culture medium. After 24 h incubation to allow cells to adhere, cells were treated with compounds 2–13 were dissolved in DMSO and diluted in DMEM at concentrations of 0.5 – 50 µM for 48 h. Cell viability was then determined by the colorimetric MTS assay using Cell Titer 96 AQueous One Solution Cell Proliferation Assay according to the manufacturer’s instruction (Promega, Madison, WI, USA). Viability was determined by measuring absorbance at 490 nm using a micro plate reader and normalizing to the viability observed under control conditions.
4.4. Determination of Apoptosis by Flow Cytometry
Apoptosis was measured by staining cells with PE Annexin V and 7-AAD. Cells were analyzed and quantified by flow cytometry (BD FACScan). Human colon cancer cells HT29 were treated with or without 5, 7-dibromoisatin derivatives (1.25 – 5.0 µM) for 24 h at 37 °C. At the end of the incubation, cells were harvested by low speed centrifugation. Cells were washed twice with cold PBS and then resuspended in binding buffer at a concentration of 1 × 106 cells/ml and stained with 7-AAD and PE Annexin V. Cells were incubated for 15 min at RT in the dark according to the manufacturer’s instructions (Promega, Madison, Wisconsin, USA). 7-AAD and Annexin V negative were identified as live cells, 7-AAD positive were identified as dead cells. Cells which were Annexin V positive and 7-AAD negative were identified as early apoptotic. Cells which were Annexin V positive and 7-AAD positive were identified as cells in late apoptosis or necrotic.
4.5. Caspase 3/7activity assay
HT-29 cells were plated at 1×104 cells per well in a black 96 well plate, and grown for 24 hours prior to treatment. Cells were exposed to DMSO, or to compounds 5, 6, 11 and 13 at 1.25 – 5 µM concentrations for 24 hours. 100µl of Apo-ONE® Caspase-3/7 Reagent was added to each well of a black 96-well plate containing 100µl of blank, control or cells in culture. The plate was gently mixed with a plate shaker at 300–500 rpm for 30 seconds. The plate was then incubated at room temperature for 2 hours. Caspase 3/7 activity was determined by measuring the fluorescence of the cleaved substrate using a microplate reader (excitation 498 nm; emission 521 nm).
4.6. Terminal Deoxynucleotide Transferase dUTP Nick End Labeling (TUNEL) assay
HT-29 cells were plated at 1×105 cells per well in a 6 well plate, and grown in 10% serum fortified media for 24 hours prior to treatment. Cells were exposed to compounds 6 and 11 at 5 µM for 24 h. Fragmented DNA of apoptotic cells were stained using an Apop Tag Red In Situ Apoptosis Detection Kit according to the manufacturer’s instructions (Chemicon, Temecula, CA), and visualized by fluorescence microscopy using appropriate filters.
4.7. Tubulin Polymerization Assay
Compounds 5, 6, 11 and 13 were tested for their anti-mitotic ability in reference to vinblastine sulfate and paclitaxel positive controls in a fluorescence-based Tubulin Polymerization Assay55. Briefly, 50 µL of tubulin reaction mix 2 mg/mL purified bovine brain tubulin (Cytoskeleton Inc., Denver, CO, USA) in 80 mM PIPES pH 6.9, 2.0 mM MgCl2, 0.5 mM EGTA, 1.0 mM GTP, and 20% glycerol) was added to duplicate wells of a half area 96-well black plate containing 5 µL of either vehicle control, paclitaxel, vinblastine sulfate, or compounds 5, 6, 11 and 13 all at a final concentration of 10 µM. The rate of polymerization was followed for 1 h at 37 °C, data points were collected at every 10 seconds, using an excitation wavelength of 360 ± 10 nm, and the fluorescence was collected at 440 ± 10 nm.
4.8. Western analysis
Antibodies used were: Pan-Akt rabbit polyclonal, phospho-Akt rabbit polyclonal, (Cell Signaling, Danvers, MA), and β-actin mouse monoclonal (Sigma, Saint Louis, MO). HT29 cells were treated with compounds 6, 11 and 13 at different concentrations for 24 hours. Plates were washed with cold PBS and the cells were lysed into lysis buffer (50 mM HEPES, 100 mM NaCl, 10 mM EDTA, 0.5 % NP40, 10% glycerol, 0.0001% Tween 20, supplemented with 0.1 mM PMSF, 0.1 mM NaVO4, 0.5 mM NaF, 5 µg/ml leupeptin, 0.1 mM DTT). The proteins were quantified according to the Bradford Assay and loaded equally onto 10% polyacrylamide gels. Proteins were electrophoresed at 150 v and transferred to nitrocellulose membranes using a semi-dry blotter (BioRad, Hercules, CA). Membranes were blocked with 5% non-fat dry milk for 2 h and incubated with primary antibody overnight at 4°C. The blots were washed 3X in TBS-Tween and incubated for 1 h in appropriate HRP-conjugated secondary antibodies (GE Healthcare, Little Chalfont, Buckinghamshire, UK). Blots were washed and developed using the SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, Rockford, IL, USA). The blots were exposed to autoradiography film.
Acknowledgement
This study was supported in part by the National Institutes of Health National Cancer Institute Grant R03-CA143999 (A. K. S.), and by Penn State Hershey Cancer Institute. The authors thank the Flow Cytometry and Solution Phase NMR Facility (Dr. Jyh-Ming Lin) at Core Research Facilities of the Pennsylvania State University, College of Medicine, Hershey, PA, for recording of NMR spectra.
References and notes
- 1.Downing KH, Nogales E. Curr. Opin. Struct. Biol. 1998;8:785. doi: 10.1016/s0959-440x(98)80099-7. [DOI] [PubMed] [Google Scholar]
- 2.Sorger PK, Dobles M, Tournebize R, Hyman AA. Curr. Opin. Cell. Biol. 1997;9:807. doi: 10.1016/s0955-0674(97)80081-6. [DOI] [PubMed] [Google Scholar]
- 3.Kim SP, JH, Koo SY, Kim JI, Kim MH, Kim JE, Jo K, Choi HG, Lee SB, Jung SH. Bioorg. Med. Chem.Lett. 2004;14:6075. doi: 10.1016/j.bmcl.2004.09.069. [DOI] [PubMed] [Google Scholar]
- 4.Lehnert M. Eur. J. Cancer. 1996;32A:927. doi: 10.1016/0959-8049(96)00069-x. [DOI] [PubMed] [Google Scholar]
- 5.Germann UA. Eur. J. Cancer. 1996;32A:927. doi: 10.1016/0959-8049(96)00057-3. [DOI] [PubMed] [Google Scholar]
- 6.Fayard E, Tintignac LA, Baudry A, Hemmings BA. J. Cell. Sci. 2005;118:5675. doi: 10.1242/jcs.02724. [DOI] [PubMed] [Google Scholar]
- 7.Vivanco I, Sawyers CL. Nat. Rev. Cancer. 2002;2:489. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 8.Sourbier C, Lindner V, Lang H, Agouni A, Schordan E, Danilin S, Rothhut S, Jacqmin D, Helwig JJ, Massfelder T. Cancer. Res. 2006;66:5130. doi: 10.1158/0008-5472.CAN-05-1469. [DOI] [PubMed] [Google Scholar]
- 9.Pandeya SN, Smitha S, Jyoti M, Sridhar SK. Acta. Pharm. 2005;55:27. [PubMed] [Google Scholar]
- 10.Cane A, Tournaire MC, Barritault D, Crumeyrolle-Arias M. Biochem. Biophys. Res. Commun. 2000;276:379. doi: 10.1006/bbrc.2000.3477. [DOI] [PubMed] [Google Scholar]
- 11.Vine KL, Locke JM, Ranson M, Benkendorff K, Pyne SG, Bremner JB. Bioorg. Med. Chem. 2007;15:931. doi: 10.1016/j.bmc.2006.10.035. [DOI] [PubMed] [Google Scholar]
- 12.Gastpar R, Goldbrunner M, Marko D, von Angerer E. J. Med. Chem. 1998;41:4965. doi: 10.1021/jm980228l. [DOI] [PubMed] [Google Scholar]
- 13.Beckers T, Reissmann T, Schmidt M, Burger AM, Fiebig HH, Vanhoefer U, Pongratz H, Hufsky H, Hockemeyer J, Frieser M, Mahboobi S. Cancer. Res. 2002;62:3113. [PubMed] [Google Scholar]
- 14.Brancale A, Silvestri R. Med. Res. Rev. 2007;27:209. doi: 10.1002/med.20080. [DOI] [PubMed] [Google Scholar]
- 15.Motzer RJ, Michaelson MD, Redman BG, Hudes GR, Wilding G, Figlin RA, Ginsberg MS, Kim ST, Baum CM, DePrimo SE, Li JZ, Bello CL, Theuer CP, George DJ, Rini BI. J. Clin. Oncol. 2006;24:16. doi: 10.1200/JCO.2005.02.2574. [DOI] [PubMed] [Google Scholar]
- 16.Prenen H, Cools J, Mentens N, Folens C, Sciot R, Schoffski P, Van Oosterom A, Marynen P, Debiec-Rychter M. Clin. Cancer. Res. 2006;12:2622. doi: 10.1158/1078-0432.CCR-05-2275. [DOI] [PubMed] [Google Scholar]
- 17.Vine KL, Locke JM, Ranson M, Pyne SG, Bremner JB. J. Med. Chem. 2007;50:5109. doi: 10.1021/jm0704189. [DOI] [PubMed] [Google Scholar]
- 18.Fahey JW, Zalcmann AT, Talalay P. Phytochemistry. 2001;56:5. doi: 10.1016/s0031-9422(00)00316-2. [DOI] [PubMed] [Google Scholar]
- 19.Drewnowski A, Gomez-Carneros C. Am. J. Clin. Nutr. 2000;72:1424. doi: 10.1093/ajcn/72.6.1424. [DOI] [PubMed] [Google Scholar]
- 20.Cinciripini PM, Hecht SS, Henningfield JE, Manley MW, Kramer BS. J. Natl. Cancer. Inst. 1997;89:1852. doi: 10.1093/jnci/89.24.1852. [DOI] [PubMed] [Google Scholar]
- 21.Keum YS, Jeong WS, Kong AN. Mutat. Res. 2004;555:191. doi: 10.1016/j.mrfmmm.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 22.Bianchini F, Vainio H. Drug. Metab. Rev. 2004;36:655. doi: 10.1081/dmr-200033468. [DOI] [PubMed] [Google Scholar]
- 23.Verhoeven DT, Goldbohm RA, van Poppel G, Verhagen H, van den Brandt PA. Cancer. Epidemiol. Biomarkers. Prev. 1996;5:733. [PubMed] [Google Scholar]
- 24.el-Bayoumy K. Nutr. Cancer. 2001;40:4. doi: 10.1207/S15327914NC401_3. [DOI] [PubMed] [Google Scholar]
- 25.Fenwick GR, Heaney RK, Mullin WJ. Crit. Rev. Food. Sci. Nutr. 1983;18:123. doi: 10.1080/10408398209527361. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Talalay P. Cancer. Res. 1994;54:1976. [PubMed] [Google Scholar]
- 27.Maheo K, Morel F, Langouet S, Kramer H, Le Ferrec E, Ketterer B, Guillouzo A. Cancer. Res. 1997;57:3649. [PubMed] [Google Scholar]
- 28.Chen YR, Wang W, Kong AN, Tan TH. J. Biol. Chem. 1998;273:1769. doi: 10.1074/jbc.273.3.1769. [DOI] [PubMed] [Google Scholar]
- 29.Yu R, Mandlekar S, Harvey KJ, Ucker DS, Kong AN. Cancer. Res. 1998;58:402. [PubMed] [Google Scholar]
- 30.Zhang Y. Mutat. Res. 2004;555:173. doi: 10.1016/j.mrfmmm.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 31.Sharma AK, Sharma A, Desai D, Madhunapantula SV, Huh SJ, Robertson GP, Amin S. J. Med. Chem. 2008;51:7820. doi: 10.1021/jm800993r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma A, Sharma AK, Madhunapantula SV, Desai D, Huh SJ, Mosca P, Amin S, Robertson GP. Clin. Cancer. Res. 2009;15:1674. doi: 10.1158/1078-0432.CCR-08-2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Madhunapantula SV, Desai D, Sharma A, Huh SJ, Amin S, Robertson GP. Mol. Cancer. Ther. 2008;7:1297. doi: 10.1158/1535-7163.MCT-07-2267. [DOI] [PubMed] [Google Scholar]
- 34.Nguyen N, Sharma A, Nguyen N, Sharma AK, Desai D, Huh SJ, Amin S, Meyers C, Robertson GP. Cancer. Prev. Res. (Phila) 2011;4:248. doi: 10.1158/1940-6207.CAPR-10-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma AK, Kline CL, Berg A, Amin S, Irby RB. Clin. Cancer. Res. 2011;17:4474. doi: 10.1158/1078-0432.CCR-10-2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bandura L, Drukala J, Wolnicka-Glubisz A, Bjornstedt M, Korohoda W. Biochem. Cell. Biol. 2005;83:196. doi: 10.1139/o04-130. [DOI] [PubMed] [Google Scholar]
- 37.Brigelius-Flohe R. Chem. Biodivers. 2008;5:389. doi: 10.1002/cbdv.200890039. [DOI] [PubMed] [Google Scholar]
- 38.Hu H, Jiang C, Li G, Lu J. Carcinogenesis. 2005;26:1374. doi: 10.1093/carcin/bgi094. [DOI] [PubMed] [Google Scholar]
- 39.Milner JA. Fed. Proc. 1985;44:2568. [PubMed] [Google Scholar]
- 40.Medina D, Morrison DG. Pathol. Immunopathol. Res. 1988;7:187. doi: 10.1159/000157115. [DOI] [PubMed] [Google Scholar]
- 41.El-Bayoumy K. Practice of Oncology. (4th edn.) 1991:1. [Google Scholar]
- 42.GF, CJ Garewal H. New York: CRC Press; 1997. p. 97. [Google Scholar]
- 43.Sharma AK, Kline C, Berg A, Amin S, Irby RB. Clin. Cancer. Res. 2011;17:4474. doi: 10.1158/1078-0432.CCR-10-2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.el-Bayoumy K. Cancer. Res. 1985;45:3631. [PubMed] [Google Scholar]
- 45.Reddy BS, Sugie S, Maruyama H, el-Bayoumy K, Marra P. Cancer. Res. 1987;47:5901. [PubMed] [Google Scholar]
- 46.Nayini J, el-Bayoumy K, Sugie S, Cohen LA, Reddy BS. Carcinogenesis. 1989;10:509. doi: 10.1093/carcin/10.3.509. [DOI] [PubMed] [Google Scholar]
- 47.el-Bayoumy K, Chae YH, Upadhyaya P, Meschter C, Cohen LA, Reddy BS. Cancer. Res. 1992;52:2402. [PubMed] [Google Scholar]
- 48.Piche A, Grim J, Rancourt C, Gomez-Navarro J, Reed JC, Curiel DT. Cancer. Res. 1998;58:2134. [PubMed] [Google Scholar]
- 49.Sakakura C, Sweeney EA, Shirahama T, Hakomori S, Igarashi Y. FEBS. Lett. 1996;379:177. doi: 10.1016/0014-5793(95)01508-6. [DOI] [PubMed] [Google Scholar]
- 50.Zhang B, Zhang D, Ren H. Zhonghua. Gan. Zang. Bing. Za Zhi. 2000;8:215. [PubMed] [Google Scholar]
- 51.Campos L, Rouault JP, Sabido O, Oriol P, Roubi N, Vasselon C, Archimbaud E, Magaud JP, Guyotat D. Blood. 1993;81:3091. [PubMed] [Google Scholar]
- 52.Bacher G, Nickel B, Emig P, Vanhoefer U, Seeber S, Shandra A, Klenner T, Beckers T. Cancer. Res. 2001;61:392. [PubMed] [Google Scholar]
- 53.Jordan MA. Curr. Med. Chem. Anticancer. Agents. 2002;2:1. doi: 10.2174/1568011023354290. [DOI] [PubMed] [Google Scholar]
- 54.Li PK, Xiao Z, Hu Z, Pandit B, Sun Y, Sackett DL, Werbovetz K, Lewis A, Johnsamuel J. Bioorg. Med. Chem. Lett. 2005;15:5382. doi: 10.1016/j.bmcl.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 55.Bonne D, Heusele C, Simon C, Pantaloni D. J. Biol. Chem. 1985;260:2819. [PubMed] [Google Scholar]