

Abstract
Densin is an abundant scaffold protein in the postsynaptic density (PSD) that forms a high-affinity complex with αCaMKII and α-actinin. To assess the function of densin, we created a mouse line with a null mutation in the gene encoding it (LRRC7). Homozygous knock-out mice display a wide variety of abnormal behaviors that are often considered endophenotypes of schizophrenia and autism spectrum disorders. At the cellular level, loss of densin results in reduced levels of α-actinin in the brain and selective reduction in the localization of mGluR5 and DISC1 in the PSD fraction, whereas the amounts of ionotropic glutamate receptors and other prominent PSD proteins are unchanged. In addition, deletion of densin results in impairment of mGluR- and NMDA receptor-dependent forms of long-term depression, alters the early dynamics of regulation of CaMKII by NMDA-type glutamate receptors, and produces a change in spine morphology. These results indicate that densin influences the function of mGluRs and CaMKII at synapses and contributes to localization of mGluR5 and DISC1 in the PSD fraction. They are consistent with the hypothesis that mutations that disrupt the organization and/or dynamics of postsynaptic signaling complexes in excitatory synapses can cause behavioral endophenotypes of mental illness.
Introduction
In excitatory synapses of the CNS, activity-dependent regulation of synaptic strength is mediated by a large protein complex called the postsynaptic density (PSD) (Kennedy, 2000). Genetic variants of PSD proteins have been associated with cognitive defects or mental illness in humans (Durand et al., 2007; Pinto et al., 2010). For example, copy number variants (CNVs) of synGAP, a prominent component of the PSD, are associated with non-syndromic mental retardation and autism spectrum disorders (ASDs) (Hamdan et al., 2009; Pinto et al., 2010); CNVs of the PSD scaffold protein shank3 have been linked to ASDs (Durand et al., 2007; Bourgeron, 2009; Pinto et al., 2010); and the 3q29 microdeletion syndrome, which involves mild mental retardation and sometimes autism, includes deletion of the gene for the PSD scaffold protein sap97 (Willatt et al., 2005). Although mental illnesses are most precisely defined in humans, a number of abnormal behaviors in rodents that are similar to those found in humans have been defined as “behavioral endophenotypes” (isolated behavioral features) associated with mental illness. These include endophenotypes associated with schizophrenia (Powell and Miyakawa, 2006) and with ASDs (Silverman et al., 2010). Mutations in PSD proteins, including synGAP and calcium/calmodulin-dependent protein kinase II (CaMKII), can produce some of these endophenotypes (Yamasaki et al., 2008; Brandon et al., 2009; Guo et al., 2009). Here we show that mice with a deletion of the PSD scaffold protein densin-180 (densin) display several behavioral endophenotypes often associated with schizophrenia and with ASDs.
Densin is sufficiently abundant in the PSD that it was among the first proteins identified by sequencing and cloning from the PSD fraction (Apperson et al., 1996). Its domain structure suggests that it functions as a scaffold protein, and it has been reported to bind directly to CaMKII, α-actinin, δ-catenin, and shank (Strack et al., 2000; Walikonis et al., 2001; Izawa et al., 2002; Quitsch et al., 2005). However, the normal physiological roles of its various associations are not known. Here we show that deletion of densin causes a reduction in the level of α-actinin globally in brain homogenate and in the PSD fraction and selectively reduces the amounts of the metabotropic glutamate receptor mGluR5 and the scaffold protein DISC1 in the PSD fraction. In contrast, the amount of CaMKII in the PSD is not reduced. At the cellular level, we show that activation of CaMKII by NMDA receptors (NMDARs) is subtly altered in neurons from the knock-out animals. Furthermore, both low-frequency- and (RS)-3,5-dihydroxyphenylglycine (DHPG)-induced long-term depression (LTD) are abnormal, and spine morphology is altered in knock-out animals. Our results support the general hypothesis that mutations that alter the organization and/or dynamics of synaptic signal transduction can result in behavioral endophenotypes associated with mental illness.
Materials and Methods
Generation of the densin knock-outs
The densin knock-out mouse line was generated by deleting exon 3 of the LRRC7 gene, which contains the transcriptional start site of the densin message. The deletion was generated by homologous recombination to introduce loxP sites, followed by introduction of Cre recombinase in utero. Clone 456C10 was selected from a CITB mouse bacterial artificial chromosome (BAC) library (Research Genetics) encoding genomic sequences of strain 129S1Sv (recently renamed 129S3Sv/ImJ) because it hybridized with a cDNA probe encoding exon 3 of the LRRC7 gene. The presence of exon 3 in the BAC clone was confirmed by PCR. The BAC DNA insertion junctions were sequenced and aligned with the known genomic sequence of the LRRC7 gene. A 9.9 kb region of clone 456C10 that included exon 3 and its surrounding introns was cloned into the pKO Scrambler 907 vector (Stratagene). The long arm of the densin targeting construct contained a 7.8 kb fragment, including intron 2, exon 3, and part of intron 3; the short arm contained a 2.1 kb fragment of intron 3. The first loxP site was inserted 4.8 kb upstream of exon 3. A hygromycin selection cassette, flanked on both sides by loxP sites, was cloned into the short arm of the targeting sequence 1.1 kb downstream from exon 3. The linearized targeting construct (25 μg) was electroporated into 1 × 107 cells per cuvette of mouse ES (CJ7) cells. Transfected ES cells were grown in the presence of hygromycin (200 μg/ml) for 7–8 d to select for homologous recombinants. Two recombinant clones with integration of both arms were identified and confirmed by PCR. One clone (2G8) exhibited a normal karyotype and was expanded for the generation of densin knock-out ES cells. Ninety-eight 129B6 blastocysts injected with ES cells from the 2G8 clone were implanted into seven pseudo-pregnant mothers. Chimeric pups exhibiting a >90% agouti coat color were used for subsequent breeding.
Eight adult male chimeras were mated to C57BL/6 EIIaCre+/+-expressing female mice (The Jackson Laboratory) developed in the laboratory of H. Westphal (NIH, Bethesda, MD; Lakso et al., 1996). F1 generation offspring were screened for mosaic Cre-recombination patterns by PCR. F1 animals exhibiting genomic mosaicism were subsequently mated to wild-type C57BL/6 mice. Segregation of the EIIaCre transgene and Cre-recombined alleles was monitored by PCR in the F2 generation. F2 generation EIIaCre−/−, densin+/− (total excision loxP 1/3 recombination pattern) males were liberally mated to wild-type C57BL/6 females to generate a large F3 population of animals heterozygous for the densin deletion. Deletion was verified by PCR. Densin knock-out mice were maintained as heterozygotes. Animals with the loxP 1 plus loxP 2/3 pattern (floxed densin, not used in this study) were bred as homozygotes. Behavioral cohorts were bred from a line outcrossed into C57BL/6 seven times. All other experiments were performed with mice that had been outcrossed twice and maintained as F2.
Genotyping of densin knock-out mice
Tissue from tail clippings or ear punches was lysed in 100 μl of Viagen direct PCR tail mix supplemented with Proteinase K overnight at 55–60°C. To genotype embryos, we prepared DNA from either the tail or leg of the embryo harvested at the time that the brain was dissected to make neuronal cultures. Samples were heated at 85°C for 40 min to inactivate Proteinase K and then centrifuged for 10 min at 16,000 × g. Template DNA was sedimented by centrifugation for 10 min at 16,000 × g and amplified by PCR in 1× Coraload (Qiagen) PCR buffer, 0.625 mm MgCl2, 0.3 mm dNTP, 1.25 U/2.5 μl of Taq polymerase (Qiagen), and 1 pm/μl of the following primers: Lox Pray Up (5′-GAGATGCTCTCA AGATAGACATG-3), Lox Pray low (5′-CTCCAATTCTGAAGCCAGTAG-3′), and Posthygro2 (5′-ACAGAACTGGCTTCTGTCCAC- 3′). The temperature was cycled as follows: 11 cycles of 95°C for 30 s, 58°C for 30 s, 72°C for 2 min; 21 cycles of 94°C for 40 s, 56°C for 30 s, 72°C for 2 min; 1 cycle of 72°C for 5 min. PCR products were fractionated on a 1.6% TBS agarose gel with ethidium bromide. A DNA band at 187 bp identifies a wild type mouse, a 257 bp identifies a knock-out mouse, and 187 and 257 bp bands indentify heterozygous mice.
Verification of densin deletion
Aliquots containing equal amounts of total protein (10 or 50 μg) measured by the bicinchoninic acid method (Pierce) with BSA as standard were fractionated by SDS-PAGE, transferred to nitrocellulose, and blocked as described below. Membranes were incubated with rabbit anti-densin antisera CT245 (1:2500) against the PDZ domain, mouse anti-densin polyclonal antibody M2 (1:2500) against the mucin homology domain, or mouse anti-densin polyclonal antibody LRR (1:1000) against the LRR domain, diluted into 20 mm Tris, 150 mm NaCl, and 0.1% Tween 20 (Apperson et al., 1996). Bound antibodies were detected with fluorescent secondary antibodies and visualized with the Odyssey Infrared Imaging System (Li-Cor Bioscience) as described below (see SDS-PAGE immunoblot analysis).
Other mouse strains
GFP line M transgenic mice (a gift from Joshua Sanes, Harvard University, Boston, MA) were maintained as homozygotes in a C57BL/6 background (Feng et al., 2000). GFP+/+ transgenics were crossed with F2 generation densin+/− mice. GFP+/− × densin+/− offspring were crossed to produce GFP+/−/densin−/− and GFP+/−/densin+/+ mice that were used for spine analysis. GluN1 knock-out mice (a gift from Peter Seeburg, Max Planck Institute, Heidelberg, Germany) were maintained as heterozygotes. GluN1+/− mutants were crossed with F2 generation densin+/− mice to produce GluN1/densin heterozygotes. GluN1+/−, densin+/− mice (F3) were subsequently crossed to produce GluN1/densin double knock-out embryos ( of embryos, 6.3%) and littermate controls for immunocytochemistry of neuronal cultures. Individual embryos were genotyped after removal of neurons to be cultured.
Fixation and Nissl staining
Densin knock-out and wild-type littermate pairs, aged 8–12 weeks, were perfused transcardially with 4% formaldehyde, 15% saturated picric acid in 0.1 m PBS for 20 min. Forebrains were dissected and postfixed overnight at 4°C, and then 50 μm coronal sections were cut with a vibratome and stored at −20°C in 50 mm phosphate buffer, 15% glucose, 30% ethylene glycol. Sections were washed in PBS (10 mm NaHPO4, 120 mm NaCl, pH 7.4), mounted on Supermount Plus slides, and dried overnight. Slides were delipidated in a series of decreasing ethanol solutions: 95% (15 min), 70% (1 min), and 50% (1 min). After a 5 min water rinse, slides were submerged in 0.5% cresyl violet, 0.125% glacial acetic acid in distilled water for 5 min, rinsed for 30 s in distilled water, and destained in 96% ethanol/0.5% acetic acid for 5 min. Slices were cleared in isopropanol (5 min), followed by 5 min in isopropanol/xylene (1:2 parts) and four rinses in xylene for 2 min each. Slides were sealed with Permount and dried at room temperature overnight. Slides were imaged at 2.5× (Plan-Neofluar 2.5×/NA 0.075) and 5× (Plan-Neofluar 5×/NA 0.15) magnification.
GFAP immunohistochemistry
Fifty-micrometer coronal sections were incubated for 10 min in a solution containing PBS, 0.5% Triton X-100, and 0.5% H2O2 to permeabilize the tissue and eliminate endogenous peroxidase activity and then incubated in blocking solution containing 0.1 m PBS and 5% normal goat serum (Invitrogen) for 10 min, followed by overnight incubation at 4°C in blocking solution plus anti-glial fibrillary acidic protein (GFAP) antibody (1:500; Millipore). Sections were washed in PBS (three times for 5 min) and incubated with biotinylated goat anti-mouse IgG (1:200; Vector Laboratories) in blocking buffer for 2 h at room temperature and then washed (three times for 5 min) in 0.1 m PBS and incubated in avidin–biotin–peroxidase complex (1:100; Vector Laboratories) in 0.1 m PBS for 30 min. Sections were developed in 20% diaminobenzidine (Vector Laboratories), mounted, dried, and coverslipped with Entillin (Thermo Fisher Scientific).
Behavioral studies
Mice were acclimated to cages in the behavioral facility for at least 2 weeks before behavioral testing. The male cohort consisted of 15 densin knock-out and 23 wild-type mice, and the female cohort consisted of nine densin knock-outs and 14 wild-type mice. Mice from both cohorts had been backcrossed seven times into a C57BL/6 background. Females were housed in groups of four. Initially, most of the males were group housed with littermates, with an equal number of wild-type and knock-out males singly housed. As the experiments progressed, most of the males were separated because they were fighting. The cohort was tested during their light cycle, except when indicated.
Open field.
Mice were placed in the corner of a plastic 50 × 50 cm square box and allowed to explore the box for 10 min. Aerial video footage was captured using Picolo (frame grabber) with Media Cruise software. The path the mouse traveled and the duration of time spent in the center quadrant (25 × 25 cm) were analyzed using Ethovision 3.0 software.
Object-place recognition.
Each mouse was acclimated (10 min) to an open box (50 × 50 cm) and then returned to its home cage (5 min). It was then allowed to explore (5 min) the open box in which two objects had been placed in opposite corners. Mice were returned to their home cages (10 min) and then reintroduced to the same box (5 min) in which one of the objects had been moved to the opposite corner while the other object remained stationary. The preference for the moved object was measured by calculating the number of investigations of each object. Investigations were defined as head movements in the direction of the object in which the tip of the nose was within 2 cm of the object.
Novel object recognition.
At 24 h after the place preference test, mice were reacclimated to the testing box (10 min) and then returned to the home cage (5 min). The mice were then exposed to the same objects used in the place preference test (5 min). After a 5 min intertrial period, one of the objects was replaced with a novel object in the same location, and the number of investigations of each object was counted.
Prepulse inhibition.
Prepulse inhibition (PPI) was assessed on a cohort of female wild-type (n = 12) and densin knock-out (n = 13) littermates (11–15 weeks old) during the dark cycle. The male cohort was unsuitable for this test because fighting has been shown to alter the PPI response in mice. Mice were restrained in a cylindrical Plexiglas tube on a platform situated in a chamber (SR Labs) in which the involuntary startle response is measured by an accelerometer underneath the platform on which the mice are resting. The equipment was operated with “Startle” software to expose the mice to the following trials: no startle noise, just the startle noise (120 db), or a prepulse (of 3 or 6 db over background) before the startle noise. Background noise was 65 db. Prepulse inhibition was calculated as follows: 100% × ((startle.pulse) − (startle.prepulse))/(startle.pulse).
Nest building.
A 2 × 2 inch square piece of cotton nesting material was placed in the wire food racks of singly caged densin knock-out and wild-type mice, at a position low enough to be easily reached (Deacon, 2006). The unshredded nesting material remaining on the rack or on the cage floor was weighed at 12 h intervals for 72 h.
Home-cage activity.
A home-cage activity monitoring system with automated behavioral detection was used to continuously track the activities of the mice over a 24 h period at 8, 10, and 12 weeks of age after acclimating to the facility for 2 weeks (Steele et al., 2007).
Clasping.
Twenty-four knock-out (15 males, 9 females) and 30 wild-type (19 males, 11 females) mice were suspended by their tails for 1 min ∼30 cm above a table top and observed for hindlimb and forelimb clasping.
Rotarod.
Mice were placed on an accelerating rotarod (∼6 to ∼50 rpm in 240 s), and the duration of time the mice remained on the rod was measured. Mice were trained on the rotarod for 2 consecutive days, two times each day separated by a 10 min rest period. Mice were tested on the third day, and the average of two trials was recorded. The maximum duration of a trial was 300 s.
Beam crossing.
Mice were placed at one end of a beam, and the time required to cross to the escape box at the other end (80 cm away) was measured by motion detectors. Mice were trained on two beams (12 and 6 mm wide) on 2 consecutive days. The mice rested for 10 min in their home cages between training sessions on the two beams. Each mouse crossed each beam three times on training days, starting with the 12 mm beam and ending with the 6 mm beam. On test day, the times for two trials in which the mice did not stop while crossing the beam were averaged.
Assessment of aggressive behavior.
During the course of the behavioral experiments, male mice housed with their littermates were monitored for aggressive behavior. Aggression was defined as chasing, biting, or initiating a fight. Often the victim was confirmed by bite marks on the back and rear.
Slice preparation and extracellular recording
Hippocampal slices were obtained from 4- to 10-month-old mice for long-term potentiation (LTP) experiments and from 3- to 4-week-old mice for LTD experiments. Mice were anesthetized with halothane and killed by cervical dislocation. Hippocampal slices (400 μm thick) were prepared by standard techniques and maintained in an interface-slice-type recording chamber (at 30°C), perfused with oxygenated (95% O2–5% CO2) artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 4.4 KCl, 25 NaHCO3, 1 NaH2PO4, 2 CaCl2, 1.2 MgSO4, and 10 glucose. After slices had recovered for at least 1 h, a bipolar, nichrome wire stimulating electrode was placed in stratum radiatum of the CA1 region to activate Schaffer collateral–commissural fiber synapses, and an extracellular glass microelectrode filled with ACSF (resistance, 5–10 MΩ) was used to record evoked field EPSPs (fEPSPs). The presynaptic fiber stimulation was adjusted to evoke fEPSPs with amplitude of ∼50% of the maximal fEPSP amplitude for each slice. fEPSPs were then elicited at 0.02 Hz during the baseline period of the experiment and after LTP or LTD induction.
Whole-cell recordings
Slices were maintained in a submerged-slice recording chamber, and inhibitory synaptic transmission was blocked with picrotoxin (100 μm) that was added to a modified ACSF (4.0 mm CaCl2, 4.0 mm MgSO4, and 2.4 mm KCl). The CA3 region of the slices was removed to prevent bursting in the absence of GABAergic inhibition. Low resistance (2–6 MΩ) patch electrodes were filled with a solution containing the following (in mm): 102 cesium gluconate, 17.5 CsCl, 10 tetraethylammonium-Cl, 5 QX314, 4.0 Mg-ATP, 0.3 Tris-GTP, and 20 HEPES, pH 7.2. EPSCs evoked by presynaptic Schaffer collateral fiber stimulation (0.2 Hz) were recorded at membrane potentials of −80 or +40 mV, and the AMPA receptor and NMDAR-mediated components of the synaptic currents were estimated by measuring EPSC amplitude 5 and 50 ms after EPSC onset, respectively. In these experiments, the intensity of presynaptic fiber stimulation was adjusted to elicit EPSCs (at −80 mV) with peak amplitudes of ∼200 pA. Statistical comparisons in these experiments were performed using ANOVAs followed by Student–Newman–Keuls tests for multiple pairwise comparisons. Miniature EPSCs (mEPSCs) were recorded at −80 mV in cells bathed in a modified ACSF containing 4.0 mm CaCl2, 2.4 mm MgSO4, 2.4 mm KCl, 100 μm picrotoxin, and 0.5–0.75 μm tetrodotoxin. mEPSCs were analyzed using a template-based event detection routine in pClamp 10 (Molecular Devices) and a threshold of 6 pA. Statistical comparisons of mEPSC amplitude and interevent interval distributions were performed using the Kolmogorov–Smirnov test.
Analysis of spines
Three GFP+/−/densin−/− and GFP+/−/densin+/+ littermate pairs, 3–6 months in age, were perfused transcardially with fixative (as described above), and 50 μm coronal sections were cut with a vibratome and mounted with Prolong Gold antifade reagent. Images were acquired with a Carl Zeiss 510 Meta confocal microscope with a 63×/NA 1.4 lens and 2× optical zoom. Images of dendrites were reconstructed from 40 0.2-μm optical sections and preprocessed with blind iterative deconvolution software (Autodeblur) from Autoquant. Spine morphology was analyzed using 3DMA spine analysis software developed in the laboratory of Brent Lindquist (Stony Brook University, Stony Brook, NY) (Koh et al., 2002). The investigator was blind to genotype during image acquisition and analysis of spine morphology.
PSD fractions
PSD fractions were prepared from seven to eight pooled wild-type or densin knock-out mice matched by age (8–16 weeks) and sex. Forebrains (excluding olfactory bulbs) were dissected from mice killed by cervical dislocation, rinsed in buffer A (0.32 m sucrose, 1 mm NaHCO3, 1 mm MgCl2, 0.5 mm CaCl2, 0.1 mm PMSF, and 1 mg/L leupeptin), and homogenized with 12 up and down strokes at 900 rpm in 14 ml of buffer A. Homogenates were diluted to 35 ml in buffer A and centrifuged at 1400 × g for 10 min. The pellet was resuspended in 35 ml of buffer A, homogenized (three strokes), and centrifuged at 710 × g for 10 min. Supernatants were combined and centrifuged at 13,800 × g for 10 min. The pellet was resuspended in 8 ml of buffer B (0.32 m sucrose, 1 mm NaHCO3), homogenized with six strokes, and layered onto a sucrose gradient (10 ml each of 0.85, 1.0, and 1.2 m sucrose in 1 mm NaH2CO3 buffer). The gradient was centrifuged for 2 h at 82,500 × g in a swinging bucket rotor. The synaptosome-enriched layer at the interface of 1.0 and 1.2 m sucrose was collected, diluted to 15 ml with solution B, and added to an equal volume of buffer B containing 1% Triton X-100. The mixture was stirred for 15 min at 4°C and centrifuged for 45 min at 36,800 × g. The pellet containing the PSD-enriched, Triton X-100-insoluble fraction was resuspended in 300 μl of 40 mm Tris, pH 8, with a 25 gauge needle and 1 ml syringe. Samples were aliquoted, frozen in liquid nitrogen, and stored at −80°C.
SDS-PAGE and immunoblot analysis
Equal amounts of protein from each sample (15–25 μg) was dissolved in SDS-PAGE sample buffer, heated at 90°C for 5 min, fractionated on 12.5% acrylamide gels, and electrically transferred to nitrocellulose membranes (Schleicher & Schuell) in 25 mm Tris, 200 mm glycine, and 20% methanol. Membranes were blocked with Odyssey blocking buffer (Li-Cor Biosciences) and then incubated in primary antibody solution: phospho-thr286CaMKII [Affinity BioReagents (ABR)], CaMKII (ABR), β-actin (Sigma), PSD-95 (ABR), α-actinin (Sigma), DISC1 (C-terminal; Sigma), mGluR5 (Millipore), α-catenin (Transduction Labs), β-catenin (Transduction Labs), Densin (Kennedy Lab), α-CaMKII (Kennedy Lab), PSD-93 (ABR), synGAP (ABR), GluN1 (ABR), GluN2A (Kennedy Lab), GluN2B (Kennedy Lab), GluA1 (Millipore), and GluA2 (Millipore). Bound antibodies were detected with IRdye700- or IRdye800-conjugated secondary antibody (Rockland) and visualized with the Odyssey Infrared Imaging System (Li-Cor Biosciences).
Cell culture and treatment
Hippocampi dissected from mice at embryonic day 15 or 16 were triturated and plated into wells of 24-well plates coated with 50 ng/ml poly-d-lysine (Sigma) and 2 ng/ml laminin (BD Biosciences) at a density of ∼50,000 neurons per well in Neurobasal medium (Invitrogen) supplemented with B-27 (Invitrogen) and glutamax-I (Invitrogen) as described previously (Brewer et al., 1993). At 16–18 DIV, cultures were pretreated for 30 min with 5 μm TTX, gently washed twice in HEPES-control salt solution (HCSS) (in mm: 120 NaCl, 5.4 KCl, 0.8 MgCl2, 1.8 CaCl2, 10 NaOH, 20 HEPES, and 5.5 glucose, pH 7.4) and then exposed to 10 μm bicuculline methiodide (Tocris Bioscience) and 10 μm glycine (Sigma) dissolved in HCSS. After treatment, the HCSS was removed, and cultures were solubilized in lysis buffer [3% SDS, 20 mm Tris-Cl, pH 7.5, 10 mm EGTA, 40 mm β-glycerophosphate, 2.5 mm MgCl2, and Protease Inhibitor Complete (Roche)]. Lysates were heated at 90°C for 5 min, and protein concentrations were determined by the bicinchoninic acid method (Pierce) using bovine serum albumin as standard.
Cell culture immunocytochemistry
Hippocampal neurons were dissociated with trypsin and mechanical trituration, plated on glass coverslips coated with poly-d,l-lysine (Sigma), and maintained as described above. After 18–21 DIV, coverslips containing neurons were rinsed in PBS and placed briefly in ice-cold methanol. The methanol was replaced with −20°C methanol, and coverslips were incubated for 10–15 min. Cells were rinsed and incubated in h-PBS (450 mm NaCl and 20 mm phosphate buffer, pH 7.4) for 15 min, blocked with 5% normal goat serum and 0.05% Triton X-100 in h-PBS for 1 h at 4°C, and then incubated overnight with primary antibodies: rabbit anti-PSD-95 (Cell Signaling Technology) and mouse anti-αCaMKII (ABR). Coverslips were washed three times (15 min/wash) in blocking buffer followed by incubation with goat anti-mouse conjugated to Alexa Fluor 568 and goat anti-rabbit conjugated to Alexa Fluor 488 secondary antibodies (Invitrogen) at room temperature for 1 h. Coverslips were washed once in blocking buffer for 15 min, twice in PBS for 15 min, and then postfixed in 2% paraformaldehyde in PBS for 10 min, followed by two washes in PBS for 10 min each. Finally, coverslips were mounted on microscope slides with a drop of Prolong antifade reagent (Invitrogen) and allowed to dry overnight. Images were acquired on a Carl Zeiss Axiovert 200M fluorescent microscope equipped with a 63×/1.4 oil objective and a high-resolution CCD camera (Axiocam MRm) controlled by Carl Zeiss AxioVision 3.1 imaging software. A fixed exposure time was set for all images in an experiment to a time that gave submaximal pixel brightness for wild-type images in that experiment. Images were analyzed with the NIH ImageJ software program. PSD-95 was used as a marker for the PSD region. Threshold for PSD-95 immunostaining was set to allow all recognizable PSD-95 puncta to be included in the creation of a mask. The PSD-95 mask was then overlaid onto the CaMKII image. Intensities of CaMKII puncta that colocalized with the PSD-95 mask were recorded for 15–20 neurons per embryo. Each genotype was analyzed at least three different times from litters dissected from three different pregnant females. Mutant animals were always compared with wild-type littermates.
Statistics
Data are presented as mean ± SEM, with n indicating the number of experiments, as stated. Statistical analyses of two groups were measured using Student's t tests (two-tailed). One-sample -tests (two-tailed) were used to determine whether datasets that were normalized to matched control values were significantly different from 100%. Statistical analyses on data containing more than two groups were performed using the one-way ANOVA test, followed by Tukey–Kramer analysis, to account for multiple comparisons. The Kolmogorov–Smirnov method was used to assess whether datasets had Gaussian distributions, as required for t tests and ANOVA analyses. In cases in which the data were not Gaussian, nonparametric tests were used as stated.
Results
Generation of densin knock-out mice
The LRRC7 gene, which encodes densin, was disrupted by loxP/Cre-mediated excision of exon 3, which contains the transcription start site. ES cells were transfected with a targeting construct that contained a LoxP site inserted into intron 2 and a hygromycin selection cassette flanked by LoxP sites inserted into intron 3. After the altered gene was incorporated into the germ line, Cre recombinase was expressed in utero to produce a mixture of three additional genotypes: a deletion of the hygromycin selection cassette, a floxed exon 3 conditional knock-out, or deletion of both exon 3 and the hygromycin cassette resulting in a full knock-out (Fig. 1A; Materials and Methods). All the studies described here were performed on the full knock-out. Deletion of exon 3 in densin knock-out mice was verified by performing PCR on genomic DNA (Fig. 1B). Immunoblots stained with antibodies specific for the N-terminal LRR domain, the mucin homology domain, or the C-terminal PDZ domain confirmed that no densin protein was expressed in the knock-out mice (Fig. 1C).
Figure 1.

Disruption of the LRRC7 gene encoding densin. A, Targeting vector and recombined floxed alleles. The targeting construct for exon 3 included a LoxP site inserted into intron 2 and a hygromycin selection cassette flanked by LoxP sites inserted into intron 3 (LoxP sites indicated by black arrowheads). Expression of Cre recombinase in utero resulted in the deletion of the hygromycin selection cassette, producing a floxed exon 3 conditional knock-out, or in the deletion of both exon 3 and the hygromycin cassette, producing a full knock-out. B, Southern blot analysis of PCR products from DNA of wild-type (Wt), heterozygous (Het), and knock-out (KO) littermates. C, Immunoblot analysis of expression of densin protein in forebrain homogenates of 6-week-old mice. Aliquots containing equal amounts of total protein were fractionated and blotted as described in Materials and Methods with antibodies raised against three distinct regions of densin: N-terminal LRR domain, mucin homology domain, and C-terminal PDZ domain.
The densin mutants are viable and born at the expected Mendelian ratio. During the first 2–6 weeks after birth, they have significantly lower body weights compared with their wild-type littermates (Fig. 2A,B) and have an ∼20% mortality rate. The disparity in body weight becomes much less dramatic by 11 weeks of age, and the mutants that survive the first 6 weeks of life appear to have a normal lifespan. In addition to their smaller body size, the knock-out mice clasp when suspended by the tail, a behavior often observed in mouse models of neurodegeneration (data not shown). Nissl-stained sections of mutant forebrains did not reveal any gross abnormalities in brain structure (Fig. 2C), and immunostaining for GFAP did not show abnormal levels of astrogliosis (Fig. 2D).
Figure 2.
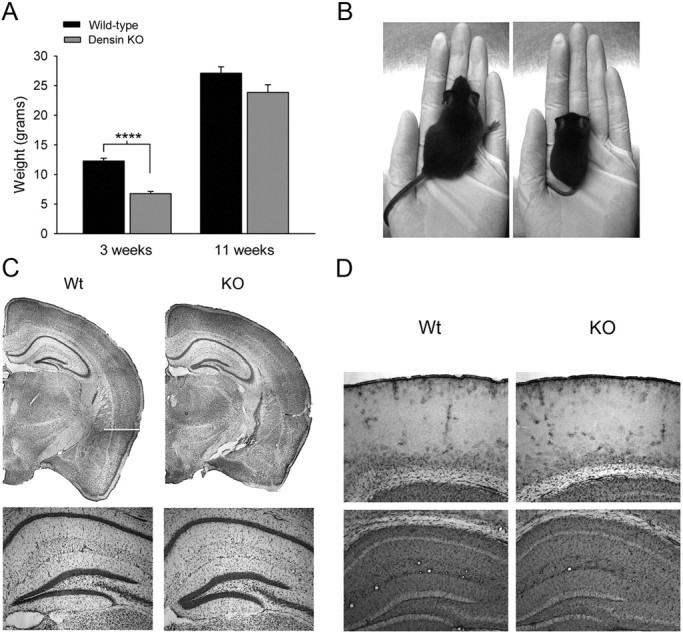
Densin knock-out mice have normal gross neuroanatomy and low body weight. A, At 3 weeks of age, densin knock-out mice had significantly lower body weights (6.8 ± 0.4 g, n = 15 mice) than wild-type mice (12.3 ± 0.5 g; n = 17 mice; ****p < 0.0001). By 11 weeks of age, the disparity in their weights was much less dramatic (wild-type, 27.1 ± 1.1 g, n = 21; densin knock-out, 23.9 ± 1.3 g; n = 19; p = 0.062). Significance was determined with two-tailed t test. Error bars represent SEM. B, Representative pictures of wild-type (left) and densin knock-out littermates (right) at 3 weeks of age. C, Representative Nissl-stained coronal sections from wild-type (left) and densin knock-out (right) mice (bregma −1.7 mm). Bottom row shows 5× images of the dentate gyrus. D, Immunohistochemical staining for GFAP is qualitatively similar in wild-type (left column) and knock-out (right column) sections. Representative images are shown of sensory cortex (top row) and the CA1 region of hippocampus (bottom row). Heterozygous, Het; Wt, wild type.
Densin knock-out mice have behavioral endophenotypes associated with mental illness
Although schizophrenia, ASDs, and other mental illnesses are distinctively human diseases, researchers working with various partial animal models of these diseases have defined the term endophenotype to mean a “heritable phenotypic indicator that reflects discrete components of pathophysiological processes more proximal to particular sets of predisposing genes than the actual clinical diagnosis” (Gottesman and Gould, 2003; Arguello and Gogos, 2006). As part of our initial characterization of the functional effects of densin deletion, we examined the performance of densin knock-out mice in behavioral tests and found that they display many behaviors that have been suggested as endophenotypes associated with human mental illness (Arguello and Gogos, 2006).
One such endophenotype is impaired short-term memory. We chose tests of short-term memory that use novelty-preference paradigms to take advantage of a rodent's natural curiosity about novel situations. Hippocampus-dependent short-term memory was assessed with a one-trial object-place recognition task in which the mice were allowed to investigate two objects for 5 min during a training trial. After a 10 min delay, a 5 min test trial was administered in which the mice were allowed to investigate the same two objects, but with one of the objects moved to a novel location. As expected, the wild-type mice investigated the moved object significantly more frequently than the stationary object (Fig. 3A). In contrast, the knock-out mice did not show a preference for the moved object, suggesting they have impaired spatial memory of the training configuration.
Figure 3.

Short-term memory, sensorimotor gating, and nest building are impaired in densin knock-out mice. A, Knock-out (KO) mice did not show increased preference for the moved object (B*) in a place preference test designed to measure hippocampus-dependent short-term spatial memory (stationary object, 15.1 ± 1.7 investigations; moved object, 13.5 ± 1.2 investigations; n = 15 mice), whereas wild-type (Wt) mice did prefer the moved object as measured by a significant increase in the number of investigations of the moved object (stationary object, 6.2 ± 0.7 investigations; moved object, 10.3 ± 1.0 investigations; n = 20 mice; *p < 0.05). Despite not showing a preference for the moved object during the testing session, densin knock-out mice investigated objects A and B more frequently on average than the wild-type mice during the training session (wild type, 8.4 ± 0.5 investigations, n = 40 trials; knock-out, 16.1 ± 0.8 investigations, n = 30 trials; ****p < 0.0001). B, Densin knock-out mice were similarly unable to discriminate between a novel object (C) and a previously viewed object in a hippocampus-independent novel object recognition task (previously viewed, 10.6 ± 1.8 investigations; novel, 11.3 ± 2.0 investigations), whereas the wild-type mice showed an increase in the number of investigations of the novel object (previously viewed, 5.3 ± 0.7 investigations; novel, 10.2 ± 1.1 investigations; n = 15 mice). Knock-out mice again showed an increased average number of investigations of objects A and B during the training session compared with wild-type mice (wild type, 6.2 ± 0.5 investigations, n = 40 trials; knock-out, 12.2 ± 1.0 investigations, n = 30 trials; p < 0.0001). C, Knock-out mice have abnormal sensorimotor gating. Startle induced by a 120 dB pulse of sound was significantly less inhibited by a prepulse (3 or 6 dB) in the knock-out (3 dB, 15 ± 5% PPI; 6 dB, 33 ± 5% PPI; n = 13 knock-out mice) compared with wild-type mice (3 dB, 42 ± 5% PPI; 6 dB, 60 ± 5% PPI; n = 12 wild types; **p < 0.01, ANOVA). D, Knock-out mice show impaired nest building. Wild-type mice shredded ∼60% of the supplied cotton within 12 h and all had fully formed nests within 48 h (black dots, n = 13 wild types). Only one of the knock-out mice removed and shredded the cotton; all others left the cotton unshredded (gray dots, n = 16 knock-out mice). Wild-type mice had significantly less unshredded cotton in their cages at all measured time points after t = 0 (p < 0.001, Mann–Whitney U test). Error bars represent SEM.
We assessed hippocampus-independent short-term memory with the one-trial object-recognition test. This test is identical to the object-place recognition test, except that a novel object is substituted in the same location for one of the training objects during the test trial. Again, wild-type mice showed a significant preference for the novel object, whereas the knock-out mice did not (Fig. 3B). Thus, the densin knock-out mice appear to have deficits in both hippocampus-dependent and -independent forms of short-term memory. Interestingly, the knock-out mice displayed hyperlocomotive behavior when interacting with the objects during the short-term memory tasks, investigating the objects twice as frequently as wild-type mice (Fig. 3A,B). In addition to short-term memory deficits, hyperactivity in response to stress or novelty has been suggested as an animal model of psychomotor agitation that often occurs in schizophrenia (Arguello and Gogos, 2006). It is unlikely that the failure of the densin knock-out mice to show a preference for novel objects resulted from a ceiling effect related to their hyperlocomotion when interacting with the objects. The highest number of average interactions recorded for the mutants, 17 in 5 min during the first exposure to objects (Fig. 3A), represents one interaction every 17.6 s, a rate that is far below the maximum possible interactions in 5 min.
PPI is a common measure of sensorimotor gating related to preattentive processing that is often deficient in patients with schizophrenia as well as other psychiatric diseases (Powell et al., 2009). In healthy subjects, a low-intensity pulse of sound preceding an acoustic startle stimulus inhibits the startle reflex. We found that a 3 or 6 dB prepulse of sound induced ∼50% greater inhibition of the startle reflex in wild-type mice than in knock-out mice (Fig. 3C). These data mean that densin deletion results in a deficit in PPI.
Deficits in nesting behavior have been observed in mouse models with schizophrenia-like features and can be induced with psychotomimetic agents (Powell and Miyakawa, 2006). These deficits are hypothesized to be related to social withdrawal, a common negative symptom in schizophrenia and autism (Powell and Miyakawa, 2006). We observed that male densin mutants had a profound deficit in nest-making behavior. We measured this behavior by providing sets of mice with identical squares of cotton. The wild-type mice shredded the cotton and made fully formed nests within 24 h. In contrast, only 1 of 16 knock-out mice shredded any of the cotton within a 72 h period (Fig. 3D).
To confirm that the knock-out mice do not have motor deficits that might confound the results of other behavioral tests, we measured their balance and coordination in the rotarod and beam-crossing tests. Densin knock-out and wild-type mice remained on an accelerating rotarod for similar lengths of time (Fig. 4A). On the beam-crossing test, the knock-out mice crossed the beam significantly more quickly than wild-type mice, consistent with the observation that the knock-out mice become hyperactive in novel or stressful situations (Fig. 4B,C). Overall, the densin mutants have similar levels of coordination, endurance, and balance to their wild-type counterparts, despite their differences in size and clasping behavior.
Figure 4.
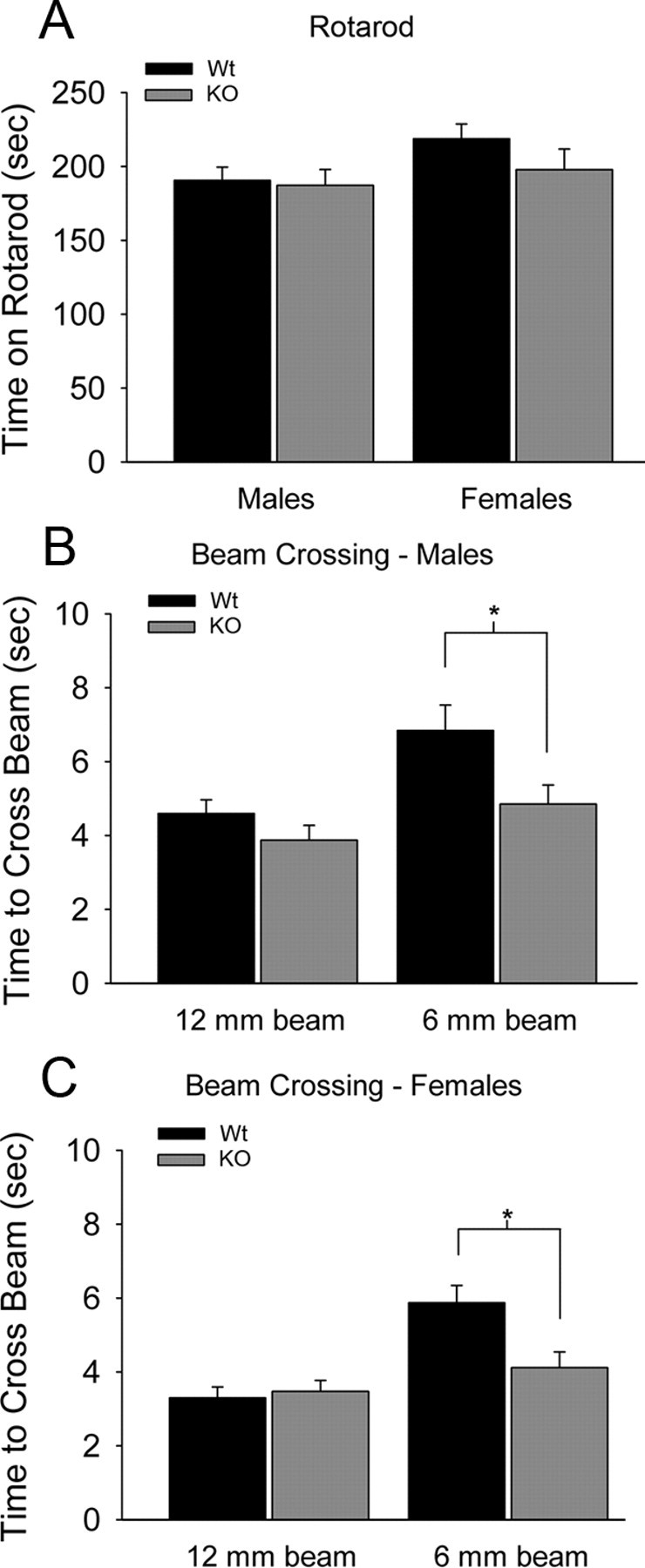
Densin knock-out mice have normal motor skills. A, Male and female knock-out (KO) mice performed as well as the wild types (Wt) on the rotarod test. Male wild-type mice stayed on the accelerating rotarod for 191 ± 9 s (n = 20 mice), whereas male knock-out mice stayed on for 187 ± 11 s (n = 15 mice). Female wild-type mice stayed on the accelerating rotarod for 219 ± 10 s (n = 11 mice), whereas female knock-out mice stayed on for 198 ± 14 s (n = 9 mice). B, Male knock-out mice crossed the 12 mm beam as fast as wild types (male wild types, 4.6 ± 0.4 s; male knock-outs, 3.9 ± 0.4 s) and crossed the 6 mm beam significantly faster than wild types (male wild types, 6.8 ± 0.7 s; male knock-outs, 4.9 ± 0.5 s; *p < 0.05, two-tailed t test). C, Similarly, female knock-out mice crossed the 12 mm beam as fast as wild types (female wild types, 3.3 ± 0.3 s; female knock-outs, 3.5 ± 0.3 s) and crossed the 6 mm beam significantly faster than wild types (female wild types, 5.9 ± 0.6 s; female knock-outs, 4.1 ± 0.4 s; *p < 0.05, two-tailed t test).
Abnormal aggression is rare in humans with schizophrenia, and it is not considered a feature of the disease. However, some mouse models with schizophrenia-like features do display abnormal aggression (Pletnikov et al., 2008). We observed that male densin knock-outs were aggressive when group-housed even with littermates. Males in nearly all cages containing densin heterozygotes or full knock-outs eventually had to be separated because of excessive fighting. Over a several hour period during behavioral experiments, we observed fighting in >82% of 11 cages containing at least one knock-out mouse and in two of three cages containing one heterozygous knock-out and one wild-type mouse. In contrast, no fighting was observed in four cages that contained only wild-type mice. We did not quantify this behavior further because the fighting occasionally resulted in injury to the mice.
Although it is not a core symptom of schizophrenia, anxiety is often comorbid with schizophrenia (Achim et al., 2011) and autism. Anxiety in mice can be measured by the open-field test. Mice have an innate fear of open space and tend to avoid the center of the open-field apparatus. Typically, they explore the outer edges of the box first and then gradually explore the center. Their level of anxiety is considered to be inversely correlated with the percentage of time spent in the center quadrant of the open field. Densin mutants spent a significantly smaller fraction of time in the center quadrant compared with wild-type mice, indicating that they have significantly higher levels of anxiety (Fig. 5A).
Figure 5.

Densin knock-out mice have increased levels of anxiety and reduced home-cage activity and are aggressive with littermates. A, Knock-out (KO) mice displayed anxiety-like behaviors in an open field. The percentage of time knock-out mice spent in the center quadrant of the open field was significantly less than wild types (Wt) during the first 2 min (wild type, 10.0 ± 1.4%; knock-out, 3.4 ± 1.3%; n = 20 wild-type mice, 15 knock-out mice; **p < 0.01), 4 min (wild type, 14.0 ± 1.8%; knock-out, 3.6 ± 1.0%; n = 17 wild-type mice, 10 knock-out mice; ***p < 0.001), and 8 min (wild type, 15.3 ± 4.0%; knock-out, 6.3 ± 1.8%; n = 8 wild-type mice, 7 knock-out mice; *p < 0.05, two-tailed t test; error bars represent SEM) of exploration in the open field. Right shows representative paths traveled by wild-type (left) and knock-out (right) mice during 10 min of exploration in an open field. The inner gray box marks the boundaries of the center quadrant. B, Densin knock-out mice are hypoactive in a home-cage setting. The heat plot shows behaviors detected by an automated home-cage monitoring system over a 24 h period at 8, 10, and 12 weeks of age (weeks 1, 2, and 3, respectively). Behaviors are expressed as the fraction of frames that the behavior was detected and normalized to wild-type values. Light blue represents behaviors for which there was a >2.5-fold decrease compared with wild-type. Bright yellow represents behaviors for which there was a >2.5-fold increase compared with wild-type. Knock-out mice spent significantly less time engaged in high-activity behaviors, such as hanging, jumping, and rearing.
Using automated behavior analysis, we tracked typical behaviors of the mice in their home cages during day and night cycles over a period of weeks (Steele et al., 2007). We found that, although the knock-out mice become hyperactive in novel situations, they are significantly more sedentary than wild-type mice in the context of a familiar environment. In fact, the densin mutants showed a threefold reduction in the amount of time engaged in high-energy behaviors, such as hanging vertically from the wire food rack and jumping (Fig. 5B). Knock-out mice spent the same amount of time sleeping as wild-type mice but showed a more than twofold increase in twitching during resting periods, suggesting that their sleep may be interrupted frequently.
In summary, densin knock-out mice have significant deficits in hippocampus-dependent and -independent short-term memory, prepulse inhibition, and nesting behavior. In addition, they display abnormal aggression and anxiety, and they become physically agitated in response to novelty. Many of these behavioral defects have been reported in other mouse models with schizophrenia and autism-like features and are considered endophenotypes related to human mental illness (Powell and Miyakawa, 2006; Silverman et al., 2010).
Loss of densin reduces the amount of α-actinin in brain homogenates and selectively reduces the levels of DISC1 and mGluR5 in the PSD fraction
Because densin interacts with a specific set of other PSD proteins, we hypothesized that its loss might alter the composition of the PSD in a way that disrupts synaptic function and thus produces behavioral abnormalities. As an initial test of this hypothesis, we isolated PSD fractions from homogenates of several sets of wild-type and densin knock-out forebrains and compared the amounts of core PSD proteins. We measured the known densin-binding proteins α-CaMKII, α-actinin, β-catenin, and δ-catenin. Only the amount of α-actinin was significantly reduced in knock-out PSD fractions (32%); it was also reduced in total brain homogenates (33%), suggesting that expression or stability of α-actinin is impaired in the absence of densin (Fig. 6). α-Actinin has been reported to bind two proteins previously implicated in schizophrenia, DISC1, and mGluR5 (Millar et al., 2003; Cabello et al., 2007). We measured their amounts and found that both DISC1 and mGluR5 are normal in brain homogenates of densin knock-out mice but significantly reduced (∼30%) in PSD fractions compared with wild type (Fig. 6). We detected no significant differences in the amounts of other core PSD proteins (β-CaMKII, PSD-95, PSD-93, and synGAP; data not shown) or of ionotropic glutamate receptor subunits (GluN1, GluN2A, GluN2B, GluA1, and GluA2; data not shown) in PSD fractions from knock-out mice. Thus, the absence of densin selectively reduces the steady-state level of α-actinin in brain and in the PSD fraction, whereas the amounts of DISC1 and mGluR5 are reduced just in the PSD fraction. The latter result was not expected and may be an indirect effect of loss of α-actinin or may be caused by disruption of other, as yet unknown, protein associations.
Figure 6.

Levels of α-actinin, DISC1, and mGluR5 are reduced in PSD fractions from densin knock-out mice. Protein levels in brain homogenates and PSD fractions from several sets of mice, each containing seven to eight pooled wild-type (WT) brains and seven to eight pooled densin knock-out (KO) brains, were determined by immunoblot. Average levels in fractions from knock-outs were normalized to those of corresponding wild types. The densin-binding protein α-actinin was reduced by 33% in brain homogenates and 32% in PSD fractions from knock-out mice compared with wild type (n = 4 subcellular fractionations). The α-actinin-binding partners DISC1 and mGluR5 were reduced by ∼30% in PSD fractions from knock-out mice but were not reduced in brain homogenates (n = 3 subcellular fractionations for DISC1; n = 5 brain homogenates and 3 PSD preparations for mGluR5). Loss of densin did not alter the levels of other known densin-interacting proteins: β-catenin, δ-catenin, or α-CaMKII. Statistical significance was determined with one-sample t tests (null hypothetical mean = 100). Error bars represent SEM. The amounts of actin or of PSD-95 (as appropriate) were quantified and used as loading controls. Bottom panels show representative bands from immunoblots of homogenates and PSD fractions. *p < 0.05.
Synaptic transmission is normal in densin knock-out neurons
We also assessed whether the synapses of densin knock-out mice display normal glutamate receptor activity. Whole-cell recordings revealed no difference in the relative contributions of AMPA receptors and NMDARs to EPSCs in CA1 pyramidal cells from wild-type and densin knock-out mice (Fig. 7A). To rule out the possibility that a proportional shift in both types of glutamate receptors had occurred in the densin mutants, we also compared the amplitude and frequency of mEPSCs in CA1 pyramidal cells and found no difference in their distribution, amplitude, or frequency between wild-type and knock-out neurons (Fig. 7B). Thus, densin is not necessary for recruitment and normal function of NMDARs at synapses, and hypofunction of NMDARs is not the underlying cause of the abnormal behavior of the densin knock-outs (Belforte et al., 2010).
Figure 7.

Evoked NMDAR currents, mEPSCs, and LTP amplitude are normal in knock-out hippocampal slices, but LTD is impaired. A, Loss of densin does not alter the ratio of NMDA/AMPA receptor currents. Top traces show representative whole-cell voltage-clamp recordings of EPSCs from wild-type (Wt) and knock-out (KO) CA1 pyramidal neurons at two different postsynaptic membrane potentials (−80 and +40 mV). The left graph shows the NMDAR-mediated EPSCs (estimated from the EPSC amplitude 50 ms after EPSC onset) normalized to the AMPA receptor-mediated component of the EPSC (estimated from the EPSC amplitude 5 ms after EPSC onset). There is no difference in EPSCs between wild-type cells (black bars; n = 3 wild-type mice, 13 cells) and knock-out cells (gray bars; n = 3 knock-out mice, 14 cells) at either membrane potential. The right graph shows the comparison of weighted mean decay time constants of NMDAR-mediated EPSCs calculated from double-exponential fits to the decay of the synaptic currents recorded at +40 mV. No difference in the decay characteristics was observed between wild-type and knock-out mice. B, Loss of densin does not affect the amplitude or frequency of mEPSCs. Top traces show representative whole-cell voltage-clamp recordings from CA1 pyramidal neurons. Cumulative amplitude (left graph) and interevent interval (right graph) distributions were similar for both genotypes (n = 3 wild-type mice, 13 cells; n = 3 knock-out mice, 13 cells). Inset graphs show averages of mEPSC amplitudes and interevent intervals. C, D, High-frequency and theta-induced forms of LTP in the CA1 region of the hippocampus are normal in knock-out mice. C, Similar levels of fEPSP potentiation were recorded 60 min after 100 Hz stimulation (2 trains, each of 1 s duration, delivered at time = 0) for both genotypes (wild-type fEPSPs were 182 ± 8% of baseline, n = 4; densin knock-out fEPSPs were 193 ± 6% of baseline, n = 4). D, The 5 Hz stimulation for 30 s (delivered at t = 0) potentiated fEPSPs to similar levels in both genotypes (wild-type fEPSPs were 159 ± 13% of baseline, n = 4 wild-type mice; knock-out fEPSPs were 161 ± 4% of baseline 45 min after 5 Hz stimulation, n = 5 knock-out mice). The trend in C and D toward smaller fEPSPs in the wild type at 5–10 min after stimuli was not statistically significant. E, Low frequency-induced LTD is impaired in knock-out hippocampal slices. Wild-type fEPSPs were depressed to 74 ± 7% of baseline (n = 7 wild-type mice) 60 min after the start of a 15 min train of 1 Hz stimulation, whereas knock-out fEPSPs returned to baseline levels (98 ± 6% of baseline, n = 8 knock-out mice). There was a significant difference in the amplitudes of the fEPSPs between the two genotypes at t = 60 (p < 0.05, two-tailed t test). F, The group 1 mGluR agonist DHPG induced a lower level of fEPSP depression in knock-out compared with wild-type slices. Bath application of DHPG depressed wild-type fEPSPs to 49 ± 12% of baseline, whereas knock-out fEPSPs were only reduced to 87 ± 5.2% of baseline during the first 5 min of drug application (p = 0.016, two-tailed t test; n = 6 wild-type, 6 knock-out mice). At 35 min after washout of DHPG (t = 45), wild-type fEPSPs were reduced to 78 ± 6.4% of baseline and knock-out fEPSPs were reduced to 92 ± 2.2% of baseline (p = 0.031, one-tailed t test). Error bars represent SEM.
Loss of densin alters the steady-state activation of CaMKII and its activation in response to synaptic activity
CaMKII is highly concentrated in the PSD fraction and in spines of cultured excitatory neurons, suggesting that one or more docking sites within the PSD mediate its postsynaptic accumulation (Kennedy et al., 1983; Kennedy, 2000). Densin and the GluN2B subunit of the NMDAR both have high-affinity binding sites for CaMKII that may play significant roles in targeting CaMKII to the PSD (Omkumar et al., 1996; Strack and Colbran, 1998; Leonard et al., 1999; Walikonis et al., 2001; Barria and Malinow, 2005; Zhou et al., 2007). To formally test the hypothesis that densin acts as a docking site for CaMKII in the PSD and examine the relative importance of the two potential docking sites, we compared the localization of CaMKII in spines of neurons lacking densin, lacking synaptic NMDARs, and those lacking both.
We generated neurons lacking synaptic NMDARs by culturing hippocampal neurons from GluN1 knock-out embryos (Fukaya et al., 2003). Engineered mice that are missing the cytosolic tails of GluN2A and GluN2B have been created (Sprengel et al., 1998); however, homozygous deletion of the tail of GluN2B results in failure of movement of all subunits of NMDARs to synapses (Sprengel et al., 1998). Deletion of the GluN1 subunit similarly causes GluN2 subunits to be retained in the endoplasmic reticulum and results in failure of movement of NMDARs to synapses (Fukaya et al., 2003). Nonetheless, excitatory synapses of neurons cultured from GluN1 knock-out embryos still form PSDs containing PSD-95. We cultured neurons lacking both densin and synaptic NMDARs from embryos bred to contain both deletions as described in Materials and Methods.
We estimated the amount of CaMKII docked at PSDs in each phenotype by measuring the integrated intensity of immunostaining for CaMKII that colocalized with PSD-95 (Fig. 8A,B). By this measure, PSDs on densin knock-out neurons did not show a significant reduction in colocalization of CaMKII with PSD-95, consistent with our finding that PSD fractions from densin knock-out mice contain normal amounts of CaMKII (Fig. 5). This result means that densin is not required for accumulation of CaMKII in PSDs. The GluN1 knock-out neurons showed only a ∼15% reduction in CaMKII staining colocalizing with PSD-95, indicating that synaptic NMDARs also are not required for accumulation of CaMKII in PSDs. However, in neurons lacking both densin and synaptic NMDARs (Densin/GluN1 double knock-out), the amount of CaMKII colocalizing with PSD-95 was reduced by ∼50%. Staining for CaMKII appeared lighter in dendritic shafts of double knock-out neurons as well as in spines (Fig. 8A, bottom right); however, the integrated intensity of all staining for CaMKII in each image divided by the total number of pixels with staining above background was not significantly reduced in the images of double knock-out neurons compared with wild-type controls. Thus, it is not clear whether the total amount of CaMKII was reduced in the double knock-out neurons or just dispersed more broadly throughout the neurons. The simplest explanation for these data is that densin and the tails of NMDAR subunits both can act as docking sites for CaMKII in the PSD and each of them can partially or completely compensate for the absence of the other in performing this function. However, when both proteins are missing, considerably less CaMKII is localized to spines, and it may be reduced in concentration in dendrites as well.
Figure 8.

Densin is not required for localization of CaMKII in spines, but loss of densin alters activity-dependent autophosphorylation of CaMKII. A, Representative images of hippocampal neurons cultured from wild-type, densin knock-out, GluN1 knock-out, and densin/GluN1 double knock-out mice, immunostained for αCaMKII (green) and PSD-95 (red). B, Loss of either densin or NMDARs (two postulated high-affinity CaMKII binding sites in the PSD) resulted in only small reductions in the intensity of CaMKII immunostaining that colocalized with PSD-95, whereas loss of both proteins resulted in a ∼50% reduction in the intensity of CaMKII immunostaining colocalizing with PSD-95. The graph shows the intensity of staining for CaMKII colocalized with PSD-95 puncta in densin knock-out, GluN1 knock-out, and densin/GluN1 double knock-out cultures normalized to wild-type levels (densin knock-out, 90.4 ± 9.1% of wild type, n = 4 densin knock-out embryos; GluN1 knock-out, 86.3 ± 1.6% of wild type, n = 4 GluN1 knock-out embryos, **p < 0.01; densin/GluN1 knock-out, 49.2 ± 7.2% of wild type, n = 3 densin/GluN1 double knock-out embryos, *p < 0.05, one-sample t test). C, Autophosphorylation of CaMKII (Thr286) in response to synaptic activity is increased in densin knock-out (KO) compared with wild-type (Wt) cultures. Top shows representative immunoblots of homogenates of hippocampal cultures that were treated with bicuculline (10 μm)/glycine (10 μm) for 0.25, 1, 3, or 5 min. Homogenates were fractionated by SDS-PAGE and stained with anti-phospho-CaMKII antibody (C, untreated control) as described under Materials and Methods. The graph shows the average intensity of phospho-CaMKII immunostaining (normalized to total CaMKII) after treatment with bicuculline/glycine, expressed as a percentage of the untreated control. The increase in phospho-CaMKII over untreated control levels is significantly greater in densin knock-out cultures compared with wild-type after treatment with bicuculline/glycine for 3 min (wild type, 147 ± 12%; densin knock-out, 200 ± 23% of control; *p < 0.05, ANOVA) and 5 min (wild type, 150 ± 23%; densin knock-out, 202 ± 23% of control; *p < 0.05, ANOVA; n = 9 wild-type and 9 densin knock-out embryos). Error bars represent SEM. D, Steady-state autophosphorylation of CaMKII is reduced in densin knock-out compared with wild-type hippocampal neurons. There was no difference in the amount of total CaMKII in wild-type and knock-out neurons (knock-out, 96 ± 7.67% of wild type, n = 9 littermate pairs). However, a significantly smaller fraction of CaMKII was phosphorylated on the threonine-286 residue of CaMKII in the knock-out compared with wild-type neurons (knock-out, 74 ± 6.3% of wild type, n = 9 littermate pairs, **p < 0.01, one-sample t test; hypothetical mean = 100).
We wondered whether the absence of densin might alter the micro-localization of CaMKII within the PSD. For example, if more CaMKII is bound to NMDARs in the absence of densin, one might predict that the rate or magnitude of activation of CaMKII would be increased in response to synaptic activity in densin knock-out neurons. To test whether this is the case, we treated hippocampal cultures with bicuculline, a GABAA receptor antagonist that causes enhanced glutamate release from excitatory synapses through disinhibition of inhibitory synapses (Ivanov et al., 2006). We found that the resulting activation of CaMKII, measured by an increase in autophosphorylation at threonine-286, was approximately twofold higher in densin knock-out neurons compared with wild type (Fig. 8C). Conversely, the basal level of autophosphorylation of threonine-286 in untreated densin knock-out neurons was reduced to ∼75% that of wild-type neurons (Fig. 8D). Together, these data indicate that densin is not required for localization of CaMKII in the PSD. However, it may be important for precise positioning of CaMKII in relation to other regulatory molecules, such that absence of densin alters both the steady-state activation of CaMKII and the dynamics of its activation in response to neuronal activity.
Long-term depression is impaired in densin knock-out neurons
We reasoned that mislocalization of mGluRs and/or of CaMKII might alter induction or stability of synaptic plasticity. We measured two commonly studied forms of LTP and found them unaffected by the densin deletion. Brief 100 Hz stimulation of Schaffer collateral fibers induced an approximate twofold increase in fEPSP amplitudes in wild-type and knock-out hippocampal slices 60 min after LTP induction (Fig. 7C). A 30 s train of 5 Hz stimulation (theta stimulation) also produced similar levels of fEPSP potentiation (∼50% increase over baseline) in wild-type and knock-out slices (Fig. 7D). Thus, LTP appears normal in the densin mutants during the first hour after induction. We observed a trend toward larger potentiation in the first 5 min after either of the induction stimuli, but the trend did not reach statistical significance. We conclude that the aberrant activation of CaMKII we observed during the first 5 min after activation of NMDARs on cultured neurons (Fig. 8C) does not significantly affect induction of LTP under conditions that produce near-maximal potentiation in slices.
In contrast, both low-frequency stimulation and DHPG-induced forms of LTD are impaired in the densin knock-out mice. A 15 min train of 1 Hz stimulation caused a 25% reduction of fEPSP amplitudes in wild-type slices but no reduction in the densin knock-out slices (Fig. 7E). This form of LTD has been shown to be dependent on activation of NMDARs (Ho et al., 2004). Bath application of DHPG, a group 1 mGluR agonist that activates mGluR1 and mGluR5, induces a non-NMDAR-dependent form of LTD (Lüscher and Huber, 2010). A 10 min application of DHPG (100 μm) induced a ∼50% transient depression in fEPSP amplitude and a stable ∼25% depression after washout of the drug in wild-type slices but only a ∼10% depression in the knock-out slices (Fig. 7F). Thus, loss of densin impairs both NMDAR-dependent and mGluR-dependent LTD, suggesting that the defect lies at a biochemical step downstream of receptor activation.
Loss of densin alters the morphology of spines on hippocampal neurons
When spine synapses are remodeled to increase or decrease synaptic strength, the number of AMPA-type receptors is increased or decreased and the actin cytoskeleton is remodeled to increase or decrease the size of the spine head. The biochemical regulatory pathways that alter the spine actin cytoskeleton are tightly linked to those that regulate surface AMPA receptors (Carlisle and Kennedy, 2005). CaMKII, along with other PSD enzymes, participates in this regulation of the actin cytoskeleton (Carlisle et al., 2008). Abnormalities in spine shape were reported in mouse models in which DISC1 isoforms were genetically deleted or knocked down with siRNA (Kvajo et al., 2008). To investigate whether loss of densin deranges the regulatory machinery that shapes spine morphology in adult mice, we first crossed densin knock-out mice with a transgenic line that sparsely expresses GFP in the CA1 region of the hippocampus (Feng et al., 2000). Spine morphology was examined in three-dimensional reconstructions of confocal images of labeled pyramidal cell dendrites from densin knock-out and wild-type mice containing the GFP label. The necks of spines were significantly more elongated in densin knock-out mice. Furthermore, when spines were categorized by morphological type, we found that the proportion of mushroom spines was increased by 31% and the number of stubby spines decreased by 22% in the knock-out mice, whereas the total number of spines was not significantly changed (Fig. 9A). The necks of densin knock-out mushroom spines were significantly longer and thinner than wild-type mushroom spines (Fig. 9B,D,E), but the average head diameter was unchanged (Fig. 9C,E). These data indicate that the defect or defects in biochemical signaling in the spine induced by loss of densin occur at positions in the regulatory network that alter steady-state spine morphology. They provide additional evidence that densin is important for proper kinetic tuning of regulatory pathways in the spine.
Figure 9.

Hippocampal dendritic spines from adult densin knock-out (KO) mice are elongated compared with spines from wild-type mice. A, Knock-out of densin resulted in a 31% increase in the number of mushroom spines (wild type, 0.22 ± 0.02; knock-out, 0.29 ± 0.02 mushroom spines/μm; *p < 0.05) and a 26% decrease in the number of stubby spines (wild type, 0.31 ± 0.03; knock-out, 0.22 ± 0.02 stubby spines/μm; *p < 0.05), whereas the total number of all spine types per micrometer of dendrite was not significantly different from wild-type levels. B, The length of mushroom spines was increased by 22% in the knock-outs compared with wild type (wild type, 0.79 ± 0.03 μm; knock-out, 0.96 ± 0.02 μm; **p < 0.01), whereas the lengths of stubby and thin spines were unchanged. C, The head diameters of mushroom, stubby, and thin spines were unchanged in densin knock-out compared with wild-type mice. D, Loss of densin resulted in a 22% decrease in the neck diameter of mushroom spines (wild type, 0.17 ± 0.01 μm; knock-out, 0.13 ± 0.01 μm; *p < 0.05). E, Representative images of dendrites in stratum radiatum of the CA1 region from wild-type and densin knock-out mice. For A–D, statistical significance was determined with two-tailed t tests on measurements of 110 mushroom, 163 stubby, 112 thin spines (385 total) from 3 wild-type mice and 187 mushroom, 154 stubby, 129 thin (475 total) from 3 densin knock-out mice. Error bars represent SEM.
Discussion
Molecular and cellular effects of deletion of densin
The domain structure of densin, which includes binding sites for several proteins, suggests that it functions as a scaffold molecule (Apperson et al., 1996; Strack et al., 2000; Walikonis et al., 2001; Izawa et al., 2002; Ohtakara et al., 2002). As such, it would be expected to influence the organization and dynamics of signaling pathways in the PSD in which it is highly concentrated. Mutation or deletion of large scaffold proteins can lead to complex behavioral or pathological phenotypes, apparently because such proteins influence more than one cellular function. A prominent example is huntingtin, the mutation of which interferes with neuronal transcription, intracellular transport, and synaptic transmission (Imarisio et al., 2008; Marcora and Kennedy, 2010) and causes the complex phenotypes associated with Huntington's disease. Another example is DISC1, which has a wide range of postulated binding partners and is located in mitochondria, the centrosome, and excitatory synapses (James et al., 2004; Brandon et al., 2009; Hayashi-Takagi et al., 2010). Mutation of DISC1 in humans produces increased risk of schizophrenia and bipolar disorder (Millar et al., 2000).
We show that loss of densin leads to reduction in the amounts of at least three other proteins in the PSD fraction. The steady-state level of the direct binding partner of densin, the cytoskeletal protein α-actinin, is reduced in brain homogenates as well as in the PSD fraction, suggesting that absence of densin causes faster degradation of α-actinin or reduced expression of the α-actinin gene. The steady-state levels of mGluR5 and DISC1 are also reduced in the PSD fraction but are not reduced in total brain homogenates. We conclude that they are expressed normally, but loss of densin specifically alters their localization to the PSD fraction. The similar amount of reduction of DISC1, mGluR5, and α-actinin in the PSD fraction suggests that a multiprotein complex containing the three proteins may be mislocated or disrupted in the absence of densin. Densin binds directly to α-actinin (Walikonis et al., 2001), which in turn binds directly to the tail of mGluR5 (Cabello et al., 2007). Thus, the mislocalization of mGluR5 could be a secondary effect of the reduced amount of α-actinin in the PSD fraction. There have been no previous reports of an association, direct or indirect, between densin and DISC1. It will be important to investigate how deletion of densin leads to reduction of DISC1 in the PSD fraction and whether this reduction is responsible for specific endophenotypes.
The C-terminal tail of densin forms a high-affinity ternary complex with α-actinin and the holoenzyme of CaMKII (Walikonis et al., 2001). Although the amount of CaMKII in the PSD fraction is not changed in the densin knock-out mice, we show that the disposition of CaMKII in the PSD is altered in a functionally significant way. The amount of CaMKII colocalizing with PSD-95 in neurons lacking both densin and synaptic NMDARs is reduced by 50% compared with a 15% reduction in neurons lacking synaptic NMDARs alone. These results suggest that densin and the NR2 subunits of the NMDAR both act as docking sites for CaMKII in the PSD and that NMDARs may compensate for the lack of densin in the densin knock-out by binding more CaMKII. Although this compensation preserves the total amount of CaMKII in the PSD, the early dynamics of activity-dependent regulation of CaMKII are altered. The basal level of active, autophosphorylated CaMKII is significantly reduced in densin knock-outs compared with wild-type neurons, and activation of CaMKII is twofold higher in the 5 min after an increase in synaptic activity produced by application of bicuculline. These data suggest that densin is important for precise positioning of CaMKII in the PSD in relation to other regulatory molecules and that disruption of positioning of CaMKII in the absence of densin causes a subtle quantitative derangement of its activity-dependent regulation.
Changes in synaptic plasticity in the densin knock-out mice
NMDAR function and NMDAR-dependent LTP are normal in hippocampal slices from densin knock-out mice. Thus, the knock-outs do not have a generalized hypofunction of NMDARs. Apparently, the derangement of activation of CaMKII by NMDARs in cultured neurons from densin knock-outs is not sufficient to alter the magnitude of tetanus-induced LTP in hippocampal slices; however, there is a trend toward greater short-term potentiation in the knock-outs during the first 5 min after tetanization.
In contrast, LTD is severely disrupted in hippocampal slices from knock-outs. This is true for both NMDAR-dependent LTD, induced by low-frequency stimulation in 3-week-old animals (Ho et al., 2004) and for mGluR-dependent LTD induced by treatment with the group 1 mGluR agonist DHPG in adults (Lüscher and Huber, 2010). Thus, it appears that a downstream step critical for synaptic depression, such as control of endocytosis of AMPA-type receptors, is compromised. It is also possible that reduction of mGluR5 itself in PSDs contributes to loss of DHPG-induced LTD. In this regard, it will be important to test whether localization or signaling through mGluR1, which is also activated by DHPG, are altered in the densin mutants.
During the remodeling of the spine that underlies synaptic plasticity, the actin cytoskeleton undergoes tightly regulated disassembly and reformation to support a larger or smaller spine head. These changes are coordinated with the insertion and removal of AMPA receptors and appear to be triggered by many of the same signaling pathways (Carlisle and Kennedy, 2005; Carlisle et al., 2008). Derangement of regulation of the spine cytoskeleton in densin knock-outs is manifested by significant changes in steady-state morphology of dendritic spines in the hippocampus. These changes, like the other changes in synaptic regulation in the knock-outs, point to subtle misregulation of signaling pathways caused by loss of the densin scaffold protein.
Behavior of densin knock-out mice
To facilitate the study of psychiatric disease mechanisms, investigators have noted that certain abnormal behaviors elicited in rodents by mutations or drug treatments appear to be related to isolated symptoms in human schizophrenia and autism (Powell and Miyakawa, 2006; Silverman et al., 2010) and have coined the term endophenotype to refer to these isolated behaviors. In humans, schizophrenia presents as a combination of symptoms, both positive (hallucinations, delusions, and severe psycho-motor agitation) and negative (blunted emotional expression, low motivation, anhedonia, and social withdrawal) (Powell and Miyakawa, 2006). It also includes cognitive deficits, such as poor working, short-term, and episodic memory and attention deficits.
We observed several endophenotypes related to schizophrenia or autism in the densin knock-outs. For example, the densin knock-outs became hyperactive when presented with novel objects and during the beam-crossing test. Baseline or novelty-induced hyperactivity are forms of psycho-motor agitation present in mouse models with schizophrenia-like features (Powell and Miyakawa, 2006). The baseline activity of the mutants in a home cage or open field was significantly reduced compared with wild-type mice. Thus, their hyperactivity is explicitly in response to high levels of novelty or stress. Densin knock-outs also had profound deficits in nesting behavior, an endophenotype related to social withdrawal (Powell and Miyakawa, 2006). Nesting deficits can be induced by psychomimetic agents and are ameliorated in some mouse models by administration of anti-psychotic medications (Kirby et al., 2010), further validating their significance as a model of negative schizophrenic symptoms. The densin knock-outs also showed significant cognitive deficits; both hippocampus-dependent and -independent forms of short-term memory were impaired.
Prepulse inhibition was reduced in the densin knock-outs. Defective prepulse inhibition is present in human schizophrenia but is not exclusively associated with it. It is also reduced in patients with autism, panic disorder, Tourette's syndrome, bipolar disorder, Huntington's disease, and obsessive-compulsive disorder (Powell et al., 2009).
Anxiety was the most consistent behavioral abnormality in the densin knock-out mice. Anxiety is not considered a core feature of schizophrenia but is frequently observed in other mouse models with schizophrenia- and autism-like features and is common in the corresponding human disorders (Mattila et al., 2010; Achim et al., 2011). Both homozygote and heterozygote densin knock-outs displayed high levels of aggression, also not a core feature of schizophrenia. Mice with a heterozygous deletion of the α-subunit of CaMKII also display abnormal defensive aggression (Chen et al., 1994), whereas mice overexpressing α-CaMKII display abnormal anxiety and offensive aggression (Hasegawa et al., 2009). Therefore, disruption of regulation of CaMKII may contribute to the anxiety and aggression displayed by the densin knock-outs.
Interestingly, mGluR5 knock-out mice show some of the same endophenotypes that we observe in the densin knock-outs, including impaired prepulse inhibition, hyperlocomotion, and short-term hippocampus-dependent memory deficits (Gray et al., 2009; Inta et al., 2010). Therefore, these particular behavioral abnormalities may be specifically related to the reduction in DHPG-induced LTD. It has been proposed that mGluR-induced LTD in the perirhinal cortex and hippocampus plays an important role in recognition of novelty (Lüscher and Huber, 2010); thus, disruption of this form of plasticity might also account for the inability of densin knock-outs to recognize novel objects or novel object place.
From 2 to 6 weeks of age, densin knock-out mice are smaller and have higher mortality rates than wild-type littermates; at later ages, these differences become insignificant. We found no abnormalities in gross brain morphology, but the possibility remains that alterations in brain circuitry, caused by developmental defects, underlie some of the abnormal behaviors.
Data emerging from experiments with mouse and human genetic studies suggest that mutation or deletion of signaling and scaffold proteins that regulate synaptic transmission at excitatory synapses may contribute to genetic vulnerability for schizophrenia and other mental illnesses. We find that deletion of the PSD scaffold protein densin, not yet associated with risk of mental illness, produces behavioral endophenotypes often associated with schizophrenia or autism spectrum disorders. Deletion of densin also results in a reduction in the PSD fraction of two proteins previously implicated in mental illnesses, mGluR5 and DISC1, and misregulation of a third, CaMKII. Our findings are consistent with the general hypothesis that small disruptions of the finely tuned regulatory machinery that adjusts the strength of excitatory synapses in response to experience can contribute to abnormal behaviors associated with mental illness.
Footnotes
This research was supported by National Institutes of Health Grants NS17660 and NS028710 (M.B.K.) and MH609197 (T.J.O.), the Gordon and Betty Moore Foundation Center for Integrative Study of Cell Biology (M.B.K. and H.J.C.), the Howard Hughes Medical Institute (A.M.-M.), National Science Foundation Grant 0543651 (T.J.O.), the McGrath Foundation (P.H.P.), and the Broad Fellows in Brain Circuitry program (K.M.G. and A.D.S.). We thank J. Sanes (Harvard University, Boston, MA) and P. Seeburg (Max Plank Institute for Medical Research, Munich, Germany) for mutant mouse strains and Sarah Cantor for help with experiments.
References
- Achim AM, Maziade M, Raymond E, Olivier D, Mérette C, Roy MA. How prevalent are anxiety disorders in schizophrenia? A meta-analysis and critical review on a significant association. Schizophr Bull. 2011;37:811–821. doi: 10.1093/schbul/sbp148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apperson ML, Moon IS, Kennedy MB. Characterization of densin-180, a new brain-specific synaptic protein of the O-sialoglycoprotein family. J Neurosci. 1996;16:6839–6852. doi: 10.1523/JNEUROSCI.16-21-06839.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arguello PA, Gogos JA. Modeling madness in mice: one piece at a time. Neuron. 2006;52:179–196. doi: 10.1016/j.neuron.2006.09.023. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, Quinlan EM, Nakazawa K. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci. 2010;13:76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeron T. A synaptic trek to autism. Curr Opin Neurobiol. 2009;19:231–234. doi: 10.1016/j.conb.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Millar JK, Korth C, Sive H, Singh KK, Sawa A. Understanding the role of DISC1 in psychiatric disease and during normal development. J Neurosci. 2009;29:12768–12775. doi: 10.1523/JNEUROSCI.3355-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EL, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Cabello N, Remelli R, Canela L, Soriguera A, Mallol J, Canela EI, Robbins MJ, Lluis C, Franco R, McIlhinney RA, Ciruela F. Actin-binding protein alpha-actinin-1 interacts with the metabotropic glutamate receptor type 5b and modulates the cell surface expression and function of the receptor. J Biol Chem. 2007;282:12143–12153. doi: 10.1074/jbc.M608880200. [DOI] [PubMed] [Google Scholar]
- Carlisle HJ, Kennedy MB. Spine architecture and synaptic plasticity. Trends Neurosci. 2005;28:182–187. doi: 10.1016/j.tins.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Carlisle HJ, Manzerra P, Marcora E, Kennedy MB. SynGAP regulates steady-state and activity-dependent phosphorylation of cofilin. J Neurosci. 2008;28:13673–13683. doi: 10.1523/JNEUROSCI.4695-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Rainnie DG, Greene RW, Tonegawa S. Abnormal fear response and aggressive behavior in mutant mice deficient for alpha-calcium-calmodulin kinase II. Science. 1994;266:291–294. doi: 10.1126/science.7939668. [DOI] [PubMed] [Google Scholar]
- Deacon RM. Assessing nest building in mice. Nat Protoc. 2006;1:1117–1119. doi: 10.1038/nprot.2006.170. [DOI] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsäter H, Sponheim E, Goubran-Botros H, Delorme R, Chabane N, Mouren-Simeoni MC, de Mas P, Bieth E, Rogé B, Héron D, Burglen L, Gillberg C, Leboyer M, Bourgeron T. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Fukaya M, Kato A, Lovett C, Tonegawa S, Watanabe M. Retention of NMDA receptor NR2 subunits in the lumen of endoplasmic reticulum in targeted NR1 knockout mice. Proc Natl Acad Sci USA. 2003;100:4855–4860. doi: 10.1073/pnas.0830996100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- Gray L, van den Buuse M, Scarr E, Dean B, Hannan AJ. Clozapine reverses schizophrenia-related behaviours in the metabotropic glutamate receptor 5 knockout mouse: association with N-methyl-d-aspartic acid receptor up-regulation. Int J Neuropsychopharmacol. 2009;12:45–60. doi: 10.1017/S1461145708009085. [DOI] [PubMed] [Google Scholar]
- Guo X, Hamilton PJ, Reish NJ, Sweatt JD, Miller CA, Rumbaugh G. Reduced expression of the NMDA receptor-interacting protein SynGAP causes behavioral abnormalities that model symptoms of schizophrenia. Neuropsychopharmacology. 2009;34:1659–1672. doi: 10.1038/npp.2008.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Gauthier J, Spiegelman D, Noreau A, Yang Y, Pellerin S, Dobrzeniecka S, Côté M, Perreau-Linck E, Perreault-Linck E, Carmant L, D'Anjou G, Fombonne E, Addington AM, Rapoport JL, Delisi LE, Krebs MO, Mouaffak F, Joober R, Mottron L, et al. Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N Engl J Med. 2009;360:599–605. doi: 10.1056/NEJMoa0805392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa S, Furuichi T, Yoshida T, Endoh K, Kato K, Sado M, Maeda R, Kitamoto A, Miyao T, Suzuki R, Homma S, Masushige S, Kajii Y, Kida S. Transgenic up-regulation of alpha-CaMKII in forebrain leads to increased anxiety-like behaviors and aggression. Mol Brain. 2009;2:6. doi: 10.1186/1756-6606-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi-Takagi A, Takaki M, Graziane N, Seshadri S, Murdoch H, Dunlop AJ, Makino Y, Seshadri AJ, Ishizuka K, Srivastava DP, Xie Z, Baraban JM, Houslay MD, Tomoda T, Brandon NJ, Kamiya A, Yan Z, Penzes P, Sawa A. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat Neurosci. 2010;13:327–332. doi: 10.1038/nn.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho OH, Delgado JY, O'Dell TJ. Phosphorylation of proteins involved in activity-dependent forms of synaptic plasticity is altered in hippocampal slices maintained in vitro. J Neurochem. 2004;91:1344–1357. doi: 10.1111/j.1471-4159.2004.02815.x. [DOI] [PubMed] [Google Scholar]
- Imarisio S, Carmichael J, Korolchuk V, Chen CW, Saiki S, Rose C, Krishna G, Davies JE, Ttofi E, Underwood BR, Rubinsztein DC. Huntington's disease: from pathology and genetics to potential therapies. Biochem J. 2008;412:191–209. doi: 10.1042/BJ20071619. [DOI] [PubMed] [Google Scholar]
- Inta D, Monyer H, Sprengel R, Meyer-Lindenberg A, Gass P. Mice with genetically altered glutamate receptors as models of schizophrenia: a comprehensive review. Neurosci Biobehav Rev. 2010;34:285–294. doi: 10.1016/j.neubiorev.2009.07.010. [DOI] [PubMed] [Google Scholar]
- Ivanov A, Pellegrino C, Rama S, Dumalska I, Salyha Y, Ben-Ari Y, Medina I. Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons. J Physiol. 2006;572:789–798. doi: 10.1113/jphysiol.2006.105510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa I, Nishizawa M, Ohtakara K, Inagaki M. Densin-180 interacts with delta-catenin/neural plakophilin-related armadillo repeat protein at synapses. J Biol Chem. 2002;277:5345–5350. doi: 10.1074/jbc.M110052200. [DOI] [PubMed] [Google Scholar]
- James R, Adams RR, Christie S, Buchanan SR, Porteous DJ, Millar JK. Disrupted in Schizophrenia 1 (DISC1) is a multicompartmentalized protein that predominantly localizes to mitochondria. Mol Cell Neurosci. 2004;26:112–122. doi: 10.1016/j.mcn.2004.01.013. [DOI] [PubMed] [Google Scholar]
- Kennedy MB. Signal-processing machines at the postsynaptic density. Science. 2000;290:750–754. doi: 10.1126/science.290.5492.750. [DOI] [PubMed] [Google Scholar]
- Kennedy MB, Bennett MK, Erondu NE. Biochemical and immunochemical evidence that the “major postsynaptic density protein” is a subunit of a calmodulin-dependent protein kinase. Proc Natl Acad Sci USA. 1983;80:7357–7361. doi: 10.1073/pnas.80.23.7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby BP, Waddington JL, O'Tuathaigh CM. Advancing a functional genomics for schizophrenia: psychopathological and cognitive phenotypes in mutants with gene disruption. Brain Res Bull. 2010;83:162–176. doi: 10.1016/j.brainresbull.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Koh IY, Lindquist WB, Zito K, Nimchinsky EA, Svoboda K. An image analysis algorithm for dendritic spines. Neural Comput. 2002;14:1283–1310. doi: 10.1162/089976602753712945. [DOI] [PubMed] [Google Scholar]
- Kvajo M, McKellar H, Arguello PA, Drew LJ, Moore H, MacDermott AB, Karayiorgou M, Gogos JA. A mutation in mouse Disc1 that models a schizophrenia risk allele leads to specific alterations in neuronal architecture and cognition. Proc Natl Acad Sci USA. 2008;105:7076–7081. doi: 10.1073/pnas.0802615105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, Westphal H. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci U S A. 1996;93:5860–5865. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard AS, Lim IA, Hemsworth DE, Horne MC, Hell JW. Calcium/calmodulin-dependent protein kinase II is associated with the N-methyl-d-aspartate receptor. Proc Natl Acad Sci USA. 1999;96:3239–3244. doi: 10.1073/pnas.96.6.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Huber KM. Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron. 2010;65:445–459. doi: 10.1016/j.neuron.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcora E, Kennedy MB. The Huntington's disease mutation impairs Huntingtin's role in the transport of NF-kB from the synapse to the nucleus. Hum Mol Genet. 2010;19:4373–4384. doi: 10.1093/hmg/ddq358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattila ML, Hurtig T, Haapsamo H, Jussila K, Kuusikko-Gauffin S, Kielinen M, Linna SL, Ebeling H, Bloigu R, Joskitt L, Pauls DL, Moilanen I. Comorbid psychiatric disorders associated with Asperger syndrome/high-functioning autism: a community- and clinic-based study. J Autism Dev Disord. 2010;40:1080–1093. doi: 10.1007/s10803-010-0958-2. [DOI] [PubMed] [Google Scholar]
- Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CA, Devon RS, St Clair DM, Muir WJ, Blackwood DH, Porteous DJ. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9:1415–1423. doi: 10.1093/hmg/9.9.1415. [DOI] [PubMed] [Google Scholar]
- Millar JK, Christie S, Porteous DJ. Yeast two-hybrid screens implicate DISC1 in brain development and function. Biochem Biophys Res Commun. 2003;311:1019–1025. doi: 10.1016/j.bbrc.2003.10.101. [DOI] [PubMed] [Google Scholar]
- Ohtakara K, Nishizawa M, Izawa I, Hata Y, Matsushima S, Taki W, Inada H, Takai Y, Inagaki M. Densin-180, a synaptic protein, links to PSD-95 through its direct interaction with MAGUIN-1. Genes Cells. 2002;7:1149–1160. doi: 10.1046/j.1365-2443.2002.00589.x. [DOI] [PubMed] [Google Scholar]
- Omkumar RV, Kiely MJ, Rosenstein AJ, Min KT, Kennedy MB. Identification of a phosphorylation site for calcium/calmodulin-dependent protein kinase II in the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem. 1996;271:31670–31678. doi: 10.1074/jbc.271.49.31670. [DOI] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Almeida J, Bacchelli E, Bader GD, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bölte S, Bolton PF, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletnikov MV, Ayhan Y, Nikolskaia O, Xu Y, Ovanesov MV, Huang H, Mori S, Moran TH, Ross CA. Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol Psychiatry. 2008;13:173–186. 115. doi: 10.1038/sj.mp.4002079. [DOI] [PubMed] [Google Scholar]
- Powell CM, Miyakawa T. Schizophrenia-relevant behavioral testing in rodent models: a uniquely human disorder? Biol Psychiatry. 2006;59:1198–1207. doi: 10.1016/j.biopsych.2006.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell SB, Zhou X, Geyer MA. Prepulse inhibition and genetic mouse models of schizophrenia. Behav Brain Res. 2009;204:282–294. doi: 10.1016/j.bbr.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quitsch A, Berhörster K, Liew CW, Richter D, Kreienkamp HJ. Postsynaptic shank antagonizes dendrite branching induced by the leucine-rich repeat protein Densin-180. J Neurosci. 2005;25:479–487. doi: 10.1523/JNEUROSCI.2699-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JL, Yang M, Lord C, Crawley JN. Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci. 2010;11:490–502. doi: 10.1038/nrn2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprengel R, Suchanek B, Amico C, Brusa R, Burnashev N, Rozov A, Hvalby O, Jensen V, Paulsen O, Andersen P, Kim JJ, Thompson RF, Sun W, Webster LC, Grant SG, Eilers J, Konnerth A, Li J, McNamara JO, Seeburg PH. Importance of the intracellular domain of NR2 subunits for NMDA receptor function in vivo. Cell. 1998;92:279–289. doi: 10.1016/s0092-8674(00)80921-6. [DOI] [PubMed] [Google Scholar]
- Steele AD, Jackson WS, King OD, Lindquist S. The power of automated high-resolution behavior analysis revealed by its application to mouse models of Huntington's and prion diseases. Proc Natl Acad Sci USA. 2007;104:1983–1988. doi: 10.1073/pnas.0610779104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack S, Colbran RJ. Autophosphorylation-dependent targeting of calcium/ calmodulin- dependent protein kinase II by the NR2B subunit of the N-methyl-d- aspartate receptor. J Biol Chem. 1998;273:20689–20692. doi: 10.1074/jbc.273.33.20689. [DOI] [PubMed] [Google Scholar]
- Strack S, Robison AJ, Bass MA, Colbran RJ. Association of calcium/calmodulin-dependent kinase II with developmentally regulated splice variants of the postsynaptic density protein densin-180. J Biol Chem. 2000;275:25061–25064. doi: 10.1074/jbc.C000319200. [DOI] [PubMed] [Google Scholar]
- Walikonis RS, Oguni A, Khorosheva EM, Jeng CJ, Asuncion FJ, Kennedy MB. Densin-180 forms a ternary complex with the α-subunit of CaMKII and α-actinin. J Neurosci. 2001;21:423–433. doi: 10.1523/JNEUROSCI.21-02-00423.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willatt L, Cox J, Barber J, Cabanas ED, Collins A, Donnai D, FitzPatrick DR, Maher E, Martin H, Parnau J, Pindar L, Ramsay J, Shaw-Smith C, Sistermans EA, Tettenborn M, Trump D, de Vries BB, Walker K, Raymond FL. 3q29 microdeletion syndrome: clinical and molecular characterization of a new syndrome. Am J Hum Genet. 2005;77:154–160. doi: 10.1086/431653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki N, Maekawa M, Kobayashi K, Kajii Y, Maeda J, Soma M, Takao K, Tanda K, Ohira K, Toyama K, Kanzaki K, Fukunaga K, Sudo Y, Ichinose H, Ikeda M, Iwata N, Ozaki N, Suzuki H, Higuchi M, Suhara T, et al. Alpha-CaMKII deficiency causes immature dentate gyrus, a novel candidate endophenotype of psychiatric disorders. Mol Brain. 2008;1:6. doi: 10.1186/1756-6606-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Takahashi E, Li W, Halt A, Wiltgen B, Ehninger D, Li GD, Hell JW, Kennedy MB, Silva AJ. Interactions between the NR2B receptor and CaMKII modulate synaptic plasticity and spatial learning. J Neurosci. 2007;27:13843–13853. doi: 10.1523/JNEUROSCI.4486-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]