

Graphical abstract

Keywords: Fragment-based lead discovery (FBLD), Chemogenomics, Serotonin 5-HT3A receptor, Histamine H4 receptor, Dual activity ligand, G-protein coupled receptor (GPCR), Ligand gated ion channel (LGIC)
Abstract
A fragment library was screened against the G protein-coupled histamine H4 receptor (H4R) and the ligand-gated ion channel serotonin 5-HT3A (5-HT3AR). Interestingly, significant overlap was found between H4R and 5-HT3AR hit sets. The data indicates that dual active H4R and 5 HT3AR fragments have a higher complexity than the selective compounds which has important implications for chemical genomics approaches. The results of our fragment-based library screening study illustrate similarities in ligand recognition between H4R and 5-HT3AR and have important consequences for selectivity profiling in ongoing drug discovery efforts on H4R and 5-HT3AR. The affinity profiles of our fragment screening studies furthermore match the chemical properties of the H4R and 5-HT3AR binding sites and can be used to define molecular interaction fingerprints to guide the in silico prediction of protein-ligand interactions and structure.
Fragment-based lead discovery (FBLD) uses low molecular weight compounds as starting points for hit and lead optimization. Compared to the drug-like compounds that are screened in typical high-throughput screening campaigns, fragments are better able to cover the corresponding chemical space. Consequently, typical fragment libraries consist of about 1000 small molecules.1 Biochemical and biophysical techniques are used to detect the low affinity fragment binding. Ligand efficiency (LE), defined as the binding energy of the ligand (ΔG in kcal mol−1) per non-H atom (Heavy Atoms, HA), is used to select the most promising hits and guide the optimization studies.2 Typical hit rates for a fragment library screen are considerably higher than for the high throughput screening of drug-like compounds.3 The higher complexity of the latter compounds drastically reduces the chances of perfect complementarity with the biological targets. Thus, fragments are particularly suited to probe the binding site of receptors,4,5 and are therefore ideal tools in chemogenomic approaches that link chemical with biological space.6 In chemogenomics studies the effect of a wide array of chemicals on a wide array of biological targets is investigated.7 The resulting two-dimensional matrix of targets versus hit compounds is useful for the discovery of ligands for novel drug targets and to have better control over the selectivity of ligands and/or drugs. Furthermore, the data can lead to a better understanding of ligand-receptor interactions.
We have screened our fragment library against the histamine H4 receptor (H4R) for which we have ongoing drug discovery programs. H4R fragment hits were grown into potent H4R ligands and fragment-merging approaches resulted in efficient scaffold hopping towards new chemical series.8,9 The H4R is considered a very promising target for treating inflammatory and allergic disorders as well in the modulation of pain and pruritis.10
Meanwhile, the same fragment library is being screened against a rapidly expanding variety of targets. Here, we describe a remarkable overlap of the fragment hit set of the H4R and the 5-HT3AR. This ligand-gated ion channel is a drug target for the treatment of irritable bowel syndrome (IBS) and chemotherapy-induced nausea and vomiting (CINV).11 Marketed drugs of 5-HT3AR include tropisteron (Navoban®) and palonosetron (Aloxi®). The results of our fragment-based library screening indicate similarities in ligand recognition between H4R and 5-HT3AR and potential selectivity issues when developing H4R or 5-HT3AR drugs. On the other side, dual activity compounds might also have clinical advantages. Next to the established role of 5HT3AR in IBS, recent findings also suggest a role of H4R in this disease. It has been found that an increased innate immune activity in the intestinal mucosa and in blood is found in subpopulations of patients with IBS.12 Mast cells and monocytes seem to be particularly important and might indicate that the H4R is also involved in this ailment.
We screened the biological activity of a diverse set of 1010 fragment-like molecules against H4R and 5-HT3AR. The compounds in this library obey general fragment library rules13: (i) heavy atoms count ⩽ 22; (ii) c log P <3; (iii) number of H-bond donors ⩽ 3; (iv) number of H-bond acceptors ⩽ 3; (v) number of rotatable bonds ⩽ 5. The fragments furthermore contain at least one ring structure and do not contain reactive functional groups.14 The structural diversity of the library was analysed, among others, by means of a scaffold classification analysis (SCA).15 In this analysis, fragments are indexed by two parameters, that is, cyclicity and complexity. Cyclicity is the ratio between ring atoms and side chain atoms (thus, if all the atoms of the molecule belong to the ring structure cyclicity equals one). In addition, the complexity was calculated as a descriptor of the size and shape of the scaffold, taking into account the smallest set of smallest rings, the number of heavy atoms, the number of bonds between the heavy atoms, and the sum of heavy atoms atomic number.15 Chemical diversity of the fragment library is furthermore confirmed by the fact that only 1.6% of the pair wise comparisons of the ECFP-4 topological fingerprints of the fragments give Tanimoto similarity values higher than 0.26.16
For the H4R fragment screen a radioligand displacement study was performed at a 10 μM fragment concentration. Hits were assigned when the fragment displaced 50% or more of the radioligand, resulting in 56 hits (hit rate: 6%). Radioligand binding was measured by displacement of [3H]histamine using membranes of HEK293 cells transiently expressing the human H4R.17 For the hit compounds, affinities were determined by subsequent radioligand displacement studies.
For the 5-HT3AR we performed a high throughput functional fragment screen18 using a fluorescent readout (Flex Station) applying a fluorescent membrane potential dye. With this screening technique we can identify compounds that have affinity for the receptor and in addition classify the hits as agonists, antagonists or inactive. From this fragment screening we identified 70 hits for the 5-HT3A receptor (hit rate: 7%). Fragments were screened at a concentration of 100 μM using stably expressed human 5-HT3AR in HEK293 cells. Binding affinities of hits were determined using radioligand binding studies measuring [3H]granisetron binding using membranes of HEK293 cells expressing the human 5-HT3AR.18
The SCA plot15 in Figure 1a shows the distribution of 5-HT3AR selective hits, H4R selective hits, and dual 5-HT3AR/H4R hits in the chemical space covered by the fragment library and demonstrates the structural diversity of the fragment hits. Interestingly, significant overlap between the H4R and 5-HT3AR hit sets occur, for example, 24% of the 5-HT3AR hits also bind H4R and 30% of the H4R hits also bind 5-HT3AR (Fig. 1b). This is ca. 10% higher than any other overlap between non-related targets that we have screened so far. In Table 1 some selective H4R ligands, selective 5-HT3AR ligands as well as compounds with affinity for both receptors are displayed. Dual hits 7, 8, 11 have comparable affinities for 5-HT3R and H4R, while dual hit 9 has 500-fold selectivity for 5-HT3R over H4R, and hit 10 has 200-fold for H4R over 5-HT3R (Table 1).
Figure 1.

(a) SCA plot showing the hit distribution for the H4R (Red), the 5-HT3AR (blue), the 5-HT3AR and H4R (green) as well as inactives, compounds which do not bind H4R or 5HT3AR (grey). Hits presented in Table 1 are labeled by their corresponding number. (b) Schematic representation of the overlap between H4R and 5-HT3AR ligands. (c) Distribution of the complexity of H4R selective fragments (red line), 5-HT3AR selective fragments (blue line), dual H4R/5-HT3AR fragments (green line), and inactives (dotted grey line).
Table 1.
Structures of fragments that bind solely H4R (1–3), solely 5-HT3AR (4–6) and both H4R and 5-HT3AR (7–11)
| # | H4R/5-HT3AR | Structure | Affinity (pKi) |
|
|---|---|---|---|---|
| H4Ra | 5-HT3ARc | |||
| 1 |  |
7.0 ± 0.1 | n.a. | |
| 2 | |
6.2 ± 0.1 | n.a. | |
| 3 | |
6.7 ± 0.0 | n.a. | |
| 4 |  |
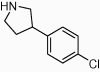 |
n.a. | 6.1 ± 0.2 |
| 5 | |
n.a. | 6.0 ± 0.0 | |
| 6 | |
n.a. | 6.1 ± 0.1 | |
| 7 |  |
6.2 ± 0.0 | 6.6 ± 0.3 | |
| 8 | |
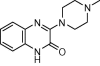 |
7.2 ± 0.0 | 7.9 ± 0.3 |
| 9 | |
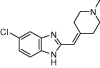 |
6.1 ± 0.1b | 8.8 ± 0.1 |
| 10 | |
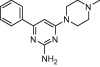 |
8.2 ± 0.1 | 5.9 ± 0.1 |
| 11 | |
6.2 ± 0.1b | 5.9 ± 0.3 | |
n.a.: Non active.
Measured by displacement of [3H]histamine binding using membranes of HEK293 cells transiently expressing the human H4R. pKi’s are calculated from at least three independent measurements as the mean ± SEM.
Determined using membranes of SK-N-MC cells transiently expressing the human H4R. pKi’s are calculated from at least three independent measurements as the mean ± SEM.
pKi: Measured by displacement of [3H]granisetron binding using membranes of HEK293 cells expressing the human 5-HT3AR. pKi’s are calculated from at least two independent measurements as the mean ± SEM.
Many of the dual H4R/5-HT3AR ligands contain a quinazoline, quinoxaline, aminopyrimidine, imidazole, or benzimidazole scaffold in combination with a positively ionizable ring system (Table 1). Figure 1c shows that most of these dual H4R/5-HT3AR fragments have a higher complexity than the H4R and 5-HT3AR selective fragments. The structural complexity of 71% of the dual 5-HT3A/H4R fragments is 0.7 or higher, while 79% of the H4R selective hits and 74% of the 5-HT3AR selective fragments is lower than 0.7. While earlier chemoinformatics analyses suggested that selective ligands are more complex in terms of pharmacophore features4 and molecular shape19, our fragment-based chemogenomics study suggests a more delicate balance between ligand complexity and target selectivity. Our fragment library screening data indicate that fragments need to have high enough complexity to hit several targets, but low enough complexity to be too specific for a single site. This is in line with the theoretical model by Hann and co-workers4 that describes probability of finding a hit when considering the complexity of the ligand. The probability of detecting a binding event is given by multiplying the probability of matching features and the probability of being able to detect low affinity binders. Our experimental data set shows that indeed the chance of finding fragment hits on two different targets favors higher complexity compounds. The relatively high complexity of the overlapping H4R and 5-HT3AR fragment hit set is furthermore a clear indication that the ligand recognition profiles of these receptors are similar. Figure 2 shows the chemical similarity of the fragment library compared to serotonin and histamine (determined by Pipeline Pilot ECFP-4 circular fingerprint20 Tanimoto similarity coefficients (Tc)). While some of the H4R selective fragments (38%) are chemically similar to histamine (i.e., ECFP-4 Tc >0.26 including 2 (Table 1)), none of the 5-HT3AR selective fragments share chemical similarity serotonin-like, and only two of the dual binders are histamine-like (including 11 see Table 1). These data show that complex H4R/5-HT3AR dual fragments are dissimilar from the (less complex) endogenous ligands of H4R and 5-HT3AR.
Figure 2.

Chemical similarity (ECFP-4 Tc) between dual and selective hits of H4R and 5-HT3AR and the endogenous ligands of H4R (histamine) and 5-HT3AR (serotonin).
The higher complexity of the dual H4R and 5-HT3AR fragments is further illustrated by the analysis of the physical-chemical distributions of fragment hits (Fig. 3). Whereas most properties are similar when comparing the selective and the dual activity fragments (see Table S1, S2 and Figure S1 for details21), the number of rings and the heavy atom count (and associated molecular weight) are higher for the dual activity hits compared to for H4R and 5-HT3AR selective fragments (Fig. 3 and Table S2).
Figure 3.

Distribution of physical-chemical properties that discriminate H4R selective fragments (red), 5-HT3AR selective fragments (blue line), dual H4R/5-HT3AR fragments (green), and inactives (grey dotted line). Distributions of other physical-chemical properties do not discriminate between the sets as shown in Figure S1
The fragment screening does not only illustrate similarities in H4R and 5-HT3AR binding profiles, but also identifies subtle differences between the properties of selective receptor ligands. Figure 3 shows that the number of H-bond donor atoms is significantly higher for the H4R selective fragments (on average 1.7 H-bond donors) than for 5-HT3AR selective fragments (on average 0.8 H-bond donors). This can be correlated with the H4R ligand pharmacophore22 that contains two H-bond donors. In the H4R binding pocket, these features are complementary to two negatively charged residues, D3.32 and E5.46 (Fig. 4).22 As a result of these strong non-hydrophobic interactions23 between ionizable H-bonding partners, many H4R ligands (including the high affinity endogenous ligand histamine) can bind the receptor with a high lipophilic efficiency,24 explaining the relatively low c log P values of H4R ligand (Fig. 3). In the 5-HT3AR binding pocket one essential glutamate H-bond interaction partner (E129) has been identified (Fig. 4).25 Ligand binding to 5-HT3AR is furthermore largely determined by aromatic interactions like π–π stacking and cation-π interactions (W183, W195, Y141, Y143, Y153, Y234, see Fig. 4)25,26, matching the requirement of a lower number of H-bond donors (and somewhat higher hydrophobicity) for 5-HT3AR ligands compared to H4R ligands. In line with the notion that the H4R binding site contains two essential H-bonding acceptor atoms and the 5-HT3AR site only one such atom, is the observation that fragment 10 possesses affinity for both 5-HT3AR and H4R, whereas the analogous fragment 3 that lacks an NH2 group, only shows affinity for the H4R.
Figure 4.

Panel A shows the predicted binding modes of the dual H4R/5-HT3AR hit 8 (green carbon atoms, see Table 1 for molecular structure) in structural models of H4R and 5-HT3AR. Parts of the backbone of transmembrane (TM) helices 3, 5, 6 and 7 (the top TM3 is not shown for clarity) in H4R and loops A, B, C and E of the extracellular ligand-binding domain (ECD) of 5-HT3AR are represented by light yellow ribbons. Important binding residues are depicted as ball-and-sticks with grey carbon atoms. Oxygen, nitrogen, and hydrogen atoms are colored red, blue, and cyan, respectively. H-bonds described in the text are depicted by black dots. The molecular interaction fingerprint (IFP)27 bit strings of 8 in H4R and 5-HT3AR are compared in panel B.
Figure 4 demonstrates how the affinity profiles from our fragment screening studies correspond with the chemical properties of the H4R and 5-HT3AR binding sites and can be used to derive molecular interaction fingerprints27 and validate structural models of protein-ligand complexes.28 In both H4R17 and 5-HT3AR models (see Supplementary data for a description of the protein modeling procedure), the positively ionizable piperazine group of the dual H4R/5-HT3AR hit 8 forms a salt bridge (D3.32 in H4R, E129 in 5-HT3AR) and makes cation-π (F7.39 in H4R, Y234 and W183 in 5-HT3AR) and aromatic π–π stacking interactions (Y3.33 and Y6.51 in H4R, Y153 in 5-HT3AR). Interestingly, while C3.36 and E5.46 are proposed to act as H-bond donor and acceptor to the carboxamide group of 10 in H4R, two water molecules (which form a conserved protein-ligand H-bond interaction network in several crystal structures of the homologous AChBP29) fulfill the same role in 5-HT3AR. The binding mode modes of 8 presented in Figure 4 do not only match the fragment-based chemogenomics analysis reported in the current study, but are also supported by earlier reported site-directed mutagenesis studies, underlining the important role of E129, W183, Y153, and Y23425,26 in ligand binding to 5-HT3AR, and the essential role of D3.32 and E5.46 in H4R-ligand interactions.22–24 The binding orientation of 8 is furthermore in line with previously experimentally validated ligand binding modes in H4R.17
Chemogenomics analyses of inter-gene family ligand promiscuity is of growing interest.30 Although GPCRs and LGICs obviously have a very different protein architecture, their ligand-binding sites can obviously bind similar (sub)structures. In this respect, the special chemical taxonomy of serotonin, that binds to several GPCRs and one ion-channel (5-HT3A) has been previously noted.31 Moreover, Mestres and co-workers have recently reported a striking cross-pharmacology between aminergic GPCRs and the 5-HT3 receptors in their in silico target profiling platform.32 Our fragment screening studies complement these findings by identifying relatively high fragment cross-reactivity between H4R and 5-HT3AR (Fig. 1b) and demonstrate that fragments are ideally suited to interrogate ligand binding sites. In the hit optimization phase, selectivity for either H4R or 5-HT3AR can be achieved, although in some cases this might proof complicated. This is illustrated by recent publications from Abbott Laboratories.33,34 These studies, that are part of their H4R drug development program, describe the in vitro and in vivo characterization of the H4R ligands 12 and 13 (A-940894). Intriguingly, both these compounds (Fig. 5) show strong inhibition at the 5-HT3AR receptor (98% inhibition at 10 μM). It is noted that these compounds contain the 2-amino-4-piperazine-pyrimidine scaffold that was also identified as binding to both H4R and 5-HT3AR in our fragment-screening (fragment 10, Table 1).
Figure 5.

Compounds in preclinical trials by Abbott.
In conclusion, the present study identifies a significant overlap between the hit fragment set for H4R and 5-HT3AR, illustrating similarities in ligand recognition and suggests that fragment-based chemogenomics analysis and molecular modeling building can be used to efficiently navigate chemical space during hit optimization in programs aimed to develop selective leads or compounds with a dual activity profile.
Acknowledgments
M.V. and I.dE. would like to acknowledge EEC grant (Neurocypres) for financial support. C.dG. acknowledges The Netherlands Organization for Scientific Research (VENI Grant 700.59.408) for financial support. This work was furthermore supported by COST Action BM0806COST. SCRL and AJT are Wellcome Trust funded (081925). There are no competing interests.
Footnotes
Supplementary data (Distributions of physical-chemical properties of fragment hits, descriptions of H4R and 5-HT3AR protein modeling procedures and experimental details of H4R and 5-HT3AR screens) associated with this article can be found, in the online version, at doi:10.1016/j.bmcl.2011.06.123.
Supplementary data
Distributions of physical-chemical properties of fragment hits, descriptions of H4R and 5-HT3AR protein modeling procedures and experimental details of H4R and 5-HT3AR screens.
References and notes
- 1.de Kloe G.E., Bailey D., Leurs R., de Esch I.J. Drug Discovery Today. 2009;14:630. doi: 10.1016/j.drudis.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 2.Hopkins A.L., Groom C.R., Alex A. Drug Discovery Today. 2004;9:430. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 3.Schuffenhauer A., Ruedisser S., Marzinzik A.L., Jahnke W., Blommers M., Selzer P., Jacoby E. Curr. Top. Med. Chem. 2005;5:751. doi: 10.2174/1568026054637700. [DOI] [PubMed] [Google Scholar]
- 4.Hann M.M., Leach A.R., Harper G. J. Chem. Inf. Comput. Sci. 2001;41:856. doi: 10.1021/ci000403i. [DOI] [PubMed] [Google Scholar]
- 5.Hajduk P.J., Huth J.R., Tse C. Drug Discovery Today. 2005;10:1675. doi: 10.1016/S1359-6446(05)03624-X. [DOI] [PubMed] [Google Scholar]
- 6.Chen I.J., Hubbard R.E.J. Comput. Aided Mol. Des. 2009;23:603. doi: 10.1007/s10822-009-9280-5. [DOI] [PubMed] [Google Scholar]
- 7.Rognan D. Br. J. Pharmacol. 2007;152:38. doi: 10.1038/sj.bjp.0707307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smits R.A., de Esch I.J., Zuiderveld O.P., Broeker J., Sansuk K., Guaita E., Coruzzi G., Adami M., Haaksma E., Leurs R. J. Med. Chem. 2008;51:7855. doi: 10.1021/jm800876b. [DOI] [PubMed] [Google Scholar]
- 9.Smits R.A., Lim H.D., Hanzer A., Zuiderveld O.P., Guaita E., Adami M., Coruzzi G., Leurs R., de Esch I.J. J. Med. Chem. 2008;51:2457. doi: 10.1021/jm7014217. [DOI] [PubMed] [Google Scholar]
- 10.Westly E. Nat. Med. 2010;16:1063. doi: 10.1038/nm1010-1063. [DOI] [PubMed] [Google Scholar]
- 11.Thompson A.J., Lummis S.C. Expert Opin. Ther. Targets. 2007;11:527. doi: 10.1517/14728222.11.4.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohman L., Simren M. Nat. Rev. Gastroenterol. Hepatol. 2010;7:163. doi: 10.1038/nrgastro.2010.4. [DOI] [PubMed] [Google Scholar]
- 13.Siegal G., Ab E., Schultz J. Drug Discovery Today. 2007;12:1032. doi: 10.1016/j.drudis.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 14.Oprea T.I. J. Comput. Aided Mol. Des. 2000;14:251. doi: 10.1023/a:1008130001697. [DOI] [PubMed] [Google Scholar]
- 15.Xu J. J. Med. Chem. 2002;45:5311. doi: 10.1021/jm010520k. [DOI] [PubMed] [Google Scholar]
- 16.Steffen A., Kogej T., Tyrchan C., Engkvist O. J. Chem. Inf. Model. 2009;49:338. doi: 10.1021/ci800326z. [DOI] [PubMed] [Google Scholar]
- 17.Lim H.D., de Graaf C., Jiang W., Sadek P., McGovern P.M., Istyastono E.P., Bakker R.A., de Esch I.J., Thurmond R.L., Leurs R. Mol. Pharmacol. 2010;77:734. doi: 10.1124/mol.109.063040. [DOI] [PubMed] [Google Scholar]
- 18.Thompson A.J., Verheij M.H.P., Leurs R., De Esch I.J.P., Lummis S.C.R. Biotechniques. 2010;49:8. doi: 10.2144/000113538. [DOI] [PubMed] [Google Scholar]
- 19.Clemons P.A., Bodycombe N.E., Carrinski H.A., Wilson J.A., Shamji A.F., Wagner B.K., Koehler A.N., Schreiber S.L. Proc. Natl. Acad. Sci. U.S.A. 2010;107:18787. doi: 10.1073/pnas.1012741107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Accelrys Software Inc. S. D., C., USA.
- 21.Distributions of physical-chemical properties of fragment hits and experimental details of H4R and 5-HT3AR screens are available in the Supplementary data
- 22.Jongejan A., Lim H.D., Smits R.A., de Esch I.J., Haaksma E., Leurs R. J. Chem. Inf. Model. 2008;48:1455. doi: 10.1021/ci700474a. [DOI] [PubMed] [Google Scholar]
- 23.Shamovsky I., de Graaf C., Alderin L., Bengtsson M., Bladh H., Borjesson L., Connolly S., Dyke H.J., van den Heuvel M., Johansson H., Josefsson B.G., Kristoffersson A., Linnanen T., Lisius A., Mannikko R., Norden B., Price S., Ripa L., Rognan D., Rosendahl A., Skrinjar M., Urbahns K. J. Med. Chem. 2009;52:7706. doi: 10.1021/jm900713y. [DOI] [PubMed] [Google Scholar]
- 24.Leeson P.D., Springthorpe B. Nat. Rev. Drug Disc. 2007;6:881. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- 25.Price K.L., Bower K.S., Thompson A.J., Lester H.A., Dougherty D., Lummis S. Biochemistry. 2008;47:6370. doi: 10.1021/bi800222n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Price K.L., Lummis S.C. J. Biol. Chem. 2004;279:23294. doi: 10.1074/jbc.M314075200. [DOI] [PubMed] [Google Scholar]
- 27.Marcou G., Rognan D. J. Chem. Inf. Model. 2007;47:195. doi: 10.1021/ci600342e. [DOI] [PubMed] [Google Scholar]
- 28.de Graaf C., Rognan D. Curr. Pharm. Des. 2009;15:4026. doi: 10.2174/138161209789824786. [DOI] [PubMed] [Google Scholar]
- 29.Hibbs R.E., Sulzenbacher G., Shi J., Talley T.T., Conrod S., Kem W.R., Taylor P., Marchot P., Bourne Y. EMBO J. 2009;28:3040. doi: 10.1038/emboj.2009.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keiser M.J., Setola V., Irwin J.J., Laggner C., Abbas A.I., Hufeisen S.J., Jensen N.H., Kuijer M.B., Matos R.C., Tran T.B., Whaley R., Glennon R.A., Hert J., Thomas K.L., Edwards D.D., Shoichet B.K., Roth B.L. Nature. 2009;462:175. doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keiser M.J., Irwin J.J., Shoichet B.K. Biochemistry. 2010;49:10267. doi: 10.1021/bi101540g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gregori-Puigjane E., Mestres J. Comb. Chem. High Throughput Screening. 2008;11:669. doi: 10.2174/138620708785739952. [DOI] [PubMed] [Google Scholar]
- 33.Liu H., Altenbach R.J., Carr T.L., Chandran P., Hsieh G.C., Lewis L.G., Manelli A.M., Milicic I., Marsh K.C., Miller T.R., Strakhova M.I., Vortherms T.A., Wakefield B.D., Wetter J.M., Witte D.G., Honore P., Esbenshade T.A., Brioni J.D., Cowart M.D. J. Med. Chem. 2008;51:7094. doi: 10.1021/jm8007618. [DOI] [PubMed] [Google Scholar]
- 34.Strakhova M.I., Cuff C.A., Manelli A.M., Carr T.L., Witte D.G., Baranowski J.L., Vortherms T.A., Miller T.R., Rundell L., McPherson M.J., Adair R.M., Brito A.A., Bettencourt B.M., Yao B.B., Wetter J.M., Marsh K.C., Liu H., Cowart M.D., Brioni J.D., Esbenshade T.A. Br. J. Pharmacol. 2009;157:44. doi: 10.1111/j.1476-5381.2009.00236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Distributions of physical-chemical properties of fragment hits, descriptions of H4R and 5-HT3AR protein modeling procedures and experimental details of H4R and 5-HT3AR screens.