

Abstract
AIM: To determine the associations between leptin and ghrelin concentrations and sustained virological response (SVR) in chronic hepatitis C patients with steatosis.
METHODS: We retrospectively assessed 56 patients infected with hepatitis C virus (HCV) genotype-1 and 40 with HCV genotype-3. Patients with decompensated cirrhosis, and those with other causes of chronic liver disease, were excluded. Serum HCV-RNA concentrations were measured before the initiation of treatment; at weeks 12 (for genotype 1 patients), 24 and 48 during treatment; and 24 wk after the end of treatment. Genotype was determined using INNO-LIPA HCV assays, and serum leptin and ghrelin concentrations were measured using enzyme-linked immunosorbent assay. Biopsy specimens were scored according to the Ishak system and steatosis was graded as mild, moderate, or severe, according to the Brunt classification.
RESULTS: Overall, SVR was positively related to the presence of genotype-3, to biopsy-determined lower histological stage of liver disease, and lower grade of steatosis. Patients ≥ 40 years old tended to be less responsive to therapy. In genotype-1 infected patients, SVR was associated with a lower grade of liver steatosis, milder fibrosis, and an absence of insulin resistance. Genotype-1 infected patients who did not achieve SVR had significantly higher leptin concentrations at baseline, with significant increases as the severity of steatosis worsened, whereas those who achieved SVR had higher ghrelin concentrations. In genotype-3 infected patients, SVR was associated only with fibrosis stage and lower homeostasis model assessment insulin resistance at baseline, but not with the degree of steatosis or leptin concentrations. Genotype-3 infected patients who achieved SVR showed significant decreases in ghrelin concentration at end of treatment. Baseline ghrelin concentrations were elevated in responders of both genotypes who had moderate and severe steatosis.
CONCLUSION: Increased serum leptin before treatment may predict non-SVR, especially in HCV genotype-1 infected patients, whereas increased ghrelin may predict SVR in genotype-1.
Keywords: Hepatitis C virus, Steatosis, Leptin, Ghrelin, Sustained virological response
INTRODUCTION
Hepatic steatosis is a histopathological feature observed in > 50% of patients with chronic hepatitis C (CHC)[1,2], but occurs less frequently in patients with chronic hepatitis B (27%-51%) and autoimmune hepatitis (16%-19%)[3,4]. Steatosis in CHC patients has been attributed to a combination of mechanisms involved in the pathogenesis of non-alcoholic fatty liver disease, as well as to the direct effect of hepatitis C virus (HCV) on hepatic lipid metabolism leading to triglyceride accumulation[5,6]. In contrast, steatosis in patients chronically infected with hepatitis B virus (HBV) is associated with host metabolic factors[7].
Leptin is an adipokine that contributes to the pathogenesis of liver steatosis[8,9]. In patients with CHC, higher serum leptin concentrations have been associated with the presence of steatosis[10]. Although no clear correlation has been observed between leptin concentrations and the extent of steatosis[11], a recent study reported that high serum leptin concentrations correlated with more severe steatosis, lower viremia, and a lower antiviral response, mainly in patients infected with HCV genotype-1, which constituted 71% of the study population[12].
Leptin, the product of the obese (ob) gene, is mainly expressed by adipose tissue, although it is expressed in other organs, including the liver[13]. Leptin plays an important role in the regulation and metabolism of body fat and may induce insulin resistance, increase fatty acid concentrations in the liver, and enhance lipid peroxidation[5,8,9]. Leptin may act as an immunomodulator, inducing the release of cytokines, such as tumor necrosis factor (TNF)-α, interferon (INF)-γ, interleukin (IL)-18, and tumor growth factor (TGF)-β1, thus promoting liver steatosis and fibrosis[8].
Ghrelin is a peptide that acts as an endogenous ligand of the growth hormone secretatog receptor[14]. Ghrelin is involved in energy metabolism, food intake, and glucose homeostasis[14,15]. Recent studies have assessed whether ghrelin acts as an independent signal of adiposity or as a downstream mediator of leptin, affecting energy balance[16].
Little is known about serum ghrelin concentrations in patients with CHC and steatosis, or on the effects of ghrelin concentration on treatment response. We therefore assessed whether pretreatment serum leptin and ghrelin concentrations differ in steatotic patients infected with HCV genotypes-1 and -3, and whether these concentrations are associated with response to antiviral treatment. We also evaluated the correlations between pretreatment serum leptin and ghrelin concentrations and liver histology and metabolic factors, as well as determining whether the impact of antiviral treatment on leptin and ghrelin concentrations differed by HCV genotype.
MATERIALS AND METHODS
Patient population
We retrospectively assessed patients with serologically, virologically, and histologically confirmed CHC, recruited between 2005 and 2008. Patients were included if they had detectable anti-HCV antibody by enzyme-linked immunosorbent assay (ELISA) III at least twice a year, detectable serum HCV-RNA by a sensitive PCR assay within 1 mo prior to the start of treatment, liver biopsy showing chronic hepatitis with steatosis within 6 mo before treatment, and elevated alanine aminotransferase (ALT) activity (> 40 IU/L and < 400 IU/L) at entry and at least once during the 6 mo before the first screening.
Patients with decompensated cirrhosis; other causes of chronic liver disease; a history of intravenous drug abuse or alcohol consumption; use of hepatotoxic drugs, herbal medications or immunosuppressive agents; diabetes; thyroid disorders; chronic renal failure; serious psychiatric disorders; HIV or HBV co-infection; or hepatocellular carcinoma, were excluded.
None of these patients had previously received antiviral treatment or steatosis-inducing therapy. The duration of HCV treatment with PEGylated INF-α and ribavirin was genotype-based. Genotype 1 patients who did not achieve undetectable HCV-RNA or a decrease in 2 logs of HCV-RNA at week 12 (early virological response or EVR), were considered non-responders but included in the study. All patients were clinically, hematologically and biochemically evaluated at weeks 2, 4, 8, 12, 24, 48, and 72 after the start of treatment.
The study protocol was approved by our institutional review board, and all patients provided written informed consent. The study conformed to the ethical guidelines of the 1975 Declaration of Helsinki.
Body composition measurements
Height and weight were determined at baseline, and body mass index (BMI), calculated as weight (in kg)/height (in m)2, was determined at baseline and at the end of treatment. Waist circumference indicative of visceral obesity was defined as > 102 cm in men and > 88 cm in women.
Laboratory and virology assays
Blood samples were obtained from all subjects after overnight fasting, and serum was obtained after centrifugation and stored in aliquots at -70°C until assayed. Routine biochemical (aspartate aminotransferase, ALT, γ-glutamyl transpeptidase, alkaline phosphatase, glucose, urea, creatinine, cholesterol, triglycerides, albumin) and hematological (hemoglobin, white blood cells, platelets) were performed using automated techniques.
Serological markers (hepatitis B surface antigen, anti-HBs, hepatitis B e antigen, anti-HBe, anti-HBc total, anti-HCV, anti-HDV, anti-HIV1,2, and anti-HAV IgM and IgG) were assayed using commercially available enzyme immunoassays (Abbott Laboratories, United States). Serum HCV-RNA levels were measured with a PCR assay (Cobas Amplicor HCV version 2, Roche Diagnostics, United States), both qualitatively (lower detection limit of 50 IU/mL) and quantitatively (range of detection 600-106 IU/mL). HCV genotype was determined using an INNO-LIPA HCV assay (Innogenetics, Belgium). Serum HCV-RNA concentrations were measured before the initiation of treatment, at weeks 12 (for genotype 1 patients), 24, and 48 during treatment, and 24 wk after the end of treatment. Serum leptin and ghrelin concentrations were measured using ELISA kits (BioVendor Laboratory, United States), at screening and at the end of treatment and expressed as ng/mL. Insulin resistance index was calculated as: insulin resistance [homeostasis model assessment insulin resistance (HOMA-IR)] = fasting insulin (mIU/L) × fasting glucose (mmol/L)/22.5.
Liver histology
Liver biopsies were obtained at baseline using the Menghini technique (mean length of biopsy cores, 1.7 cm). Liver tissue was fixed in 10% neutral formalin and paraffin-embedded sections were stained with hematoxylin-eosin and Masson trichrome stains. Liver biopsies were scored by an experienced liver pathologist using the Ishak scoring system. Steatosis was quantified as the percentage of hepatocytes that contained fat droplets and was graded using a three-tier scale: grade 1/mild (1%-33%), 2/moderate (33%-66%), and 3/severe (> 66%). Patients with histopathological findings of steatohepatitis, including hepatocellular ballooning or perisinusoidal fibrosis in zone 3, were excluded.
Statistical analysis
All continuous variables are presented as mean ± SD or medians ± interquartile ranges (75th-25th percentiles) if they deviated from normality. The association between each genotype and continuous variables was determined by ANOVA (using post-hoc Scheffe’s test when the omnibus test was significant) or Student’s t-test. The association between each genotype and categorical variables was determined using the χ2 test. Multiple logistic regression analyses were used to determine the association between response to therapy and leptin concentrations, ghrelin concentrations, hepatic steatosis, fibrosis, and HOMA-IR, with results presented as odds ratios and 95% CI. These analyses were performed separately for each genotype and for measurements at baseline and at the end of follow up. For reasons of multicollinearity, it was not possible to include steatosis and fibrosis in the same model. Therefore, fibrosis and steatosis were alternatively introduced (one at a time) into the core model. Mixed effect models were used to examine the relationship between baseline and end of follow-up leptin and ghrelin concentrations among responders and non-responders. There were no significant changes in weight or fat composition in the study population during treatment; therefore, there was no need to adjust leptin and ghrelin concentrations for BMI after treatment. All reported probability values (P-values) were based on two-sided tests, with significance set at 0.05. All statistical analyses were performed using the SAS statistical package (Version 9.1, SAS Institute Inc., NC).
RESULTS
Of the 154 treatment-naive patients with CHC screened, 96 fulfilled our enrollment criteria and completed treatment. The study population consisted of 60 men and 36 women, all of Caucasian origin; of these, 56 were infected with HCV genotype-1 and 40 with HCV genotype-3.
HCV genotypes and response to treatment
All patients received combination therapy with PEGylated INF a-2b or a-2a, plus weight-adjusted ribavirin, for 24 (genotype-3) or 48 (genotype-1) weeks. All tolerated treatment well and completed treatment without any major side effects, significant reductions in drug dose, and significant changes in BMI (reduction more than 2 kg/m2). Patients infected with HCV genotype-1 who did not achieve sustained virological response (SVR) stopped therapy, whereas those infected with HCV genotype-3 were treated for 24 wk without measurements of HCV-RNA at week 12, according to currently approved treatment guidelines. Of the 96 patients, 62 (64.6%) achieved SVR, whereas the remaining 34 (35.4%) did not.
Clinical, virological, and histological data of patients of both genotypes who did and did not attain SVR are presented in Table 1. Of the 56 patients with genotype 1, 32 (57.1%) achieved SVR, compared with 30 of 40 patients (75%) with genotype-3; thus SVR was significantly related to infection with genotype-3 (P < 0.05). In addition, SVR was significantly associated with lower histological stage of liver disease (P < 0.001) and lower grade of steatosis in liver biopsy (P = 0.001). Patients ≥ 40 years old tended to be less responsive to therapy (P = 0.05) (Table 1). Further analysis by genotype showed that, in genotype-1 infected patients, SVR was associated with a lower grade of liver steatosis (P = 0.0001), mild fibrosis (P = 0.001), and absence of insulin resistance (P = 0.01) (Table 2). In genotype-3 infected patients, SVR was associated with stage of fibrosis (P = 0.04) and lower HOMA-IR at baseline (P = 0.01), but not to degree of steatosis (Table 2).
Table 1.
Clinical, virological, and histological data of the study population (mean ± SD) n (%)
| SVR (n = 62) | Non-SVR (n = 34) | P-value | |
| Age | 35.01 ± 1.57 | 39.21 ± 2.07 | 0.050 |
| Sex (male/female) | 36/26 | 21/13 | |
| BMI | 24.700 ± 0.179 | 25.530 ± 0.637 | 0.060 |
| Genotype-1 | 32 (57.1) | 24 (42.9) | |
| Genotype-3 | 30 (75) | 10 (25) | |
| Stage (0-6) | 1.661 ± 0.095 | 2.412 ± 0.141 | < 0.001 |
| Grade (0-18) | 5.371 ± 0.136 | 5.265 ± 0.204 | NS |
| Steatosis (1-3) | 1.565 ± 0.082 | 2.060 ± 0.126 | 0.001 |
NS: Non-significant; SVR: Sustained virological response; BMI: Body mass index.
Table 2.
Distribution of 56 genotype-1 and 40 genotype-3-infected patients by demographic, host and viral factors with respect to response to hepatitis C virus therapy (mean ± SD) n (%)
| Variable |
Genotype 1 (n = 56) |
Genotype 3 (n = 40) |
||||
| Responders(n = 32) | Non-responders(n = 24) | P value | Responders(n = 30) | Non-responders(n = 10) | P-value | |
| Gender | 0.70 | 0.47 | ||||
| Male | 15 (46.9) | 10 (41.7) | 13 (43.3) | 6 (60.0) | ||
| Female | 17 (53.1) | 14 (58.3) | 17 (56.7) | 4 (40.0) | ||
| Age (yr) | 0.17 | 0.15 | ||||
| ≤ 40 | 23 (71.9) | 13 (54.2) | 29 (96.7) | 8 (80.0) | ||
| > 40 | 9 (28.1) | 11 (45.8) | 1 (3.3) | 2 (20.0) | ||
| Body mass index (kg/m2) | 0.28 | 0.42 | ||||
| < 25 | 18 (56.3) | 10 (41.7) | 23 (76.7) | 6 (60.0) | ||
| 25-28 | 14 (43.7) | 14 (58.3) | 7 (23.3) | 4 (40.0) | ||
| Grade of hepatic steatosis | 0.0001 | 0.72 | ||||
| Mild | 22 (68.7) | 5 (20.8) | 12 (40.0) | 3 (30.0) | ||
| Moderate | 10 (31.3) | 12 (50.0) | 13 (43.3) | 4 (40.0) | ||
| Severe | 0 (0.0) | 7 (29.2) | 5 (16.7) | 3 (30.0) | ||
| Fibrosis score | 0.001 | 0.04 | ||||
| 1 | 17 (53.1) | 3 (12.5) | 16 (53.3) | 1 (10.0) | ||
| 2 | 11 (34.4) | 8 (33.3) | 11 (36.7) | 7 (70.0) | ||
| 3-4 | 4 (12.5) | 13 (54.2) | 3 (10.0) | 2 (20.0) | ||
| 5-6 | 0 | 0 | 0 | 0 | ||
| HOMA-IR | 0.01 | 0.01 | ||||
| < 2 | 11 (34.4) | 4 (16.7) | 19 (63.3) | 1 (10.0) | ||
| 2-3 | 10 (31.2) | 2 (8.3) | 10 (33.3) | 7 (70.0) | ||
| > 3 | 11 (34.4) | 18 (75.0) | 1 (3.3) | 2 (20.0) | ||
| Leptin_baseline (ng/mL) | 37.66 ± 10.39 | 49.67 ± 13.44 | 0.001 | 24.33 ± 7.98 | 28.20 ± 9.72 | 0.22 |
| Ghrelin_baseline (ng/mL) | 0.286 ± 0.167 | 0.190 ± 0.119 | 0.02 | 0.778 ± 0.654 | 0.564 ± 0.324 | 0.18 |
| Leptin end of follow-up (ng/mL) | 26.69 ± 11.77 | 41.50 ± 16.24 | < 0.0001 | 23.27 ± 9.54 | 26.70 ± 6.82 | 0.30 |
| Ghrelin end of follow-up (ng/mL) | 0.456 ± 0.254 | 0.239 ± 0.213 | 0.001 | 0.420 ± 0.321 | 0.450 ± 0.261 | 0.79 |
HOMA-IR: Homeostasis model assessment insulin resistance.
Leptin, ghrelin and response to treatment
Baseline leptin concentrations did not differ between patients who did and did not attain SVR, but were significantly lower after successful treatment in patients who attained SVR (P = 0.01) (Table 3). Among patients infected with HCV genotype-1, non-responders had significantly higher serum leptin concentrations, both at baseline (P = 0.001) and at the end of follow-up (P < 0.0001) than those who attained SVR (Table 2). Using mixed effect model analysis, we observed a statistically significant difference between baseline and follow-up leptin concentrations among genotype-1 infected patients who achieved SVR (P = 0.001), as well as a borderline significant difference among non-responders (P = 0.06) (Table 4).
Table 3.
Leptin and ghrelin concentrations at baseline and end of follow-up in patients infected with hepatitis C virus genotypes-1 and -3 by response to therapy, as well as mixed effect model derived estimates of differences in mean scores (mean ± SD)
| Variable | Responders | Non-responders | P-value for the adjusted difference1 |
| Leptin | |||
| Baseline (ng/mL) | 30.99 (9.185) | 38.43 (11.58) | NS |
| End of Follow-up (ng/mL) | 23.76 (8.055) | 34.10 (11.53) | 0.01 |
| Ghrelin | |||
| Baseline (ng/mL) | 0.532 (0.410) | 0.377 (0.223) | 0.01 |
| End of Follow-up (ng/mL) | 0.438 (0.288) | 0.345 (0.237) | 0.05 |
Adjusted for both leptin and ghrelin concentrations. NS: Non-significant.
Table 4.
Leptin and ghrelin concentrations at baseline and at end of follow-up by response to treatment in patients with hepatitis C virus genotypes-1 and -3, as well as mixed effect model derived estimates of the differences in mean scores (mean ± SD)
| Variable | Baseline | End of follow-up | P-value for the adjusted difference1 |
| Genotype 1 SVRs | |||
| Leptin (ng/mL) | 37.66 (10.39) | 26.69 (11.77) | 0.001 |
| Ghrelin (ng/mL) | 0.286 (0.167) | 0.456 (0.254) | 0.001 |
| Genotype 1 non-SVRs | |||
| Leptin (ng/mL) | 49.67 (13.44) | 43.58 (16.17) | 0.060 |
| Ghrelin (ng/mL) | 0.190 (0.119) | 0.239 (0.213) | 0.320 |
| Genotype 3 SVRs | |||
| Leptin (ng/mL) | 24.33 (7.98) | 23.27 (9.54) | 0.510 |
| Ghrelin (ng/mL) | 0.778 (0.654) | 0.420 (0.321) | 0.001 |
| Genotype 3 non-SVRs | |||
| Leptin (ng/mL) | 28.20 (9.72) | 26.70 (6.82) | 0.610 |
| Ghrelin (ng/mL) | 0.564 (0.324) | 0.450 (0.261) | 0.470 |
Adjusted for both leptin and ghrelin concentrations.
Among patients infected with HCV genotype-3, however, there were no significant differences in leptin concentrations at baseline and at end of follow-up between those who did and did not achieve SVR (Table 2). Using mixed effect model analysis, leptin remained unchanged, both in responders (P = 0.51) and non-responders (P = 0.61) (Table 4).
Overall, patients who achieved SVR had higher serum ghrelin concentrations, both at baseline (P = 0.01) and at the end of follow up (P = 0.05), than patients who did not achieve SVR (Table 3). Genotype-1 infected patients who achieved SVR had statistically significant higher ghrelin concentrations at baseline (P = 0.02) and at the end of treatment (P = 0.001) than nonresponders (Table 2), with responders showing significantly higher ghrelin concentrations at end of treatment than at baseline (P = 0.001) (Table 4). In contrast, ghrelin concentrations in genotype-3 infected patients did not differ between responders and nonresponders, both at baseline and at the end of treatment (Table 2). Mixed effect model analysis showed that ghrelin concentrations were significantly lower at the end of treatment than at baseline in patients who achieved SVR (P = 0.001), but not in non-responders (P = 0.47) (Table 4).
Leptin, ghrelin and steatosis
Overall, steatosis grade at baseline was higher in non-responders than in patients who achieved SVR, with steatosis grade at baseline being significantly greater as leptin concentrations increased, a difference more obvious in patients with moderate and severe steatosis (P = 0.05). This correlation was also observed in genotype-1 non-responders, but not in genotype-3 non-responders or in patients of either genotype who achieved SVR (Figure 1).
Figure 1.
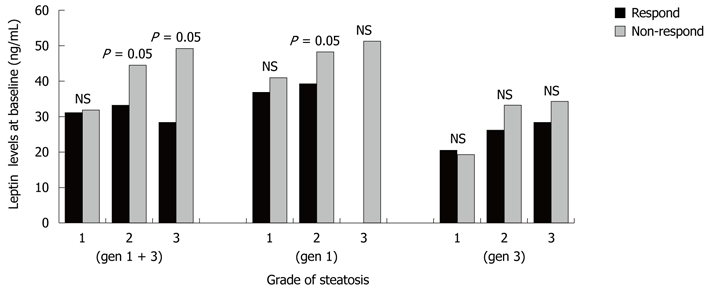
Correlation between leptin concentrations and steatosis in patients infected with hepatitis C virus genotypes-1 and -3 who did and did not achieve sustained virological response following treatment with PEGylated interferon a-2b or a-2a plus ribavirin. NS: Not significant.
Ghrelin concentration at baseline was higher in responders of both genotypes with moderate (P = 0.05) and severe (P = 0.001) steatosis. In non-responders, however, there was no significant correlation between the grade of steatosis and ghrelin concentrations. In genotype-1 infected patients, both in responders and non-responders, ghrelin concentrations decreased significantly as the grade of steatosis increased (P = 0.01), and responders with genotype-3 and moderate or severe steatosis had significantly higher serum concentrations of ghrelin (P = 0.01) (Figure 2). A strong correlation between the severity of steatosis and higher viral load at baseline was observed in patients infected with HCV genotype-3 (P = 0.01), but not in those infected with HCV genotype-1 (P = NS) (Figure 3).
Figure 2.

Correlation between ghrelin concentrations levels and steatosis in patients infected with hepatitis C virus genotypes11 and -3 who did and did not achieve sustained virological response following treatment with PEGylated interferon a-2b or a-2a plus ribavirin. NS: Not significant.
Figure 3.
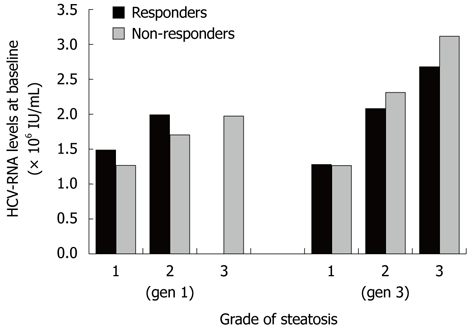
Correlation between viral load and steatosis in patients infected with hepatitis C virus genotypes-1 and -3 who did and did not achieve sustained virological response following treatment with PEGylated interferon a-2b or a-2a plus ribavirin. HCV: Hepatitis C virus.
Multivariate analysis
Using multivariate logistic regression analysis, we found that higher leptin concentrations at baseline were significantly associated with non-response to therapy in patients infected with HCV genotype-1, but not HCV genotype-3. There were no significant associations between response to treatment and ghrelin concentrations in patients of either genotype. Furthermore, HCV genotype-1 infected with moderate or severe steatosis, as well as those with more severe fibrosis, were less likely to respond to therapy (Table 5). In patients with genotype-3, however, there were no significant associations between response to treatment and steatosis, fibrosis, or leptin or ghrelin concentrations, both at baseline and at end of treatment. Only patients with higher levels of HOMA (IR) seemed less likely to respond to therapy (Table 6).
Table 5.
Multiple logistic regression derived odds ratios and 95% confidence intervals for response to hepatitis C virus therapy among patients infected with hepatitis C virus genotype-1 with respect to hepatic steatosis, and leptin and ghrelin concentrations at baseline and at end of follow-up
| Variable | Category or increment | ORs | 95% CIs | P-value |
| At baseline | ||||
| Hepatic steatosis | 1 level more | 0.12 | 0.02-0.66 | 0.01 |
| Leptin | 10 ng/mL more | 0.43 | 0.22-0.83 | 0.01 |
| Ghrelin | 0.1 ng/mL more | 1.11 | 0.67-2.01 | 0.63 |
| HOMA-IR | 1 level more | 1.34 | 0.42-4.31 | 0.63 |
| Alternatively introduced variables | ||||
| Fibrosis | 1 level more | 0.36 | 0.13 -0.96 | 0.04 |
| Leptin | 10 ng/mL more | 0.45 | 0.24-0.86 | 0.02 |
| Ghrelin | 0.1 ng/mL more | 1.35 | 0.82-2.45 | 0.26 |
| HOMA-IR | 1 level more | 0.96 | 0.33-2.79 | 0.94 |
| At end of follow-up | ||||
| Hepatic steatosis | 1 level more | 0.13 | 0.02-0.89 | 0.04 |
| Leptin | 10 ng/mL more | 0.38 | 0.18-0.81 | 0.01 |
| Ghrelin | 0.1 ng/mL more | 1.22 | 0.74-2.01 | 0.41 |
| HOMA-IR | 1 level more | 1.98 | 0.54-7.30 | 0.30 |
| Alternatively introduced variables | ||||
| Fibrosis | 1 level more | 0.30 | 0.10-0.93 | 0.04 |
| Leptin | 10 ng/mL more | 0.34 | 0.16-0.72 | 0.01 |
| Ghrelin | 0.1 ng/mL more | 1.22 | 0.74-1.82 | 0.47 |
| HOMA-IR | 1 level more | 1.28 | 0.43-3.79 | 0.66 |
CI: Confidence interval; HOMA-IR: Homeostasis model assessment insulin resistance; ORs: Odds ratios.
Table 6.
Multiple logistic regression derived odds ratios and 95% CIs for response to hepatitis C virus therapy among patients infected with hepatitis C virus genotype-3 with respect to hepatic steatosis, and leptin and ghrelin concentrations at baseline and at end of follow-up
| Variable | Category or increment | ORs | 95% CIs | P-value |
| At baseline | ||||
| Hepatic steatosis | 1 level more | 1.04 | 0.19-5.55 | 0.97 |
| Leptin | 10 ng/mL more | 0.64 | 0.18-2.24 | 0.49 |
| Ghrelin | 0.1 ng/mL more | 1.11 | 0.90-1.49 | 0.32 |
| HOMA-IR | 1 level more | 0.13 | 0.02-0.68 | 0.02 |
| Alternatively introduced variables | ||||
| Fibrosis | 1 level more | 0.16 | 0.02-0.11 | 0.06 |
| Leptin | 10 ng/mL more | 1.22 | 0.37-4.08 | 0.74 |
| Ghrelin | 0.1 ng/mL more | 1.22 | 0.90-1.65 | 0.16 |
| HOMA-IR | 1 level more | 0.16 | 0.03-0.83 | 0.03 |
| At end of follow-up | ||||
| Hepatic steatosis | 1 level more | 1.77 | 0.39-7.95 | 0.46 |
| Leptin | 10 ng/mL more | 0.40 | 0.10-1.55 | 0.18 |
| Ghrelin | 0.1 ng/mL more | 1.00 | 0.74-1.22 | 0.76 |
| HOMA-IR | 1 level more | 0.10 | 0.02-0.59 | 0.01 |
| Alternatively introduced variables | ||||
| Fibrosis | 1 level more | 0.40 | 0.08-1.52 | 0.16 |
| Leptin | 10 ng/mL more | 0.70 | 0.22-2.18 | 0.54 |
| Ghrelin | 0.1 ng/mL more | 1.00 | 0.74-1.35 | 0.85 |
| HOMA-IR | 1 level more | 0.14 | 0.03-0.71 | 0.02 |
CI: Confidence interval; HOMA-IR: Homeostasis model assessment insulin resistance; ORs: Odds ratios.
DISCUSSION
The mechanism by which HCV induces steatosis remains unclear. Steatosis in patients infected with the non-3 genotype has been associated with increased BMI, visceral obesity, increased cholesterol and triglyceride concentrations, insulin resistance, metabolic syndrome, diabetes, alcohol consumption, and increased sensitivity of the liver to oxidative stress or cytokine-mediated injury[17,18].
Leptin is a putative link between HCV infection and steatosis[19]. Although a high incidence of hyperleptinemia has been observed in HCV infected patients with liver steatosis[10,20], the underlying mechanism promoting this effect remains undefined. Leptin may increase insulin resistance and fatty acid concentrations in the liver, leading to enhanced lipid peroxidation and promoting steatosis[5]. Leptin may also induce the release of cytokines, such as TNF-α, INF-γ, IL-18, and TGF-β1, which are involved in the pathogenesis of both liver steatosis and fibrosis[8]. In steatosis, activated hepatic stellate cells, but not quiescent cells, can express leptin[21].
Our results clearly show that, in HCV infected patients with liver steatosis, serum leptin levels tend to increase as the grade of steatosis worsens. This finding is significant, especially in genotype-1 patients, suggesting that leptin increases during infection as a part of the host immune response, and may contribute to the development of steatosis. Although steatosis is more common and more severe in patients infected with HCV genotype-3[17,18], we found that leptin concentrations were not correlated with either the grade of steatosis or response to treatment. Structural and nonstructural proteins of HCV genotype-3 may directly cause steatosis by provoking oxidative stress[6,22,23]. Alternatively, the viral core protein may target microsomal triglyceride transfer protein activity, modifying very low density lipoprotein assembly in, and secretion by hepatocytes[24,25]. The core protein may also affect the cytoplasmic domain of members of the TNF receptor family or act directly on the mitochondria, leading to increased oxidative stress and lipid peroxidation[26]. In patients infected with HCV genotype-3, we found that the grade of steatosis was correlated with higher viral load at baseline, in agreement with the direct “steatogenic” effect of this genotype[6].
Although ghrelin is important in food intake, energy balance, and the regulation of the growth hormone releasing mechanism[15], its role in hepatic disease has not been extensively evaluated to date. Increased serum ghrelin concentrations have been reported in patients with cirrhosis and hepatocellular carcinoma, suggesting that this adipokine may be involved in the anorexia-cachexia syndrome during the terminal stages of liver diseases[27]. Data on ghrelin concentrations in patients with CHC are limited[28].
We found that genotype-1 responders had higher serum ghrelin concentrations at baseline than non-responders, and that its concentration increased significantly in the former at the end of treatment, indicating that ghrelin may prevent or reduce steatosis by negatively regulating leptin. This may enhance the likelihood of SVR, since responders also have lower baseline leptin concentrations. In genotype-3 infected patients, however, ghrelin may be considered an independently acting factor, based on our finding that responders with moderate and severe steatosis had high ghrelin concentrations at baseline and that these concentrations were reduced significantly after treatment. In contrast, no significant differences were observed in non-responders and there were no correlations with leptin concentrations.
Our findings are in accordance with previous reports, which found that steatosis was an independent negative predictor of response to antiviral therapy[29-32]. We also found that genotype 1 patients with elevated leptin concentrations before treatment had a lower likelihood of achieving SVR, irrespective of their viral load. This observation is in keeping with the role of leptin as a suppressor of cytokine signaling 3 in the liver[33], or as a factor that inhibits INF signaling.
In conclusion, our results suggest that the extent of hepatic steatosis, in addition to the stage of fibrosis and the viral genotype, may affect the likelihood of SVR in CHC patients. Leptin appears to contribute to the pathogenesis of steatosis, and we found that elevated serum leptin concentration may be an independent predictor of SVR in HCV genotype-1 infected patients. Increase ghrelin concentrations after successful treatment in genotype 1 patients indicate that this peptide plays a role in the achievement of SVR. Further investigations are needed to determine whether ghrelin acts as a downstream mediator of leptin and to assess the influence of ghrelin on liver steatosis and HCV infection.
COMMENTS
Background
Steatosis is a frequent histopathological feature in patients with chronic hepatitis C (CHC). Leptin and ghrelin are involved in body fat regulation and metabolism. Higher serum leptin concentrations have been associated with steatosis, but less is known about leptin and ghrelin concentrations in patients with CHC and steatosis or the effect of these peptides on response to treatment.
Research frontiers
Leptin is a putative link between hepatitis C virus (HCV) infection and steatosis in HCV genotype-1 infected patients; however, the underlying mechanism remains undefined. Increased ghrelin concentrations have been reported in patients with cirrhosis and hepatocellular carcinoma, but its role in hepatic steatosis has not been extensively evaluated. Steatosis is an independent negative predictor of response to antiviral therapy. Our results clearly show that, in HCV infected patients with steatosis, serum leptin levels tend to increase as the grade of steatosis worsens. Non-responding genotype-1 infected patients have elevated leptin at baseline and genotype-1 and -3 responders have higher ghrelin concentrations at baseline. In genotype-3 infected patients, neither the degree of steatosis nor leptin concentration had any effect on response to treatment.
Innovations and breakthroughs
Several studies have highlighted the importance of steatosis and hyperleptinemia in the achievement of sustained virological response (SVR), but this study is the first to find genotype-dependent associations between the degree of steatosis and leptin and ghrelin concentrations. These findings show the significance of baseline leptin and ghrelin concentrations in the achievement of SVR, as well as the impact of antiviral treatment on serum leptin and ghrelin levels.
Applications
These findings suggest that serum leptin concentrations may be an independent negative predictor of SVR in HCV genotype-1 infected patients with steatosis; the role of ghrelin requires be further investigation.
Terminology
Leptin, the ob gene product, is expressed mainly by adipose tissue, although it is expressed in other organs, including the liver. Leptin is important for body fat regulation and metabolism. Ghrelin, a peptide that acts as an endogenous ligand for the growth hormone secretatog receptor, is involved in energy metabolism, food intake and glucose homeostasis.
Peer review
The research study has an important outcome and could be further strengthened by exploring the existing data for an effect of sex on these parameters, if any.
Footnotes
Peer reviewer: Rashmi, Kaul, PhD, Assistant Professor, Department of Biochemistry and Microbiology, Oklahoma State University-Center for Health Sciences, 1111 W 17th St, Tulsa, OK 74107, United States
S- Editor Tian L L- Editor Stewart GJ E- Editor Zheng XM
References
- 1.Hwang SJ, Luo JC, Chu CW, Lai CR, Lu CL, Tsay SH, Wu JC, Chang FY, Lee SD. Hepatic steatosis in chronic hepatitis C virus infection: prevalence and clinical correlation. J Gastroenterol Hepatol. 2001;16:190–195. doi: 10.1046/j.1440-1746.2001.02407.x. [DOI] [PubMed] [Google Scholar]
- 2.Hourigan LF, Macdonald GA, Purdie D, Whitehall VH, Shorthouse C, Clouston A, Powell EE. Fibrosis in chronic hepatitis C correlates significantly with body mass index and steatosis. Hepatology. 1999;29:1215–1219. doi: 10.1002/hep.510290401. [DOI] [PubMed] [Google Scholar]
- 3.Monto A. Hepatitis C and steatosis. Semin Gastrointest Dis. 2002;13:40–46. [PubMed] [Google Scholar]
- 4.Lefkowitch JH, Schiff ER, Davis GL, Perrillo RP, Lindsay K, Bodenheimer HC, Balart LA, Ortego TJ, Payne J, Dienstag JL. Pathological diagnosis of chronic hepatitis C: a multicenter comparative study with chronic hepatitis B. The Hepatitis Interventional Therapy Group. Gastroenterology. 1993;104:595–603. doi: 10.1016/0016-5085(93)90432-c. [DOI] [PubMed] [Google Scholar]
- 5.Adinolfi LE, Gambardella M, Andreana A, Tripodi MF, Utili R, Ruggiero G. Steatosis accelerates the progression of liver damage of chronic hepatitis C patients and correlates with specific HCV genotype and visceral obesity. Hepatology. 2001;33:1358–1364. doi: 10.1053/jhep.2001.24432. [DOI] [PubMed] [Google Scholar]
- 6.Lonardo A, Adinolfi LE, Loria P, Carulli N, Ruggiero G, Day CP. Steatosis and hepatitis C virus: mechanisms and significance for hepatic and extrahepatic disease. Gastroenterology. 2004;126:586–597. doi: 10.1053/j.gastro.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 7.Tsochatzis E, Papatheodoridis GV, Manesis EK, Chrysanthos N, Kafiri G, Archimandritis AJ. Hepatic steatosis in chronic hepatitis B develops due to host metabolic factors: a comparative approach with genotype 1 chronic hepatitis C. Dig Liver Dis. 2007;39:936–942. doi: 10.1016/j.dld.2007.07.151. [DOI] [PubMed] [Google Scholar]
- 8.Giannini E, Barreca T, Testa R. Leptin in nonalcoholic steatohepatitis: is it one of the “hits”? Am J Gastroenterol. 2001;96:2519–2520. doi: 10.1111/j.1572-0241.2001.04078.x. [DOI] [PubMed] [Google Scholar]
- 9.Uygun A, Kadayifci A, Yesilova Z, Erdil A, Yaman H, Saka M, Deveci MS, Bagci S, Gulsen M, Karaeren N, et al. Serum leptin levels in patients with nonalcoholic steatohepatitis. Am J Gastroenterol. 2000;95:3584–3589. doi: 10.1111/j.1572-0241.2000.03297.x. [DOI] [PubMed] [Google Scholar]
- 10.Romero-Gómez M, Castellano-Megias VM, Grande L, Irles JA, Cruz M, Nogales MC, Alcón JC, Robles A. Serum leptin levels correlate with hepatic steatosis in chronic hepatitis C. Am J Gastroenterol. 2003;98:1135–1141. doi: 10.1111/j.1572-0241.2003.07450.x. [DOI] [PubMed] [Google Scholar]
- 11.Gwak GY, Kim TH, Yu SJ, Yoon JH, Yong JJ, Park SC, Lee HS. Lack of association between serum leptin levels and hepatic steatosis, fibrosis or response to antiviral therapy in Korean chronic hepatitis C patients. Hepatogastroenterology. 2007;54:844–848. [PubMed] [Google Scholar]
- 12.Eguchi Y, Mizuta T, Yasutake T, Hisatomi A, Iwakiri R, Ozaki I, Fujimoto K. High serum leptin is an independent risk factor for non-response patients with low viremia to antiviral treatment in chronic hepatitis C. World J Gastroenterol. 2006;12:556–560. doi: 10.3748/wjg.v12.i4.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahima RS, Flier JS. Leptin. Annu Rev Physiol. 2000;62:413–437. doi: 10.1146/annurev.physiol.62.1.413. [DOI] [PubMed] [Google Scholar]
- 14.Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–660. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- 15.Wren AM, Small CJ, Ward HL, Murphy KG, Dakin CL, Taheri S, Kennedy AR, Roberts GH, Morgan DG, Ghatei MA, et al. The novel hypothalamic peptide ghrelin stimulates food intake and growth hormone secretion. Endocrinology. 2000;141:4325–4328. doi: 10.1210/endo.141.11.7873. [DOI] [PubMed] [Google Scholar]
- 16.Cummings DE, Foster KE. Ghrelin-leptin tango in body-weight regulation. Gastroenterology. 2003;124:1532–1535. doi: 10.1016/s0016-5085(03)00350-0. [DOI] [PubMed] [Google Scholar]
- 17.Myers RP, Messous D, Poynard T, Imbert-Bismut F. Association between leptin, metabolic factors and liver histology in patients with chronic hepatitis C. Can J Gastroenterol. 2007;21:289–294. doi: 10.1155/2007/876076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Czaja AJ, Carpenter HA, Santrach PJ, Moore SB. Host- and disease-specific factors affecting steatosis in chronic hepatitis C. J Hepatol. 1998;29:198–206. doi: 10.1016/s0168-8278(98)80004-4. [DOI] [PubMed] [Google Scholar]
- 19.Ellidokuz E, Cömlekçi A, Ellidokuz H, Akpinar H, Gökçe C, Tankurt E, Sagol O, Simsek I, Gönen O. The role of serum leptin levels in chronic hepatitis C with steatosis. Hepatogastroenterology. 2003;50 Suppl 2:cclxix–cclxxii. [PubMed] [Google Scholar]
- 20.Manolakopoulos S, Bethanis S, Liapi C, Stripeli F, Sklavos P, Margeli A, Christidou A, Katsanika A, Vogiatzakis E, Tzourmakliotis D, et al. An assessment of serum leptin levels in patients with chronic viral hepatitis: a prospective study. BMC Gastroenterol. 2007;7:17. doi: 10.1186/1471-230X-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clouston AD, Jonsson JR, Purdie DM, Macdonald GA, Pandeya N, Shorthouse C, Powell EE. Steatosis and chronic hepatitis C: analysis of fibrosis and stellate cell activation. J Hepatol. 2001;34:314–320. doi: 10.1016/s0168-8278(00)00096-9. [DOI] [PubMed] [Google Scholar]
- 22.Matos CA, Perez RM, Pacheco MS, Figueiredo-Mendes CG, Lopes-Neto E, Oliveira EB, Lanzoni VP, Silva AE, Ferraz ML. Steatosis in chronic hepatitis C: relationship to the virus and host risk factors. J Gastroenterol Hepatol. 2006;21:1236–1239. doi: 10.1111/j.1440-1746.2006.04308.x. [DOI] [PubMed] [Google Scholar]
- 23.Rubbia-Brandt L, Leandro G, Spahr L, Giostra E, Quadri R, Malé PJ, Negro F. Liver steatosis in chronic hepatitis C: a morphological sign suggesting infection with HCV genotype 3. Histopathology. 2001;39:119–124. doi: 10.1046/j.1365-2559.2001.01208.x. [DOI] [PubMed] [Google Scholar]
- 24.Rubbia-Brandt L, Quadri R, Abid K, Giostra E, Malé PJ, Mentha G, Spahr L, Zarski JP, Borisch B, Hadengue A, et al. Hepatocyte steatosis is a cytopathic effect of hepatitis C virus genotype 3. J Hepatol. 2000;33:106–115. doi: 10.1016/s0168-8278(00)80166-x. [DOI] [PubMed] [Google Scholar]
- 25.Perlemuter G, Sabile A, Letteron P, Vona G, Topilco A, Chrétien Y, Koike K, Pessayre D, Chapman J, Barba G, et al. Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: a model of viral-related steatosis. FASEB J. 2002;16:185–194. doi: 10.1096/fj.01-0396com. [DOI] [PubMed] [Google Scholar]
- 26.Lai MM. Hepatitis C virus proteins: direct link to hepatic oxidative stress, steatosis, carcinogenesis and more. Gastroenterology. 2002;122:568–571. doi: 10.1053/gast.2002.31474. [DOI] [PubMed] [Google Scholar]
- 27.Tacke F, Brabant G, Kruck E, Horn R, Schöffski P, Hecker H, Manns MP, Trautwein C. Ghrelin in chronic liver disease. J Hepatol. 2003;38:447–454. doi: 10.1016/s0168-8278(02)00438-5. [DOI] [PubMed] [Google Scholar]
- 28.Watanabe H, Saito T, Karasawa T, Kudo S, Nakano K, Ito JI, Sugahara K, Saito K, Togashi H, Kawata S. Reduction of serum ghrelin concentration during interferon-alpha therapy in patients with chronic hepatitis C. Hepatol Res. 2005;33:14–18. doi: 10.1016/j.hepres.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 29.Szanto P, Grigorescu M, Dumitru I, Serban A. Steatosis in hepatitis C virus infection. Response to anti-viral therapy. J Gastrointestin Liver Dis. 2006;15:117–124. [PubMed] [Google Scholar]
- 30.Thomopoulos KC, Theocharis GJ, Tsamantas AC, Siagris D, Dimitropoulou D, Gogos CA, Labropoulou-Karatza C. Liver steatosis is an independent risk factor for treatment failure in patients with chronic hepatitis C. Eur J Gastroenterol Hepatol. 2005;17:149–153. doi: 10.1097/00042737-200502000-00004. [DOI] [PubMed] [Google Scholar]
- 31.Guidi M, Muratori P, Granito A, Muratori L, Pappas G, Lenzi M, Bianchi FB. Hepatic steatosis in chronic hepatitis C: impact on response to anti-viral treatment with peg-interferon and ribavirin. Aliment Pharmacol Ther. 2005;22:943–949. doi: 10.1111/j.1365-2036.2005.02679.x. [DOI] [PubMed] [Google Scholar]
- 32.Reddy KR, Govindarajan S, Marcellin P, Bernstein D, Dienstag JL, Bodenheimer H, Rakela J, Messinger D, Schmidt G, Ackrill A, et al. Hepatic steatosis in chronic hepatitis C: baseline host and viral characteristics and influence on response to therapy with peginterferon alpha-2a plus ribavirin. J Viral Hepat. 2008;15:129–136. doi: 10.1111/j.1365-2893.2007.00901.x. [DOI] [PubMed] [Google Scholar]
- 33.Walsh MJ, Jonsson JR, Richardson MM, Lipka GM, Purdie DM, Clouston AD, Powell EE. Non-response to antiviral therapy is associated with obesity and increased hepatic expression of suppressor of cytokine signalling 3 (SOCS-3) in patients with chronic hepatitis C, viral genotype 1. Gut. 2006;55:529–535. doi: 10.1136/gut.2005.069674. [DOI] [PMC free article] [PubMed] [Google Scholar]