

Abstract
AIM: To confirm the role of sex-determining region Y-box 7 (Sox7) in aspirin-mediated growth inhibition of COX-independent human colorectal cancer cells.
METHODS: The cell survival percentage was examined by MTT (Moto-nuclear cell direc cytotoxicity) assay. SOX7 expression was assessed by using reverse transcription-polymerase chain reaction and Western blotting. SB203580 was used to inhibit the p38MAPK signal pathway. SOX7 promoter activity was detected by Luciferase reporter assay.
RESULTS: SOX7 was upregulated by aspirin and was involved in aspirin-mediated growth inhibition of SW480 human colorectal cancer cells. The p38MAPK pathway played a role in aspirin-induced SOX7 expression, during which the AP1 transcription factors c-Jun and c-Fos upregulated SOX7 promoter activities.
RESULTS: SOX7 is upregulated by aspirin and is involved in aspirin-mediated growth inhibition of human colorectal cancer SW480 cells.
Keywords: SOX7, Aspirin, p38MAPK, Colorectal cancer, SB203580
INTRODUCTION
In Western Europe and the United States, colorectal cancer is the second most common fatal cancer next to lung cancer. Aspirin is believed to have a chemopreventive role in colorectal cancer based on considerable observational data, which show that the rates of colorectal cancers and adenomas are 40%-50% lower in aspirin users[1,2]. Aspirin is a nonsteroidal anti-inflammatory drug (NSAID). There have been indications that use of NSAIDs can lead to the regression of colorectal adenomas[3]. In several rodent models of colorectal cancer, the NSAIDs indomethacin, aspirin, sulindac and piroxicam were shown to be able to reduce tumor growth[4]. However, the molecular mechanisms underlying the cancer preventive effects of NSAIDs are not well understood, and this has been an active issue of research interest.
Commonly, prevention of cancer may be implemented through several means, including cell cycle arrest, induction of apoptosis and inhibition of angiogenesis. One most widely accepted mechanism for the anticancer effect of NSAIDs is the reduction of prostaglandin synthesis by inhibiting COX activity[5-7]. However, the importance of COX inhibition for the anti-proliferative effects of NSAIDs is controversial at the present time, since NSAIDs also manifest growth inhibitory effects against colon cancer cell lines that do not express COX-1 or COX-2 enzymes[8-11]. A common mechanism of NSAID action appears to be the induction of apoptosis, although several in vitro studies in CRC cells have proposed that different molecular pathways may be affected by distinct types of NSAIDs[9,12-14]. To date, little is known about the COX-independent molecular targets of NSAID action in cancer cells.
The mitogen-activated protein kinase (MAPK) superfamily, including the extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 MAPK, is involved in mediating the processes of cell growth and death[15,16]. JNK and p38 MAPK pathways are activated in response to chemicals and environmental stress[17], while the ERK cascade, activated by mitogenic stimuli, is critical for proliferation and survival[18]. Recent evidence indicates that the MAPK family proteins are important mediators of apoptosis induced by stressful stimuli[18,19]. JNK and p38 MAPK are collectively termed stress-activated protein kinases because they are activated by a variety of stress-related stimuli and also activated by chemotherapy drugs[20,21]. Activation of p38 MAPK distorts mitochondrial function via an increase in the ratio of Bax and anti-apoptotic (Bcl-2) members leading to an increased mitochondrial membrane permeability, the release of cytochrome c, and the activation of caspases[22]. Once activated, p38 regulates multiple cellular processes, including transcription, translation, cell cycle progression, and apoptosis. Schwenger et al[23] previously showed that sodium salicylate, the active component of aspirin, activated p38 to induce apoptosis. Also, aspirin activates the p38MAPK pathway, leading to the degradation of cyclin D1, nucleolar translocation of RelA, and apoptosis[24].
Sox genes encode transcription factors that possess strong homology to the high-mobility group (HMG box), which are homologous to sex-determining region of Y-chromosome in the HMG box. There are at least 30 Sox members expressed in many different cell types and tissues, and at multiple stages during development[25]. Sex-determining region Y-box 7 (Sox7), together with Sox17 and Sox18, belongs to the Sox group F subfamily. Sox7 encodes an HMG box transcription factor and has been implicated in parietal endoderm differentiation[26]. Our previous study demonstrated that the expression of SOX7 mRNA was frequently down-regulated in human colorectal cancer cell lines and in primary colorectal tumor tissues, and restoration of SOX7 induced colorectal cancer cell apoptosis, inhibited cell proliferation and colony formation[27].
Despite these available data, the mechanistic function of aspirin in inhibiting COX2 negative colorectal cancer cells awaits further investigation. In this study, we have examined the role of SOX7 in aspirin-mediated growth inhibition of COX2 negative SW480 cells, and we found that SOX7 is regulated by aspirin and the p38 MAPK pathway in SW480 cells. Our study has disclosed the involvement of SOX7 in aspirin-mediated growth inhibition of COX2 negative cancer cells, providing a new insight into the mechanism by which aspirin inhibits COX2 negative colorectal cancer.
MATERIALS AND METHODS
Cell lines and reagents
Human colorectal cancer cell line SW480 and human embryonic kidney HEK-293T cells were cultured in appropriate media with 10% FBS (fetal bovine serum), 100 U/mL penicillin and 100 μg/mL streptomycin, and kept in a humidified atmosphere of 20 mL/L CO2. Genomic DNA was extracted using the standard Proteinase-K method. Total RNA was extracted by using the Trizol reagent (TAKARA).
Drug treatment
Aspirin (Sigma, St. Louis, MO) was dissolved in 1 mol/L Tris-HCl (pH 7.5) to a stock concentration of 1 mol/L and the pH adjusted to 7.2 with 4 mol/L HCl. SW480 cells were treated for 24, 48 and 72 h by adding various volumes of stock to obtain final concentrations of 1, 2 and 5 mmol/L of aspirin. Control cells were treated with an equivalent volume of Tris-HCl (pH 7.2).
The p38 inhibitor SB203580 (Sigma, St. Louis, MO) was dissolved in dimethyl sulfoxide (DMSO) to a stock concentration of 10 mmol/L. SW480 cells were treated with 10 μmol/L SB203580 for 30 h. Control cells were treated with an equivalent volume of DMSO.
MTT assay
Cell proliferation was assessed by the MTT (Moto-nuclear cell direc cytotoxicity assay) [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide] assay. SW480 cells were plated at 1 × 103 cells/well on 96-well plates. After treatment with aspirin or transfection, 20 μL of MTT (5 mg/mL) was added to each well; the samples were incubated for 4 h at 37 °C and then subcultured in the medium with 100 μL DMSO. The absorbance of each well was determined at 492 nm. Survival percentage (%) was calculated relative to the control.
Plasmid constructs and transfection
SOX7 gene promoter (GenBank accession NM_031439) was cloned by PCR from the genome derived from normal human colorectal tissue using the following primers: 5’-CCCAAGCTTCTGCCGACTTTCATTCAGTAGGTG-3’ (sense) and 5’-CCGCTCGAGGTAGGCTCCCAGCAGCGAAG-3’ (antisense). To generate SOX7 promoter-luciferase (pGL3-SOX7-luc) construct, the PCR production was subcloned into the pGL3 enhancer vector via HindIII and XholI sites. Short interfering RNA (siRNA) targeting SOX7 sequence (ACGCCGAGCTGTCGGATGG) was synthesized[27]. An oligonucleotide that represents the siRNA was cloned into the pSliencer4.1-CMV neo-vector (Ambion) between BamHI and HindIII sites following the manufacturer’s instructions. c-Fos and c-Jun were gifts from Dr. Shizuo Akira (Osaka University, Japan); p38α was a gift from Dr. Roger David (University of Mass. Medical School); and SOX7 expression plasmid was previously constructed in this laboratory[27]. Plasmids were transfected using Lipofectamine™2000 (Invitrogen) follow the manufacturer’s instructions.
Luciferase reporter assay
Reporter gene assays were done as previously described[27]. Briefly, 5 × 104 cells were seeded in 24-well tissue culture plates 24 h before transfection. The pGL3-SOX7-luc reporter vector was transfected at 500 ng/well and the Renilla luciferase control plasmid pREP7-RLuc was cotransfected at 50 ng/well as an internal control reporter. After treatment with indicated doses of aspirin or cotransfection with c-Fos and c-Jun expression vectors for 24 h, cells were washed and lysed in passive lysis buffer (Promega) and the transfection efficiency was normalized to the paired Renilla luciferase activity by using the Dual Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions.
Reverse transcription-polymerase chain reaction
For cDNA synthesis, 1 μg of total RNA was reverse transcribed using the RT-Systems supplied by Promega. Quantitative real-time reverse transcription-polymerase chain reaction (RT-PCR) was carried out on an ABI Prism 7000 Sequence Detection System (Applied Biosystems), and SYBR Green (TOYOBO) was used as a double-stranded DNA-specific fluorescent dye. The PCR primer sequences were as follows: SOX7: 5’-ACCAACGGGTCCCACAGA-3’(sense) and 5’-CCACTCAAGGCACAAGAAGG-3’(antisense)[27]; β-actin: 5’-TCGTGCGTGACATTAAGGAG-3’ (sense) and 5’-ATGCCAGGGTACATGGTGGT-3’ (antisense)[28].
Western blotting assay
Western blotting was performed as described previously[29]. The primary antibodies used were the mouse anti-SOX7 (1:1 000, R&D system) and mouse anti-β-actin (1:10 000, Sigma).
RESULTS
Aspirin and SOX7 inhibit the growth of SW480 human colorectal cancer cells
We first demonstrated that aspirin treatment results in a profound concentration- and time-dependent reduction in the proliferation rate of SW480 cells (Figure 1A). Our previous experiments showed that SOX7 was frequently down-regulated in human colorectal cancer cell lines including SW480[27]. In order to investigate the relationship between SOX7 and aspirin, we tested the effect of SOX7 on the growth of SW480 cells, and we found that restoration of SOX7 inhibits SW480 cell proliferation (Figure 1B and C).
Figure 1.
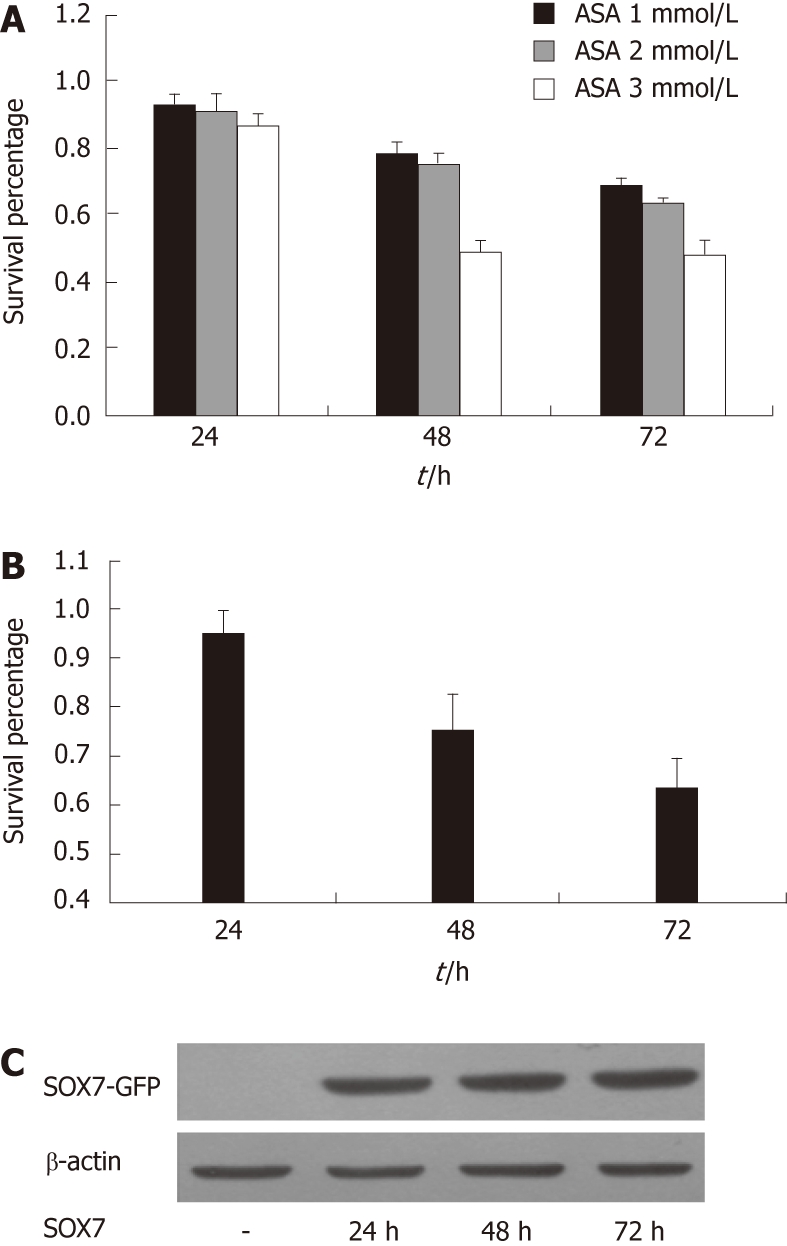
Aspirin and SOX7 inhibit the growth of SW480 cells. A: Dose- and time-dependent effects of aspirin on the growth rates of SW480 human colon cancer cells. SW480 cells were treated for 24, 48 or 72 h in aspirin-containing culture medium; 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay was used to estimate the survival percentage compared with untreated cells; B: Effect of SOX7 expression on cell proliferation. MTT assay was used to estimate the proliferation at different time points after transfection of SOX7 expression vector; C: Western blotting was used to verify the expression of SOX7 after transfection of SOX7 expression vector at indicated time points. MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide. ASA: Aspirin.
SOX7 is induced by aspirin in SW480 colorectal cancer cells
Since both aspirin and SOX7 were able to inhibit the growth of SW480 cells, we sought to examine whether SOX7 can be induced by aspirin in SW480 cells. Our reporter gene experiments demonstrated that aspirin upregulated the activities of SOX7 promoter, and a dose of 1 mmol/L aspirin was sufficient to induce SOX7 promoter activity (Figure 2A). This dose was then used in the later experiments throughout the study. RT-PCR and Western blot assays showed that the mRNA (Figure 2B) and protein (Figure 2C) levels of SOX7 were both upregulated by 1 mmol/L aspirin. These data indicate that the SOX7 expression is induced by aspirin in SW480 cells.
Figure 2.
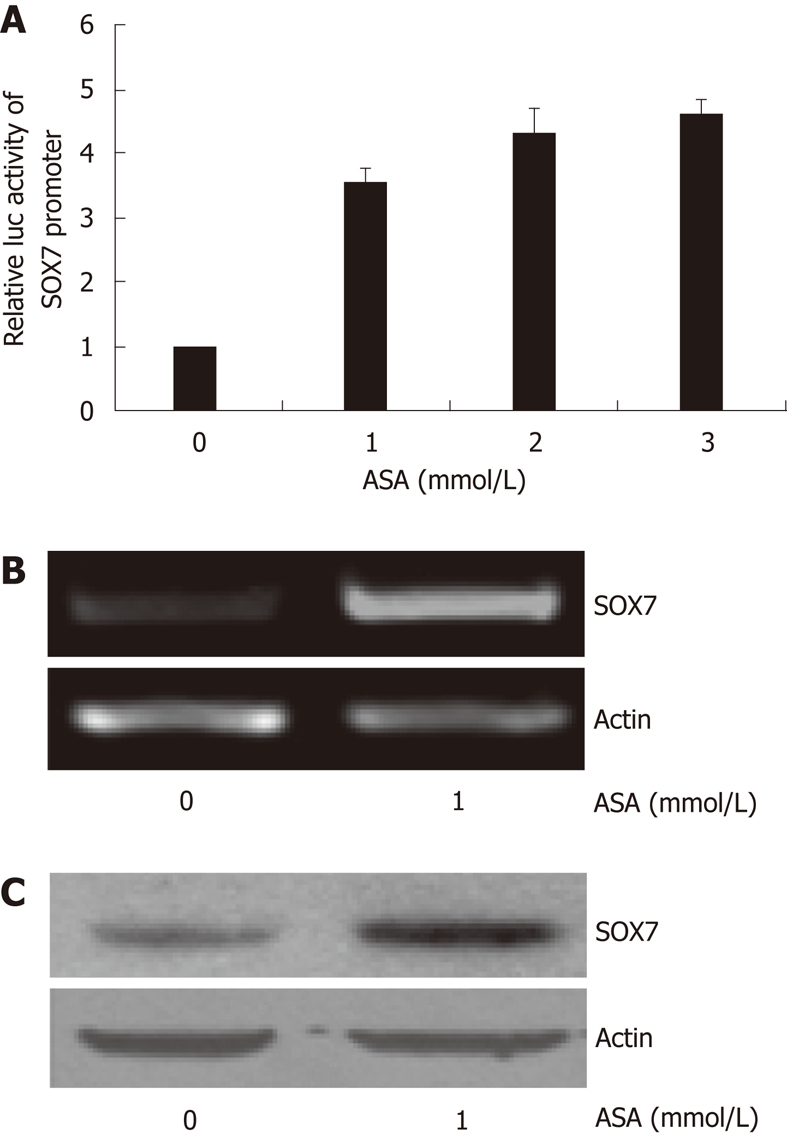
Aspirin upregulates SOX7 expression in SW480 cells. A: SW480 cells were transfected with pGL3-SOX7-luc plasmid, and relative luciferase activity was determined after treatment with different doses of aspirin for 24 h; B and C: Reverse transcription-polymerase chain reaction (B) and Western blotting (C) detected the expression of SOX7 after treatment with aspirin for 30 h; β-actin was used as the internal reference. ASA: Aspirin.
SOX7 is involved in aspirin-mediated growth inhibition of human colorectal cancer cells
The above data indicate that both aspirin and SOX7 inhibit the growth of SW480 cells, and aspirin upregulates the expression of SOX7 in SW480 cells. We next intended to determine whether SOX7 played a role in aspirin-inhibited growth of SW480 cells. First, we constructed a SOX7 siRNA plasmid and tested the interference efficiency of this plasmid. As shown in Figure 3A, transfection of SW480 cells with SOX7 siRNA plasmid resulted in a significant reduction in the SOX7 protein expression induced by aspirin, confirming the effective interference of this SOX7-siRNA. We then transfected SOX7 siRNA plasmid into SW480 cells, and we found that interference of SOX7 expression restored the growth rate of the aspirin-inhibited SW480 cells (Figure 3B). This indicated that SOX7 is involved in aspirin-mediated growth inhibition of human colorectal cancer SW480 cells.
Figure 3.
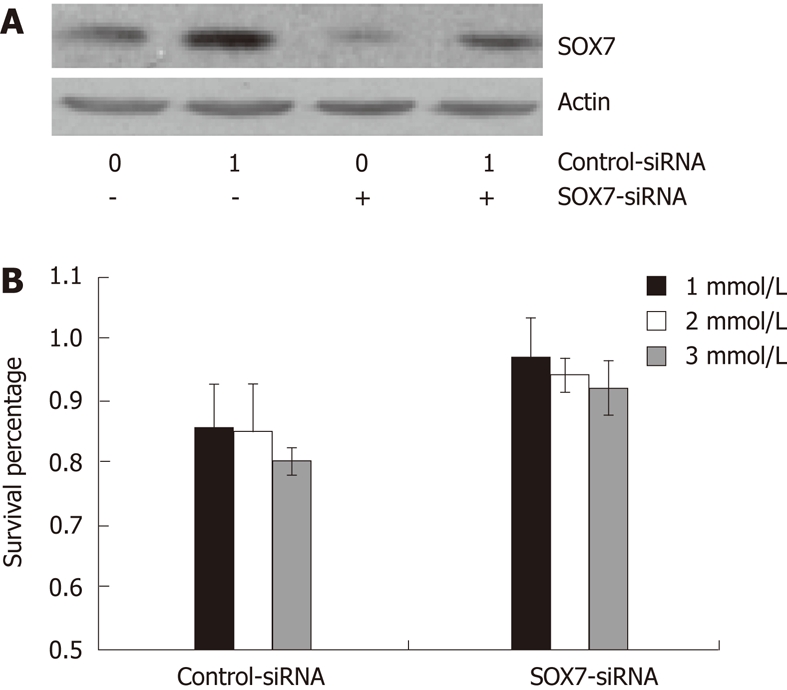
Knockdown of SOX7 counteracts the growth inhibitory effect of aspirin in SW480 cells. A: Western blotting verification of the interfering efficiency of SOX7 Short interfering RNA in aspirin-treated SW480 cells. β-actin was used as the internal reference; B: Effect of SOX7 knockdown on SW480 cell proliferation after treatment with different doses of aspirin for 48 h by moto-nuclear cell direc cytotoxicity assay assay. MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide.
Aspirin induces SOX7 expression through the p38MAPK pathway in SW480 colorectal cancer cells
We and others have shown that the p38 MAPK pathway can be activated by aspirin (data not shown)[24]. We thus sought to examine whether the p38 MAPK pathway is involved in aspirin-induced SOX7 expression. We demonstrated that SOX7 mRNA is significantly upregulated upon the overexpression of p38α in SW480 cells (Figure 4A). In contrast, in HEK-293T cells that express relatively high levels of SOX7, inhibition of p38 by SB203580 reduced the mRNA level of SOX7 (Figure 4B). We further examined the effects of the p38 inhibitor SB203580 on aspirin-induced SOX7 expression, and we discovered that chemical inhibition of p38 substantially abrogates the aspirin-induced upregulation of SOX7. We also found that upon the inhibition of the p38 MAPK pathway by SB203580, the upregulation of SOX7 by aspirin is counteracted at both mRNA and protein levels (Figure 4C and D). In addition, our data show that the AP1 transcription factors c-Jun and c-Fos upregulate SOX7 promoter activities in SW480 cells (Figure 4E).
Figure 4.
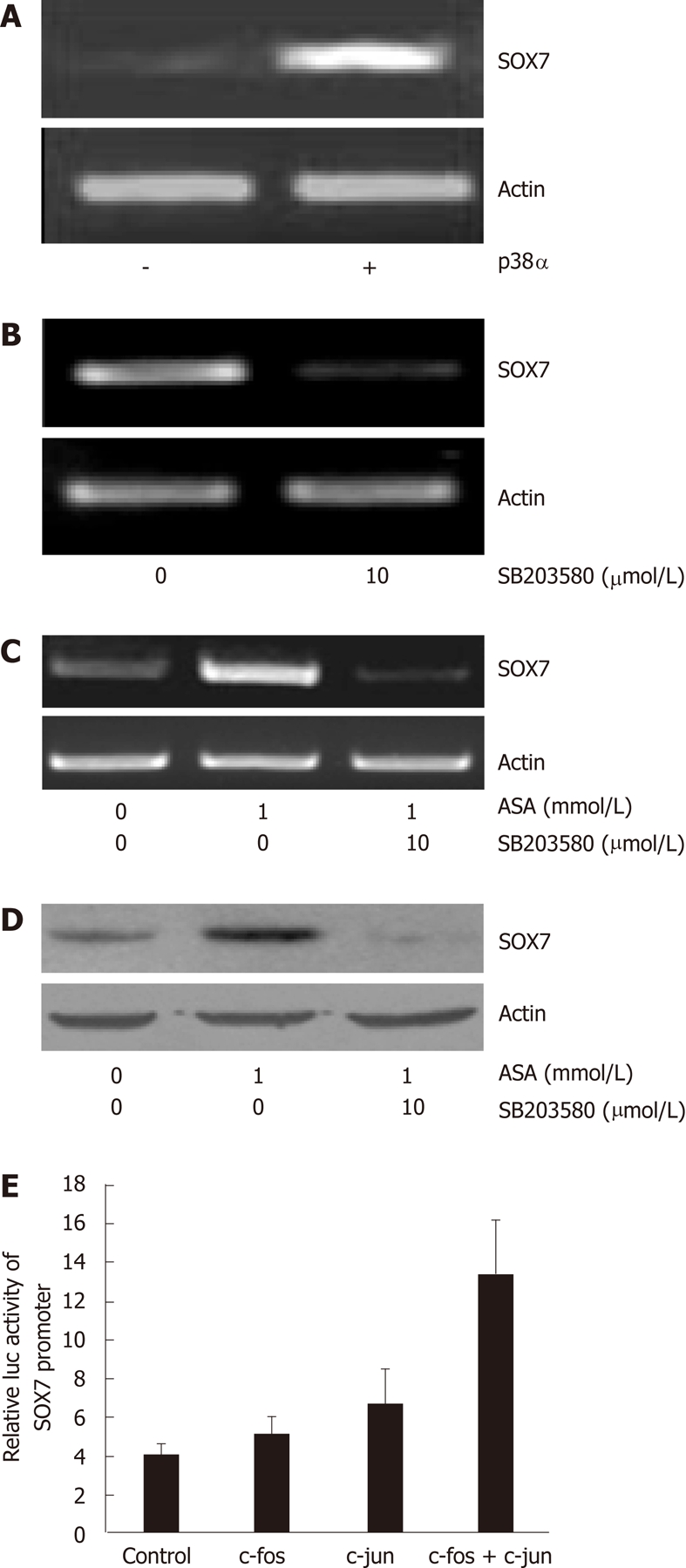
Role of the p38MAPK pathway in aspirin-induced SOX7 expression. A: HEK 293T cells were transfected with p38α expression vector; reverse transcription-polymerase chain reaction (RT-PCR) was used to detect SOX7 mRNA level 30 h after transfection; B: SW480 cells were treated with 10 μmol/L SB203580 for 30 h and SOX7 mRNA level was measured by RT-PCR; C: SW480 cells were treated with aspirin or aspirin combined with SB203580 for 30 h and SOX7 mRNA level was measured by RT-PCR; D: Cells were treated as in Figure 4 and SOX7 protein level was detected by Western blot; E: SW480 cells were transfected with pGL3-SOX7-luc plasmid and various expression vectors as indicated for 24 h and relative luciferase activity was examined. ASA: Aspirin.
DISCUSSION
The primary purpose of this study was to shed insights into the complex mechanistic action of aspirin in colorectal cancer inhibition and prevention. The results of the study show that SOX7 is upregulated by aspirin, and that SOX7 is involved in aspirin-mediated growth inhibition of human colorectal cancer SW480 cells. Moreover, we provided evidence that aspirin induces SOX7 expression through the p38 MAPK pathway.
Recently, aspirin and related NSAIDs have attracted considerable research attention as the compounds that might be of potential benefit in the chemoprevention of cancer[2]. However, the molecular mechanisms by which aspirin and related NSAIDs inhibit tumor formation and growth have largely remained unsolved; and hence the utilization of these compounds in cancer therapeutics is still a substantially disputed issue. One potential mechanism underlying the action of NSAIDs involves the inhibition of COX activity[5]; but there is evidence that in COX negative colorectal cancer SW480 cells, aspirin is also able to inhibit the growth of the cells[8]. Goel et al[30] reported that aspirin could act through COX-independent mechanisms that resulted in an increased expression of DNA mismatch repair proteins and subsequent apoptosis in SW480 cells. Our previous data suggested that the SOX7 gene may play a crucial role in colorectal cancer development as a tumor suppressor[27]. The data from the present study show that SOX7 is upregulated by aspirin in SW480 cells and that it is involved in aspirin-mediated growth inhibition of SW480 cells (Figures 2 and 3), implying that SOX7 may be a target for aspirin in COX-independent cancer cells.
The p38 MAPK pathway plays pivotal roles in regulation of inflammation, proliferation, and cell death. Activation of the p38 pathway is generally mediated by conditions of cell stress and culminates in phosphorylation of p38 on a conserved regulatory domain by the upstream kinases MKK3 and MKK6[31]. In COX negative colorectal cancer SW480 cells, aspirin can activate the p38 MAPK pathway[24]. However, the mechanisms that are involved in this process need to be future investigated. Moreover, in the process of mouse preimplantation development, inhibition of the p38 MAP kinase pathway resulted in the suppression of SOX7 expression, implying that SOX7 may be a target gene of the p38 MAPK pathway[32]. Our experiments in this study indeed prove that the chemical inhibition of p38 substantially abrogates the upregulation of SOX7 upon aspirin treatment (Figure 4). These results indicate that SOX7 is regulated by aspirin and the p38 MAPK pathway in COX negative colorectal cancer cells.
The AP1 transcription complex consists of homodimers and heterodimers of the members from the fos (c-fos, fosB, fra-1 and fra-2) and jun (c-jun, junB and junD) families, and the complex activates target genes by binding with high affinity to particular DNA cis-elements, i.e., the 12-O-tetradecanoylphorbol-13-acetate (TPA) response elements (TRE)[33]. The AP1 activity is regulated at the transcriptional and post-translational levels by several external stimuli, mainly involving MAPK cascades[34]. Our Luciferase reporter assays showed that SOX7 promoter activity was moderately upregulated upon the overexpression of c-fos or c-jun, but markedly upregulated upon the simultaneous overexpression of both c-fos and c-jun (Figure 4E), indicating that c-fos and c-jun may form a dimer to regulate SOX7 promoter, though the details need to be further studied.
In summary, the experimental data presented in this report demonstrate that SOX7 is upregulated by aspirin and is involved in aspirin-mediated growth inhibition of human colorectal cancer SW480 cells. The regulation of SOX7 by aspirin is implemented through the p38MAPK pathway. Overall, this study will help to advance our understanding of the mechanisms of action of aspirin in inhibiting COX-independent colorectal cancer cells.
COMMENTS
Background
Colorectal cancer (CRC) is one of the most prevalent cancers worldwide. Aspirin has been implicated to prevent CRC. The p38MAPK pathway is activated in aspirin-mediated apoptosis in a number of cancer cells. The sex-determining region Y-box 7 (Sox7) is a member of the high mobility group (HMG) transcription factor family, essential for embryonic development and endoderm differentiation.
Research frontiers
Aspirin is a nonsteroidal anti-inflammatory drug (NSAID), which has been implicated in preventing human colorectal cancer (CRC). However, the molecular mechanisms underlying the cancer preventive effects of NSAIDs are not well understood, and this has been an active issue of research interest. One most widely accepted mechanism for the anticancer effect of NSAIDs is the reduction of prostaglandin synthesis by inhibiting COX activity. To date, little is known about the COX-independent molecular targets of NSAID action in cancer cells.
Innovations and breakthroughs
The experimental data presented in this report demonstrate that SOX7 is up-regulated by aspirin and is involved in aspirin-mediated growth inhibition of COX-independent human colorectal cancer SW480 cells. The regulation of SOX7 by aspirin is implemented through the p38MAPK pathway. Overall, this study will help to advance our understanding towards the mechanisms of action of aspirin in inhibiting COX-independent colorectal cancer cells.
Applications
By understanding how SOX7 is involved in aspirin-mediated growth inhibition of COX-independent human colorectal cancer cells, this study may represent a future gene target strategy for patients with COX-independent colorectal cancer.
Terminology
SOX7 is an HMG box transcription factor and has been implicated in parietal endoderm differentiation; recent reports have referred to the role of SOX7 in tumor suppression. mitogen-activated protein kinase (MAPK) is the mitogen-activated protein kinase superfamily, which includes the extracellular signal-regulated kinase, c-Jun N-terminal kinase, and p38 MAPK, and is involved in mediating the processes of cell growth and death. Recent evidence indicates that the MAPK family proteins are important mediators of apoptosis induced by stressful stimuli.
Peer review
The authors examined the role of SOX7 in aspirin-mediated growth inhibition of COX2 negative SW480 cells, and found that SOX7 was regulated by aspirin and the p38 MAPK pathway in SW480 cells. The present results disclose the involvement of SOX7 in aspirin-mediated growth inhibition of COX2 negative cancer cells, providing a new insight to the mechanism as to how aspirin inhibits COX2 negative colorectal cancer.
Footnotes
Supported by The National Natural Science Foundation of China, No. 31071149; the Fundamental Research Funds for the Central Universities, No. 09ZDQD01 and No. 10QNJJ014; and the National Science Foundation of China, No. J0830627-2
Peer reviewer: Dr. Ondrej Slaby, Department of Comprehensive Cancer Care, Masaryk Memorial Cancer Institute, Zluty kopec 7, Brno 65653, Czech Republic
S- Editor Wu X L- Editor Logan S E- Editor Xiong L
References
- 1.García-Rodríguez LA, Huerta-Alvarez C. Reduced risk of colorectal cancer among long-term users of aspirin and nonaspirin nonsteroidal antiinflammatory drugs. Epidemiology. 2001;12:88–93. doi: 10.1097/00001648-200101000-00015. [DOI] [PubMed] [Google Scholar]
- 2.Jänne PA, Mayer RJ. Chemoprevention of colorectal cancer. N Engl J Med. 2000;342:1960–1968. doi: 10.1056/NEJM200006293422606. [DOI] [PubMed] [Google Scholar]
- 3.García Rodríguez LA, Huerta-Alvarez C. Reduced incidence of colorectal adenoma among long-term users of nonsteroidal antiinflammatory drugs: a pooled analysis of published studies and a new population-based study. Epidemiology. 2000;11:376–381. doi: 10.1097/00001648-200007000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Reddy BS, Rao CV, Rivenson A, Kelloff G. Inhibitory effect of aspirin on azoxymethane-induced colon carcinogenesis in F344 rats. Carcinogenesis. 1993;14:1493–1497. doi: 10.1093/carcin/14.8.1493. [DOI] [PubMed] [Google Scholar]
- 5.Gupta RA, Dubois RN. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer. 2001;1:11–21. doi: 10.1038/35094017. [DOI] [PubMed] [Google Scholar]
- 6.Plummer SM, Holloway KA, Manson MM, Munks RJ, Kaptein A, Farrow S, Howells L. Inhibition of cyclo-oxygenase 2 expression in colon cells by the chemopreventive agent curcumin involves inhibition of NF-kappaB activation via the NIK/IKK signalling complex. Oncogene. 1999;18:6013–6020. doi: 10.1038/sj.onc.1202980. [DOI] [PubMed] [Google Scholar]
- 7.Brown JR, DuBois RN. COX-2: a molecular target for colorectal cancer prevention. J Clin Oncol. 2005;23:2840–2855. doi: 10.1200/JCO.2005.09.051. [DOI] [PubMed] [Google Scholar]
- 8.Lai MY, Huang JA, Liang ZH, Jiang HX, Tang GD. Mechanisms underlying aspirin-mediated growth inhibition and apoptosis induction of cyclooxygenase-2 negative colon cancer cell line SW480. World J Gastroenterol. 2008;14:4227–4233. doi: 10.3748/wjg.14.4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanif R, Pittas A, Feng Y, Koutsos MI, Qiao L, Staiano-Coico L, Shiff SI, Rigas B. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem Pharmacol. 1996;52:237–245. doi: 10.1016/0006-2952(96)00181-5. [DOI] [PubMed] [Google Scholar]
- 10.Chan TA. Nonsteroidal anti-inflammatory drugs, apoptosis, and colon-cancer chemoprevention. Lancet Oncol. 2002;3:166–174. doi: 10.1016/s1470-2045(02)00680-0. [DOI] [PubMed] [Google Scholar]
- 11.Yin H, Xu H, Zhao Y, Yang W, Cheng J, Zhou Y. Cyclooxygenase-independent effects of aspirin on HT-29 human colon cancer cells, revealed by oligonucleotide microarrays. Biotechnol Lett. 2006;28:1263–1270. doi: 10.1007/s10529-006-9084-9. [DOI] [PubMed] [Google Scholar]
- 12.Shiff SJ, Rigas B. Nonsteroidal anti-inflammatory drugs and colorectal cancer: evolving concepts of their chemopreventive actions. Gastroenterology. 1997;113:1992–1998. doi: 10.1016/s0016-5085(97)99999-6. [DOI] [PubMed] [Google Scholar]
- 13.Kopp E, Ghosh S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science. 1994;265:956–959. doi: 10.1126/science.8052854. [DOI] [PubMed] [Google Scholar]
- 14.Stark LA, Din FV, Zwacka RM, Dunlop MG. Aspirin-induced activation of the NF-kappaB signaling pathway: a novel mechanism for aspirin-mediated apoptosis in colon cancer cells. FASEB J. 2001;15:1273–1275. [PubMed] [Google Scholar]
- 15.Bode AM, Dong Z. Targeting signal transduction pathways by chemopreventive agents. Mutat Res. 2004;555:33–51. doi: 10.1016/j.mrfmmm.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 16.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 17.Whitmarsh AJ, Davis RJ. Analyzing JNK and p38 mitogen-activated protein kinase activity. Methods Enzymol. 2001;332:319–336. doi: 10.1016/s0076-6879(01)32212-7. [DOI] [PubMed] [Google Scholar]
- 18.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 19.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 20.Miyoshi N, Uchida K, Osawa T, Nakamura Y. A link between benzyl isothiocyanate-induced cell cycle arrest and apoptosis: involvement of mitogen-activated protein kinases in the Bcl-2 phosphorylation. Cancer Res. 2004;64:2134–2142. doi: 10.1158/0008-5472.can-03-2296. [DOI] [PubMed] [Google Scholar]
- 21.Lin HJ, Chao PD, Huang SY, Wan L, Wu CJ, Tsai FJ. Aloe-emodin suppressed NMDA-induced apoptosis of retinal ganglion cells through regulation of ERK phosphorylation. Phytother Res. 2007;21:1007–1014. doi: 10.1002/ptr.2138. [DOI] [PubMed] [Google Scholar]
- 22.Mandal C, Dutta A, Mallick A, Chandra S, Misra L, Sangwan RS, Mandal C. Withaferin A induces apoptosis by activating p38 mitogen-activated protein kinase signaling cascade in leukemic cells of lymphoid and myeloid origin through mitochondrial death cascade. Apoptosis. 2008;13:1450–1464. doi: 10.1007/s10495-008-0271-0. [DOI] [PubMed] [Google Scholar]
- 23.Schwenger P, Bellosta P, Vietor I, Basilico C, Skolnik EY, Vilcek J. Sodium salicylate induces apoptosis via p38 mitogen-activated protein kinase but inhibits tumor necrosis factor-induced c-Jun N-terminal kinase/stress-activated protein kinase activation. Proc Natl Acad Sci USA. 1997;94:2869–2873. doi: 10.1073/pnas.94.7.2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thoms HC, Dunlop MG, Stark LA. p38-mediated inactivation of cyclin D1/cyclin-dependent kinase 4 stimulates nucleolar translocation of RelA and apoptosis in colorectal cancer cells. Cancer Res. 2007;67:1660–1669. doi: 10.1158/0008-5472.CAN-06-1038. [DOI] [PubMed] [Google Scholar]
- 25.Pevny LH, Lovell-Badge R. Sox genes find their feet. Curr Opin Genet Dev. 1997;7:338–344. doi: 10.1016/s0959-437x(97)80147-5. [DOI] [PubMed] [Google Scholar]
- 26.Wegner M. From head to toes: the multiple facets of Sox proteins. Nucleic Acids Res. 1999;27:1409–1420. doi: 10.1093/nar/27.6.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Huang S, Dong W, Li L, Feng Y, Pan L, Han Z, Wang X, Ren G, Su D, et al. SOX7, down-regulated in colorectal cancer, induces apoptosis and inhibits proliferation of colorectal cancer cells. Cancer Lett. 2009;277:29–37. doi: 10.1016/j.canlet.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y, Gao Y, Zhang G, Huang S, Dong Z, Kong C, Su D, Du J, Zhu S, Liang Q, et al. DNMT3a plays a role in switches between doxorubicin-induced senescence and apoptosis of colorectal cancer cells. Int J Cancer. 2011;128:551–561. doi: 10.1002/ijc.25365. [DOI] [PubMed] [Google Scholar]
- 29.Wang X, Pan L, Feng Y, Wang Y, Han Q, Han L, Han S, Guo J, Huang B, Lu J. P300 plays a role in p16(INK4a) expression and cell cycle arrest. Oncogene. 2008;27:1894–1904. doi: 10.1038/sj.onc.1210821. [DOI] [PubMed] [Google Scholar]
- 30.Goel A, Chang DK, Ricciardiello L, Gasche C, Boland CR. A novel mechanism for aspirin-mediated growth inhibition of human colon cancer cells. Clin Cancer Res. 2003;9:383–390. [PubMed] [Google Scholar]
- 31.Ben-Levy R, Hooper S, Wilson R, Paterson HF, Marshall CJ. Nuclear export of the stress-activated protein kinase p38 mediated by its substrate MAPKAP kinase-2. Curr Biol. 1998;8:1049–1057. doi: 10.1016/s0960-9822(98)70442-7. [DOI] [PubMed] [Google Scholar]
- 32.Maekawa M, Yamamoto T, Tanoue T, Yuasa Y, Chisaka O, Nishida E. Requirement of the MAP kinase signaling pathways for mouse preimplantation development. Development. 2005;132:1773–1783. doi: 10.1242/dev.01729. [DOI] [PubMed] [Google Scholar]
- 33.Malnou CE, Brockly F, Favard C, Moquet-Torcy G, Piechaczyk M, Jariel-Encontre I. Heterodimerization with different Jun proteins controls c-Fos intranuclear dynamics and distribution. J Biol Chem. 2010;285:6552–6562. doi: 10.1074/jbc.M109.032680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen W, Bowden GT. Activation of p38 MAP kinase and ERK are required for ultraviolet-B induced c-fos gene expression in human keratinocytes. Oncogene. 1999;18:7469–7476. doi: 10.1038/sj.onc.1203210. [DOI] [PubMed] [Google Scholar]