

Abstract
Structural alterations provoked by mutations or genetic variations in the gene sequence of G protein-coupled receptors may lead to abnormal function of the receptor molecule and, ultimately, to disease. While some mutations lead to changes in domains involved in agonist binding, receptor activation or coupling to effectors, others may cause misfolding and lead to retention/degradation of the protein molecule by the quality control system of the cell. Several strategies, including genetic, chemical and pharmacological approaches have been shown to rescue function of trafficking-defective misfolded G protein-coupled receptors. Among these, pharmacological strategies offer the most promising therapeutic tool to promote proper trafficking of misfolded proteins to the plasma membrane. Pharmacological chaperones or “pharmacoperones,” are small compounds that permeate the plasma membrane, enter cells, and bind selectively to misfolded proteins and correct folding allowing routing of the target protein to the plasma membrane, where the receptor may bind and respond to agonist stimulation. In this review we describe new therapeutic opportunities based on misfolding of otherwise functional human gonadotropin-releasing hormone receptors. This particular receptor is highly sensitive to single changes in chemical charge and its intracellular traffic is delicately balanced between expression at the plasma membrane or retention/degradation in the endoplasmic reticulum; it is, therefore, a particularly instructive model to understand both protein routing and the molecular mechanisms whereby pharmacoperones rescue misfolded intermediates or conformationally defective receptors.
Keywords: Gonadotropin-releasing hormone, gonadotropin releasing hormone receptor, G protein-coupled receptors, mutant rescue, pharmacoperones, protein misfolding
I. INTRODUCTION
The heptahelical G protein-coupled receptors (GPCRs) constitute a large and functionally diverse superfamily of membrane proteins whose primary function is to transduce extracellular stimuli into the intracellular environment through the activation of one or more signal transduction pathways mediated by G proteins and other interacting proteins. This system of cellular communication is so effective, that the ligands that recognize and activate these receptors are highly variable in chemical structure and include photons, odorants, pheromones, hormones, lipids, and neurotransmitters that vary in size from small biogenic amines to peptides to large proteins (Rosenbaum, Rasmussen, & Kobilka, 2009; Ulloa-Aguirre & Conn, 1998, 2009; Ulloa-Aguirre, Stanislaus, Janovick, & Conn, 1999). Estimates range from 1–5% of the genome in mammals encodes for this superfamily of receptors, which currently constitute the single most important source of therapeutic targets for many diseases (Takeda, Kadowaki, Haga, Takaesu, & Mitaku, 2002). In fact, 60–70% of all approved drugs derived their benefits by selective targeting to these receptors, which at least 50% are thought to be the target for endogenous ligands (P. J. Conn, Christopoulos, & Lindsley, 2009).
As with other protein molecules, structural alterations provoked by mutations or genetic variations in the gene sequence of GPCRs may lead to abnormal function of the receptor and, eventually, to disease, depending on the location and the nature of the substitution or modification. Mutations in these receptors are known to be responsible for a large number of disorders, including cancers, heritable obesity and endocrine disease, which underlines their importance as therapeutic targets. These structural alterations may provoke either gain- or loss-of-function of the affected receptor (Milligan, 2003; Ulloa-Aguirre & Conn, 1998). Loss-of-function mutations usually alter domains involved in specific functions of the receptor, such as agonist binding, receptor activation, interaction with accessory/scaffold proteins or with coupled effectors, or sequences that dictate proper folding and intracellular trafficking of the receptor to the cell surface plasma membrane (PM). It is becoming well recognized that mutations of GPCRs frequently lead to misfolding and subsequent retention/degradation by the quality control system (QCS) of the cell. Further, misfolding can result in protein molecules that retain intrinsic function yet become misrouted and, for reasons of mislocation only, cease to function normally and result in disease. Recognition of this latter concept immediately presents the therapeutic opportunity to correct misrouting and rescue mutants, thereby restoring function and, potentially, curing disease (Bernier, Bichet, & Bouvier, 2004; Bernier et al., 2006; Ulloa-Aguirre, Janovick, Leanos-Miranda, & Conn, 2003). In fact, there is now a wealth of information demonstrating that complete functional rescue of misfolded mutant receptors both in vitro and in vivo is possible by using small nonpeptide molecules called pharmacological chaperones or pharmacoperones; these compounds, often designed originally as receptor antagonists, have proved to serve as effective molecular templates, promoting correct folding and allowing the mutants to pass the scrutiny of the cellular QCS and be expressed at the cell surface plasma membrane (Bernier et al., 2006; P. M. Conn & Janovick, 2009a; Loo & Clarke, 2007; Nakamura, Yasuda, Hirota, & Shimizu, 2010; Ulloa-Aguirre, Janovick, Brothers, & Conn, 2004). Thus, the endoplasmic reticulum QCS represents a potential site for a variety of therapeutic interventions in an array of diseases characterized by conformationally aberrant proteins. This chapter summarizes updated information on the gonadotropin-releasing hormone (GnRH) receptor type I (GnRHR) misfolding, which has proved to be a valuable paradigm for the development of drugs potentially useful in regulating GPCR trafficking in health and disease.
II. THE ENDOPLASMIC RETICULUM QUALITY CONTROL SYSTEM
Synthesis and processing of secretory and membrane proteins occurs in the endoplasmic reticulum (ER) and Golgi apparatus. Similar to other proteins synthesized by the cell, G protein-coupled receptors are subjected to a stringent quality control system that checks the integrity and correct folding of the newly synthesized protein into a three-dimensional protein structure determined by its amino acid sequence. The three-dimensional protein structure is stabilized by several noncovalent interactions involving hydrogen bonds, electrostatic interactions, and hydrophobic interactions as well as by covalent bonds between cysteine side chains that form disulfide bridges (P. L. Clark, 2004; Englander, Mayne, & Krishna, 2007; Ulloa-Aguirre & Conn, 2009; Ulloa-Aguirre, Janovick, Brothers et al., 2004). By monitoring the structural and folding correctness of newly synthesized proteins, the ER QCS prevents accumulation of defective, misfolded proteins that may easily aggregate in a highly crowded environment and interfere with normal cell function (Ellgaard & Helenius, 2003; Hartl & Hayer-Hartl, 2009; Helenius, 2001; Sitia & Braakman, 2003; Trombetta & Parodi, 2003). Due to the dense packing of thousands of macromolecules within the cells, protein folding should be a highly effective process; in fact, folding of a protein is not randomly performed but rather follows pathways that limit the number of possible conformations searching for the native fold (Ellis, 2007). The mechanisms that operate at the ER to identify and sort proteins according to their maturation status include specialized folding factors, escort proteins, retention factors, enzymes, and members of major molecular chaperone families (Brooks, 1999; Ellis, 2007; Hartl & Hayer-Hartl, 2002; Morello, Petaja-Repo, Bichet, & Bouvier, 2000). Molecular chaperones are ER-resident proteins that assist in folding or assembly of the polypeptide for efficient ER export, preventing aggregation or incorrect interactions between misfolded proteins and other molecules (Broadley & Hartl, 2009; Ellis, 2007; Leandro & Gomes, 2008; Ni & Lee, 2007). The QCS is not protein-specific and although the steric character of the protein backbone restricts the spectrum of protein shapes that are recognized by the stringent quality control mechanisms, some features displayed by proteins, including unwanted hydrophobic surfaces, unpaired cysteines, immature glycans, and particular sequence motifs, have been identified as important for low affinity chaperoneprotein association (Angelotti, Daunt, Shcherbakova, Kobilka, & Hurt, 2010; Dong, Filipeanu, Duvernay, & Wu, 2007; Dunham & Hall, 2009; Ulloa-Aguirre & Conn, 2009). Proteins that do not fulfill the criteria of the ER QCS or whose aberrant structure cannot be corrected by molecular chaperones, are submitted to degradation through the polyubiquitination/proteasome pathway (Fig. 1) (Ulloa-Aguirre, Janovick, Brothers et al., 2004; Werner, Brodsky, & McCracken, 1996). Correctly folded proteins, however, are allowed to enter the pathway leading to their final destination within the cell (e.g. the plasma membrane) after processing at the Golgi is completed. In this scenario, it is easy to understand why the scrutiny by the ER QCS relies on conformational features of the protein rather than on functional criteria, so even minor alterations in the secondary or tertiary structure of a protein may lead to intracellular retention and/or degradation (Angelotti et al., 2010; Ulloa-Aguirre, Janovick, Brothers et al., 2004). Because of this, point mutations resulting in protein sequence variations may result in misfolded and disease-causing proteins that accumulate or that are rapidly degraded making the protein unable to reach their functional destination within the cell (Hartl & Hayer-Hartl, 2009). Examples of diseases caused by conformational defects of proteins include neurodegenerative diseases, familial hypercholesterolemia, cystic fibrosis, retinitis pigmentosa, and diabetes insipidus, among others (Bernier, Lagace, Bichet, & Bouvier, 2004; Ulloa-Aguirre, Janovick, Brothers et al., 2004).
Figure 1.
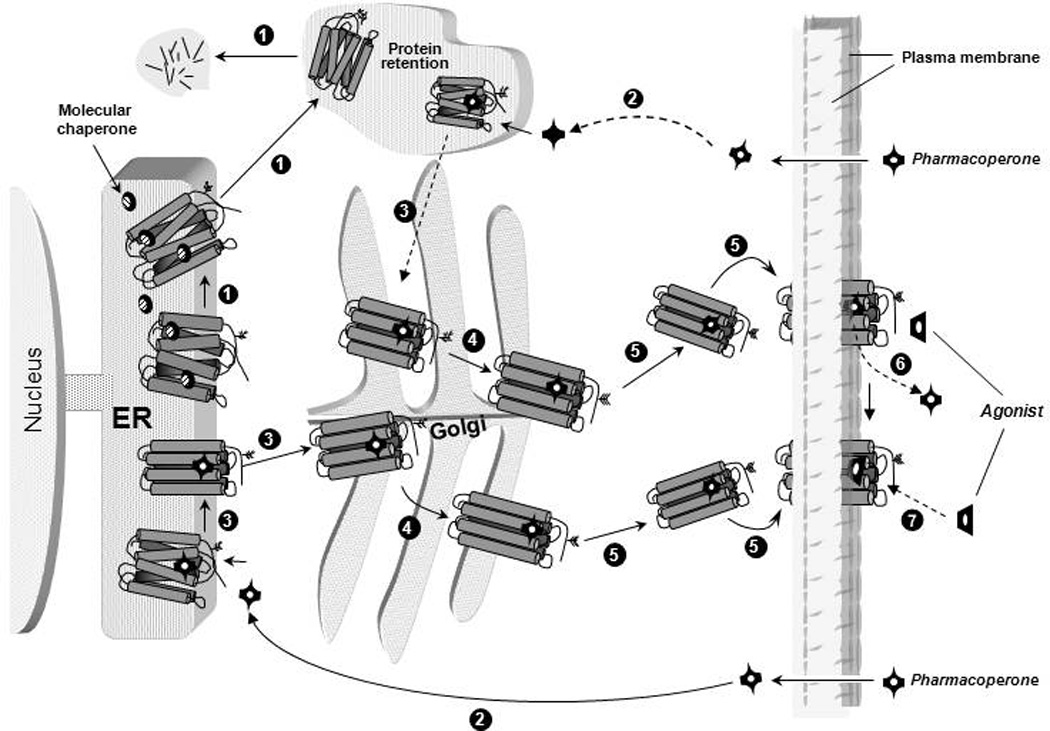
Quality control system in the endoplasmic reticulum (ER). Newly synthesized proteins are translocated to the lumen of the ER where folding is facilitated or corrected by molecular chaperones (oval structures); when folding fails, misfolded proteins are retained in the ER and targeted for degradation through the polyubiquitination/proteasome pathway (step1 in black circles). After diffusing into the cell (step 2) pharmacoperones (black crossed structures) diffuse into the cell and selectively bind to the misfolded protein to influence folding (step 3) promoting correct routing to the Golgi complex for further processing (e.g. glycosylation) (step 4). Previously synthesized misfolded proteins, retained by the QCS, may be still rescued by pharmacoperones (steps 2 and 3 at the top). Mature processed proteins are then delivered to the cell surface plasma membrane (step 5), where the pharmacoperone can dissociate from the target protein (step 6) allowing the receptor to interact with agonist (step 7).
It has also become clear that variable (but significant) amounts of even some wild-type (WT) GPCRs are expressed inefficiently (i.e. retained in the ER), apparently as a result of misfolding (Andersson, D'Antona, Kendall, Von Heijne, & Chin, 2003; Cook, Zhu, & Hinkle, 2003; Kato & Touhara, 2009; Lu, Echeverri, & Moyer, 2003; Lu, Staszewski, Echeverri, Xu, & Moyer, 2004; Petaja-Repo et al., 2001; Petaja-Repo, Hogue, Laperriere, Walker, & Bouvier, 2000; Pietila et al., 2005; Wuller et al., 2004), suggesting that this level of post-translational control may itself provide another level of potential therapeutic intervention (P. M. Conn & Janovick, 2009a, 2009b; P. M. Conn & Ulloa-Aguirre, 2010). This effective “inefficiency” may impact on cellular information networking; in fact, recent understanding of the unfolded protein response [UPR, (Nardai, Vegh, Prohaszka, & Csermely, 2006; Rab et al., 2007; Tuusa, Markkanen, Apaja, Hakalahti, & Petaja-Repo, 2007)] suggests that there is the potential for information transfer as a result of protein retention by the ER. In principle, the (endogenous) chaperoning system could mediate the fraction of the newly synthesized receptor that traverses to the plasma membrane and may contribute to the physiological changes in levels of GPCRs that are observed in different physiological settings.
Several in vitro approaches to correct folding and promote trafficking of proteins from the ER to the PM have been described. These include physical methods (e.g. incubation at reduced temperatures) (Brown, Hong-Brown, & Welch, 1997b; Ulloa-Aguirre, Janovick, Brothers et al., 2004), genetic modifications (e.g. addition or deletion of specific sequences into the conformationally defective protein) (Dunham & Hall, 2009; Katada, Tanaka, & Touhara, 2004; Krautwurst, Yau, & Reed, 1998; Lin et al., 1998; Maya-Nunez, Janovick, & Conn, 2000; Maya-Nunez et al., 2002; Schulein, Zuhlke, Krause, & Rosenthal, 2001; Wetzel et al., 1999), manipulation of the ER and/or post-ER mechanisms that influence GPCR export (e.g. over expression of molecular chaperones or introduction of cell-penetrating peptides that modify cytosolic Ca2+ stores affecting function of Ca2+-dependent chaperones) (Brothers, Janovick, & Conn, 2006; P. M. Conn, Ulloa-Aguirre, Ito, & Janovick, 2007; Gorbatyuk et al.; Morello et al., 2001; Oueslati et al., 2007), and the use of non-specific chemical stabilizers (chemical chaperones) (e.g. osmolytes such as trimethylamine N-oxide, glycerol or heavy water) (Arakawa, Ejima, Kita, & Tsumoto, 2006; Brown, Hong-Brown, & Welch, 1997a; Brown et al., 1997b ; Gekko & Timasheff, 1981; Leandro & Gomes, 2008; Loo & Clarke, 2007; Robben, Sze, Knoers, & Deen, 2006; Sampedro & Uribe, 2004) and pharmacological chaperones (“pharmacoperones”). Genetic approaches, albeit effective, are probably impractical as therapeutic intervention because, if it were possible to access the gene sequence, the primary error could be directly corrected. Chemical chaperones require high concentrations for effective folding of mutant proteins and hence are too toxic for in vivo applications; in addition, since they are nonspecific they might potentially increase secretion or intracellular retention of many different proteins in various cellular compartments leading to inappropriate changes in the levels and/or secretion of many proteins, thereby compromising cell function and/or local homeostasis (Castro-Fernandez, Maya-Nunez, & Conn, 2005).
Pharmacoperones appear to be among the most promising therapeutic approaches to treat conformational diseases (Arakawa et al., 2006; Bernier, Lagace et al., 2004; P. M. Conn & Janovick, 2009a; P. M. Conn & Ulloa-Aguirre, 2010; P. M. Conn et al., 2007; Loo & Clarke, 2007; Nakamura et al., 2010; Ulloa-Aguirre et al., 2003). Pharmacoperones are small, often lipophilic compounds that enter cells and serve as specific molecular templates, promoting correct folding, and allowing the mutant proteins to pass the QCS and be expressed at the cellular loci where they may function (Arakawa et al., 2006; Bernier, Bichet et al., 2004; Bernier, Lagace et al., 2004; P. M. Conn & Ulloa-Aguirre, 2010; Loo & Clarke, 2007; Ulloa-Aguirre et al., 2003). Frequently, such molecules were initially identified as peptidomimetics from high throughput screens for antagonists or agonists and may come from diverse chemical classes. Because such peptidomimetics interact with proteins to which they are selectively targeted, it has been the first place where many investigators have started in the search for agents that bind to and stabilize misfolded proteins, including GPCRs. Among particular PM proteins that lead to conformational diseases and that may benefit from protein rescue are mutants of the chloride channel cystic fibrosis transmembrane conductance regulator, which causes cystic fibrosis (Amaral, 2006; Dormer et al., 2001; Galietta et al., 2001; Zhang et al., 2003), as well as mutants of the GnRHR, V2R, rhodopsin, and melanocortin-4 receptor, which lead to congenital hypogonadotropic hypogonadism (Ulloa-Aguirre et al., 2003; Ulloa-Aguirre, Janovick, Leanos-Miranda, & Conn, 2004), nephrogenic diabetes insipidus (Bernier, Lagace et al., 2004; Bichet, 2006; Morello & Bichet, 2001), retinitis pigmentosa (Noorwez et al., 2003; Noorwez et al., 2004; Noorwez, Ostrov, McDowell, Krebs, & Kaushal, 2008), and early-onset obesity (Tao, 2010). In the case of particular GPCRs [e.g. the GnRHR, the vasopressin V2 receptor (V2R), the melanocortin-4 receptor, and rhodopsin], this approach has succeeded with a striking number of different misfolded mutants, supporting the view that pharmacoperones will become a powerful ammunition in our arsenal of therapeutic options (Bernier et al., 2006; P. M. Conn & Ulloa-Aguirre, 2010; P. M. Conn et al., 2007; Mendes, van der Spuy, Chapple, & Cheetham, 2005; Noorwez et al., 2004; Noorwez et al., 2008; Tao, 2010).
III. MISFOLDING OF GPCRS AND DISEASE
As mentioned previously, mutations in GPCRs may cause misrouting of otherwise functional proteins and lead to disease (Table 1). This is the case with the X-linked nephrogenic diabetes insipidus, the autosomal dominant form of retinitis pigmentosa, some forms of morbid obesity, and hypogonadotropic hypogonadism due to mutations in the GnRHR. Mutations in the V2R gene cause X-linked nephrogenic diabetes insipidus, a disease characterized by an inability to concentrate urine despite normal or elevated plasma concentrations of the antidiuretic hormone arginine vasopressin (Bernier et al., 2006; Bichet, 2006; Fujiwara & Bichet, 2005; Hermosilla et al., 2004). A number (nearly 70%) of V2R mutants causing X-linked diabetes insipidus are unable to reach the cell surface membrane and respond to agonist stimulation (Bernier et al., 2006; P. M. Conn et al., 2007). In retinitis pigmentosa, ER trapping of misfolded mutant rhodopsin eventually leads to rod photoreceptor degeneration (Mendes et al., 2005; Saliba, Munro, Luthert, & Cheetham, 2002) followed by cone degeneration. Trafficking-defective mutants of the glycoprotein hormone receptors (luteinizing hormone, follicle-stimulating hormone, and thyrotropin receptors) have been described as a cause of Leydig cell hypoplasia (Gromoll et al., 2002; Martens et al., 2002), ovarian failure (Aittomaki et al., 1995; Tranchant et al.), and congenital hypothyroidism (Biebermann et al., 1997; Calebiro et al., 2005), respectively. The melanocortin-1 receptor, has been found to be mutated in patients with skin and hair abnormalities, and increased susceptibility to skin cancers (Beaumont et al., 2005; Beaumont et al., 2007); among the 60 or so mutants described, at least four display decreased PM expression (Beaumont et al., 2005). Misfolding and intracellular retention of mutants from two other melanocortin receptors, the melanocortin-3 and melanocortin-4 receptors associated with regulation of fat deposition and energy homeostasis, have been detected in patients with morbid obesity (Tao, 2010; Tao & Segaloff, 2003). Mutations that provoke trapping of the endothelin-B receptor have been detected in a subset of patients with Hirschsprung’s disease or aganglionic megacolon (Fuchs et al., 2001; Tanaka et al., 1998), while mutations in the calcium-sensing receptor leading to intracellular retention of the abnormal receptor have been found in patients with familial hypocalciuric hypercalcemia (D'Souza-Li et al., 2002). Intracellular retention of the chemokine receptor 5 at the ER has also been observed in a subset of subjects with resistance to HIV infection (Rana et al., 1997) and mutations leading to receptor misfolding of the human GnRHR (hGnRHR) cause congenital hypogonadotropic hypogonadism (HH) (Beranova et al., 2001; Ulloa-Aguirre, Janovick, Leanos-Miranda et al., 2004).
Table 1.
Loss of function diseases or abnormalities caused by GPCR misfolding. Reproduced from Conn & Ulloa-Aguirre (2010) with permission from Cell Press, Cambridge, MA.
| Disease or Abnormality | GPCR | Refs |
|---|---|---|
| Retinitis pigmentosa | Rhodopsin | (Mendes et al., 2005) |
| Nephrogenic diabetes insipidus | V2Ra | (Bichet, 2006; Fujiwara & Bichet, 2005; Hermosilla et al., 2004) |
| Hypogonadotropic hypogonadism | GnRHR | (Ulloa-Aguirre & Conn, 2009) |
| Familial hypocalciuric hypercalcemia | CaR | (Huang & Breitwieser,2007) |
| Male pseudohermaphroditism | LHR | (Huhtaniemi & Themmen, 2005) |
| Hypergonadotropic hypogonadism | ||
| Ovarian dysgenesis | FSHR | (Huhtaniemi & Themmen, 2005) |
| Congenital hypothyroidism | TSHR | (Calebiro et al., 2005) |
| Hirschsprung’s disease | E-BR | (Amiel et al., 2008) |
| Red head color and fair skin (RHC phenotype and propensity to skin cancer) | MC1R | (Beaumont et al., 2005; Beaumont et al., 2007) |
| Familial glucocorticoid deficiency | MC2R | (A. J. Clark, Metherell, Cheetham, & Huebner, 2005) |
| Obesity | MC3R, MC4R | (Tao, 2005) |
| Resistance to HIV-1 infection | CCR5 | (Reiche, Bonametti, Voltarelli, Morimoto, & Watanabe, 2007) |
V2R: Vasopressin Type-2 receptor; GnRHR: Gonadotropin-releasing hormone receptor; CaR: Calcium-sensing receptor; LHR: Lutropin (luteinizing hormone) receptor; FSHR: Follitropin (follicle-stimulating hormone) receptor; TSHR: Thyrotropin receptor; E-BR: Endothelin-B receptor; MC1R: Melanocortin-1 receptor; MC2R: Melanocortin-2 receptor [or adrenocorticotropin (ACTH) receptor]; MC3R: Melanocortin-3 receptor; MC4R: Melanocortin-4 receptor; CCR5: Chemokine receptor-5.
IV. MUTATIONS IN THE HUMAN GNRHR
A. Structural features of the GnRHR
The GnRHR is a GPCR that has already been a focus of drug development. This receptor belongs to the rhodopsin/β-adrenergic-like family of GPCRs (family A) (Millar et al., 2004; Ulloa-Aguirre & Conn, 1998). Its natural ligand is gonadotropin-releasing hormone, a decapeptide produced by the hypothalamus and released in synchronized pulses to the anterior pituitary to regulate pubertal development, sexual maturation and reproductive competence (P. M. Conn & Crowley, 1994; P. M. Conn, Huckle, Andrews, & McArdle, 1987; Knobil, 1974; Santen & Bardin, 1973; Ulloa-Aguirre & Timossi, 2000). The GnRHR is coupled to the trimeric Gq/11 protein localized in the cytoplasm and associated with the intracellular domains of the receptor, whose activation by agonists stimulates the effector enzyme phospholipase-Cβ, leading to phosphatidylinositol 4,5-biphosphate hydrolysis and formation of the second messengers inositol 1,4,5-triphosphate and diacylglycerol. The former messenger diffuses through the cytoplasm, promoting the release of intracellular calcium and the rapid release of both gonadotropins (P. M. Conn et al., 1987). These second messengers also activate different species of protein kinase C, which in concert with Ca2+ regulates gonadotropin secretion and mRNA expression of several genes, including gonadotropins subunit genes. Protein kinase C activates mitogen-activated protein kinases (MAPKs) cascade, which regulate a number of transcriptional responses to GnRH in a cell context-dependent manner (Naor, 2009).
Unlike other members of the family A of GPCRs, the human GnRHR exhibits several unique features including the reciprocal exchange of the conserved Asp and Asn residues in the transmembrane helix (TM)-2 and -7, the replacement of Thr with Ser in the Asp-Arg-Tyr motif located in the junction of the helix-3 and the IL-2, and the lack of the carboxyl-terminal extension (Ctail) into the cytosol (Millar et al., 2004) (Fig. 2). Fish, reptiles, birds, and the primate type II GnRHR (McArdle, Davidson, & Willars, 1999; Millar, 2003) contain a carboxyl extension, which is associated with differential physiological receptor regulation (Blomenrohr et al., 1999; Heding et al., 1998; Lin et al., 1998; McArdle et al., 1999); when the piscine sequence is added to the GnRHR, it dramatically increases PME levels of this receptor (Janovick, Ulloa-Aguirre, & Conn, 2003). Another feature of the human GnRHR is the presence of the amino acid residue Lys at position 191 in the extracellular loop (EL) 2, which is frequently Glu or Gly in non-primate mammals (Janovick et al., 2006; Ulloa-Aguirre, Janovick, Miranda, & Conn, 2006); in rat and mice GnRHRs an orthologous amino acid is absent, resulting in a structure that is one residue smaller and conferring the rodent GnRHR increased cell surface plasma membrane expression (Arora, Chung, & Catt, 1999). The mechanisms by which the presence of Lys191 limits the number of wild-type hGnRHR molecules exported from the ER to the PM involves primarily formation of the Cys14–Cys200 bridge, which stabilizes the human GnRHR in a conformation compatible with ER export (Janovick et al., 2006; Jardon-Valadez, Ulloa-Aguirre, & Pineiro, 2008; Ulloa-Aguirre et al., 2006). In fact, Cys14–Cys200 bridge formation and PME expression of the hGnRHR are both increased by deleting (primate-specific) Lys191; on the contrary, in rat or mouse GnRHRs that lack Lys191, the bridge is non-essential for receptor expression (Janovick et al., 2006). Nevertheless, the observation that addition of this particular residue to the rat sequence does not modify PM expression, indicates that other changes are required for its effects. A strategy based on identification of amino acids that both (a) co-evolved with Lys191 and (b) were thermodynamically unfavorable substitutions, identified motifs in multiple domains of the human GnRHR that control the destabilizing influence of Lys191 on the Cys14–Cys200 bridge, resulting in diminished PME (Fig. 3A) (Janovick et al., 2006; Ulloa-Aguirre et al., 2006). The data showed a novel and underappreciated means of posttranslational control by altering interaction with the QCS and provided a biochemical explanation of the basis of disease-causing mutations of this receptor.
Figure 2.
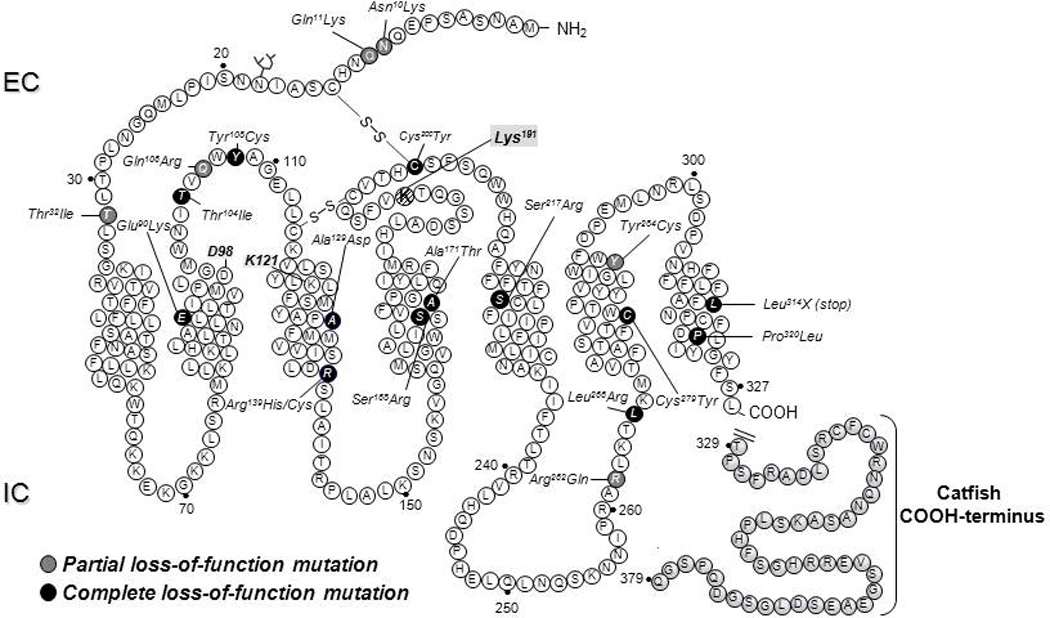
Location of the inactivating mutations in the human GnRHR. Black ovals are mutations that provoke complete loss-of-function of the receptor, whereas grey ovals are mutations that lead to partial loss-of-function. Also shown is the location of Lys191 at the second extracellular loop (hatched circle) and the sequence of the COOH-terminus of the catfish GnRHR (grey-shaded circles), which is a targetting sequence employed in vitro to promote trafficking of GnRHRs to the plasma membrane. EC, extracellular space; IC, intracellular space.
Figure 3.
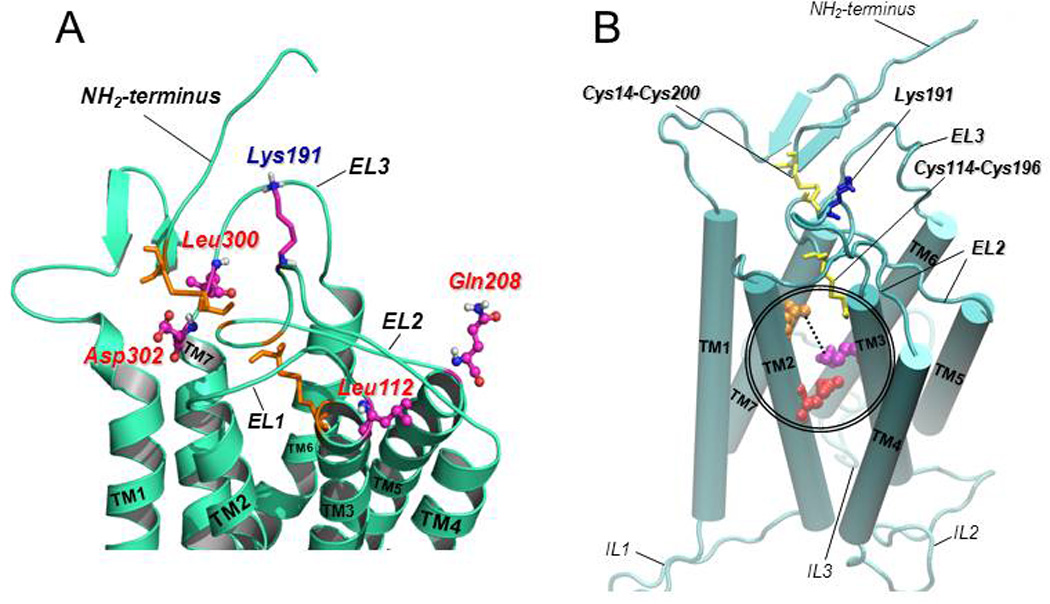
A: Predicted structure of the upper-third portion of the hGnRHR based on homology modeling with the structure of bovine rhodopsin (Jardon-Valadez et al., 2008). The coiled structures represent the antiparallel α-helices of transmembrane domains 1 to 7 connected by the extracellular loops (EL) of the receptor (curved cords). Disulfide bonds between Cys14 (at the NH2-terminus) and Cys200 (at the EL2), and between Cys114 (at the COOH-terminal end of the EL1) and Cys196 (at the EL2) are shown as orange sticks. The location of the amino acid residues that represent a motif of four non-contiguous residues at positions 112 (Leu, at the EL1), 208 (Gln, at the EL2), 300 (Leu, at the EL3), and 302 (Asp, at the EL3) that presumably control the destabilizing role of Lys191 (shown as purple, blue and grey sticks, at the EL2) on the association of the NH2-terminus and the EL2 and subsequent formation of the Cys14–Cys200 bridge are shown in colored circles and sticks. B: Predicted model of the hGnRHR showing the seven transmembrane helices (displayed as rods) connected by the extracellular (EL) and intracellular (IL) loops (Jardon-Valadez et al., 2008). Cys14–Cys200 and Cys114–Cys196 disulfide bridges are shown as yellow sticks; Lys 191 is represented by blue sticks. Glu90 (at the TM2; red spheres) forms a salt bridge with Lys121 (at the TM3; purple spheres) which is eliminated by the Glu90Lys mutation. Pharmacoperones act to stabilize the Glu90Lys mutant by bridging residues Asp98 (at the extracellular face of TM1; orange spheres) and Lys121 (discontinuous line).
B. Mutations in the human GnRHR
Loss of function mutations in the hGnRHR, result in inability to respond to GnRH (Beranova et al., 2001; de Roux & Milgrom, 2001; de Roux et al., 1997). Gonadotropin-releasing hormone receptor mutations can lead to partial or complete forms of HH, resulting in decreased or apulsatile gonadotropin release and reproductive failure (de Roux et al., 1999; Ulloa-Aguirre, Janovick, Leanos-Miranda et al., 2004). To date, 21 inactivating mutations (including two leading to missing of large sequences) in the GNRHR have been described as a cause of HH (Fig. 2). Seven homozygous and 12 heterozygous combinations of human GnRHR mutants are expressed by individuals exhibiting either partial or complete forms of HH (Beranova et al., 2001; Leanos-Miranda, Ulloa-Aguirre, Janovick, & Conn, 2005; Ulloa-Aguirre, Janovick, Leanos-Miranda et al., 2004). Although in vitro expression of a number of these mutated GnRHRs in heterologous systems has shown that these mutations appear to alter several functions of the molecule, including ligand binding, receptor activation or interaction with coupled effectors (Bedecarrats & Kaiser, 2007; Bedecarrats, Linher, & Kaiser, 2003; Meysing et al., 2004), protein misfolding and resultant misrouting is a mechanisms that, by itself, may lead to loss of function of the hGnRHR (Janovick, Maya-Nunez, & Conn, 2002; Leanos-Miranda, Janovick, & Conn, 2002; Maya-Nunez et al., 2002; Ulloa-Aguirre, Janovick, Leanos-Miranda et al., 2004). In fact, it has been shown that the majority (~90%) of the hGnRHR mutants whose function has been examined to date (19 mutants) are trafficking-defective receptors as disclosed by mutational studies and/or response to pharmacological chaperones (Maya-Nunez et al., 2002; Ulloa-Aguirre et al., 2003; Ulloa-Aguirre, Janovick, Leanos-Miranda et al., 2004). Because reproductive failure is not life-threatening, it is likely that many cases (particularly partial HH forms) go undiagnosed and, individual mutants, if severe in the phenotype, are not passed to progeny. Such ER-retained mutants frequently show a change in residue charge compared with the WT receptor (e.g. the Glu90Lys mutant), or gain (e.g. the Tyr108Cys mutant) or loss of either Cys (an amino acid known to form bridges associated with the formation of third order structure of proteins) (e.g. the Cys200Tyr mutant) or Pro (an amino acid associated with a forced turn in the protein sequence and frequently seen as the first residue of a helix, presumably due to its structural rigidity) (e.g. the Pro320Leu mutant) residues (Fig. 2).
Structural features, Cys and salt bridges, of the hGnRHR may explain the mechanism(s) whereby naturally-occurring mutations in this receptor lead to defective intracellular trafficking and HH. For some hGnRHR mutants causing HH, these mechanisms have been already dilucidated:
Cys200Tyr: This mutation prevents formation of the disulfide bridge Cys14–Cys200 required in the hGnRHR to pass the QCS (Ulloa-Aguirre et al., 2006). In this regard, mutants that lacked either the Cys14–Cys200 bridge, Lys191, or both have been examined. The markedly reduced expression and function of the naturally occurring Cys200Tyr mutant or of the laboratory-manufacutred mutants Cys14Ala or Cys14Ser mutants lacking the Cys14–Cys200 bridge, was restored closely to hGnRHR WT levels by pharmacoperones and/or by deleting Lys191 (Janovick et al., 2006; Jardon-Valadez et al., 2009; Leanos-Miranda et al., 2002). Further, deletion of Lys191 resulted in changes in the dynamic behavior of the mutants as disclosed by molecular dynamics simulations: the distance between the sulfur or oxygen-sulfur groups of Cys (or Ser)14 and Cys200 was shorter and more stable, and the conformation of the NH2-terminus and the EL2 exhibited less fluctuations than when Lys191 was present (Jardon-Valadez et al., 2009).
Ser168Lys and Ser217Lys: These substitutions are located in TMs 4 and 5, respectively, at the hGnRHR (Fig. 2) and they represent thermodynamically unfavored substitutions that twist the corresponding α-helices, moving the EL2 away from the NH2-terminal, preventing formation of the Cys14–Cys200 bridge (Janovick et al., 2006; Ulloa-Aguirre et al., 2006). The mutant proteins never pass the cellular QCS and both are completely refractory to rescue by genetic or pharmacologic approaches.
Glu90Lys: This was the first loss-of-function mutation in the hGnRHR in which misfolding was identified as the underlying defect (Maya-Nunez et al., 2002). The Glu→Lys substitution at this position prevents the formation of a Glu90-Lys121 salt bridge destabilizing the interaction between TM2 and TM3 (Fig. 3B) (Janovick et al., 2009; Jardon-Valadez et al., 2008) required to pass the QCS (Janovick et al., 2009). Consequently, the Glu90Lys mutant is retained in the ER (Brothers, Cornea, Janovick, & Conn, 2004). Interesting, pharmacoperone drugs rescue the Glu90Lys mutant by forming a surrogate TM2–TM3 bridge (see below) and, when removed, unveiled constitutive activity of the rescued receptor (Janovick & Conn, 2010; Janovick et al., 2009). Deletion of Lys191, which also rescue function of this mutant (Maya-Nunez et al., 2002), additionally produced constitutive activity. These findings indicate that by requiring the intact TM2–TM3 salt bridge for correct trafficking, the cell protects itself from plasma membrane expression of a constitutively active receptor. Collaterally, this suggests that the search for constitutively activated receptors should include misrouted mutants.
Tyr108Cys: This substitution provokes formation of an aberrant disulfide bridge between Cys108 and Cys200 (Maya-Nunez et al., 2011), provoking gross receptor distortion, which precludes its plasma membrane expression. Function of this mutant can be partially rescued by deleting Lys191 or by pharmacoperone treatment; complete rescue is possible only when both strategies are combined.
V. RESCUE OF MISFOLDED HGNRHR MUTANTS WITH PHARMACOPERONES
Understanding the structure and mechanism of GnRH action has already led to pharmaceutical development of useful drugs for the treatment of cancer and disorders of reproduction. Beyond this, however, the GnRHR-ligand system is a particularly good model to understand both protein routing and the mechanism of rescue by pharmacoperones. Among the reasons for these views are the following:
The GnRHR is one of the smallest GPCRs (328 amino acids in the human and most non-rodent mammals; 327 in rat and mouse sequences); it may be close to the “limit” size, containing only the bare essentials required for ligand binding and signal transduction. There are technical advantages for working with such small proteins, since these require fewer primers for synthesis and for sequencing than do larger GPCRs (typically twice the size of the GnRHR). In fact, over 15 years, we have successfully created and characterized a library of hundreds of useful naturally occurring or laboratory manufactured GnRHR mutants and epitope and fluorescently tagged chimeras that have been extremely useful in studying receptor routing (Brothers, Janovick, & Conn, 2003; Janovick et al., 2009; Jardon-Valadez et al., 2009; Ulloa-Aguirre, Janovick, Brothers et al., 2004). In addition, naturally-occurring mutants of the GnRHR system (see above) are frequently located in similar regions (i.e. associated with export motifs) as those reported for other GPCRs (Ulloa-Aguirre & Conn, 2009). The relatively small size of the GnRHR also presents fewer domains to consider in identification of important structural motifs and because the size of hydrophobic domains is relatively constant, the ratio of these to nonhydrophobic regions is relatively high in the GnRHR due to the short amino and carboxyl tails (Jardon-Valadez et al., 2008). As in the case of the V2R and rhodopsin, the relatively small size of the GnRHR has allowed us to understand a great deal of its structure, including the mechanism of action of several mutants (P. M. Conn et al., 2007) (see below). The size of these receptors, may explain why they are the most frequenly affected among the GPCRs superfamily by mutations leading to ER trapping and disease (Tan, Brady, Nickols, Wang, & Limbird, 2004).
The physiology of the system mediated by the GnRHR is well-characterized in many animal models and provides a basis for understanding processing differences now known to occur in different species (Knobil & Neill, 1994; Millar, Pawson, Morgan, Rissman, & Lu, 2008; Ulloa-Aguirre & Timossi, 2000). We have taken advantage of the large number of GnRHR sequences available (going back to the mating factors in yeast, and in flies, fish, reptiles, birds, pre-primate vertebrates and primates) and have been able to determine how changes in routing were impacted by sequence changes [i.e. natural mutations (P. M. Conn, Janovick, Brothers, & Knollman, 2006; P. M. Conn, Knollman, Brothers, & Janovick, 2006)]. Among primate receptors, for example, we identified specializations that make these receptors able to decode frequency-modulated as well as amplitude-modulated signals (Janovick, Brothers, Knollman, & Conn, 2007). We also noted structural changes among particular animals that appear to be explained by reproductive specializations (Janovick et al., 2006; Knollman, Janovick, Brothers, & Conn, 2005; Ulloa-Aguirre et al., 2006). The marsupial opossum and guinea pig, for example, are outliers in the GnRHR structure associated with birth from a pouch, rather than a vaginal birth, and a lengthened luteal phase, compared to other rodents (respectively). The hGnRHR, unlike the rat and mouse counterparts, appears “balanced” in its distribution between the PM and ER (P. M. Conn, Janovick et al., 2006; P. M. Conn, Knollman et al., 2006); in fact, about 50% of the WT hGnRHR (in cells transfected with the corresponding sequence) is retained in the ER and can be “rescued” by pharmacoperones. Although at first consideration this appears to be an inefficient use of newly synthesized protein, the strong and convergent evolutionary pressure for this suggests a regulatory advantage (P. M. Conn, Janovick et al., 2006; P. M. Conn, Knollman et al., 2006; Ulloa-Aguirre et al., 2006). This system offers the ability to examine the evolution of the QCS system since these receptors have been cloned from a wide range of animals [fish, birds, reptiles, many mammals and multiple primates (Janovick, Ulloa-Aguirre et al., 2003)].
A great deal of information is also available regarding the cellular mechanism of action of the GnRHR (Jennes, Ulloa-Aguirre, Janovick, & Conn, 2008; Knobil, 1974; Lim et al., 2009; Naor, 2009). In addition, we have available substantive information on the mechanism of misfolded ER (P. M. Conn, Janovick et al., 2006; P. M. Conn, Knollman et al., 2006) mutant interactions with pharmacoperones (P. M. Conn et al., 2007; Ulloa-Aguirre et al., 2003) and the molecular basis of the dominant-negative effect at the ER (P. M. Conn, Janovick et al., 2006; P. M. Conn, Knollman et al., 2006), access to multiple drug classes of pharmacoperones for the GnRHR and multiple drugs within each class (Janovick, Goulet et al., 2003) with sufficient quantities to enable in vivo studies. Other studies (Janovick, Brothers, Cornea et al., 2007) indicate that the mutant receptor that is already trapped in the ER can be freed by pharmacoperones; this observation increases the potential therapeutic reach of this approach since pharmacoperones do not need to be present at the precise moment of receptor synthesis.
The ability of different GnRHR peptidomimetics to rescue defective hGnRHR mutants causing HH has been extensively analyzed (Ashton, Sisco, Kieczykowski et al., 2001; Ashton, Sisco, Yang, Lo, Yudkovitz, Cheng et al., 2001; Ashton, Sisco, Yang, Lo, Yudkovitz, Gibbons et al., 2001; Janovick, Goulet et al., 2003; Janovick et al., 2009; Leanos-Miranda et al., 2002; Leanos-Miranda et al., 2005). The peptidomimetics assessed as potential pharmacoperones came from four different chemical classes: indoles, quinolones, thienopyrimidinediones, and erythromycin-derived macrolides (Fig. 4) which were originally developed as GnRH peptidomimetic antagonists (Ashton, Sisco, Kieczykowski et al., 2001; Ashton, Sisco, Yang, Lo, Yudkovitz, Cheng et al., 2001; Ashton, Sisco, Yang, Lo, Yudkovitz, Gibbons et al., 2001; Janovick, Goulet et al., 2003; Sasaki et al., 2003). These particular pharmacoperones were selected for study as potential pharmacoperones considering their predicted ability to permeate the cell membrane and specifically bind the GnRHR with a known affinity, rather than for their originally described actions as antagonists (P.M. Conn). In fact, rescue of misfolded GPCRs might also be achieved by agonists of the natural ligand (Leskela, Markkanen, Pietila, Tuusa, & Petaja-Repo, 2007).
Figure 4.
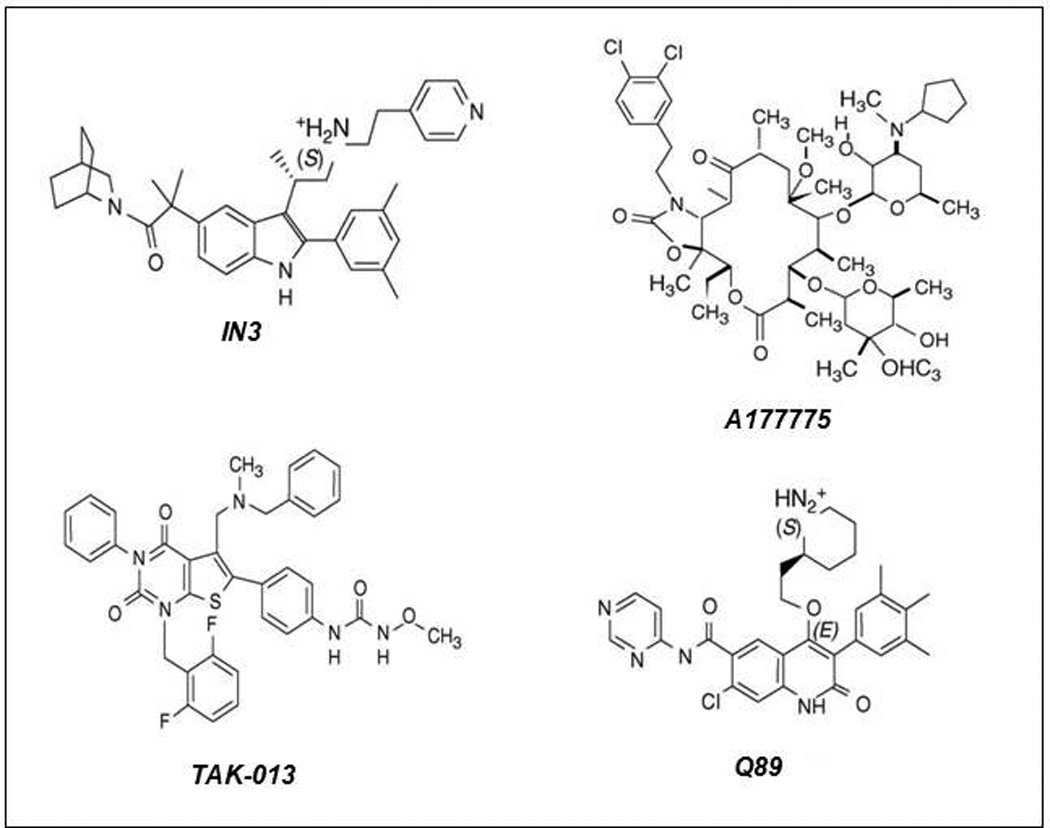
The structure of four representative pharmacoperones from different chemical clases. IN3: ((2S)-2-[5-[2-(2-azabicyclo[2.2.2]oct-2-yl)-1,1-dimethyl-2-oxoethyl]-2-(3,5-dimethylphenyl)-1H-indol-3-yl]- N-(2-pyridin-4-ylethyl)propan-1-amine (Merck and Company, Rahway, NJ, USA); Q89: (7-chloro-2-oxo-4-{2-[(2S)-piperidin-2-yl]ethoxy}-N-pyrimidin-4-yl-3-(3,4,5-trimethylphenyl)-1,2-dihydroquinoline-6-carboxamide) (Merck and Company); TAK-013: (N-{4-[5-{[benzyl(methyl)amino]methyl}-1-(2,6-difluorobenzyl)-2,4-dioxo-3-phenyl-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-N’-methoxyurea) (Takeda Chemical Industries, Ltd., Osaka 532–8686, Japan); A177775: A-177775.0 [3′-N-desmethyl-3′-N-cyclopentyl-11-deoxy-11-[carboxy-(3,4-dichlorophenylethylamino)]-6-O-methyl-erythromycin A 11,12-(cyclic carbamate)] (Abbott Laboratories, Abbott Park, IL, USA)
The first pharmacoperone tested (In3, Fig. 4) belongs to the indole class and provided the first proof of principle for rescue misfolded human GnRHRs (P. M. Conn, Leanos-Miranda, & Janovick, 2002; Janovick et al., 2002; Leanos-Miranda et al., 2002). Further studies then examined the efficacy of chemically distinct drugs (indoles, quinolones, and erythromycin-derived macrolides) as pharmacoperons for a palette of misfolded mutant GnRHRs (Janovick, Goulet et al., 2003). These studies demonstrated that all but three [S168R, S217R, and L314X(stop)] of the 17 hGnRHR mutants tested were partially or completely rescued with pharmacoperones (P. M. Conn & Janovick, 2009a, 2009b; P. M. Conn et al., 2002; Janovick, Goulet et al., 2003; Leanos-Miranda et al., 2002). The efficacy of these drugs (measured by the ability of a fixed dose of the pharmacoperone to rescue receptor function in terms of inositol phosphate production in response to agonist) was proportional to the binding affinity of the pharmacoperone for the Wt receptor and, in general, there was a lack of rescue specificity for the different drugs (i.e. all effective agents rescued virtually the same mutants) (Janovick, Goulet et al., 2003), an expected finding considering that these molecules were originally designed as GnRH peptidomimetics and thus would presumably compete with the natural ligand for receptor occupancy (Finch, Sedgley, Armstrong, Caunt, & McArdle) (see below). Accordingly, mutants that rescued poorly with one pharmacoperone class, rescued poorly with all (Janovick, Goulet et al., 2003). All peptidomimetics studied with an IC50 value (for Wt GnRHR) ≤ 2.3 nM had measurable efficacy in rescuing GnRHR mutants, and within a single chemical class this ability correlated to these IC50 values. Among the most effective pharmacoperones tested, the indole In30 seems to be the most potent based on the IC50 value (0.2 nM) for the Wt GnRHR, followed by the quinolone Q89 (IC50 0.3 nM), In3 (IC50 0.6 nM), and the erythromycin macrolides A177775 (IC50 17.7 nM) and A2222509 (IC50 20 nM) (Janovick, Goulet et al., 2003). As mentioned above, the S168R and S217R GnRHRs are mutants in which the thermodynamic changes leading to receptor distortion are too severe to allow stabilization by pharmacoperones (Ulloa-Aguirre et al., 2006). Accordingly, even though these two mutants are not rescued by any of these compounds, their failure to route correctly is attributable to severe misfolding, and probably not to an intrinsic inability to potentially participate in particular receptor functions.
VI. MECHANISM OF ACTION OF PHARMACOPERONES
Desirable characteristics of molecules that could potentially function as pharmacoperones for misfolded proteins include: i. Ability to reach physiological concentrations; ii. Capacity to permeate the cell surface membrane; iii. Ability to localize and intervene at the ER and/or post-ER compartments where the misfolded protein is synthesized or retained; iv. Ability to remain undegraded long enough to stabilize the target mutant; v. Specificity for the target protein; and vi. Ability to bind reversibly to the target mutant so that they may dissociate from the target molecule after its localization at the correct cellular destination (e.g. the plasma membrane) or alternatively not to compete with the natural ligand binding site. Pharmacoperones can correct folding of defective proteins allowing that the mutant could escape the ER QCS and traffic to the PM or interfering with its aggregation or degradation. The mechanism(s) by which pharmacoperones stabilize and rescue PM expression of the target receptor is still speculative. In fact, current information is mostly based on theoretical predictions of protein structure and drug interactions (Janovick et al., 2009; Nowak, Cuny, Choi, Lansbury, & Ray, 2010; Wuller et al., 2004). The mechanisms proposed to explain the ability of pharmacoperones to stabilize misfolded proteins include: a. Binding and enhancement of the stability of the native or native-like state of the target protein for which they have higher affinity that for intermediate, immature forms (i.e. non-native structures) and b. Binding to the less folded, non-native folding intermediates and act as a scaffold for subsequent folding, increasing the rate at which these intermediates are converted to the native form. This would prevent the protein from being recognized by the ER QCS as defective, allowing it to escape from degradation and promoting its transport to the Golgi apparatus for further processing (Arakawa et al., 2006; Noorwez et al., 2004).
Since pharmacoperones are often, but not exclusively, peptidomimetic antagonists, they must be removed after rescue and insertion into the PM for receptor function, so that the rescued PM-expressed receptor can be activated by its cognate ligand. Accommodating the need for exposing the rescued mutant to pharmacoperones in in vivo conditions would probably necessitate pulsatile administration of the pharmacoperone. A combined strategy employing pharmacological (blockage of protein synthesis or intracellular transport followed by exposure to pharmacoperones), biochemical and morphological approaches was used to determine whether pharmacoperones need to be present at the time of synthesis to function or whether a previously misfolded/retained protein molecule could be stabilized in a correct conformation by exposure to pharmacoperones after synthesis (Janovick, Brothers, Cornea et al., 2007). Studies were performed in stably and transiently transfected cells using 12 mutants and 10 pharmacoperones selected from different chemical classes (indoles, quinolones and erythromycin macrolides). The data indicated that previously synthesized mutant GnRHRs that are misfolded and retained by the QCS, are still sensitive to rescue by pharmacoperones even when these are not present at the time and after the synthesis is complete. This observation strongly suggests that whether the misfolded protein is being synthesized at the time of drug administration need not to be considered in determining the pattern of pharmacoperone administration in vivo. Nevertheless, considering the possibility that the half-life of ER-retained mutants can be short (Robben, Knoers, & Deen, 2005) or that some mutants may be prone to form stable aggregates (Illing, Rajan, Bence, & Kopito, 2002; Saliba et al., 2002), which may block rescue by pharmacoperones, these would need to be present for as protracted a period as possible, whenever optimal rescue is desired.
As noted before, the exact mechanisms whereby pharmacoperones promote correct folding are still uncertain. In the case of naturally occurring hGnRHR mutants, we have shown that one particular mutant, the Glu90Lys mutant is completely rescued by genetic or pharmacological approaches (Leanos-Miranda et al., 2002; Maya-Nunez et al., 2002). Using site directed mutagenesis, confocal microscopy, ligand docking, and computer modeling, we have shown that a number of different chemical classes of pharmacoperones [indoles and quinolones, and the distinctly different erythromycin macrolide, A177775, and TAK-013 (a thieno[2,3-b]pyrimidine-2,4-dione; Fig. 4)] act to stabilize the Glu90Lys mutant by bridging residues Asp98 (at the extracellular face of TM1) and Lys121 (at the TM3) (P. M. Conn & Janovick, 2009b; Janovick et al., 2009; Jardon-Valadez et al., 2008; Millar et al., 2004; Soderhall, Polymeropoulos, Paulini, Gunther, & Kuhne, 2005) (Fig. 3B). This pharmacoperone-mediated bridge serves as a surrogate or supplementary bond for the naturally occurring and highly conserved Glu90-Lys121 salt bridge, leading to stabilization between TM2 and TM3 (Janovick et al., 2009; Jardon-Valadez et al., 2008), which apparently represents a structural requirement for passage of the GnRHR through the cellular QCS and to the plasma membrane. Since GnRH includes as contact points Asp98 and Lys121, it was not surprising that competitors of GnRH may interact at or near the ligand binding site, which resides in the lateral plane of the PM, a region bearing a high percentage of hydrophobic residues (Jardon-Valadez et al., 2008). In fact, analysis of the linear sequences of both Glu90 and Lys121 show that they are hydrophobic regions with a modest number of ionic or polar groups: for Glu90, for example, LLE90TLIVMPLD98 and around Lys121 is VLSYLK121LFSM. The identification of this conserved ionic site strongly suggests that the antagonists tested to date as pharmacoperones to rescue misfolded hGnRHRs were all chosen on the basis of this preferential ionpair and/or polar interaction with the charged residues. The finding that all pharmacoperones tested to date rescue most of the hGnRHR mutants, no matter the distribution of the mutations throughout the sequence of the receptor (Fig. 2), indicates that the Glu90-Lys121 bridge represents an additional core that, once stabilized, yields a structure that overcomes the scrutiny of the QCS. For the hGnRHR this core might stabilize the orientation of, and relation between TMs 2 and 3, as the Cys14–Cys200 bridge does with the second extracellular loop and the amino-terminus, and indirectly the TMs 4 and 5.
VII. THE DOMINANT-NEGATIVE EFFECT OF HGNRHR MUTANTS AND RECEPTOR RESCUE
Although the GnRHR was one of the first GPCRs shown to oligomerize upon agonist activation at the PM as part of normal receptor function (P. M. Conn, Rogers, Stewart, Niedel, & Sheffield, 1982; Cornea, Janovick, Maya-Nunez, & Conn, 2001), the finding of oligomerization in the ER-Golgi complex at the time of protein synthesis and routing to the cell surface has emerged as a new and important concept for GPCRs function (Angers, Salahpour, & Bouvier, 2002; Bouvier, 2001; Bulenger, Marullo, & Bouvier, 2005; Milligan, 2007). Constitutive oligomerization at the ER has been demonstrated for a number of GPCRs, including the receptors for GABAB (Kaupmann et al., 1998; Margeta-Mitrovic, 2002; Margeta-Mitrovic, Jan, & Jan, 2000), melatonin (Ayoub et al., 2002), dopamine D2 (Guo, Shi, & Javitch, 2003), vasopressin (Terrillon, Barberis, & Bouvier, 2004) and serotonin (Herrick-Davis, Grinde, & Mazurkiewicz, 2004) as well as for the δ-opioid receptor (McVey et al., 2001), the β2-adrenergic receptor (Angers et al., 2000; Mercier, Salahpour, Angers, Breit, & Bouvier, 2002; Salahpour et al., 2004) and the follicle-stimulating hormone receptor (Thomas et al., 2007). Among the functions of homo- and hetero-oligomerization at the ER include effective quality control of protein folding prior to export to the PM (Angers et al., 2002; Bouvier, 2001; Bulenger et al., 2005), which could be effected through hiding exposed hydrophobic surfaces or retention sequences that would otherwise signal an improperly folded receptor and be recognized as a misfolded structure by the QCS of the cell. For example, inhibiting homodimerization of the β2-adrenergic receptor, leads to ER retention and perturb cell surface targeting (Salahpour et al., 2004). In the case of the GABAB receptor, heterodimerization between GABABR1 and GABABR2 is apparently obligatory for cell surface expression of a functional receptor (Kaupmann et al., 1998; Margeta-Mitrovic et al., 2000; White et al., 1998); formation of a coilcoil domain between the Ctail of GABAB receptor subtypes masks an ER retention signal located in the Ctail of the GABABR1 thereby promoting the ER export of the heterodimer to the PM (Margeta-Mitrovic et al., 2000). A similar role in receptor outward trafficking has been shown for the α1D- and α1B-adrenergic receptors (Hague, Uberti, Chen, Hall, & Minneman, 2004) and the β2-adrenergic receptor (Uberti, Hague, Oller, Minneman, & Hall, 2005). Although intracellular association of GPCRs as homoor hetero-dimers could lead, in principle, to cell surface targeting, it also may provoke intracellular retention of the complex (a dominant negative effect) (Benkirane, Jin, Chun, Koup, & Jeang, 1997; Brothers et al., 2004; Le Gouill et al., 1999; Zarinan et al., 2010; Zhu & Wess, 1998). In fact it has been extensively demonstrated that misfolded mutants of several GPCRs interfere with the cell surface expression of their corresponding WT counterparts through their association in the ER and misrouting of the resulting complex (Brothers et al., 2004; Gehret, Bajaj, Naider, & Dumont, 2006; Lee et al., 2000; Zarinan et al., 2010; Zhu & Wess, 1998).
In the case of the hGnRHR, evidence has been provided that non-functional misfolded hGnRHRs inhibit expression of the WT receptor (Knollman et al., 2005; Leanos-Miranda, Ulloa-Aguirre, Ji, Janovick, & Conn, 2003). The inhibition varies depending on the particular hGnRHRs coexpressed and the ratio of hGnRHR mutant to WT hGnRHR cDNA co-transfected (Leanos-Miranda et al., 2003). Confocal microscopy of fluorescently labeled WT hGnRHR has shown that the dominant negative effect of misfolded mutants results from WT receptor trapping in the ER by mislocalized mutants (Brothers et al., 2004). Pharmacologic chaperones restore correct folding, rescuing mutants and WT receptor from these oligomers. Studies on a palette of WT and mutant rodent and human GnRHRs have allowed identification of a critical residue involved in altered routing and the transdominant inhibitory effect of mutant hGnRHRs (Knollman et al., 2005). Rat WT GnRHR retains the ability to oligomerize (since human and mouse mutants exert a dominant negative effect effect on rat WT sequence) but, unlike human or mouse receptors, escapes the dominant negative effect provoked by rat GnRHR mutants because these mutants route to the plasma membrane with higher efficiency than mouse or human mutants. The difference in both routing and the dominant negative effect appears mediated primarily by the presence of Ser216 in the rat GnRHR. In the hGnRHR, the homologous amino acid is also Ser (in position 217) and the efficiency in routing is mitigated by the primate-unique insertion of Lys191 that, alone, dramatically decreases routing of the receptor. In fact, the dominant-negative effect of naturally occurring hGnRHR mutants does not occur with genetically modified receptors bearing deletion of Lys191 or a carboxyl-terminus targeting sequence (Janovick, Ulloa-Aguirre et al., 2003). Thus, the dominant negative effect of human receptor mutants lies in the presence of Lys191 (along with Ser217), which greatly increases the susceptibility of disturbing the conformation of this receptor with single charge changes in the primary sequence, and makes the hGnRHR more susceptible to defective trafficking by disease-related point mutations.
Since the transdominant inhibitory effect of mutants associated with HH may potentially worsen the disease in heterozygous individuals, the function of mutant hGnRHR pairs associated with compound heterozygous patients showing complete or partial forms of HH and the response to pharmacoperone rescue has been investigated (Leanos-Miranda et al., 2005). Coexpression of each pair of mutants in COS-7 cells resulted in either an active predominant effect [i.e. the combination of mutants yielded similar responses to agonist stimulation as did the more active of the two mutants transfected individually (e.g. Gln106Arg/Leu266Arg and Ala171Thr/Gln106Arg mutant hGnRHR pairs)], an additive effect (e.g. Arg262Gln/Gln106Arg, and Asn10Lys/Gln106Arg mutant GnRHR pairs), or a dominant negative effect [e.g. Leu314X(stop)/Gln106Arg, Gln106Arg+Ser217Arg/Arg262Gln, and Leu314X(stop)/Arg262Gln mutant GnRHR pairs]. For all combinations, addition of a pharmacoperone increased both agonist binding and effector coupling. Although effective, the net ability to rescue with a pharmacoperone was unpredictable because responses could be either similar or higher or lower than those exhibited by the less affected mutant. Thus, depending on the genotype, partial or full restoration of receptor function in response to pharmacological chaperones may be achievable goals in patients with loss-of-function mutations in the hGnRHR gene. Pharmacoperones might either correct folding or provoke refolding of the mutant receptors, allowing the possibility that one or both of the mutants may escape the QCS and traffic to the PM or interfere with aggregation and degradation of the mutant receptors. The observations that a synthetic α2-adrenergic receptor helix 6-derived peptide inhibited dimerization of this receptor (Hebert et al., 1996), that co-transfection of WT follicle-stimulating hormone receptor fragments specifically rescued WT FSHR expression from the transdominant inhibition by mutant receptors (Zarinan et al., 2010), and that aggregation of secretory proteins may be inhibited by cell-permeant synthetic ligands (Rivera et al., 2000) further support the latter possibility. These data concurrently suggest that in vivo use of such strategies could be highly effective in overriding the dominant negative effect of a mutation on the WT receptor, as well as in rescue of the mutant itself.
VIII. Conclusion
In this chapter we review the conceptual and developmental history of pharmacoperone drugs. Among the reasons that mutations result in disease is that conformationally-defective proteins are misrouted and do not reach their site of physiological action. Some mutants exacerbate mutational disease by binding nascent WT proteins and causing them, also, to become misrouted as part of a mutant-WT complex. Pharmacoperones (from pharmacological chaperones) are small, target specific, cell permeating molecules that bind to misfolded/misrouted mutants allowing them to refold and to sub serve the requirements of the cellular quality control system and route correctly. When pharmacoperones are used to rescue mutants and return them to their site of physiological action, they frequently regain activity. For this reason pharmacoperones offer an alternative to correction of mutational defects by genetic engineering and may prove to be more facile and advantageous.
A number of compounds obtained from high-throughput screening strategies currently are under study for their potential application as pharmacoperones to treat diseases caused by misfolded GPCRs, including HH due to point mutations in the human GnRHR (Noorwez et al., 2008; Fan et al., 2009; Ostrov, 2009; Conn, 2010), as well as to improve function of the WT receptor by increasing the number of available functional membrane receptors. Further studies employing combined strategies that include mutagenesis, functional studies, and computational modeling are warranted to define for novel pharmacoperones (agonists, antagonists, partial agonists, inverse agonists, and allosteric modulators) that may stabilize the correctly routed form of the receptor protein allowing the endogenous agonist to interact with the receptor without the necessity of removing the drug after rescue.
Acknowledgments
This work was supported by National Institutes of Health Grants DK85040, RR030229, TW/HD-00668 and P51RR000163 (PMC), and grant 86881 from CONACyT, Mexico (AU). Alfredo Ulloa-Aguirre is a recipient of a Research Career Development Award from the Fundación IMSS, México.
Non-standard abbreviations
- Ctail
carboxyl terminal extension found in piscine GnRHR
- ER
endoplasmic reticulum
- GnRH
gonadotropin-releasing hormone
- GnRHR
gonadotropin-releasing hormone receptor
- GPCR
G protein-coupled receptor
- HH
hypogonadotropin hypogonadism
- PM
plasma membrane
- QCS
quality control system
- TM
transmembrane helix
- WT
wild-type
Footnotes
Conflict of Interest Statement
The authors have no conflicts of interest to declare.
References
- Aittomaki K, Lucena JL, Pakarinen P, Sistonen P, Tapanainen J, Gromoll J, et al. Mutation in the follicle-stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure. Cell. 1995;82(6):959–968. doi: 10.1016/0092-8674(95)90275-9. [DOI] [PubMed] [Google Scholar]
- Amaral MD. Therapy through chaperones: sense or antisense? Cystic fibrosis as a model disease. J Inherit Metab Dis. 2006;29(2–3):477–487. doi: 10.1007/s10545-006-0251-x. [DOI] [PubMed] [Google Scholar]
- Amiel J, Sproat-Emison E, Garcia-Barcelo M, Lantieri F, Burzynski G, Borrego S, et al. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet. 2008;45(1):1–14. doi: 10.1136/jmg.2007.053959. [DOI] [PubMed] [Google Scholar]
- Andersson H, D'Antona AM, Kendall DA, Von Heijne G, Chin CN. Membrane assembly of the cannabinoid receptor 1: impact of a long N-terminal tail. Mol Pharmacol. 2003;64(3):570–577. doi: 10.1124/mol.64.3.570. [DOI] [PubMed] [Google Scholar]
- Angelotti T, Daunt D, Shcherbakova OG, Kobilka B, Hurt CM. Regulation of G-protein coupled receptor traffic by an evolutionary conserved hydrophobic signal. Traffic. 2010;11(4):560–578. doi: 10.1111/j.1600-0854.2010.01033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angers S, Salahpour A, Bouvier M. Dimerization: an emerging concept for G protein-coupled receptor ontogeny and function. Annu Rev Pharmacol Toxicol. 2002;42:409–435. doi: 10.1146/annurev.pharmtox.42.091701.082314. [DOI] [PubMed] [Google Scholar]
- Angers S, Salahpour A, Joly E, Hilairet S, Chelsky D, Dennis M, et al. Detection of beta 2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET) Proc Natl Acad Sci U S A. 2000;97(7):3684–3689. doi: 10.1073/pnas.060590697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa T, Ejima D, Kita Y, Tsumoto K. Small molecule pharmacological chaperones: From thermodynamic stabilization to pharmaceutical drugs. Biochim Biophys Acta. 2006;1764(11):1677–1687. doi: 10.1016/j.bbapap.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Arora KK, Chung HO, Catt KJ. Influence of a species-specific extracellular amino acid on expression and function of the human gonadotropin-releasing hormone receptor. Mol Endocrinol. 1999;13(6):890–896. doi: 10.1210/mend.13.6.0291. [DOI] [PubMed] [Google Scholar]
- Ashton WT, Sisco RM, Kieczykowski GR, Yang YT, Yudkovitz JB, Cui J, et al. Orally bioavailable, indole-based nonpeptide GnRH receptor antagonists with high potency and functional activity. Bioorg Med Chem Lett. 2001;11(19):2597–2602. doi: 10.1016/s0960-894x(01)00512-1. [DOI] [PubMed] [Google Scholar]
- Ashton WT, Sisco RM, Yang YT, Lo JL, Yudkovitz JB, Cheng K, et al. Substituted indole-5-carboxamides and -acetamides as potent nonpeptide GnRH receptor antagonists. Bioorg Med Chem Lett. 2001;11(13):1723–1726. doi: 10.1016/s0960-894x(01)00274-8. [DOI] [PubMed] [Google Scholar]
- Ashton WT, Sisco RM, Yang YT, Lo JL, Yudkovitz JB, Gibbons PH, et al. Potent nonpeptide GnRH receptor antagonists derived from substituted indole-5-carboxamides and - acetamides bearing a pyridine side-chain terminus. Bioorg Med Chem Lett. 2001;11(13):1727–1731. doi: 10.1016/s0960-894x(01)00275-x. [DOI] [PubMed] [Google Scholar]
- Ayoub MA, Couturier C, Lucas-Meunier E, Angers S, Fossier P, Bouvier M, et al. Monitoring of ligand-independent dimerization and ligand-induced conformational changes of melatonin receptors in living cells by bioluminescence resonance energy transfer. J Biol Chem. 2002;277(24):21522–21528. doi: 10.1074/jbc.M200729200. [DOI] [PubMed] [Google Scholar]
- Beaumont KA, Newton RA, Smit DJ, Leonard JH, Stow JL, Sturm RA. Altered cell surface expression of human MC1R variant receptor alleles associated with red hair and skin cancer risk. Hum Mol Genet. 2005;14(15):2145–2154. doi: 10.1093/hmg/ddi219. [DOI] [PubMed] [Google Scholar]
- Beaumont KA, Shekar SN, Newton RA, James MR, Stow JL, Duffy DL, et al. Receptor function, dominant negative activity and phenotype correlations for MC1R variant alleles. Hum Mol Genet. 2007;16(18):2249–2260. doi: 10.1093/hmg/ddm177. [DOI] [PubMed] [Google Scholar]
- Bedecarrats GY, Kaiser UB. Mutations in the human gonadotropin-releasing hormone receptor: insights into receptor biology and function. Semin Reprod Med. 2007;25(5):368–378. doi: 10.1055/s-2007-984743. [DOI] [PubMed] [Google Scholar]
- Bedecarrats GY, Linher KD, Kaiser UB. Two common naturally occurring mutations in the human gonadotropin-releasing hormone (GnRH) receptor have differential effects on gonadotropin gene expression and on GnRH-mediated signal transduction. J Clin Endocrinol Metab. 2003;88(2):834–843. doi: 10.1210/jc.2002-020806. [DOI] [PubMed] [Google Scholar]
- Benkirane M, Jin DY, Chun RF, Koup RA, Jeang KT. Mechanism of transdominant inhibition of CCR5-mediated HIV-1 infection by ccr5delta32. J Biol Chem. 1997;272(49):30603–30606. doi: 10.1074/jbc.272.49.30603. [DOI] [PubMed] [Google Scholar]
- Beranova M, Oliveira LM, Bedecarrats GY, Schipani E, Vallejo M, Ammini AC, et al. Prevalence, phenotypic spectrum, and modes of inheritance of gonadotropin-releasing hormone receptor mutations in idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2001;86(4):1580–1588. doi: 10.1210/jcem.86.4.7395. [DOI] [PubMed] [Google Scholar]
- Bernier V, Bichet DG, Bouvier M. Pharmacological chaperone action on G-protein-coupled receptors. Curr Opin Pharmacol. 2004;4(5):528–533. doi: 10.1016/j.coph.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Bernier V, Lagace M, Bichet DG, Bouvier M. Pharmacological chaperones: potential treatment for conformational diseases. Trends Endocrinol Metab. 2004;15(5):222–228. doi: 10.1016/j.tem.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Bernier V, Morello JP, Zarruk A, Debrand N, Salahpour A, Lonergan M, et al. Pharmacologic chaperones as a potential treatment for X-linked nephrogenic diabetes insipidus. J Am Soc Nephrol. 2006;17(1):232–243. doi: 10.1681/ASN.2005080854. [DOI] [PubMed] [Google Scholar]
- Bichet DG. Nephrogenic diabetes insipidus. Nephrol Ther. 2006;2(6):387–404. doi: 10.1016/j.nephro.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Biebermann H, Schoneberg T, Krude H, Schultz G, Gudermann T, Gruters A. Mutations of the human thyrotropin receptor gene causing thyroid hypoplasia and persistent congenital hypothyroidism. J Clin Endocrinol Metab. 1997;82(10):3471–3480. doi: 10.1210/jcem.82.10.4286. [DOI] [PubMed] [Google Scholar]
- Blomenrohr M, Heding A, Sellar R, Leurs R, Bogerd J, Eidne KA, et al. Pivotal role for the cytoplasmic carboxyl-terminal tail of a nonmammalian gonadotropin-releasing hormone receptor in cell surface expression, ligand binding, and receptor phosphorylation and internalization. Mol Pharmacol. 1999;56(6):1229–1237. doi: 10.1124/mol.56.6.1229. [DOI] [PubMed] [Google Scholar]
- Bouvier M. Oligomerization of G-protein-coupled transmitter receptors. Nat Rev Neurosci. 2001;2(4):274–286. doi: 10.1038/35067575. [DOI] [PubMed] [Google Scholar]
- Broadley SA, Hartl FU. The role of molecular chaperones in human misfolding diseases. FEBS Lett. 2009;583(16):2647–2653. doi: 10.1016/j.febslet.2009.04.029. [DOI] [PubMed] [Google Scholar]
- Brooks DA. Introduction: molecular chaperones of the ER: their role in protein folding and genetic disease. Semin Cell Dev Biol. 1999;10(5):441–442. doi: 10.1006/scdb.1999.0314. [DOI] [PubMed] [Google Scholar]
- Brothers SP, Cornea A, Janovick JA, Conn PM. Human loss-of-function gonadotropin-releasing hormone receptor mutants retain wild-type receptors in the endoplasmic reticulum: molecular basis of the dominant-negative effect. Mol Endocrinol. 2004;18(7):1787–1797. doi: 10.1210/me.2004-0091. [DOI] [PubMed] [Google Scholar]
- Brothers SP, Janovick JA, Conn PM. Unexpected effects of epitope and chimeric tags on gonadotropin-releasing hormone receptors: implications for understanding the molecular etiology of hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2003;88(12):6107–6112. doi: 10.1210/jc.2003-031047. [DOI] [PubMed] [Google Scholar]
- Brothers SP, Janovick JA, Conn PM. Calnexin regulated gonadotropin-releasing hormone receptor plasma membrane expression. J Mol Endocrinol. 2006;37(3):479–488. doi: 10.1677/jme.1.02142. [DOI] [PubMed] [Google Scholar]
- Brown CR, Hong-Brown LQ, Welch WJ. Correcting temperature-sensitive protein folding defects. J Clin Invest. 1997a;99(6):1432–1444. doi: 10.1172/JCI119302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CR, Hong-Brown LQ, Welch WJ. Strategies for correcting the delta F508 CFTR protein-folding defect. J Bioenerg Biomembr. 1997b;29(5):491–502. doi: 10.1023/a:1022491124939. [DOI] [PubMed] [Google Scholar]
- Bulenger S, Marullo S, Bouvier M. Emerging role of homo- and heterodimerization in G-protein-coupled receptor biosynthesis and maturation. Trends Pharmacol Sci. 2005;26(3):131–137. doi: 10.1016/j.tips.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Calebiro D, de Filippis T, Lucchi S, Covino C, Panigone S, Beck-Peccoz P, et al. Intracellular entrapment of wild-type TSH receptor by oligomerization with mutants linked to dominant TSH resistance. Hum Mol Genet. 2005;14(20):2991–3002. doi: 10.1093/hmg/ddi329. [DOI] [PubMed] [Google Scholar]
- Castro-Fernandez C, Maya-Nunez G, Conn PM. Beyond the signal sequence: protein routing in health and disease. Endocr Rev. 2005;26(4):479–503. doi: 10.1210/er.2004-0010. [DOI] [PubMed] [Google Scholar]
- Clark AJ, Metherell LA, Cheetham ME, Huebner A. Inherited ACTH insensitivity illuminates the mechanisms of ACTH action. Trends Endocrinol Metab. 2005;16(10):451–457. doi: 10.1016/j.tem.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Clark PL. Protein folding in the cell: reshaping the folding funnel. Trends Biochem Sci. 2004;29(10):527–534. doi: 10.1016/j.tibs.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8(1):41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PM. Recue of gonadotropin hormone receptor mutants. 7,695,917. US. 2010
- Conn PM, Crowley WF., Jr Gonadotropin-releasing hormone and its analogs. Annu Rev Med. 1994;45:391–405. doi: 10.1146/annurev.med.45.1.391. [DOI] [PubMed] [Google Scholar]
- Conn PM, Huckle WR, Andrews WV, McArdle CA. The molecular mechanism of action of gonadotropin releasing hormone (GnRH) in the pituitary. Recent Prog Horm Res. 1987;43:29–68. doi: 10.1016/b978-0-12-571143-2.50007-1. [DOI] [PubMed] [Google Scholar]
- Conn PM, Janovick JA. Drug development and the cellular quality control system. Trends Pharmacol Sci. 2009a;30(5):228–233. doi: 10.1016/j.tips.2009.02.002. [DOI] [PubMed] [Google Scholar]
- Conn PM, Janovick JA. Trafficking and quality control of the gonadotropin releasing hormone receptor in health and disease. Mol Cell Endocrinol. 2009b;299(2):137–145. doi: 10.1016/j.mce.2008.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PM, Janovick JA, Brothers SP, Knollman PE. 'Effective inefficiency': cellular control of protein trafficking as a mechanism of post-translational regulation. J Endocrinol. 2006;190(1):13–16. doi: 10.1677/joe.1.06771. [DOI] [PubMed] [Google Scholar]
- Conn PM, Knollman PE, Brothers SP, Janovick JA. Protein folding as posttranslational regulation: evolution of a mechanism for controlled plasma membrane expression of a G protein-coupled receptor. Mol Endocrinol. 2006;20(12):3035–3041. doi: 10.1210/me.2006-0066. [DOI] [PubMed] [Google Scholar]
- Conn PM, Leanos-Miranda A, Janovick JA. Protein origami: therapeutic rescue of misfolded gene products. Mol Interv. 2002;2(5):308–316. doi: 10.1124/mi.2.5.308. [DOI] [PubMed] [Google Scholar]
- Conn PM, Rogers DC, Stewart JM, Niedel J, Sheffield T. Conversion of a gonadotropin-releasing hormone antagonist to an agonist. Nature. 1982;296(5858):653–655. doi: 10.1038/296653a0. [DOI] [PubMed] [Google Scholar]
- Conn PM, Ulloa-Aguirre A. Trafficking of G-protein-coupled receptors to the plasma membrane: insights for pharmacoperone drugs. Trends Endocrinol Metab. 2010;21(3):190–197. doi: 10.1016/j.tem.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PM, Ulloa-Aguirre A, Ito J, Janovick JA. G protein-coupled receptor trafficking in health and disease: lessons learned to prepare for therapeutic mutant rescue in vivo. Pharmacol Rev. 2007;59(3):225–250. doi: 10.1124/pr.59.3.2. [DOI] [PubMed] [Google Scholar]
- Cook LB, Zhu CC, Hinkle PM. Thyrotropin-releasing hormone receptor processing: role of ubiquitination and proteasomal degradation. Mol Endocrinol. 2003;17(9):1777–1791. doi: 10.1210/me.2003-0073. [DOI] [PubMed] [Google Scholar]
- Cornea A, Janovick JA, Maya-Nunez G, Conn PM. Gonadotropin-releasing hormone receptor microaggregation. Rate monitored by fluorescence resonance energy transfer. J Biol Chem. 2001;276(3):2153–2158. doi: 10.1074/jbc.M007850200. [DOI] [PubMed] [Google Scholar]
- D'Souza-Li L, Yang B, Canaff L, Bai M, Hanley DA, Bastepe M, et al. Identification and functional characterization of novel calcium-sensing receptor mutations in familial hypocalciuric hypercalcemia and autosomal dominant hypocalcemia. J Clin Endocrinol Metab. 2002;87(3):1309–1318. doi: 10.1210/jcem.87.3.8280. [DOI] [PubMed] [Google Scholar]
- de Roux N, Milgrom E. Inherited disorders of GnRH and gonadotropin receptors. Mol Cell Endocrinol. 2001;179(1–2):83–87. doi: 10.1016/s0303-7207(01)00471-3. [DOI] [PubMed] [Google Scholar]
- de Roux N, Young J, Brailly-Tabard S, Misrahi M, Milgrom E, Schaison G. The same molecular defects of the gonadotropin-releasing hormone receptor determine a variable degree of hypogonadism in affected kindred. J Clin Endocrinol Metab. 1999;84(2):567–572. doi: 10.1210/jcem.84.2.5449. [DOI] [PubMed] [Google Scholar]
- de Roux N, Young J, Misrahi M, Genet R, Chanson P, Schaison G, et al. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. N Engl J Med. 1997;337(22):1597–1602. doi: 10.1056/NEJM199711273372205. [DOI] [PubMed] [Google Scholar]
- Dong C, Filipeanu CM, Duvernay MT, Wu G. Regulation of G protein-coupled receptor export trafficking. Biochim Biophys Acta. 2007;1768(4):853–870. doi: 10.1016/j.bbamem.2006.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormer RL, Derand R, McNeilly CM, Mettey Y, Bulteau-Pignoux L, Metaye T, et al. Correction of delF508-CFTR activity with benzo(c)quinolizinium compounds through facilitation of its processing in cystic fibrosis airway cells. J Cell Sci. 2001;114(Pt 22):4073–4081. doi: 10.1242/jcs.114.22.4073. [DOI] [PubMed] [Google Scholar]
- Dunham JH, Hall RA. Enhancement of the surface expression of G protein-coupled receptors. Trends Biotechnol. 2009;27(9):541–545. doi: 10.1016/j.tibtech.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4(3):181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- Ellis RJ. Protein misassembly: macromolecular crowding and molecular chaperones. Adv Exp Med Biol. 2007;594:1–13. doi: 10.1007/978-0-387-39975-1_1. [DOI] [PubMed] [Google Scholar]
- Englander SW, Mayne L, Krishna MM. Protein folding and misfolding: mechanism and principles. Q Rev Biophys. 2007;40(4):287–326. doi: 10.1017/S0033583508004654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch AR, Sedgley KR, Armstrong SP, Caunt CJ, McArdle CA. Trafficking and signalling of gonadotrophin-releasing hormone receptors: an automated imaging approach. Br J Pharmacol. 159(4):751–760. doi: 10.1111/j.1476-5381.2009.00413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs S, Amiel J, Claudel S, Lyonnet S, Corvol P, Pinet F. Functional characterization of three mutations of the endothelin B receptor gene in patients with Hirschsprung's disease: evidence for selective loss of Gi coupling. Mol Med. 2001;7(2):115–124. [PMC free article] [PubMed] [Google Scholar]
- Fujiwara TM, Bichet DG. Molecular biology of hereditary diabetes insipidus. J Am Soc Nephrol. 2005;16(10):2836–2846. doi: 10.1681/ASN.2005040371. [DOI] [PubMed] [Google Scholar]
- Galietta LJ, Springsteel MF, Eda M, Niedzinski EJ, By K, Haddadin MJ, et al. Novel CFTR chloride channel activators identified by screening of combinatorial libraries based on flavone and benzoquinolizinium lead compounds. J Biol Chem. 2001;276(23):19723–19728. doi: 10.1074/jbc.M101892200. [DOI] [PubMed] [Google Scholar]
- Gehret AU, Bajaj A, Naider F, Dumont ME. Oligomerization of the yeast alpha-factor receptor: implications for dominant negative effects of mutant receptors. J Biol Chem. 2006;281(30):20698–20714. doi: 10.1074/jbc.M513642200. [DOI] [PubMed] [Google Scholar]
- Gekko K, Timasheff SN. Mechanism of protein stabilization by glycerol: preferential hydration in glycerol-water mixtures. Biochemistry. 1981;20(16):4667–4676. doi: 10.1021/bi00519a023. [DOI] [PubMed] [Google Scholar]
- Gorbatyuk MS, Knox T, LaVail MM, Gorbatyuk OS, Noorwez SM, Hauswirth WW, et al. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc Natl Acad Sci U S A. 2010;107(13):5961–5966. doi: 10.1073/pnas.0911991107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromoll J, Schulz A, Borta H, Gudermann T, Teerds KJ, Greschniok A, et al. Homozygous mutation within the conserved Ala-Phe-Asn-Glu-Thr motif of exon 7 of the LH receptor causes male pseudohermaphroditism. Eur J Endocrinol. 2002;147(5):597–608. doi: 10.1530/eje.0.1470597. [DOI] [PubMed] [Google Scholar]
- Guo W, Shi L, Javitch JA. The fourth transmembrane segment forms the interface of the dopamine D2 receptor homodimer. J Biol Chem. 2003;278(7):4385–4388. doi: 10.1074/jbc.C200679200. [DOI] [PubMed] [Google Scholar]
- Hague C, Uberti MA, Chen Z, Hall RA, Minneman KP. Cell surface expression of alpha1D-adrenergic receptors is controlled by heterodimerization with alpha1B-adrenergic receptors. J Biol Chem. 2004;279(15):15541–15549. doi: 10.1074/jbc.M314014200. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295(5561):1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16(6):574–581. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- Hebert TE, Moffett S, Morello JP, Loisel TP, Bichet DG, Barret C, et al. A peptide derived from a beta2-adrenergic receptor transmembrane domain inhibits both receptor dimerization and activation. J Biol Chem. 1996;271(27):16384–16392. doi: 10.1074/jbc.271.27.16384. [DOI] [PubMed] [Google Scholar]
- Heding A, Vrecl M, Bogerd J, McGregor A, Sellar R, Taylor PL, et al. Gonadotropin-releasing hormone receptors with intracellular carboxyl-terminal tails undergo acute desensitization of total inositol phosphate production and exhibit accelerated internalization kinetics. J Biol Chem. 1998;273(19):11472–11477. doi: 10.1074/jbc.273.19.11472. [DOI] [PubMed] [Google Scholar]
- Helenius A. Quality control in the secretory assembly line. Philos Trans R Soc Lond B Biol Sci. 2001;356(1406):147–150. doi: 10.1098/rstb.2000.0759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermosilla R, Oueslati M, Donalies U, Schonenberger E, Krause E, Oksche A, et al. Disease-causing V(2) vasopressin receptors are retained in different compartments of the early secretory pathway. Traffic. 2004;5(12):993–1005. doi: 10.1111/j.1600-0854.2004.00239.x. [DOI] [PubMed] [Google Scholar]
- Herrick-Davis K, Grinde E, Mazurkiewicz JE. Biochemical and biophysical characterization of serotonin 5-HT2C receptor homodimers on the plasma membrane of living cells. Biochemistry. 2004;43(44):13963–13971. doi: 10.1021/bi048398p. [DOI] [PubMed] [Google Scholar]
- Huang Y, Breitwieser GE. Rescue of calcium-sensing receptor mutants by allosteric modulators reveals a conformational checkpoint in receptor biogenesis. J Biol Chem. 2007;282(13):9517–9525. doi: 10.1074/jbc.M609045200. [DOI] [PubMed] [Google Scholar]
- Huhtaniemi IT, Themmen AP. Mutations in human gonadotropin and gonadotropin-receptor genes. Endocrine. 2005;26(3):207–217. doi: 10.1385/ENDO:26:3:207. [DOI] [PubMed] [Google Scholar]
- Illing ME, Rajan RS, Bence NF, Kopito RR. A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteasome system. J Biol Chem. 2002;277(37):34150–34160. doi: 10.1074/jbc.M204955200. [DOI] [PubMed] [Google Scholar]
- Janovick JA, Brothers SP, Cornea A, Bush E, Goulet MT, Ashton WT, et al. Refolding of misfolded mutant GPCR: Post-translational pharmacoperone action in vitro. Mol Cell Endocrinol. 2007;272(1–2):77–85. doi: 10.1016/j.mce.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janovick JA, Brothers SP, Knollman PE, Conn PM. Specializations of a G-protein-coupled receptor that appear to aid with detection of frequency-modulated signals from its ligand. Faseb J. 2007;21(2):384–392. doi: 10.1096/fj.06-6901com. [DOI] [PubMed] [Google Scholar]
- Janovick JA, Conn PM. Salt bridge integrates GPCR activation with protein trafficking. Proc Natl Acad Sci U S A. 2010;107(9):4454–4458. doi: 10.1073/pnas.0914261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janovick JA, Goulet M, Bush E, Greer J, Wettlaufer DG, Conn PM. Structure-activity relations of successful pharmacologic chaperones for rescue of naturally occurring and manufactured mutants of the gonadotropin-releasing hormone receptor. J Pharmacol Exp Ther. 2003;305(2):608–614. doi: 10.1124/jpet.102.048454. [DOI] [PubMed] [Google Scholar]
- Janovick JA, Knollman PE, Brothers SP, Ayala-Yanez R, Aziz AS, Conn PM. Regulation of G protein-coupled receptor trafficking by inefficient plasma membrane expression: molecular basis of an evolved strategy. J Biol Chem. 2006;281(13):8417–8425. doi: 10.1074/jbc.M510601200. [DOI] [PubMed] [Google Scholar]
- Janovick JA, Maya-Nunez G, Conn PM. Rescue of hypogonadotropic hypogonadism-causing and manufactured GnRH receptor mutants by a specific protein-folding template: misrouted proteins as a novel disease etiology and therapeutic target. J Clin Endocrinol Metab. 2002;87(7):3255–3262. doi: 10.1210/jcem.87.7.8582. [DOI] [PubMed] [Google Scholar]
- Janovick JA, Patney A, Mosely R, Goulet M, Altman M, Rush T, et al. Molecular mechanism of action of pharmacoperone rescue of misrouted GPCR mutants: the GnRH receptor. Molec Endocrinol. 2009;23(2):157–168. doi: 10.1210/me.2008-0384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janovick JA, Ulloa-Aguirre A, Conn PM. Evolved regulation of gonadotropin-releasing hormone receptor cell surface expression. Endocrine. 2003;22(3):317–327. doi: 10.1385/ENDO:22:3:317. [DOI] [PubMed] [Google Scholar]
- Jardon-Valadez E, Aguilar-Rojas A, Maya-Nunez G, Leanos-Miranda A, Pineiro A, Conn PM, et al. Conformational Effects of Lys191 in the Human Gonadotrophin-Releasing Hormone Receptor (hGnRHR). Mutagenesis and Molecular Dynamics Simulations Studies. J Endocrinol. 2009 doi: 10.1677/JOE-08-0527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardon-Valadez E, Ulloa-Aguirre A, Pineiro A. Modeling and molecular dynamics simulation of the human gonadotropin-releasing hormone receptor in a lipid bilayer. J Phys Chem B. 2008;112(34):10704–10713. doi: 10.1021/jp800544x. [DOI] [PubMed] [Google Scholar]
- Jennes L, Ulloa-Aguirre A, Janovick JA, Conn PM. GnRH and the GnRHRIn. In: Pfaff DW, editor. Hormones, Brain and Behavior. 2nd Edition. San Diego, CA: Academic Press; 2008. (pp. In press). [Google Scholar]
- Katada S, Tanaka M, Touhara K. Structural determinants for membrane trafficking and G protein selectivity of a mouse olfactory receptor. J Neurochem. 2004;90(6):1453–1463. doi: 10.1111/j.1471-4159.2004.02619.x. [DOI] [PubMed] [Google Scholar]
- Kato A, Touhara K. Mammalian olfactory receptors: pharmacology, G protein coupling and desensitization. Cell Mol Life Sci. 2009;66(23):3743–3753. doi: 10.1007/s00018-009-0111-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaupmann K, Malitschek B, Schuler V, Heid J, Froestl W, Beck P, et al. GABA(B)-receptor subtypes assemble into functional heteromeric complexes. Nature. 1998;396(6712):683–687. doi: 10.1038/25360. [DOI] [PubMed] [Google Scholar]
- Knobil E. On the control of gonadotropin secretion in the rhesus monkey. Recent Prog Horm Res. 1974;30(0):1–46. doi: 10.1016/b978-0-12-571130-2.50005-5. [DOI] [PubMed] [Google Scholar]
- Knobil E, Neill J, editors. The Physiology of Reproduction. Vol. 1 & 2. New York: Raven Press; 1994. [Google Scholar]
- Knollman PE, Janovick JA, Brothers SP, Conn PM. Parallel regulation of membrane trafficking and dominant-negative effects by misrouted gonadotropin-releasing hormone receptor mutants. J Biol Chem. 2005;280(26):24506–24514. doi: 10.1074/jbc.M501978200. [DOI] [PubMed] [Google Scholar]
- Krautwurst D, Yau KW, Reed RR. Identification of ligands for olfactory receptors by functional expression of a receptor library. Cell. 1998;95(7):917–926. doi: 10.1016/s0092-8674(00)81716-x. [DOI] [PubMed] [Google Scholar]
- Le Gouill C, Parent JL, Caron CA, Gaudreau R, Volkov L, Rola-Pleszczynski M, et al. Selective modulation of wild type receptor functions by mutants of G-protein-coupled receptors. J Biol Chem. 1999;274(18):12548–12554. doi: 10.1074/jbc.274.18.12548. [DOI] [PubMed] [Google Scholar]
- Leandro P, Gomes CM. Protein misfolding in conformational disorders: rescue of folding defects and chemical chaperoning. Mini Rev Med Chem. 2008;8(9):901–911. doi: 10.2174/138955708785132783. [DOI] [PubMed] [Google Scholar]
- Leanos-Miranda A, Janovick JA, Conn PM. Receptor-misrouting: an unexpectedly prevalent and rescuable etiology in gonadotropin-releasing hormone receptor-mediated hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2002;87(10):4825–4828. doi: 10.1210/jc.2002-020961. [DOI] [PubMed] [Google Scholar]
- Leanos-Miranda A, Ulloa-Aguirre A, Janovick JA, Conn PM. In vitro coexpression and pharmacological rescue of mutant gonadotropin-releasing hormone receptors causing hypogonadotropic hypogonadism in humans expressing compound heterozygous alleles. J Clin Endocrinol Metab. 2005;90(5):3001–3008. doi: 10.1210/jc.2004-2071. [DOI] [PubMed] [Google Scholar]
- Leanos-Miranda A, Ulloa-Aguirre A, Ji TH, Janovick JA, Conn PM. Dominant-negative action of disease-causing gonadotropin-releasing hormone receptor (GnRHR) mutants: a trait that potentially coevolved with decreased plasma membrane expression of GnRHR in humans. J Clin Endocrinol Metab. 2003;88(7):3360–3367. doi: 10.1210/jc.2003-030084. [DOI] [PubMed] [Google Scholar]
- Lee SP, O'Dowd BF, Ng GY, Varghese G, Akil H, Mansour A, et al. Inhibition of cell surface expression by mutant receptors demonstrates that D2 dopamine receptors exist as oligomers in the cell. Mol Pharmacol. 2000;58(1):120–128. doi: 10.1124/mol.58.1.120. [DOI] [PubMed] [Google Scholar]
- Leskela TT, Markkanen PM, Pietila EM, Tuusa JT, Petaja-Repo UE. Opioid receptor pharmacological chaperones act by binding and stabilizing newly synthesized receptors in the endoplasmic reticulum. J Biol Chem. 2007 doi: 10.1074/jbc.M610896200. [DOI] [PubMed] [Google Scholar]
- Lim S, Pnueli L, Tan JH, Naor Z, Rajagopal G, Melamed P. Negative feedback governs gonadotrope frequency-decoding of gonadotropin releasing hormone pulse-frequency. PLoS One. 2009;4(9):e7244. doi: 10.1371/journal.pone.0007244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Janovick JA, Brothers S, Blomenrohr M, Bogerd J, Conn PM. Addition of catfish gonadotropin-releasing hormone (GnRH) receptor intracellular carboxyl-terminal tail to rat GnRH receptor alters receptor expression and regulation. Mol Endocrinol. 1998;12(2):161–171. doi: 10.1210/mend.12.2.0056. [DOI] [PubMed] [Google Scholar]
- Loo TW, Clarke DM. Chemical and pharmacological chaperones as new therapeutic agents. Expert Rev Mol Med. 2007;9(16):1–18. doi: 10.1017/S1462399407000361. [DOI] [PubMed] [Google Scholar]
- Lu M, Echeverri F, Moyer BD. Endoplasmic reticulum retention, degradation, and aggregation of olfactory G-protein coupled receptors. Traffic. 2003;4(6):416–433. doi: 10.1034/j.1600-0854.2003.00097.x. [DOI] [PubMed] [Google Scholar]
- Lu M, Staszewski L, Echeverri F, Xu H, Moyer BD. Endoplasmic reticulum degradation impedes olfactory G-protein coupled receptor functional expression. BMC Cell Biol. 2004;5:34. doi: 10.1186/1471-2121-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margeta-Mitrovic M. Assembly-dependent trafficking assays in the detection of receptor-receptor interactions. Methods. 2002;27(4):311–317. doi: 10.1016/s1046-2023(02)00088-9. [DOI] [PubMed] [Google Scholar]
- Margeta-Mitrovic M, Jan YN, Jan LY. A trafficking checkpoint controls GABA(B) receptor heterodimerization. Neuron. 2000;27(1):97–106. doi: 10.1016/s0896-6273(00)00012-x. [DOI] [PubMed] [Google Scholar]
- Martens JW, Lumbroso S, Verhoef-Post M, Georget V, Richter-Unruh A, Szarras-Czapnik M, et al. Mutant luteinizing hormone receptors in a compound heterozygous patient with complete Leydig cell hypoplasia: abnormal processing causes signaling deficiency. J Clin Endocrinol Metab. 2002;87(6):2506–2513. doi: 10.1210/jcem.87.6.8523. [DOI] [PubMed] [Google Scholar]
- Maya-Nunez G, Janovick JA, Aguilar-Rojas A, Jardon-Valadez E, Leanos-Miranda A, Zarinan T, et al. Biochemical mechanism of pathogenesis of human gonadotropin-releasing hormone receptor mutants Thr104Ile and Tyr108Cys associated with familial hypogonadotropic hypogonadism. [Research Paper] Mol Cell Endocrinol. 2011 doi: 10.1016/j.mce.2011.01.016. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya-Nunez G, Janovick JA, Conn PM. Combined modification of intracellular and extracellular loci on human gonadotropin-releasing hormone receptor provides a mechanism for enhanced expression. Endocrine. 2000;13(3):401–407. doi: 10.1385/ENDO:13:3:401. [DOI] [PubMed] [Google Scholar]
- Maya-Nunez G, Janovick JA, Ulloa-Aguirre A, Soderlund D, Conn PM, Mendez JP. Molecular basis of hypogonadotropic hypogonadism: restoration of mutant (E(90)K) GnRH receptor function by a deletion at a distant site. J Clin Endocrinol Metab. 2002;87(5):2144–2149. doi: 10.1210/jcem.87.5.8386. [DOI] [PubMed] [Google Scholar]
- McArdle CA, Davidson JS, Willars GB. The tail of the gonadotrophin-releasing hormone receptor: desensitization at, distal to, G protein-coupled receptors. Mol Cell Endocrinol. 1999;151(1–2):129–136. doi: 10.1016/s0303-7207(99)00024-6. [DOI] [PubMed] [Google Scholar]
- McVey M, Ramsay D, Kellett E, Rees S, Wilson S, Pope AJ, et al. Monitoring receptor oligomerization using time-resolved fluorescence resonance energy transfer and bioluminescence resonance energy transfer. The human delta -opioid receptor displays constitutive oligomerization at the cell surface, which is not regulated by receptor occupancy. J Biol Chem. 2001;276(17):14092–14099. doi: 10.1074/jbc.M008902200. [DOI] [PubMed] [Google Scholar]
- Mendes HF, van der Spuy J, Chapple JP, Cheetham ME. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol Med. 2005;11(4):177–185. doi: 10.1016/j.molmed.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Mercier JF, Salahpour A, Angers S, Breit A, Bouvier M. Quantitative assessment of beta 1- and beta 2-adrenergic receptor homo- and heterodimerization by bioluminescence resonance energy transfer. J Biol Chem. 2002;277(47):44925–44931. doi: 10.1074/jbc.M205767200. [DOI] [PubMed] [Google Scholar]
- Meysing AU, Kanasaki H, Bedecarrats GY, Acierno JS, Jr, Conn PM, Martin KA, et al. GNRHR mutations in a woman with idiopathic hypogonadotropic hypogonadism highlight the differential sensitivity of luteinizing hormone and follicle-stimulating hormone to gonadotropin-releasing hormone. J Clin Endocrinol Metab. 2004;89(7):3189–3198. doi: 10.1210/jc.2003-031808. [DOI] [PubMed] [Google Scholar]
- Millar RP. GnRH II and type II GnRH receptors. Trends Endocrinol Metab. 2003;14(1):35–43. doi: 10.1016/s1043-2760(02)00016-4. [DOI] [PubMed] [Google Scholar]
- Millar RP, Lu ZL, Pawson AJ, Flanagan CA, Morgan K, Maudsley SR. Gonadotropin-releasing hormone receptors. Endocr Rev. 2004;25(2):235–275. doi: 10.1210/er.2003-0002. [DOI] [PubMed] [Google Scholar]
- Millar RP, Pawson AJ, Morgan K, Rissman EF, Lu ZL. Diversity of actions of GnRHs mediated by ligand-induced selective signaling. Front Neuroendocrinol. 2008;29(1):17–35. doi: 10.1016/j.yfrne.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G. Constitutive activity and inverse agonists of G protein-coupled receptors: a current perspective. Mol Pharmacol. 2003;64(6):1271–1276. doi: 10.1124/mol.64.6.1271. [DOI] [PubMed] [Google Scholar]
- Milligan G. G protein-coupled receptor dimerisation: molecular basis and relevance to function. Biochim Biophys Acta. 2007;1768(4):825–835. doi: 10.1016/j.bbamem.2006.09.021. [DOI] [PubMed] [Google Scholar]
- Morello JP, Bichet DG. Nephrogenic diabetes insipidus. Annu Rev Physiol. 2001;63:607–630. doi: 10.1146/annurev.physiol.63.1.607. [DOI] [PubMed] [Google Scholar]
- Morello JP, Petaja-Repo UE, Bichet DG, Bouvier M. Pharmacological chaperones: a new twist on receptor folding. Trends Pharmacol Sci. 2000;21(12):466–469. doi: 10.1016/s0165-6147(00)01575-3. [DOI] [PubMed] [Google Scholar]
- Morello JP, Salahpour A, Petaja-Repo UE, Laperriere A, Lonergan M, Arthus MF, et al. Association of calnexin with wild type and mutant AVPR2 that causes nephrogenic diabetes insipidus. Biochemistry. 2001;40(23):6766–6775. doi: 10.1021/bi002699r. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Yasuda D, Hirota N, Shimizu T. Specific ligands as pharmacological chaperones: The transport of misfolded G-protein coupled receptors to the cell surface. IUBMB Life. 2010;62(6):453–459. doi: 10.1002/iub.344. [DOI] [PubMed] [Google Scholar]
- Naor Z. Signaling by G-protein-coupled receptor (GPCR): studies on the GnRH receptor. Front Neuroendocrinol. 2009;30(1):10–29. doi: 10.1016/j.yfrne.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Nardai G, Vegh EM, Prohaszka Z, Csermely P. Chaperone-related immune dysfunction: an emergent property of distorted chaperone networks. Trends Immunol. 2006;27(2):74–79. doi: 10.1016/j.it.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Ni M, Lee AS. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007;581(19):3641–3651. doi: 10.1016/j.febslet.2007.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noorwez SM, Kuksa V, Imanishi Y, Zhu L, Filipek S, Palczewski K, et al. Pharmacological chaperone-mediated in vivo folding and stabilization of the P23H-opsin mutant associated with autosomal dominant retinitis pigmentosa. J Biol Chem. 2003;278(16):14442–14450. doi: 10.1074/jbc.M300087200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noorwez SM, Malhotra R, McDowell JH, Smith KA, Krebs MP, Kaushal S. Retinoids assist the cellular folding of the autosomal dominant retinitis pigmentosa opsin mutant P23H. J Biol Chem. 2004;279(16):16278–16284. doi: 10.1074/jbc.M312101200. [DOI] [PubMed] [Google Scholar]
- Noorwez SM, Ostrov DA, McDowell JH, Krebs MP, Kaushal S. A high-throughput screening method for small-molecule pharmacologic chaperones of misfolded rhodopsin. Invest Ophthalmol Vis Sci. 2008;49(7):3224–3230. doi: 10.1167/iovs.07-1539. [DOI] [PubMed] [Google Scholar]
- Nowak RJ, Cuny GD, Choi S, Lansbury PT, Ray SS. Improving binding specificity of pharmacological chaperones that target mutant superoxide dismutase-1 linked to familial amyotrophic lateral sclerosis using computational methods. J Med Chem. 2010;53(7):2709–2718. doi: 10.1021/jm901062p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oueslati M, Hermosilla R, Schonenberger E, Oorschot V, Beyermann M, Wiesner B, et al. Rescue of a nephrogenic diabetes insipidus-causing vasopressin V2 receptor mutant by cell-penetrating peptides. J Biol Chem. 2007 doi: 10.1074/jbc.M611530200. [DOI] [PubMed] [Google Scholar]
- Petaja-Repo UE, Hogue M, Laperriere A, Bhalla S, Walker P, Bouvier M. Newly synthesized human delta opioid receptors retained in the endoplasmic reticulum are retrotranslocated to the cytosol, deglycosylated, ubiquitinated, and degraded by the proteasome. J Biol Chem. 2001;276(6):4416–4423. doi: 10.1074/jbc.M007151200. [DOI] [PubMed] [Google Scholar]
- Petaja-Repo UE, Hogue M, Laperriere A, Walker P, Bouvier M. Export from the endoplasmic reticulum represents the limiting step in the maturation and cell surface expression of the human delta opioid receptor. J Biol Chem. 2000;275(18):13727–13736. doi: 10.1074/jbc.275.18.13727. [DOI] [PubMed] [Google Scholar]
- Pietila EM, Tuusa JT, Apaja PM, Aatsinki JT, Hakalahti AE, Rajaniemi HJ, et al. Inefficient maturation of the rat luteinizing hormone receptor. A putative way to regulate receptor numbers at the cell surface. J Biol Chem. 2005;280(28):26622–26629. doi: 10.1074/jbc.M413815200. [DOI] [PubMed] [Google Scholar]
- Rab A, Bartoszewski R, Jurkuvenaite A, Wakefield J, Collawn JF, Bebok Z. Endoplasmic reticulum stress and the unfolded protein response regulate genomic cystic fibrosis transmembrane conductance regulator expression. Am J Physiol Cell Physiol. 2007;292(2):C756–C766. doi: 10.1152/ajpcell.00391.2006. [DOI] [PubMed] [Google Scholar]
- Rana S, Besson G, Cook DG, Rucker J, Smyth RJ, Yi Y, et al. Role of CCR5 in infection of primary macrophages and lymphocytes by macrophage-tropic strains of human immunodeficiency virus: resistance to patient-derived and prototype isolates resulting from the delta ccr5 mutation. J Virol. 1997;71(4):3219–3227. doi: 10.1128/jvi.71.4.3219-3227.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiche EM, Bonametti AM, Voltarelli JC, Morimoto HK, Watanabe MA. Genetic polymorphisms in the chemokine and chemokine receptors: impact on clinical course and therapy of the human immunodeficiency virus type 1 infection (HIV-1) Curr Med Chem. 2007;14(12):1325–1334. doi: 10.2174/092986707780597934. [DOI] [PubMed] [Google Scholar]
- Rivera VM, Wang X, Wardwell S, Courage NL, Volchuk A, Keenan T, et al. Regulation of protein secretion through controlled aggregation in the endoplasmic reticulum. Science. 2000;287(5454):826–830. doi: 10.1126/science.287.5454.826. [DOI] [PubMed] [Google Scholar]
- Robben JH, Knoers NV, Deen PM. Characterization of vasopressin V2 receptor mutants in nephrogenic diabetes insipidus in a polarized cell model. Am J Physiol Renal Physiol. 2005;289(2):F265–F272. doi: 10.1152/ajprenal.00404.2004. [DOI] [PubMed] [Google Scholar]
- Robben JH, Sze M, Knoers NV, Deen PM. Rescue of vasopressin V2 receptor mutants by chemical chaperones: specificity and mechanism. Mol Biol Cell. 2006;17(1):379–386. doi: 10.1091/mbc.E05-06-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459(7245):356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salahpour A, Angers S, Mercier JF, Lagace M, Marullo S, Bouvier M. Homodimerization of the beta2-adrenergic receptor as a prerequisite for cell surface targeting. J Biol Chem. 2004;279(32):33390–33397. doi: 10.1074/jbc.M403363200. [DOI] [PubMed] [Google Scholar]
- Saliba RS, Munro PM, Luthert PJ, Cheetham ME. The cellular fate of mutant rhodopsin: quality control, degradation and aggresome formation. J Cell Sci. 2002;115(Pt 14):2907–2918. doi: 10.1242/jcs.115.14.2907. [DOI] [PubMed] [Google Scholar]
- Sampedro JG, Uribe S. Trehalose-enzyme interactions result in structure stabilization and activity inhibition. The role of viscosity. Mol Cell Biochem. 2004;256–257(1–2):319–327. doi: 10.1023/b:mcbi.0000009878.21929.eb. [DOI] [PubMed] [Google Scholar]
- Santen RJ, Bardin CW. Episodic luteinizing hormone secretion in man. Pulse analysis, clinical interpretation, physiologic mechanisms. J Clin Invest. 1973;52(10):2617–2628. doi: 10.1172/JCI107454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S, Cho N, Nara Y, Harada M, Endo S, Suzuki N, et al. Discovery of a thieno[2,3-d]pyrimidine-2,4-dione bearing a p-methoxyureidophenyl moiety at the 6-position: a highly potent and orally bioavailable non-peptide antagonist for the human luteinizing hormone-releasing hormone receptor. J Med Chem. 2003;46(1):113–124. doi: 10.1021/jm020180i. [DOI] [PubMed] [Google Scholar]
- Schulein R, Zuhlke K, Krause G, Rosenthal W. Functional rescue of the nephrogenic diabetes insipidus-causing vasopressin V2 receptor mutants G185C and R202C by a second site suppressor mutation. J Biol Chem. 2001;276(11):8384–8392. doi: 10.1074/jbc.M007045200. [DOI] [PubMed] [Google Scholar]
- Sitia R, Braakman I. Quality control in the endoplasmic reticulum protein factory. Nature. 2003;426(6968):891–894. doi: 10.1038/nature02262. [DOI] [PubMed] [Google Scholar]
- Soderhall JA, Polymeropoulos EE, Paulini K, Gunther E, Kuhne R. Antagonist and agonist binding models of the human gonadotropin-releasing hormone receptor. Biochem Biophys Res Commun. 2005;333(2):568–582. doi: 10.1016/j.bbrc.2005.05.142. [DOI] [PubMed] [Google Scholar]
- Takeda S, Kadowaki S, Haga T, Takaesu H, Mitaku S. Identification of G protein-coupled receptor genes from the human genome sequence. FEBS Lett. 2002;520(1–3):97–101. doi: 10.1016/s0014-5793(02)02775-8. [DOI] [PubMed] [Google Scholar]
- Tan CM, Brady AE, Nickols HH, Wang Q, Limbird LE. Membrane trafficking of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2004;44:559–609. doi: 10.1146/annurev.pharmtox.44.101802.121558. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Moroi K, Iwai J, Takahashi H, Ohnuma N, Hori S, et al. Novel mutations of the endothelin B receptor gene in patients with Hirschsprung's disease and their characterization. J Biol Chem. 1998;273(18):11378–11383. doi: 10.1074/jbc.273.18.11378. [DOI] [PubMed] [Google Scholar]
- Tao YX. Molecular mechanisms of the neural melanocortin receptor dysfunction in severe early onset obesity. Mol Cell Endocrinol. 2005;239(1–2):1–14. doi: 10.1016/j.mce.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Tao YX. The melanocortin-4 receptor: physiology, pharmacology, and pathophysiology. Endocr Rev. 2010;31(4):506–543. doi: 10.1210/er.2009-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao YX, Segaloff DL. Functional characterization of melanocortin-4 receptor mutations associated with childhood obesity. Endocrinology. 2003;144(10):4544–4551. doi: 10.1210/en.2003-0524. [DOI] [PubMed] [Google Scholar]
- Terrillon S, Barberis C, Bouvier M. Heterodimerization of V1a and V2 vasopressin receptors determines the interaction with beta-arrestin and their trafficking patterns. Proc Natl Acad Sci U S A. 2004;101(6):1548–1553. doi: 10.1073/pnas.0305322101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RM, Nechamen CA, Mazurkiewicz JE, Muda M, Palmer S, Dias JA. Follice-stimulating hormone receptor forms oligomers and shows evidence of carboxyl-terminal proteolytic processing. Endocrinology. 2007;148(5):1987–1995. doi: 10.1210/en.2006-1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tranchant T, Durand G, Gauthier C, Crepieux P, Ulloa-Aguirre A, Royere D, et al. Preferential beta-arrestin signalling at low receptor density revealed by functional characterization of the human FSH receptor A189 V mutation. Mol Cell Endocrinol. 2011;331(1):109–118. doi: 10.1016/j.mce.2010.08.016. [DOI] [PubMed] [Google Scholar]
- Trombetta ES, Parodi AJ. Quality control and protein folding in the secretory pathway. Annu Rev Cell Dev Biol. 2003;19:649–676. doi: 10.1146/annurev.cellbio.19.110701.153949. [DOI] [PubMed] [Google Scholar]
- Tuusa JT, Markkanen PM, Apaja PM, Hakalahti AE, Petaja-Repo UE. The Endoplasmic Reticulum Ca(2+)-pump SERCA2b Interacts with G Protein-coupled Receptors and Enhances their Expression at the Cell Surface. J Mol Biol. 2007 doi: 10.1016/j.jmb.2007.02.108. [DOI] [PubMed] [Google Scholar]
- Uberti MA, Hague C, Oller H, Minneman KP, Hall RA. Heterodimerization with beta2-adrenergic receptors promotes surface expression and functional activity of alpha1D-adrenergic receptors. J Pharmacol Exp Ther. 2005;313(1):16–23. doi: 10.1124/jpet.104.079541. [DOI] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Conn PM. G Protein-coupled receptors and the G protein family. In: Conn PM, editor. Handbook of Physiology. Section 7: The endocrine system. New York: Oxford University Press; 1998. pp. 87–124. Vol. 1: Cellular Endocrinology. [Google Scholar]
- Ulloa-Aguirre A, Conn PM. Targeting of G protein-coupled receptors to the plasma membrane in health and disease. Front Biosci. 2009;14:973–994. doi: 10.2741/3290. [DOI] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Janovick JA, Brothers SP, Conn PM. Pharmacologic rescue of conformationally-defective proteins: implications for the treatment of human disease. Traffic. 2004;5(11):821–837. doi: 10.1111/j.1600-0854.2004.00232.x. [DOI] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Janovick JA, Leanos-Miranda A, Conn PM. Misrouted cell surface receptors as a novel disease aetiology and potential therapeutic target: the case of hypogonadotropic hypogonadism due to gonadotropin-releasing hormone resistance. Expert Opin Ther Targets. 2003;7(2):175–185. doi: 10.1517/14728222.7.2.175. [DOI] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Janovick JA, Leanos-Miranda A, Conn PM. Misrouted cell surface GnRH receptors as a disease aetiology for congenital isolated hypogonadotrophic hypogonadism. Hum Reprod Update. 2004;10(2):177–192. doi: 10.1093/humupd/dmh015. [DOI] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Janovick JA, Miranda AL, Conn PM. G-protein-coupled receptor trafficking: understanding the chemical basis of health and disease. ACS Chem Biol. 2006;1(10):631–638. doi: 10.1021/cb600360h. [DOI] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Stanislaus D, Janovick JA, Conn PM. Structure-activity relationships of G protein-coupled receptors. Arch Med Res. 1999;30(6):420–435. doi: 10.1016/s0188-0128(99)00041-x. [DOI] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Timossi C. Biochemical and functional aspects of gonadotrophin-releasing hormone and gonadotrophins. Reprod Biomed Online. 2000;1(2):48–62. doi: 10.1016/s1472-6483(10)61901-3. [DOI] [PubMed] [Google Scholar]
- Werner ED, Brodsky JL, McCracken AA. Proteasome-dependent endoplasmic reticulum-associated protein degradation: an unconventional route to a familiar fate. Proc Natl Acad Sci U S A. 1996;93(24):13797–13801. doi: 10.1073/pnas.93.24.13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel CH, Oles M, Wellerdieck C, Kuczkowiak M, Gisselmann G, Hatt H. Specificity and sensitivity of a human olfactory receptor functionally expressed in human embryonic kidney 293 cells and Xenopus Laevis oocytes. J Neurosci. 1999;19(17):7426–7433. doi: 10.1523/JNEUROSCI.19-17-07426.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JH, Wise A, Main MJ, Green A, Fraser NJ, Disney GH, et al. Heterodimerization is required for the formation of a functional GABA(B) receptor. Nature. 1998;396(6712):679–682. doi: 10.1038/25354. [DOI] [PubMed] [Google Scholar]
- Wuller S, Wiesner B, Loffler A, Furkert J, Krause G, Hermosilla R, et al. Pharmacochaperones post-translationally enhance cell surface expression by increasing conformational stability of wild-type and mutant vasopressin V2 receptors. J Biol Chem. 2004;279(45):47254–47263. doi: 10.1074/jbc.M408154200. [DOI] [PubMed] [Google Scholar]
- Zarinan T, Perez-Solis MA, Maya-Nunez G, Casas-Gonzalez P, Conn PM, Dias JA, et al. Dominant negative effects of human follicle-stimulating hormone receptor expression-deficient mutants on wild-type receptor cell surface expression. Rescue of oligomerization-dependent defective receptor expression by using cognate decoys. Mol Cell Endocrinol. 2010;321(2):112–122. doi: 10.1016/j.mce.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XM, Wang XT, Yue H, Leung SW, Thibodeau PH, Thomas PJ, et al. Organic solutes rescue the functional defect in delta F508 cystic fibrosis transmembrane conductance regulator. J Biol Chem. 2003;278(51):51232–51242. doi: 10.1074/jbc.M309076200. [DOI] [PubMed] [Google Scholar]
- Zhu X, Wess J. Truncated V2 vasopressin receptors as negative regulators of wild-type V2 receptor function. Biochemistry. 1998;37(45):15773–15784. doi: 10.1021/bi981162z. [DOI] [PubMed] [Google Scholar]