

Abstract
The processes regulating cortical surface area expansion during development and evolution are unknown. We show that loss of function of all fibroblast growth factor receptors (FgfRs) expressed at the earliest stages of cortical development causes severe deficits in surface area growth by embryonic day 12.5 (E12.5) in the mouse. In FgfR mutants, accelerated production of neurons led to severe loss of radial progenitors and premature termination of neurogenesis. Nevertheless, these mutants showed remarkably little change in cortical layer structure. Birth-dating experiments indicated that a greater proportion of layer fates was generated during early neurogenic stages, revealing that FgfR activity normally slows the temporal progression of cortical layer fates. Electroporation of a dominant-negative FgfR at E11.5 increased cortical neurogenesis in normal mice—an effect that was blocked by simultaneous activation of the Notch pathway. Together with changes in the expression of Notch pathway genes in FgfR mutant embryos, these findings indicate that Notch lies downstream of FgfR signaling in the same pathway regulating cortical neurogenesis and begin to establish a mechanism for regulating cortical surface expansion.
Introduction
In early cerebral cortical development most neuroepithelial progenitors undergo symmetric, self-replicative division, leading to a geometric expansion of the progenitor pool. However, the onset of neurogenesis at ∼E11.5 in the mouse (Takahashi et al., 1996) slows this expansion and begins to deplete neuronal progenitors as cortical neurons are born. Therefore, the rate of neurogenesis during this phase of development could play a crucial role in cortical surface area expansion. Species differences in cortical surface area may be determined by variations in these mechanisms, yet the factors controlling this process are largely unknown (Rakic, 1995; Haydar et al., 2003; Götz and Barde, 2005; Kriegstein et al., 2006).
During the neurogenic phase, daughters of radial (apical) progenitor cells in the dorsal telencephalic ventricular zone (VZ) migrate to the subventricular zone (SVZ), where they may divide again as intermediate neuronal precursors (INPs) before migrating to the cortical plate (Haubensak et al., 2004; Noctor et al., 2004; Englund et al., 2005). The progression of a VZ precursor to an INP and on to postmitotic cortical neuron is marked by sequential changes in transcription factor expression, where Pax6 (VZ), Tbr2 (SVZ), and Tbr1 (cortical neuron) mark each stage (Englund et al., 2005). Some VZ precursors also give rise directly to cortical neurons without going through an INP stage (Miyata et al., 2001; Noctor et al., 2004). After the generation of layer 6, the remaining layers form in the order 5-4-3-2—a process reflected by the sequential expression of the transcription factors ZFPM2 (layer 6 and subplate), Ctip2 (layer 5), Cux1 (layer 2/3), and Satb2 (layer 2/3) in neuronal precursors (Nieto et al., 2004; Arlotta et al., 2005; Chen et al., 2005; Molyneaux et al., 2007; Britanova et al., 2008).
Notch signaling is well known for regulating neuronal differentiation in vertebrates and invertebrates (Yoon et al., 2004; Louvi and Artavanis-Tsakonas, 2006; Kageyama et al., 2008; Coumailleau et al., 2009). In the telencephalon, Notch activation through the canonical effector C-promoter binding factor 1 (CBF1) pathway suppresses proneural genes such as Ngn2 and Mash1 to maintain the radial glial lineage (Mizutani et al., 2007). Differentiating neuroblasts express the Notch ligands Delta-like (Dll) and Jagged, which in turn activate Notch signaling and prevent neuronal differentiation in adjacent cells—a process known as lateral inhibition, which functions to prevent the untimely loss of the progenitor pool. Crucially, however, the upstream control mechanisms that set the stage for Notch signaling and cortical neurogenesis remain obscure.
Fibroblast growth factors are secreted signaling molecules that play multiple roles in the CNS and control the size of the cerebral cortex (Vaccarino et al., 1999b; Shin et al., 2004) as well as its patterning (Fukuchi-Shimogori and Grove, 2001; O'Leary and Nakagawa, 2002; Garel et al., 2003; Hébert and Fishell, 2008). Several Fgf ligands, including Fgf2, 3, 7, 8, 10, 15, 17, and 18, are expressed in the rostral telencephalic midline and early cortical primordium (Vaccarino et al., 1999b; Garel et al., 2003; Storm et al., 2003; Cholfin and Rubenstein, 2007), and have been demonstrated to regulate neurogenesis (Raballo et al., 2000; Borello et al., 2008). Among the four Fgf receptors (FgfRs), only FgfR1–3 are expressed in the developing CNS (Ford-Perriss et al., 2001). Recent studies have demonstrated that FgfRs regulate CNS growth, including the hippocampus (Ohkubo et al., 2004) and the cerebral cortex (Kang et al., 2009; Thomson et al., 2009; Stevens et al., 2010), but the cellular and molecular mechanisms of FgfR function in the process of neurogenesis and cortical surface area expansion have been unclear.
Here we have assessed overall FgfR function at the onset of cortical development by conditional genetic deletion of FgfRs in the dorsal telencephalon by E10.0 using Emx1-Cre mice. Together with in utero electroporation and in vitro clonal culture studies, we find that FgfR signaling promotes early surface area expansion in cortical development, acting through Notch to regulate the cellular decision between cortical neurogenesis and stem cell renewal. Remarkably, cortical layer development was intact in FgfR mutant embryos despite large changes in surface area growth and marked shortening of the neurogenic period.
Materials and Methods
Mice.
Mice carrying floxed FgfR1 (Pirvola et al., 2002) and FgfR2 (Yu et al., 2003) alleles were bred with a null FgfR3 line (Deng et al., 1996) and an Emx1Cre line (Iwasato et al., 2004) to generate mice lacking FgfR1, FgfR2, and FgfR3 in the Emx1 lineage (Emx1; TKO). A total of 16 Emx1;TKO embryos were obtained in an approximately Mendelian ratio (1/16), suggesting no embryonic lethality. No phenotype was observed in Emx1+/+;FgfR1f/f;FgfR2 f/f;FgfR3+/− embryos, and therefore these as well as Emx1+/+;FgfR1f/f;FgfR2f/f;FgfR3+/+ embryos served as control littermates. Comparison of Emx1+/c embryos (n > 10) with wild type littermates revealed no phenotype with respect to cortical development, suggesting that the presence of a single cre allele (making Emx1 heterozygous due to the ‘knock-in’ mutation) does not significantly contribute to the Emx1;TKO phenotype. Noon of the day of vaginal plug detection was designated E0.5. All experimental procedures involving animals were performed in accordance with the policies of the Yale Institutional Animal Care and Use Committee.
Tissue preparation.
Embryos were collected at defined developmental stages, and their brains immediately dissected in diethylpyrocarbonate (DEPC)-treated PBS and fixed in 4% PFA overnight at 4°C. On the following day brains were equilibrated overnight in 25% sucrose/DEPC-PBS and cryoprotected in OCT. Sections (25 μm) were produced using a Leica cryostat, stored at −80°C, and then processed for either immunohistochemistry or RNA in situ hybridization.
Immunohistochemistry and in situ hybridization.
Immunohistochemistry was performed as described previously (Smith et al., 2006). Primary antibodies were detected using Alexa Fluor-conjugated secondary antibodies from Invitrogen. Primary antibodies and dilutions are shown in Table 1. The immunostaining for the Notch intracellular domain (NICD, mAb D3B8, Cell Signaling Technology) and Hes1 used an antigen retrieval step by incubating for 5 min in 0.01 m Na citrate, pH 6.0, at 95°C with biotin/streptavidin detection [biotinylated goat anti-rabbit (1:1000) or donkey anti-guinea pig (1:200), Vector Labs; Streptavidin Alexa Fluor 594 (1:400), Invitrogen]. Detection of BrdU, chlorodeoxyuridine (CldU), and iododeoxyuridine (IdU) was performed as previously described (Raballo et al., 2000). In situ hybridization was performed using alkaline phosphatase detection of digoxigenin-labeled riboprobes as previously described (Grove et al., 1998; Rash and Grove, 2007). Fluorescence 3D optical sectioning and bright-field imaging used a Zeiss Apotome or Axioplan microscope and Axiovision 4.8 software.
Table 1.
Primary antibodies used in this study
| Antibody | Species | Source | Dilution |
|---|---|---|---|
| Tbr1 | rb | Millipore | 1:1000 |
| Tbr2 | rb | Millipore | 1:1000 |
| Phosphohistone H3 | rt | Sigma | 1:1000 |
| Satb2 | rb | Victor Tarabykin | 1:1000 |
| Ctip2 | rt | Abcam | 1:1000 |
| Pax6 | rb | Millipore | 1:1000 |
| βIII Tubulin | ms | Promega | 1:2000 tissue 1:4000 in vitro |
| Ki67 | rb | Vector | 1:1000 |
| BrdU | rt | Accurate | 1:400; 1:250 for CldU labeling |
| BrdU/IdU | ms | BD Biosciences | 1:100 (for IdU labeling) |
| Activated Caspase 3 | rb | Cell Signaling | 1:500 |
| β-Galactisidase | ck | Abcam | 1:500 |
| Hes1 | gp | Ryoichiro Kageyama | 1:250 |
| ZfpM2 | gt | Santa Cruz | 1:250 |
| Cux1 | rb | Santa Cruz | 1:100 |
| GFP | ck | Abcam | 1:1000 |
| NICD val 1744 | rb (monoclonal) | Cell Signaling | 1:1000 |
| Nestin | ms | Millipore | 1:2000 tissue 1:4000 in vitro |
| BLBP | rb | Millipore | 1:1000 |
| Par3 | rb | Millipore | 1:500 |
rb, Rabbit; rt, rat; ms, mouse; ck, chicken; gp, guinea pig; gt, goat.
BrdU, CldU, and IdU labeling.
Intraperitoneal injections of BrdU, CldU, or IdU (Sigma) were administered at 50 mg/kg to pregnant dams. For Tbr1+ neuron birth dating and progenitor cell cycle reentry experiments, a pulse of BrdU was administered at E10.5, E11.5, or E12.5, 24 h before embryo fixation. For dual labeling of early- and late-born cohorts of cortical neurons, pulses of CldU at E12.5 and IdU at E16.5 were given, and the animals killed at E18.5.
Expression constructs and in utero electroporation.
Enhanced green fluorescent protein (eGFP) was subcloned into pBluescript (Stratagene), downstream of the BLBP (brain lipid-binding protein) promoter and an IRES to create pBLBP-eGFP. A truncated FgfR1 (SW2) (Werner et al., 1993) was subcloned into pBLBP-eGFP following the BLBP promoter. pBLBP-eGFP, pBLBP-SW2, or pUbiC-NICD-Myc (provided by J.J.B., details upon request) plasmid DNA was prepared using an Endofree Maxi kit (Qiagen) and 0.5 μl was electroporated at a concentration of 1 μg/μl.
All electroporation experiments used CD1 dams (Charles River) anesthetized by isoflurane and prepared for surgery according to Yale Institutional Animal Care and Use Committee rules. Embryos were electroporated at E11.5 as described previously (Fukuchi-Shimogori and Grove, 2001), except that we used a BTX ECM 830 (Harvard Apparatus) square electroporator delivering 27 V via gold paddle electrodes.
Cell culture and clonal analysis.
Electroporation of pBLBP-eGFP alone (n = 6) or together with SW2 (n = 4) plasmid DNA was performed at E11.5, followed by 2 d in utero. Cortical explants containing eGFP+ cells were isolated and dissociated in DMEM with B27 supplement and 5 ng/ml FGF2, and cultured at a density of 500,000 cells/cm2 for 3 d. Cultured cells were then fixed with 4% PFA for 30 min at 4°C and stained for Nestin, βIII tubulin, and eGFP. The eGFP+ clones were imaged blind with respect to gene expression and then scored for clone size and the number of cells expressing Nestin or βIII tubulin. The cell type fraction of the total number of eGFP+ cells (n = 523 for pBLBP-eGFP; n = 731 for SW2) across all clones (n = 133 for pBLBP-eGFP controls and n = 302 for SW2) and the average within each clone was calculated and statistically assessed using a two-tailed Student's t test.
Morphometric analysis, densitometry, and cell population quantification.
Morphometric and cell population estimates were performed using a Zeiss Axioskop 2 Mot Plus microscope operating Stereoinvestigator and Neurolucida systems (MicroBrightField). The rostrocaudal axes of cortical hemispheres were sampled in their entirety using regular section intervals. Cortical volume was estimated by planimetry. Nuclear profiles of cell populations were randomly sampled using the optical fractionator probe. Cortical and VZ surface areas were estimated by tracing contours of cortical marginal zone or ventricular surface in Neurolucida and computing a product of average contour length, average measured section thickness, and the total number of cortical sections. Total cortical surface area included the neocortical and hippocampal primordia, bounded by the piriform cortex (laterally) and the corticoseptal boundary, cortical hem, or dentate gyrus, depending on the age and rostrocaudal level. VZ surface area encompassed the region bounded by the pallial-subpallial boundary and the corticoseptal boundary, cortical hem or dentate gyrus, depending on the age and rostrocaudal level.
For electroporation experiments, all counts were done double blind using Z-stack images with a depth of 12 μm acquired with a Zeiss Apotome running Axiovision 4.8. Ctip2+ and Satb2+ cells were counted double blind using multiple 250-μm-wide sampling sectors encompassing the whole cortical wall in the rostral and caudal cortex, bounded by and excluding the subplate and marginal zone. Cux1+ and ZFPM2+ cells were counted in 250 μm cortical columns in the lateral neocortex and a simple ratio computed. CldU/Cux1 and CldU/ZFPM2 double-positive cells were scored as CldU positive if the majority of the nucleus showed labeling.
Densitometry for NICD or Hes1 signal intensity was performed in 12 μm Z-stacks in NIH ImageJ. Individual nuclei of eGFP+ cells were outlined with an oval and the 8-bit red channel average signal intensity was measured. Background NICD or Hes1 signal intensity was measured from the remaining cells that did not express eGFP, and a ratio was computed, representing the departure from nearby cells signal level.
Statistical analysis.
A Student's t test or factorial ANOVA assessing the effect of either genotype alone or interaction of age * genotype was performed using Statview 4.0 on an iMac G4 (Apple) and used to compute significance levels for total cortical wall volume and number and density of each cell population. Error bars represent SE and a significance threshold of p < 0.05 was used.
Results
Fgf signaling in the Emx1 lineage is necessary for embryonic expansion of the cerebral cortex
Knock-out of FgfR signaling in the cerebral cortical primordium was achieved by targeted inactivation of FgfR1 and FgfR2 via Emx1Cre-mediated recombination in mice null for FgfR3. The Emx1Cre line (Iwasato et al., 2004) drives recombination in the dorsal telencephalon by E10.0 (Fig. 1A,B). Since FgfR4 is not expressed in the brain during cortical development (Ford-Perriss et al., 2001; Kang et al., 2009), this should produce embryos lacking functional FgfRs within the Emx1 lineage. −FgfR signaling was assessed using in situ hybridization for downstream Fgf target genes in the Ets family; at E11.5 we found an almost complete loss of Pea3 expression in Emx1;TKO embryos (Fig. 1C,D). In contrast, CoupTF1, which is normally repressed by FgfR signaling (Fukuchi-Shimogori and Grove, 2003; Garel et al., 2003), was strongly upregulated in the majority of the cortical primordium (Fig. 1E,F).
Figure 1.

Impaired surface area expansion in Emx1;TKO embryos. A, B, Recombination of Emx1Cre in R26R mice; green shows β-Gal+ cells within the Pax6+ cortical VZ (red) at E10.0. B is a higher-magnification view of the boxed region in A. C–F, Emx1;TKO embryos show a near complete loss of Pea3 mRNA expression in the cortical primordium (C, D), while CoupTF1 is upregulated (E, F). G–I, Developmental analysis from E11.5 to E18.5 showing severely reduced cortical volume and surface area after E11.5 (G). Cortical wall surface area and volume estimates are plotted in H and I. For cortical plate surface area across all ages from E11.5 to E18.5, a factorial ANOVA showed a main effect of genotype (F = 16.94; p = 0.0021) and age (F = 25.49; p < 0.0001) (n = 10 Emx1;TKOs and 11 littermate controls). For cortical wall volume a factorial ANOVA showed a main effect of genotype (F = 109.5; p < 0.0001), age (F = 395.8; p < 0.0001), and interaction of genotype * age (F = 50.2; p < 0.0001). LV, Lateral ventricle; NP, nasal process; CP, cortical primordium; MGE, medial ganglionic eminence; LGE, lateral ganglionic eminence; SE, septum; STR, striatum. Scale bar, 1 mm.
At E18.5, the dorsal telencephalon of Emx1;TKOs showed a 52% reduction in total volume and a 46% reduction in surface area (Fig. 1G–I). At all ages examined, the caudal neocortex and hippocampus showed a severe volume and surface area reduction similar to the rostral neocortex (Fig. 2A,B,E,F), suggesting that FgfR signaling is important both rostrally and caudally. To understand the developmental mechanisms of these defects, we quantified total volume and surface area of neocortex and hippocampus from E11.5 to E18.5 by unbiased stereological methods. At E11.5 the size of the cortical primordium of Emx1;TKOs appeared similar to controls, however, just 1 d later Emx1;TKO embryos showed a 50.2% reduction in surface area (Fig. 1G,H). Interestingly, the growth rates for cortical volume and surface area were similar for control and Emx1;TKOs after E12.5. For example, cortical volume exhibited a 13.7-fold increase from E12.5 to E18.5 in wild type mice and an 11.2-fold increase in Emx1;TKOs (Fig. 1H,I). These data indicate a major requirement for FgfR signaling in cortical surface area expansion between E11.5 and E12.5. As expected, ventral telencephalic development was essentially unaffected in the mutants (Figs. 1G, 2A,B,I,J).
Figure 2.
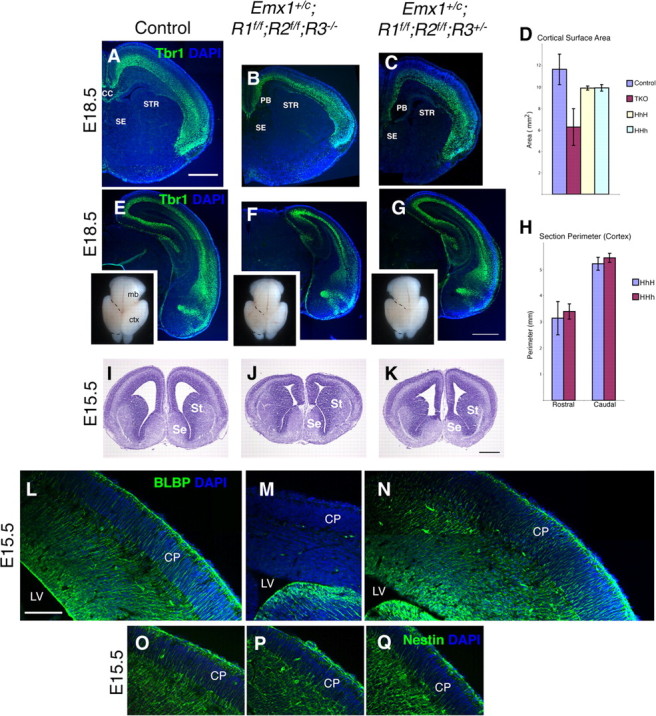
Substantial rescue of the Emx1;TKOs phenotype by restoration of one allele of FgfR2 or FgfR3. A–K, Tbr1-immunostained E18.5 coronal sections (A–C, E–G) (insets in E–G show whole brains; dorsal view) and cresyl violet-stained E15.5 sections (I–K) showing severely reduced cortical surface area in Emx1;TKOs compared with control littermates, and extent of rescue by restoration of one copy of FgfR3. The septum and striatum were similar in size in all embryos examined. D, H, Total cortical surface area (D) and rostral vs caudal section perimeter measurements (H) showed that restoration of one copy of FgfR2 or FgfR3 rescued the Emx1;TKO phenotype to a similar extent both rostrally and caudally. L–Q, Radial glia completely lacked BLBP expression in Emx1;TKOs (L, M), with rescue by the presence of a single copy of FgfR3 (N), although the radial glial scaffold appeared intact as assessed by Nestin staining (O–Q). ctx, Cerebral cortex; mb, midbrain; ST, striatum; CP, cortical plate; CC, corpus callosum; PB, Probst bundle; HhH, Emx1+/c; FgfR1f/f; FgfR2+/f; FgfR3−/−; HHh, Emx1+/c; FgfR1f/f; FgfR2f/f; FgfR3+/−. Scale bars: A–K, 1 mm; L–Q, 250 μm.
We next examined an allelic series that compared FgfR1/FgfR2 double mutants, FgfR3 single mutants, Emx1;TKOs, and the effects of restoring one allele of either FgfR2 or FgfR3. We found that the most severe defects were always present in the Emx1;TKOs (n = 16 Emx1;TKOs), and the presence of one copy of either FgfR3 or FgfR2 substantially rescued defects observed in Emx1;TKOs both rostrally and caudally (Fig. 2A,K; n > 9). Furthermore, restoring one allele of FgfR2 or FgfR3 did not result in differential rescue of rostral versus caudal cortical area (Fig. 2H), suggesting that FgfR2 and FgfR3 appear to work equivalently in these aspects of early cortical development.
We next investigated whether changes in cell death or proliferation might explain the phenotype of Emx1;TKO mice. Apoptotic cells expressing activated Caspase3 appeared to be slightly increased in Emx1;TKOs at E11.5 and E13.5, but the total number was insufficient to explain the loss in cortical volume (Fig. 3A–D). The density of phosphohistone H3 (PH3)+ cells, which are actively undergoing mitosis, and BrdU incorporation rates were also similar, suggesting that the overall rate of proliferation was not significantly altered (Fig. 3E–H).
Figure 3.
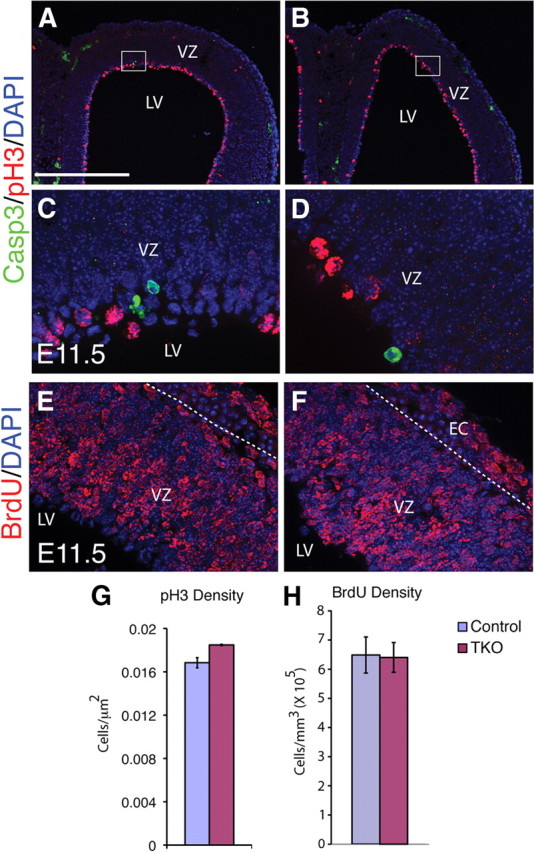
Caspase 3-mediated cell death and proliferation appear unchanged in Emx1;TKOs at E11.5. A–D, Isolated Caspase 3 (Casp-3)+ cells (green) were detectable in control (A, C) and Emx1;TKO embryos (B, D); however, they were extremely rare. C and D are high-power images of the boxed regions in A and B. In these brains we estimated 952 Casp-3+ cells per cortical hemisphere (Emx1;TKO) vs 624 cells in wild type. E–H, Positive cells were often found far from the lateral ventricles, in the SVZ or preplate, but some were also observed in the VZ. PH3 (red) labels mitotic cells primarily at the VZ surface at E11.5 in both control and Emx1;TKO embryos, and the number was only modestly different (G). BrdU incorporation at E10.5 was imaged and quantified after 1 d survival (E, F, H), and was nearly identical in both control and Emx1;TKO embryos (H). EC, ectoderm. Scale bars: A, B, 500 μm; C–F, 62.5 μm.
Precocious neuron production in Emx1;TKO mice curtails early expansion of the cortical primordium
The severe reduction in cortical plate volume observed in Emx1;TKOs was accompanied by a 52% reduction in the total number of cortical neurons immunoreactive for Tbr1 at E18.5, by the end of neurogenesis (Fig. 4F,M,N). Unexpectedly however, at E12.5 and E13.5, the rostrolateral telencephalon showed increased preplate/cortical plate thickness as assessed by βIII-tubulin and Tbr1 immunohistochemistry (by ∼50%; arrowheads in Fig. 4A,B,G,H). Therefore we quantified the overall number and density of cortical neurons produced at different stages of development.
Figure 4.
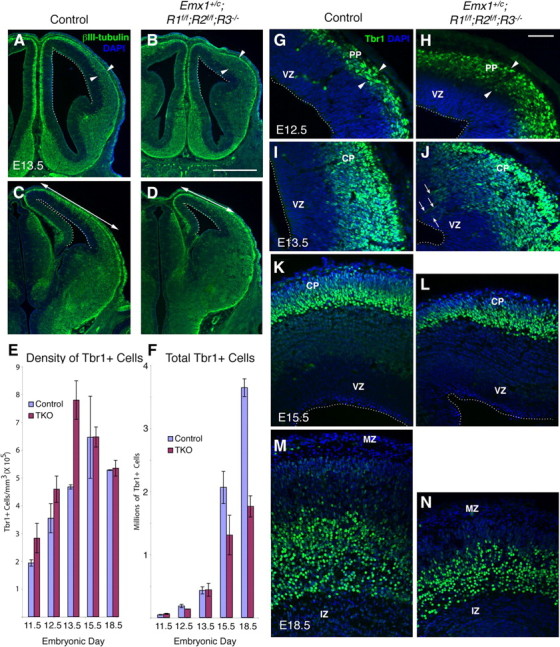
Early excess of cortical neurons and later deficit in Emx1;TKOs. βIII tubulin (A–D) and Tbr1 (G–N) immunostaining showing excess early cortical neurons at E12.5 (G, H) and E13.5 (I, J), and increased preplate/cortical plate thickness (arrowheads in A, B) in mutants compared with control littermates. By E15.5 (K, L) thinning of the cortical plate begins and the final number of Tbr1+ neurons is decreased by 51.7% in Emx1;TKOs at E18.5 (M, N). Stereological estimates of Tbr1+ cell density and total number are plotted in E and F, respectively. A factorial ANOVA showed a main effect of genotype (F = 45.7; p < 0.0001), age (F = 245.1; p < 0.0001), and interaction of genotype * age (F = 29.6; p < 0.0001) (n = 10 Emx1;TKOs and 11 littermate controls) on the total number of Tbr1+ neurons, and a main effect of genotype (F = 7.88; p < 0.017) and age (F = 16.18; p = 0.0001) on Tbr1+ cell density. PP, Preplate; MZ, marginal zone; IZ, intermediate zone. Scale bars: A–D, 1 mm; G–N, 125 μm.
Compared with littermate controls, we found a significantly increased density of Tbr1+ neurons in the preplate and cortical plate of Emx1;TKO mutants at E11.5, E12.5, and E13.5 (Fig. 4E,G–J). The total Tbr1 neuron number was similar among Emx1;TKOs and littermate controls at E13.5 despite the smaller cortical wall volume of the Emx1;TKOs (Fig. 4F). However, by E15.5, Emx1;TKOs began to show a deficit in the total number of these cells (Fig. 4K,L), with the greatest losses evident at E18.5 (Fig. 4F,M,N). This developmental analysis indicates an early overproduction of cortical neurons and a reduction in late embryogenesis.
Increased production of Tbr1+ neurons and Tbr2+ intermediate precursors in the VZ
Many excitatory cortical neurons are derived from Tbr2+ INPs in the SVZ, which in turn are generated in the VZ by asymmetric division of Pax6+ radial glial precursors (Noctor et al., 2001; Kowalczyk et al., 2009). Other excitatory cortical neurons are derived through direct neurogenesis in the VZ (Miyata et al., 2001; Noctor et al., 2004; Englund et al., 2005). To address whether early overproduction of cortical neurons is due to direct or indirect neurogenesis, or both, we first assessed the rates of Tbr2+ INP production in the VZ and SVZ. Stereological measurements indicated a substantial increase in the density of Tbr2+ cells in Emx1;TKOs at E12.5 and E13.5, followed by a sharp decrease at E18.5 (Fig. 5A–F,I). The excess Tbr2+ cells were present in the main body of the SVZ as well as in the VZ (Fig. 5A,B insets, C,D). The total Tbr2+ cell number was similar among Emx1;TKOs and littermate controls at E12.5–E13.5 despite the smaller cortical wall volume of the Emx1;TKOs (Fig. 5J). However, at E15.5 and after, Emx1;TKOs showed an increasingly severe deficit in number of Tbr2+ cells (Fig. 5G,H,J).
Figure 5.
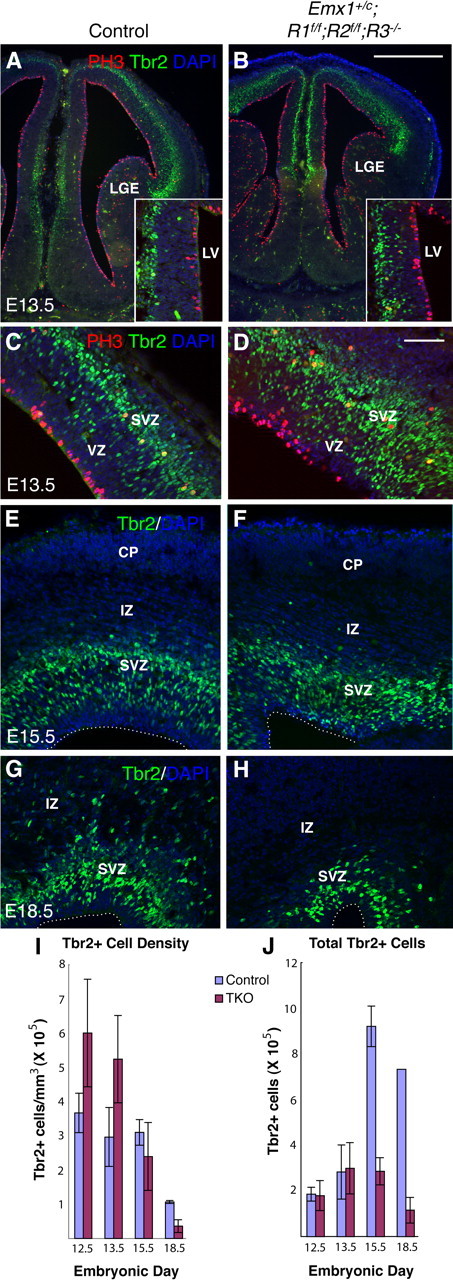
Emx1;TKO embryos show precocious production of INPs followed by SVZ depletion. A–H, Tbr2 immunohistochemistry revealing overproduction of INPs at E12.5–E13.5 (A–D) and increased Tbr2+ cell density in cingulate (inset in A, B) and lateral cortex (C, D) in Emx1;TKOs compared with littermate controls. Tbr2+ cells began to be depleted at E15.5 (E, F), showing an 84.2% deficit by E18.5 (G, H). Stereological quantification in A, J. A factorial ANOVA showed a main effect of genotype (F = 25.27; p = 0.0015), age (F = 9.78; p = 0.0067, n = 7), and an age * genotype interaction (F = 9.07; p = 0.0083) on the total number of Tbr2+ cells (n = 7 Emx1;TKOs and 8 littermate controls). Scale bars: A, B, 1 mm; C–H, 125 μm.
Emx1;TKO VZ volume was quantified using planimetry of the Pax6+ domain, and was only 52.8% that of control embryos (3.69 ± 0.36 × 108 μm3 vs 1.95 ± 0.053 × 108 μm3). In control littermates, only a tiny fraction of all Tbr1+ cells were found in the VZ between E11.5 and E13.5. In Emx1;TKO embryos we observed a 4.4-fold increase in the density of Tbr1+ cells in the VZ (436.4 ± 60.8%; Figs. 4I,J, 6A,B). The increase in density of Tbr2+ cells in the VZ was twofold (96.3 ± 26.5%; Fig. 5A–D). Together, these results suggest that the excess Tbr1+ cells in the Emx1;TKO cortical plate were derived both from Tbr2+ intermediate precursors as well as from increased Tbr1+ cortical neuron differentiation directly from cells in the VZ.
Figure 6.
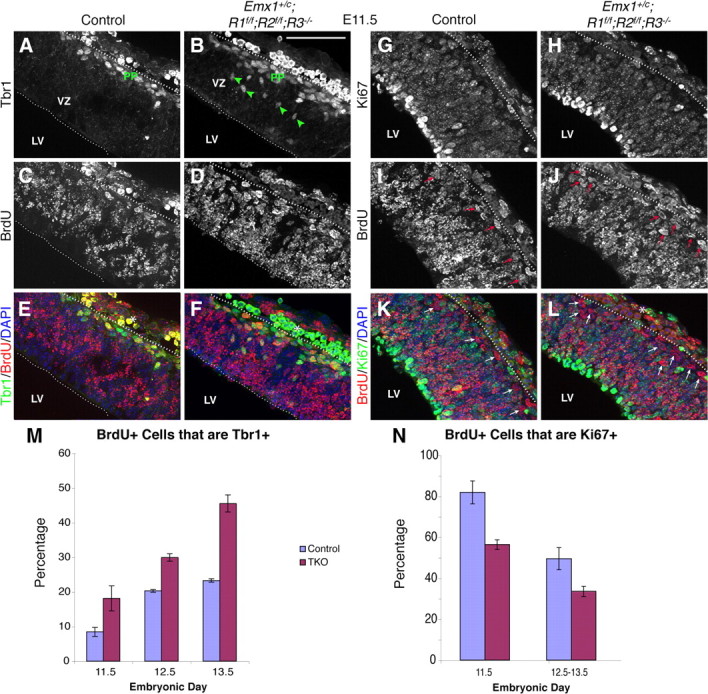
Increased cortical neurogenesis and decreased progenitor self-renewal in Emx1;TKO embryos. Dotted lines demarcate the cortical wall; analysis excluded the overlying head ectoderm and mesoderm. Tbr1/BrdU immunostaining (A–F) and Ki67/BrdU immunostaining (G–L) at E11.5, following BrdU incorporation at E10.5, with stereological quantification (M, N). The fraction of BrdU+ cells that colabeled for Tbr1 (E, F, yellow cells) was significantly increased in Emx1;TKOs as plotted in (M) for E11.5 through E13.5. A factorial ANOVA revealed effects of genotype (F = 53.898; p < 0.0001), age (F = 45.91; p < 0.0001) and interaction of genotype*age (F = 4.69; p = 0.045, n = 7 Emx1;TKOs and 7 control littermates). Green arrows in A and B indicate Tbr1+ cells in the VZ; PP, preplate. The fraction of BrdU+ cells that colabeled for Ki67, representing cells that reenter the cell cycle (K, L, yellow cells) was significantly decreased in Emx1;TKOs across age as plotted in (N) for E11.5–E13.5. A factorial ANOVA showed a main effect of genotype (F = 17.06; p = 0.006) and age (F = 21.90; p = 0.0017, n = 5 Emx1;TKOs, 6 control littermates). The number of cells that stained for BrdU only, representing cells that are likely postmitotic, was increased in Emx1;TKOs (compare I, J with K, L, arrows). Asterisks, Green and yellow cells in the ectoderm/mesenchyme are mainly red blood cells that cross-react with the Tbr1 antibody. Scale bar, 125 μm.
Increased cortical neurogenesis and decreased self-renewal of VZ precursors in embryos lacking FgfR signaling
Because we did not find evidence for decreased proliferation of progenitors in the VZ, we focused on an alternative hypothesis—namely, that early self-renewal of radial glial progenitors in the VZ is decreased in favor of neuronal differentiation. To quantify the number of neurons and progenitors generated at defined times in cortical development, we labeled mitotically active precursors using a pulse of BrdU at E10.5, E11.5, or E12.5 and assessed their fate 24 h later. Incorporation of BrdU in dividing precursors was combined with either Tbr1 or Ki67 staining to determine 1) rates of differentiation of Tbr1+ cells, and 2) cell cycle reentry/exit, respectively. At each age, we found a significant increase in the relative number of Tbr1+/BrdU+ cells (up to 50% at E13.5), indicating elevated production of Tbr1+ neurons in Emx1;TKOs (Fig. 6A–F,M). In concert, the proportion of BrdU+ cells that were BrdU+/Ki67+ at E11.5 changed from 81.9% (±5.6) in control embryos to 56.4% (±2.3) in Emx1;TKOs. At E12.5–E13.5, a similar decrease in the proportion of BrdU+/Ki67+ cells was found, from 49.5% (±5.37) in control embryos to 33.6% (±2.5) in Emx1;TKOs (Fig. 6G–L,N). Correspondingly, the number of BrdU+/Ki67− cells was increased in the cortical plate of Emx1;TKO embryos, confirming excess exit of precursors from the mitotic cycle (Fig. 6J,L, arrows).
Precocious differentiation of excitatory neurons in the absence of FgfR signaling depletes radial progenitors in the VZ
To investigate the dynamics of radial progenitors and committed neuronal precursors in the VZ, we assessed the expression of Pax6 and Ngn2—transcription factors that characterize radial glial cells and committed neuronal progenitors, respectively. The number of Pax6+ progenitors in the dorsal telencephalon was decreased by at least 50% in Emx1;TKOs from E12.5 to E15.5 (Fig. 7A–D,M). However, by E18.5, the total number of Pax6+ cells in the cortical wall of Emx1;TKO was depleted by ∼79% compared with control embryos (Fig. 7E,F,M). Correspondingly, the Emx1;TKO VZ showed an increasingly severe deficit in ventricular surface area (−50.6% at E12.5; −65.3% at E18.5, p = 0.0005 by factorial ANOVA; n = 7 Emx1;TKO, 8 control littermates) (Fig. 7N) and a progressive decrease in VZ thickness from E13.5 onward. In control embryos, the Pax6+ fraction of cells in the neocortical wall decreased between E12.5 and E18.5; in Emx1;TKOs this fraction decreased more rapidly (Fig. 7O). This depletion of Pax6+ progenitors did not disrupt the radial glial scaffold, although the density of Nestin+ radial fibers traversing the cortical plate at E15.5 appeared somewhat decreased (Fig. 2O,P).
Figure 7.
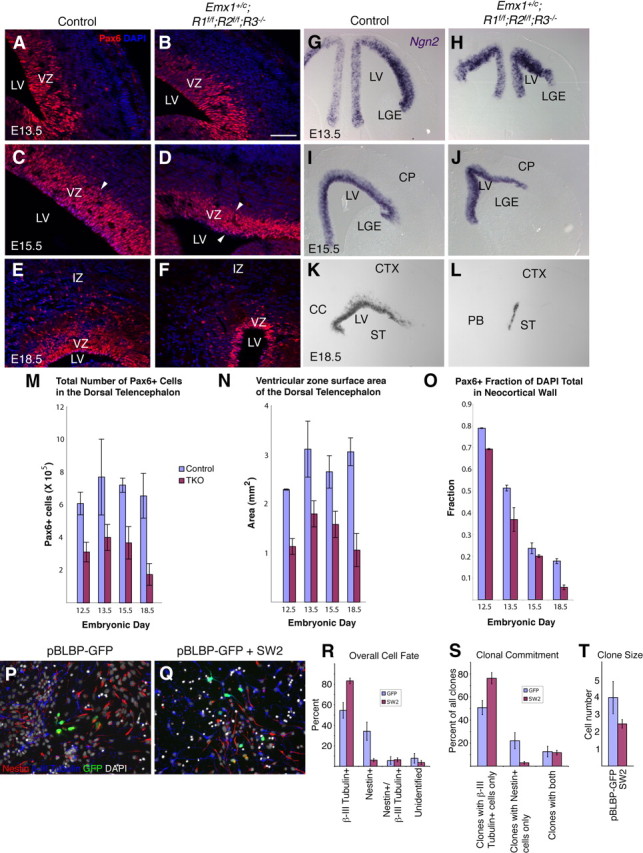
Depletion of VZ progenitors leads to an early termination of cortical neurogenesis. Pax6 immunostaining in control (A, C, E) or Emx1;TKO (B, D, F) embryos showing a progressive decrease in VZ thickness in Emx1;TKOs, confirmed by stereological estimates of the total number of Pax6+ cells in the dorsal telencephalon (neocortex and hippocampus) (M). A factorial ANOVA showed a main effect of genotype for decreased Pax6+ cells: F = 21.24; p = 0.0025 (n = 7 Emx1;TKOs and 8 littermate controls). VZ surface area, estimated using Neurolucida, was also strongly reduced as plotted in N (factorial ANOVA: F = 36.21; p = 0.0005). The fraction of Pax6+ cells in the DAPI total measured in 250-μm-wide sectors of dorsolateral neocortex showed a more rapid decrease in Emx1;TKOs than in controls (factorial ANOVA: genotype, F = 301.86; p < 0.0001) (O). Ngn2 in situ hybridization (G–L) labels the cortical VZ, showing an increase in staining at E13.5 and severe depletion by E18.5. Radial glia electroporated in utero at E11.5 with pBLBP-eGFP alone (P) or together with dominant-negative FgfR (SW2) (Q) and cultured for 3 d in vitro showed that attenuated FgfR signaling increases the number of eGFP+/βIII tubulin+ neurons (R) as well as the proportion of clones exclusively composed of eGFP+/βIII tubulin+ cells (S) (t test; p = 0.003), reduces clone size (T) and decreases the proportion of clones composed only of eGFP+/Nestin+ cells (S) (t test; p = 0.005). A two-way ANOVA revealed a statistically significant interaction of neurogenesis rate and clone size; F = 5.12, p = 0.0378 (n = 4 control eGFP; n = 6 SW2). Arrowheads indicate thickness of the VZ in C and D. Scale bars: A–F, P, Q, 125 μm; G–L, 500 μm.
In parallel to the decrease in Pax6+ cells of the VZ, we observed an increase in Ngn2-expressing cells at E13.5 within the VZ (Fig. 7G,H), suggesting an early increase in committed neuronal precursors. However, Ngn2 staining was drastically curtailed by E18.5, indicating a premature end to the neurogenic period (Fig. 7K,L). Expression of Par3, a gene implicated in promoting progenitor self-renewal (Costa et al., 2008), did not appear to be substantially altered at the ventricular surface at E13.5 (data not shown).
Increased neuronal differentiation in clones of progenitors receiving dominant-negative FgfR
To more directly analyze the role of FgfRs in neuronal differentiation of radial progenitors in the cortical VZ, we electroporated a dominant-negative FgfR construct, SW2 (Werner et al., 1993; Shin et al., 2004) driven by the BLBP promoter, together with the control plasmid pBLBP-eGFP at E11.5 and cultured eGFP+ cells, assessing clonal fate after 3 d in vitro. In this experiment, cell lineage as well as the proportion of neurogenic vs proliferative divisions could be measured (Fig. 7P–T). We quantified the average clonal size and the proportion of eGFP+ cells expressing βIII tubulin or Nestin, and found a substantially increased proportion of neurons in clones deriving from radial glial cells receiving the dominant-negative FgfR construct, SW2, compared with those receiving the control plasmid alone (Fig. 7P–R). Furthermore, radial glial cells receiving SW2 generated an increased number of clones committed to generate only neurons and suppressed the development of radial glial cell-only clones (Fig. 7S). A lower clonal size correlated with increased neuronal fate (Fig. 7T), indicating that SW2 caused an increase in radial progenitor cell cycle exit and neuronal differentiation.
Accelerated cortical layer development in Emx1;TKOs
Because progenitors in both the VZ and SVZ were depleted more rapidly in Emx1;TKOs during the neurogenic phase, we hypothesized that the laminar structure of the cerebral cortex might be altered. Cortical plate thickness was significantly reduced at E18.5, although less dramatically than cortical surface area (Fig. 8). We assessed the relative proportion of cells that had acquired a specific layer fate after immunostaining for layer-specific markers and quantification in 250 μm sectors within the dorsolateral neocortex. Quite surprisingly, we observed no significant difference in the ratio of Cux1+ to ZFPM2+ neurons, marking layers 2/3 and 6/SP, respectively (Fig. 8A–C). Immunohistochemistry for Ctip2 and Satb2, marking neurons of layers 5 and 2/3, respectively, further showed that Emx1;TKOs retained similar cortical layer structure as control littermates (Fig. 8D–F).
Figure 8.
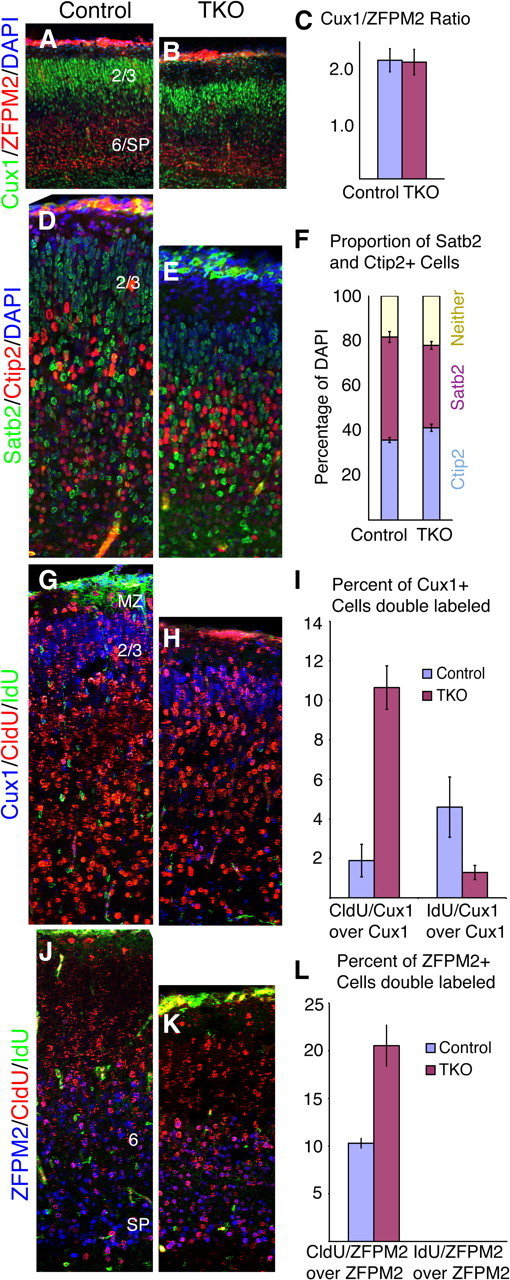
Cortical layer development in Emx1;TKO embryos. Control (A, D, G, J) or Emx1;TKO (B, E, H, K) embryos processed for the indicated layer markers show similar layer structure. Overall layer order appeared normal, as was the ratio of Cux1 and ZFPM2+ cells (A–C) (n = 3 Emx1;TKOs and 3 littermate controls; t test: p = 0.92). There was a modest imbalance toward an increase in Ctip2 in Emx1;TKO embryos, reflecting greater early (deep layer) cortical neurogenesis (D–F) (n = 3 controls, 3 Emx1;TKOs; two-way ANOVA assessing interaction of genotype * layer population; p = 0.05). However, both Cux1+ (G–I) and ZFPM2+ (J–L) cells were more likely to have been born on E12.5 (CldU+) than E16.5 (IdU+) in Emx1;TKO embryos, whereas fewer Cux1+ cells were born at E16.5 in Emx1;TKO embryos compared with littermate controls (G–I) (two-way ANOVA assessing interaction of genotype * Cux1+ birthdate: F = 33.49; p = 0.0004; n = 3 Emx1;TKOs, n = 3 control littermates). As expected, of over 7500 cells counted, no ZFPM2+ cell was IdU+ in either controls or Emx1;TKOs (J–L). SP, Subplate; 2/3, cortical layers 2/3; 6, cortical layer 6. Scale bar, 125 μm.
We next assessed the schedule of cortical neurogenesis in Emx1;TKO embryos by labeling dividing precursors at E12.5 and E16.5 with CldU and IdU, respectively, and examining the cortex at E18.5. Most CldU+ cells were observed in deep cortical layers in control mice, whereas the IdU+ cells were seen in upper layers, migrating through the intermediate zone or still resident in the VZ/SVZ (Fig. 8G,H; data not shown). In Emx1;TKOs we found an increased density of CldU+ cells (born on E12.5) in the cortical plate and an almost complete disappearance of IdU+ cells (born on E16.5) in the cortical wall (Fig. 8H,K), suggesting a shortened neurogenic period. Together, these findings suggest increased immediate differentiation of E12.5 VZ precursors in the absence of FgfR, rather than continued self-renewal.
To quantify the birthdate of neurons in different cortical layers, we performed triple immunolabeling for CldU/IdU/Cux1 or CldU/IdU/ZFPM2. In Emx1;TKOs we found a dramatic (5-fold) increase in the proportion of upper layer Cux1+ neurons colabeled with CldU, and a substantial decrease IdU colabeling (Fig. 8G–I). Expectedly, an increased proportion of ZFPM2+ neurons was labeled with CldU, but none with IdU, reflecting increased neurogenesis at E12.5 (Fig. 8J–L). These results demonstrate that a greater share of cortical layer fates is generated during the early neurogenic period in Emx;TKOs.
Notch acts downstream of FgfR signaling in cortical neurogenesis
Like FgfR, Notch signaling also inhibits neuronal differentiation and promotes radial glial identity in the cortical primordium (Gaiano et al., 2000; Faux et al., 2001; Wahl et al., 2007; Shimojo et al., 2008). To determine whether Notch activity could be affected in Emx1;TKO mutants, we first examined the production of cleaved (active) Notch (NICD). Immunohistochemistry using an antibody specific for NICD reveals puncta mostly corresponding to NICD-containing nuclei (Fig. 9A). In E11.5–E12.5 Emx1;TKO mutants there was decreased NICD staining in the cortical VZ and each cell on average appeared to show fewer puncta compared with controls (Fig. 9B). In situ hybridization for Notch1 indicated a decrease in Notch1 mRNA in Emx1;TKOs, potentially explaining the decreased NICD staining (Fig. 9C–F).
Figure 9.
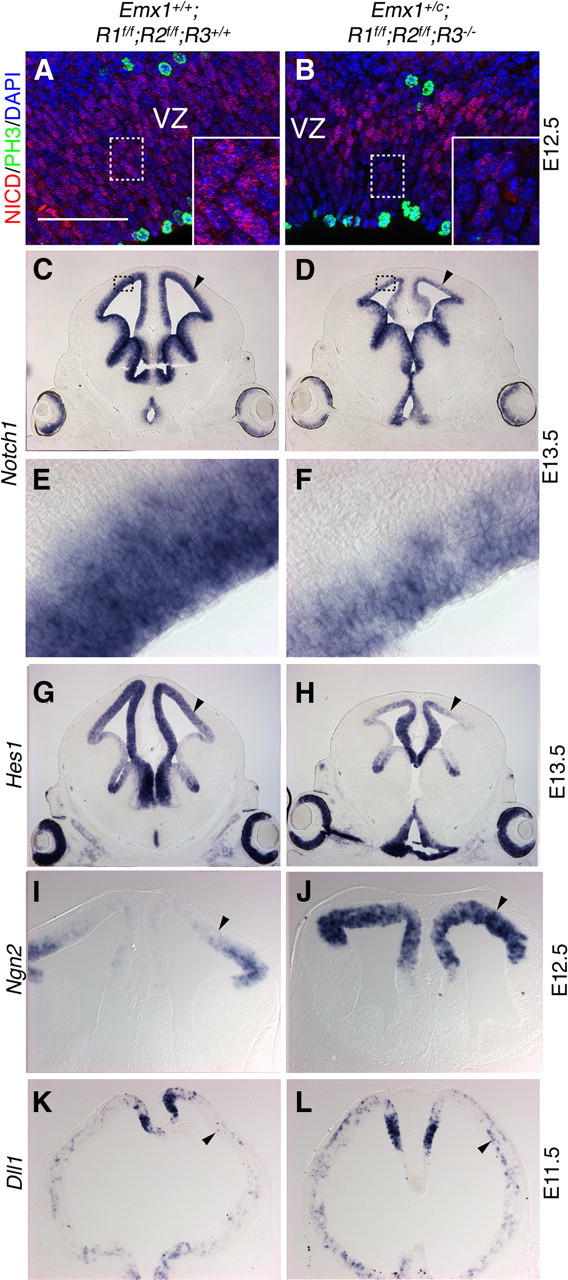
FgfR signaling regulates Notch and proneural gene expression in cortical neurogenesis. NICD staining of intracellular puncta was reduced in Emx1;TKO embryos (A, B), suggesting that Notch production and/or cleavage is impaired. Insets (A, B) are high-magnification images of the boxed regions. In situ hybridization for Notch1 demonstrated decreased expression in the cortical VZ of Emx1;TKO embryos at E13.5 (C–F). E, F, High-power images of the boxed regions in C, D. Analysis of Notch downstream targets showed decreased Hes1 (G, H), increased Ngn2 (I, J), and increased Dll1 (K, L) mRNA production as early as E11.5 in the cortical VZ (arrowheads) during the period of increased early neurogenesis in Emx1;TKOs. (M, N). Scale bars: A, B, E, F, 125 μm; C, D, G, H, 2 mm; I–L, 1 mm.
Next we assessed the expression of Notch target genes Hes1, Ngn2, Mash1, and Dll1 by in situ hybridization. In Emx1;TKOs we found substantially decreased expression of Hes1 in the cortical VZ at E13.5 (Fig. 9G,H), indicating decreased Notch downstream activity. Corresponding to decreased Hes1 expression, we detected increased expression of Ngn2, Dll1, and Mash1, which are inhibited by Hes gene products, in the dorsal telencephalon at E11.5–E12.5 (Fig. 9I–L; data not shown). BLBP, a marker of radial glia and direct downstream target of Notch signaling (Anthony et al., 2005), was completely absent from cortical radial glia in Emx1;TKOs at E15.5, but restoration of one copy of FgfR3 rescued BLBP expression (Fig. 2L–N).
To directly test the relation between FgfR and Notch signaling, we used in utero electroporation of the SW2 dominant-negative FgfR construct at E11.5 to decrease FgfR activity and assess NICD and Hes1 expression in vivo. In all electroporations, pBLBP-eGFP was coelectroporated to visualize radial glial progenitors and their progeny (Fig. 10). Using densitometric measurements of the intensity of NICD and Hes1 protein in eGFP+ cells, we found that SW2 electroporation decreased both NICD and Hes1 expression, suggesting that physiological FgfR signaling maintains Notch activity cell autonomously (Fig. 10A–F).
Figure 10.
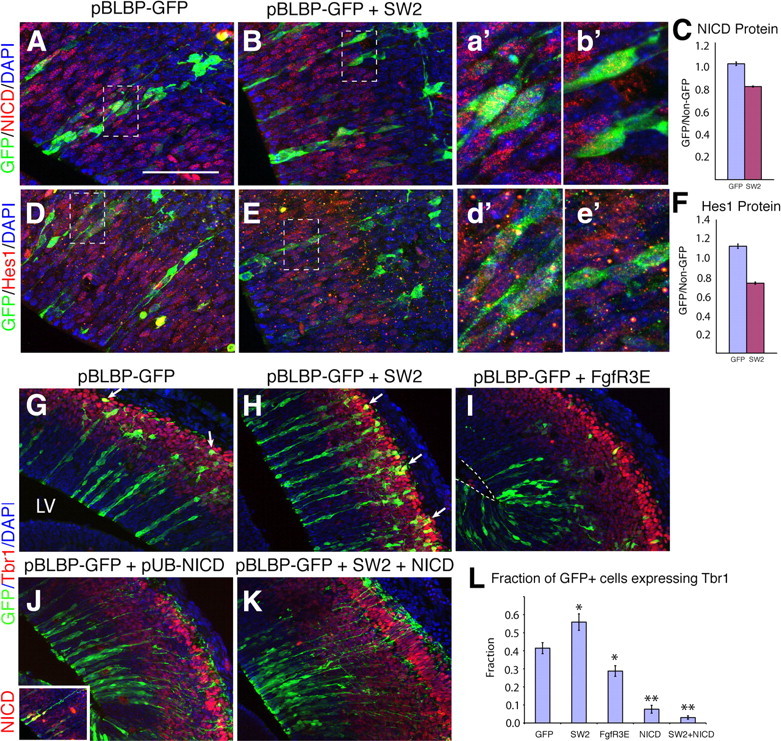
Notch is downstream of FgfR signaling in cortical neurogenesis. A–E, In utero electroporation at E11.5 of pBLBP-eGFP, alone (A, D) or together with a dominant-negative FgfR (SW2) (Werner et al., 1993; Shin et al., 2004) (B, E), was coupled with immunocytochemical detection and densitometric measurements of NICD (A, B) or Hes1 (D, E) proteins to determine cell autonomous effects of attenuating FgfR signaling. a′, b′, d′, and e′ are higher-magnification views of A, B, D, and E, respectively. Both NICD and Hes1 protein levels were significantly reduced in eGFP+ cells compared with nearby cells (C, F). G–L, Tbr1 immunostaining in eGFP+ cells electroporated with pBLBP-eGFP alone (G) or together with SW2 (H), constitutively active FgfR (FgfR3E) (I), constitutively active Notch (pUB-NICD) (J), or SW2 + NICD (K), demonstrating that SW2 increased cortical neurogenesis and NICD virtually abolished it. However, SW2 was unable to reverse the effects of NICD, suggesting that FgfR signaling is upstream of NICD in cortical neurogenesis. Overexpression of NICD was confirmed in NICD-electroporated eGFP+ cells by double immunostaining (inset in J). The number of pBLBP-eGFP+ cells (radial glia and their progeny) coexpressing the neuronal marker, Tbr1 is quantified in (L). *p < 0.05, **p < 0.0005 compared with pBLBP-eGFP alone, Student's t test. Scale bars: A, B, D, E, 125 μm; a′, b′, d′, e′, 40 μm; G–K, 250 μm; J inset, 400 μm.
We further used in utero electroporation to determine whether the FGF and Notch pathways could interact in regulating neurogenesis in vivo. Electroporation of SW2 caused excess genesis of Tbr1+ neurons (Fig. 10G,H,L), similar to Emx1;TKO embryos. This confirms a likely cell-autonomous role for FgfR signaling in inhibiting neurogenesis. Conversely, electroporation of constitutively active forms of FgfR (FgfR3E or FgfR3M) (Iwata et al., 2001) caused a reduction in neurogenesis and retention of eGFP+ cells within the VZ (Fig. 10I,L). Electroporation of a plasmid containing active Notch (NICD) caused a similar but more severe effect; most of these cells remained in the VZ and did not acquire Tbr1 expression (Fig. 10J,L). Thus, activation of FgfR caused a partial phenocopy of NICD activity. To determine whether Notch and FgfR signaling are in the same or separate pathways controlling cortical neurogenesis, we coelectroporated the SW2 and NICD constructs. If the pathways were separate, an intermediate phenotype is expected. However, if one phenotype overrides the other, then an epistatic relationship exists and the two pathways can be linked. We observed no significant difference between the NICD and the SW2/NICD coelectroporated groups (Fig. 10J–L), indicating that NICD blocked the effect of SW2 and hence Notch signaling lies downstream of FgfR activity.
Discussion
One of the major unanswered questions in cortical development is what causes progenitors to cease self-renewing and begin producing cortical neurons, and whether such a mechanism may regulate cortical surface area (Rakic, 1995). Our findings provide a partial answer, linking one of the major embryonic signaling systems (Fgfs) to the Notch pathway—a known regulator of neurogenesis.
We found that the loss of FgfR signaling decreased cortical surface area, duration of neurogenesis and the final number of cortical neurons, but with little change in cortical layer structure. This stands in contrast with many other demonstrations of altered rates of neurogenesis, where the cortical plate shows various forms of dysplasia and cellular heterotopias, suggesting interference with cell adhesion and migration (Chenn and Walsh, 2003; Louvi et al., 2004; Imayoshi et al., 2010).
In Emx1;TKO mutants, radial glial stem cells in the early cortical VZ differentiated into cortical neurons, rather than continuing to self-renew. This increased rate of differentiation began to deplete VZ progenitors during the cortical expansion phase, before E12.5, leading to an early neurogenic peak, but curtailment of overall neurogenic potential. These changes stunted the tangential growth of the cortical scaffold and caused a premature decrease in ventricle size and a final decrease in the surface area (and, to a lesser degree, thickness) of the cortical plate. Thus, FgfR signaling normally tempers the early rate of cortical neurogenesis so that VZ precursor expansion may physically enlarge the surface of the cortical scaffold, lengthening the neurogenic period and ultimately achieving a larger cortical structure. Despite equally severe rostral and caudal surface area deficits, caudalization of the Emx1;TKO cortical primordium, likely due to the loss of rostralizing FgfR signaling (Fukuchi-Shimogori and Grove, 2001; Garel et al., 2003), was suggested by a rostral shift in the expression domain of CoupTF1. Thus, overall Fgf signaling appears to carry two separate activities—one regulating area patterning and the other promoting cortical surface area growth.
The schedule of cortical plate surface area growth is severely stunted in Emx1;TKO embryos between E11.5 and E12.5, suggesting that during this time, most available progenitor divisions in the VZ were neurogenic. Indeed, an in vitro clonal analysis showed that radial precursors expressing a dominant-negative FgfR were more likely to stop the process of self-renewal and generated an increased number of neurons, further suggesting that FgfR acts in a cell-autonomous fashion. However, ventricular surface area did grow between E12.5 and E13.5, suggesting that loss of FgfR signaling does not completely halt self-renewal. Indeed other factors undoubtedly contribute in this process. For example Wnt ligands secreted from the cortical hem (Grove et al., 1998) might be able to partially compensate for the loss of FgfR signaling and it remains unknown how this signaling system responds to loss of FgfR signaling. Future work should examine how such pathways may be deregulated.
FgfR signaling slows the temporal progression of cortical layer fates
Our findings reveal that FgfR signaling promotes cortical surface area expansion without causing major alterations in cortical layer structure. This implies that FgfRs are major regulators of neurogenesis in that their action occurs largely before, and independently of, layer commitment. That the cortical laminar pattern can be determined with little dependence on surface area growth parallels the diversity of the cortical structure in mammalian species, as the cortical surface varies 1000-fold while thickness and layer pattern are much more conserved (Rakic, 1995). Cortical development normally relies on the sequential production of each laminar cell type. In Emx1;TKO embryos the cortical structure probably also follows an ‘inside-out’ pattern of development, but a much greater proportion of the cortical layers is derived from neurons that were born early in the process, suggesting that the process of layer determination can be influenced by the initial size of the stem cell pool. These results further suggest that FgfR signaling by delaying neurogenesis also slows the specification of cortical layers, a process that is apparently independent of birthdate, and could be determined at the stem cell level, within neuronal progenitors, or perhaps within early postmitotic neurons, during radial migration. Such mechanisms may maintain normal layer structure in larger, more slowly-developing cortices such as those in carnivores and primates.
Notch is downstream of FgfR signaling in cortical neurogenesis
Like FgfR signaling, Notch is also required for the maintenance of radial glial stem cells and the repression of neurogenesis in the cortical primordium, and indeed, the two pathways have been suggested to interact in some way in vitro (Faux et al., 2001; Yoon et al., 2004). Our results suggest a clear hierarchy in this interaction, as Emx1;TKO embryos showed a decrease in production of active NICD, Notch1 mRNA, and the Notch downstream gene Hes1, together with an upregulation of Ngn2, Dll1 and Mash1. The complete loss of BLBP, a direct target of Notch (Anthony et al., 2005), in cortical radial glial cells of Emx1;TKO embryos underscores the cooperation of FgfR and Notch signaling in maintaining radial glial precursor identity.
Downregulation of FgfR signaling in normal embryos using dominant-negative FgfR (SW2) produced measurable, cell autonomous changes in NICD and Hes1 expression correlating with increased neurogenesis, mimicking the Emx1;TKO. The simultaneous activation of Notch revealed a crucial connection—namely that Notch activation overrides changes due to manipulation of FgfR activity. Together with a partial phenocopy of NICD activation by constitutively active FgfR (FgfR3E), these data indicate that FgfR and Notch cooperate to maintain radial glial self-renewal in the cortical VZ, and that Notch lies downstream of FgfR.
Mutations in the Notch pathway have produced a range of defects not limited to changes in self-renewal and differentiation, but also including cell adhesion and migrational defects leading to cortical dysmorphology (Hatakeyama et al., 2004; Imayoshi et al., 2010). The Emx1;TKO embryos recapitulate the self-renewal defects but not the others. This might be due to incomplete knockdown of the Notch pathway or, alternatively, may indicate that FgfRs selectively engage a restricted self-renewal function for Notch. Although the downregulation of Notch message in the VZ of Emx1;TKO embryos and the cell-autonomous downregulation of NICD within cells with attenuated FgfR signaling may suggest that FgfR signaling promotes Notch transcription directly, we cannot exclude an effect more downstream in the Notch pathway (i.e., at the level of Hes genes, CBF1, or Rbpj). For example, loss of FgfR signaling in newly committed INPs might have a further effect on neurogenesis via Notch downstream effectors, as a CBF1-independent role for Notch signaling in INPs has also been reported (Mizutani et al., 2007). However, the early excess of INPs observed in Emx1;TKOs may be attributed to increased differentiation of radial glial precursors and need not be due to a separate role for FgfR in INPs.
Diversity of function in the FgfR signaling system
The ablation of FgfR1 and FgfR2 driven by the hGFAP-Cre line beginning from E13.5, with or without a concomitant loss of FgfR3, results in decreased progenitor self-renewal and excess production of INPs (Kang et al., 2009; Stevens et al., 2010) but produces only a modest decrease in cortical wall volume and surface area. The comparatively mild surface area phenotype of hGFAP;TKOs or hGFAP;FgfR1;FgfR2 DKOs (Kang et al., 2009; Stevens et al., 2010) contrasts with the strong surface area deficit observed already at E12.5 in Emx1;TKO mutants. This further indicates that the major cortical expansion phase driven by FgfR signaling is already completed before E13.5. Indeed, Fgf ligands and receptors are more highly expressed in the telencephalon at early stages of development (Vaccarino et al., 1999a,b; Fukuchi-Shimogori and Grove, 2001; Grove and Fukuchi-Shimogori, 2003; Rash and Grove, 2007; Sahara and O'Leary, 2009).
The highly localized sources of Fgf ligands and their potentially different roles add complexity to the story, as they exhibit preferential activity for certain FgfRs. In particular, Fgf2 increases the initial number of cortical stem cells (Vaccarino et al., 1999b; Raballo et al., 2000) and Fgf8 promotes neural stem cell self-renewal (Borello et al., 2008). As Fgf2 binds all FgfR expressed in the telencephalon and Fgf8 binds predominantly FgfR2 and FgfR3, these two ligands may exert mutually compensatory roles in cerebral cortical growth. However, Fgf15 was shown to increase cortical neurogenesis, opposing Fgf8 function (Borello et al., 2008), and Fgf10 knock-out embryos showed delayed cortical neurogenesis, accumulation of undifferentiated radial glial progenitors, and elevated production of Tbr2+ and Tbr1+ cells (Sahara and O'Leary, 2009). Because our mutant embryos likely represent a near-complete loss of FgfR signaling in the cortical primordium, it is possible that the opposing action of some Fgf ligands in the brain may occur indirectly or be transduced primarily through noncanonical pathways.
Our analysis of the full allelic series of Fgf receptor mutants shows substantial rescue of the Emx1;TKO phenotype by only a single copy of either FgfR2 or FgfR3. This indicates a high degree of sensitivity to Fgf activity, permitting a broad range of small changes to incrementally modify local areal expansion rates and local cortical thickness. The highly variable spatiotemporal expression patterns of telencephalic Fgf ligands, including Fgf8, 17, 18, 15, 10, 3, 2, and receptors provide multiple points of regulation available to mammals for modification of the cortex over the course of evolution. Indeed it remains to be seen what role each Fgf ligand may have in these processes. We speculate that this underlying molecular structure is sufficient to explain much of the diversity in the size of the cerebral cortex and its constituent functional areas in different species. Further evolutionary studies of cortical development, as well as studies of pathological conditions relating to cortical growth, should pursue possible mechanistic origins of surface area and cortical structure diversity in the Fgf signaling pathway.
Footnotes
This work was supported by NIH Grants R01 MH067715 (F.M.V.) and T32 MH18268 (B.G.R.) and by a Brown-Coxe postdoctoral fellowship (B.G.R.). We thank Dr. Takuji Iwasato for the Emx1-Cre mice, Dr. Yas Ohkubo for plasmid construction, Dr. Karen Smith for teaching stereology, Shawna Ellis and Catherine Levine for excellent technical help, Allyson Vermaak for excellent surgical assistance, Dr. Natalina Salmaso for help with statistical analysis, Dr. Ryoichiro Kageyama for the Hes1 antibody, Dr. Tomoko Iwata for the gift of dominant active FgfR3 plasmids, and Drs. Nenad Sestan, Karen Smith, and Hanna Stevens for helpful discussions.
References
- Anthony TE, Mason HA, Gridley T, Fishell G, Heintz N. Brain lipid-binding protein is a direct target of Notch signaling in radial glial cells. Genes Dev. 2005;19:1028–1033. doi: 10.1101/gad.1302105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45:207–221. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Borello U, Cobos I, Long JE, McWhirter JR, Murre C, Rubenstein JL. FGF15 promotes neurogenesis and opposes FGF8 function during neocortical development. Neural Dev. 2008;3:17. doi: 10.1186/1749-8104-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britanova O, de Juan Romero C, Cheung A, Kwan KY, Schwark M, Gyorgy A, Vogel T, Akopov S, Mitkovski M, Agoston D, Sestan N, Molnár Z, Tarabykin V. Satb2 is a postmitotic determinant for upper-layer neuron specification in the neocortex. Neuron. 2008;57:378–392. doi: 10.1016/j.neuron.2007.12.028. [DOI] [PubMed] [Google Scholar]
- Chen B, Schaevitz LR, McConnell SK. Fezl regulates the differentiation and axon targeting of layer 5 subcortical projection neurons in cerebral cortex. Proc Natl Acad Sci U S A. 2005;102:17184–17189. doi: 10.1073/pnas.0508732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenn A, Walsh CA. Increased neuronal production, enlarged forebrains and cytoarchitectural distortions in beta-catenin overexpressing transgenic mice. Cereb Cortex. 2003;13:599–606. doi: 10.1093/cercor/13.6.599. [DOI] [PubMed] [Google Scholar]
- Cholfin JA, Rubenstein JL. Patterning of frontal cortex subdivisions by Fgf17. Proc Natl Acad Sci U S A. 2007;104:7652–7657. doi: 10.1073/pnas.0702225104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa MR, Wen G, Lepier A, Schroeder T, Götz M. Par-complex proteins promote proliferative progenitor divisions in the developing mouse cerebral cortex. Development. 2008;135:11–22. doi: 10.1242/dev.009951. [DOI] [PubMed] [Google Scholar]
- Coumailleau F, Fürthauer M, Knoblich JA, González-Gaitán M. Directional Delta and Notch trafficking in Sara endosomes during asymmetric cell division. Nature. 2009;458:1051–1055. doi: 10.1038/nature07854. [DOI] [PubMed] [Google Scholar]
- Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 2005;25:247–251. doi: 10.1523/JNEUROSCI.2899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faux CH, Turnley AM, Epa R, Cappai R, Bartlett PF. Interactions between fibroblast growth factors and Notch regulate neuronal differentiation. J Neurosci. 2001;21:5587–5596. doi: 10.1523/JNEUROSCI.21-15-05587.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford-Perriss M, Abud H, Murphy M. Fibroblast growth factors in the developing central nervous system. Clin Exp Pharmacol Physiol. 2001;28:493–503. doi: 10.1046/j.1440-1681.2001.03477.x. [DOI] [PubMed] [Google Scholar]
- Fukuchi-Shimogori T, Grove EA. Neocortex patterning by the secreted signaling molecule FGF8. Science. 2001;294:1071–1074. doi: 10.1126/science.1064252. [DOI] [PubMed] [Google Scholar]
- Fukuchi-Shimogori T, Grove EA. Emx2 patterns the neocortex by regulating FGF positional signaling. Nat Neurosci. 2003;6:825–831. doi: 10.1038/nn1093. [DOI] [PubMed] [Google Scholar]
- Gaiano N, Nye JS, Fishell G. Radial glial identity is promoted by Notch1 signaling in the murine forebrain. Neuron. 2000;26:395–404. doi: 10.1016/s0896-6273(00)81172-1. [DOI] [PubMed] [Google Scholar]
- Garel S, Huffman KJ, Rubenstein JL. Molecular regionalization of the neocortex is disrupted in Fgf8 hypomorphic mutants. Development. 2003;130:1903–1914. doi: 10.1242/dev.00416. [DOI] [PubMed] [Google Scholar]
- Götz M, Barde YA. Radial glial cells defined and major intermediates between embryonic stem cells and CNS neurons. Neuron. 2005;46:369–372. doi: 10.1016/j.neuron.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Grove EA, Fukuchi-Shimogori T. Generating the cerebral cortical area map. Annu Rev Neurosci. 2003;26:355–380. doi: 10.1146/annurev.neuro.26.041002.131137. [DOI] [PubMed] [Google Scholar]
- Grove EA, Tole S, Limon J, Yip L, Ragsdale CW. The hem of the embryonic cerebral cortex is defined by the expression of multiple Wnt genes and is compromised in Gli-3 deficient mice. Development. 1998;125:2315–2325. doi: 10.1242/dev.125.12.2315. [DOI] [PubMed] [Google Scholar]
- Hatakeyama J, Bessho Y, Katoh K, Ookawara S, Fujioka M, Guillemot F, Kageyama R. Hes genes regulate size, shape and histogenesis of the nervous system by control of the timing of neural stem cell differentiation. Development. 2004;131:5539–5550. doi: 10.1242/dev.01436. [DOI] [PubMed] [Google Scholar]
- Haubensak W, Attardo A, Denk W, Huttner WB. Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: a major site of neurogenesis. Proc Natl Acad Sci U S A. 2004;101:3196–3201. doi: 10.1073/pnas.0308600100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydar TF, Ang E, Jr, Rakic P. Mitotic spindle rotation and mode of cell division in the developing telencephalon. Proc Natl Acad Sci U S A. 2003;100:2890–2895. doi: 10.1073/pnas.0437969100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hébert JM, Fishell G. The genetics of early telencephalon patterning: some assembly required. Nat Rev Neurosci. 2008;9:678–685. doi: 10.1038/nrn2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imayoshi I, Sakamoto M, Yamaguchi M, Mori K, Kageyama R. Essential roles of Notch signaling in maintenance of neural stem cells in developing and adult brains. J Neurosci. 2010;30:3489–3498. doi: 10.1523/JNEUROSCI.4987-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasato T, Nomura R, Ando R, Ikeda T, Tanaka M, Itohara S. Dorsal telencephalon-specific expression of Cre recombinase in PAC transgenic mice. Genesis. 2004;38:130–138. doi: 10.1002/gene.20009. [DOI] [PubMed] [Google Scholar]
- Iwata T, Li CL, Deng CX, Francomano CA. Highly activated Fgfr3 with the K644M mutation causes prolonged survival in severe dwarf mice. Hum Mol Genet. 2001;10:1255–1264. doi: 10.1093/hmg/10.12.1255. [DOI] [PubMed] [Google Scholar]
- Kageyama R, Ohtsuka T, Shimojo H, Imayoshi I. Dynamic Notch signaling in neural progenitor cells and a revised view of lateral inhibition. Nat Neurosci. 2008;11:1247–1251. doi: 10.1038/nn.2208. [DOI] [PubMed] [Google Scholar]
- Kang W, Wong LC, Shi SH, Hébert JM. The Transition from Radial Glial to Intermediate Progenitor Cell Is Inhibited by FGF Signaling during Corticogenesis. J Neurosci. 2009;29:14571–14580. doi: 10.1523/JNEUROSCI.3844-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk T, Pontious A, Englund C, Daza RA, Bedogni F, Hodge R, Attardo A, Bell C, Huttner WB, Hevner RF. Intermediate neuronal progenitors (basal progenitors) produce pyramidal-projection neurons for all layers of cerebral cortex. Cereb Cortex. 2009;19:2439–2450. doi: 10.1093/cercor/bhn260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein A, Noctor S, Martínez-Cerdeño V. Patterns of neural stem and progenitor cell division may underlie evolutionary cortical expansion. Nat Rev Neurosci. 2006;7:883–890. doi: 10.1038/nrn2008. [DOI] [PubMed] [Google Scholar]
- Louvi A, Artavanis-Tsakonas S. Notch signalling in vertebrate neural development. Nat Rev Neurosci. 2006;7:93–102. doi: 10.1038/nrn1847. [DOI] [PubMed] [Google Scholar]
- Louvi A, Sisodia SS, Grove EA. Presenilin 1 in migration and morphogenesis in the central nervous system. Development. 2004;131:3093–3105. doi: 10.1242/dev.01191. [DOI] [PubMed] [Google Scholar]
- Miyata T, Kawaguchi A, Okano H, Ogawa M. Asymmetric inheritance of radial glial fibers by cortical neurons. Neuron. 2001;31:727–741. doi: 10.1016/s0896-6273(01)00420-2. [DOI] [PubMed] [Google Scholar]
- Mizutani K, Yoon K, Dang L, Tokunaga A, Gaiano N. Differential Notch signalling distinguishes neural stem cells from intermediate progenitors. Nature. 2007;449:351–355. doi: 10.1038/nature06090. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci. 2007;8:427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- Nieto M, Monuki ES, Tang H, Imitola J, Haubst N, Khoury SJ, Cunningham J, Gotz M, Walsh CA. Expression of Cux-1 and Cux-2 in the subventricular zone and upper layers II-IV of the cerebral cortex. J Comp Neurol. 2004;479:168–180. doi: 10.1002/cne.20322. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR. Neurons derived from radial glial cells establish radial units in neocortex. Nature. 2001;409:714–720. doi: 10.1038/35055553. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martínez-Cerdeño V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- Ohkubo Y, Uchida AO, Shin D, Partanen J, Vaccarino FM. Fibroblast growth factor receptor 1 is required for the proliferation of hippocampal progenitor cells and for hippocampal growth in mouse. J Neurosci. 2004;24:6057–6069. doi: 10.1523/JNEUROSCI.1140-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary DD, Nakagawa Y. Patterning centers, regulatory genes and extrinsic mechanisms controlling arealization of the neocortex. Curr Opin Neurobiol. 2002;12:14–25. doi: 10.1016/s0959-4388(02)00285-4. [DOI] [PubMed] [Google Scholar]
- Pirvola U, Ylikoski J, Trokovic R, Hébert JM, McConnell SK, Partanen J. FGFR1 Is Required fot he Development of the Auditory Sensory Epithelium. Neuron. 2002;35:671–680. doi: 10.1016/s0896-6273(02)00824-3. [DOI] [PubMed] [Google Scholar]
- Raballo R, Rhee J, Lyn-Cook R, Leckman JF, Schwartz ML, Vaccarino FM. Basic Fibroblast Growth Factor (Fgf2) is necessary for cell proliferation and neurogenesis in the developing cerebral cortex. J Neurosci. 2000;20:5012–5023. doi: 10.1523/JNEUROSCI.20-13-05012.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P. A small step for the cell, a giant leap for mankind: a hypothesis of neocortical expansion during evolution. Trends Neurosci. 1995;18:383–388. doi: 10.1016/0166-2236(95)93934-p. [DOI] [PubMed] [Google Scholar]
- Rash BG, Grove EA. Patterning the dorsal telencephalon: a role for sonic hedgehog? J Neurosci. 2007;27:11595–11603. doi: 10.1523/JNEUROSCI.3204-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahara S, O'Leary DD. Fgf10 regulates transition period of cortical stem cell differentiation to radial glia controlling generation of neurons and basal progenitors. Neuron. 2009;63:48–62. doi: 10.1016/j.neuron.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojo H, Ohtsuka T, Kageyama R. Oscillations in notch signaling regulate maintenance of neural progenitors. Neuron. 2008;58:52–64. doi: 10.1016/j.neuron.2008.02.014. [DOI] [PubMed] [Google Scholar]
- Shin DM, Korada S, Raballo R, Shashikant CS, Simeone A, Taylor JR, Vaccarino F. Loss of glutamatergic pyramidal neurons in frontal and temporal cortex resulting from attenuation of FGFR1 signaling is associated with spontaneous hyperactivity in mice. J Neurosci. 2004;24:2247–2258. doi: 10.1523/JNEUROSCI.5285-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KM, Ohkubo Y, Maragnoli ME, Rasin MR, Schwartz ML, Sestan N, Vaccarino FM. Midline radial glia translocation and corpus callosum formation require FGF signaling. Nat Neurosci. 2006;9:787–797. doi: 10.1038/nn1705. [DOI] [PubMed] [Google Scholar]
- Stevens HE, Smith KM, Maragnoli ME, Fagel D, Borok E, Shanabrough M, Horvath TL, Vaccarino FM. Fgfr2 is required for the development of the medial prefrontal cortex and its connections with limbic circuits. J Neurosci. 2010;30:5590–5602. doi: 10.1523/JNEUROSCI.5837-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm EE, Rubenstein JL, Martin GR. Dosage of Fgf8 determines whether cell survival is positively or negatively regulated in the developing forebrain. Proc Natl Acad Sci U S A. 2003;100:1757–1762. doi: 10.1073/pnas.0337736100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Nowakowski RS, Caviness VS. The leaving or Q fraction of the murine cerebral proliferative epithelium: a general model of neocortical neuronogenesis. J Neurosci. 1996;16:6183–6196. doi: 10.1523/JNEUROSCI.16-19-06183.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson RE, Kind PC, Graham NA, Etherson ML, Kennedy J, Fernandes AC, Marques CS, Hevner RF, Iwata T. Fgf receptor 3 activation promotes selective growth and expansion of occipitotemporal cortex. Neural Dev. 2009;4:4. doi: 10.1186/1749-8104-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccarino FM, Schwartz ML, Raballo R, Rhee J, Lyn-Cook R. Fibroblast Growth Factor signaling regulates growth and morphogenesis at multiple steps during brain development. In: Pedersen RA, Shatten G, editors. Current topics in developmental biology. San Diego: Academic; 1999a. pp. 179–200. [DOI] [PubMed] [Google Scholar]
- Vaccarino FM, Schwartz ML, Raballo R, Nilsen J, Rhee J, Zhou M, Doetschman T, Coffin JD, Wyland JJ, Hung YT. Changes in cerebral cortex size are governed by fibroblast growth factor during embryogenesis. Nat Neurosci. 1999b;2:246–253. doi: 10.1038/6350. [DOI] [PubMed] [Google Scholar]
- Wahl MB, Deng C, Lewandoski M, Pourquié O. FGF signaling acts upstream of the NOTCH and WNT signaling pathways to control segmentation clock oscillations in mouse somitogenesis. Development. 2007;134:4033–4041. doi: 10.1242/dev.009167. [DOI] [PubMed] [Google Scholar]
- Werner S, Weinberg W, Liao X, Peters KG, Blessing M, Yuspa SH, Weiner RL, Williams LT. Targeted expression of a dominant-negative FGF receptor mutant in the epidermis of transgenic mice reveals a role of FGF in keratinocyte organization and differentiation. EMBO J. 1993;12:2635–2643. doi: 10.1002/j.1460-2075.1993.tb05924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon K, Nery S, Rutlin ML, Radtke F, Fishell G, Gaiano N. Fibroblast growth factor receptor signaling promotes radial glial identity and interacts with Notch1 signaling in telencephalic progenitors. J Neurosci. 2004;24:9497–9506. doi: 10.1523/JNEUROSCI.0993-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130:3063–3074. doi: 10.1242/dev.00491. [DOI] [PubMed] [Google Scholar]