

Abstract
Both traditional and purpose designed nonsteroidal anti-inflammatory drugs (NSAIDs), selective for inhibition of cyclooxygenase (COX) -2 alleviate pain and inflammation but confer a cardiovascular hazard, attributable to inhibition of COX-2 derived prostacyclin (PGI2). Deletion of microsomal PGE synthase–1 (mPGES-1), the dominant enzyme that converts the COX derived intermediate product, PGH2, to form PGE2, modulates inflammatory pain in rodents. By contrast with COX-2 deletion or inhibition, PGI2 formation is augmented in mPGES-1−/− mice an effect which may confer cardiovascular benefit, yet undermine the analgesic potential of inhibitors of this enzyme. This review will consider the cardiovascular biology of mPGES1, and the complex challenge of developing inhibitors of this enzyme.
Keywords: Prostaglandin, prostacyclin, PGE synthase–1, cyclooxygenase, cardiovascular, inflammation
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) alleviate pain and reduce fever by inhibiting prostaglandin (PG) G/H synthases (commonly known as cyclooxygenases or COXs), pivotal enzymes in the biosynthetic cascade that leads to formation of prostanoids (Figure 1). Metabolism of arachidonic acid by COXs yields the intermediate prostaglandin (PG) endoperoxide product, PGH2 that is further metabolized to prostanoids by terminal synthases. The analgesic efficacy of NSAIDs is largely attributable to suppression of formation of COX-2 derived PGE2 and PGI2 and their serious gastrointestinal (GI) adverse effects result largely from suppression of COX-1 derived PGE2 and PGI2 in gastroduodenal epithelium and COX-1 derived TxA2 formed by platelets. These observations prompted the development of purpose designed (pd) NSAIDs selective for inhibition of COX-2 and indeed, they proved less likely to cause serious GI adverse events than the traditional (t) NSAIDs like ibuprofen and naproxen, that inhibited both COX-1 and COX-2. Several tNSAIDs, such as diclofenac and meloxicam, also inhibit preferentially COX-2 at their therapeutic doses. Despite their diminished propensity to cause GI complications, COX-2 inhibitors were shown to increase the risk of myocardial infarction(MI), stroke, systemic and pulmonary hypertension, congestive heart failure and sudden cardiac death(Garcia Rodriguez et al. 2008; Grosser et al. 2006; Grosser et al. 2010). This increased cardiovascular hazard is attributable to suppression of COX-2 derived prostaglandins, particularly, PGI2 (Grosser et al. 2006; Grosser et al. 2010). Expression of the cardiovascular hazard in an individual patient is likely modulated by drug selectivity, potency and exposure, underlying baseline cardiovascular risk and concomitant therapies, such as low dose aspirin(Garcia Rodriguez et al. 2008; Grosser et al. 2006; Grosser et al. 2010).
Fig. 1. Biosynthetic/response pathway for prostanoids.

Arachidonic acid is released from membrane phospholipids upon physiological or inflammatory stimulation, and then catalyzed by cyclooxygenases(COXs) to form the intermediate product, PGH2. Terminal isomerases, PGI2 synthase (PGIS), thromboxane synthase (TxS), microsomal (m) or cytosolice (c) PGE2 synthase(PGES), mPGES (microsomal PGE2 synthase), hematopoietic(h) - or lipocalin(l)- type PGD2 synthase(PGDS) and PGF2α synthase (PGFS), convert PGH2 to form PGI2, TxA2, PGE2, PGD2 and PGF2α, respectively. Prostanoids exert their action mainly via corresponding G-protein coupled receptors, for example, PGI2 via I prostane (P) rececptor, IP, TxA2 via TP (two TP isomers, α and β are spliced from same gene), and PGE2 via four receptors (EP1, EP2, EP3 and EP4). Among the three identified PGESs, mPGES-1 in the dominant source of PGE2 formation in inflammation.
The withdrawal from the market and failure of regulatory approval of several pd NSAIDs selective for inhibition of COX-2 prompted interest in microsomal (m)-PGE synthase (PGES) -1 as an alternative drug target(Jakobsson et al. 1999; Samuelsson et al. 2007; Thoren et al. 2003) This enzyme is a member of the MAPEG (membrane-associated proteins in eicosanoid and glutathione metabolism) superfamily and was experimentally identified by Jakobsson et al as microsome-associated and 152 amino acids in length and as exhibiting glutathione-dependent PGE synthase activity(0.25 micromol/min/mg)(Jakobsson et al. 1999). The human PGES-1 gene is localized to chromosome 9q34.3, contains 3 exons and spans 14.8 kb (Forsberg et al. 2000). The human mPGES-1 sequence shows roughly 80% similarity to the enzyme in mouse, rat or cow. Electron crystallography demonstrated that microsomal PGES-1 constitutes a trimer in two-dimensional crystals(Jegerschold et al. 2008; Xing et al. 2009) (Figure 2). Human mPGES-1 inhibitors may not be pharmacologically effective in animals, and such species differences in inhibitor binding efficiency may be attributable to some key residues- Thr-131, Leu-135, and Ala-138 in human mPGES-1(Pawelzik et al. 2010). These residues are situated in transmembrane helix 4, lining the entrance to the cleft between two subunits in the protein trimer, and regulate inhibitor access to the enzyme. For detail illustration see reference (Pawelzik et al. 2010).
Fig. 2.
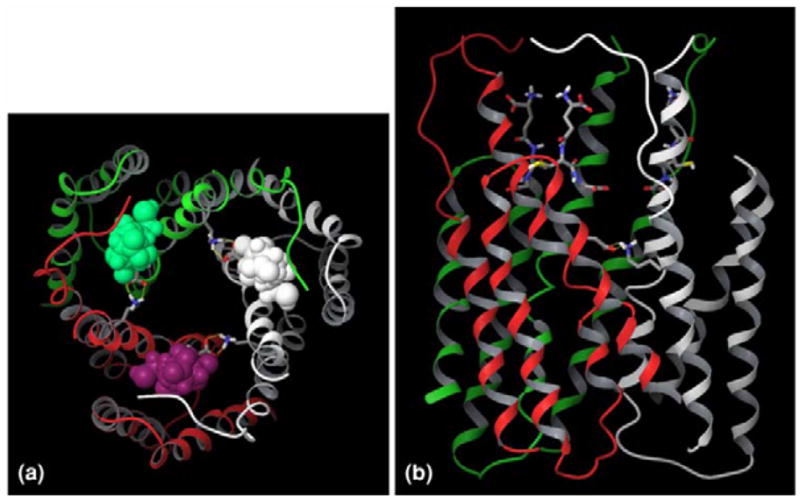
Overall structure of human mPGES-1 trimer. The three mPGES-1 monomers are colored in grey, red and green, respectively. Three glutathione molecules are complexed with mPGES-1 trimer at the intermonomeric interfaces, and they are represented by space-filling model in (a) and stick model in (b). (a) Top view from the cytoplasmic side of the membrane. (b) Side view from the membrane. Top and bottom are respectively the cytoplasmic and luminal phase. This figure is reproduced with permission from J Comput Aided Mol Des. 2009;23(1):13–24.
Two other PGE synthases have also been identified, mPGES-2(Murakami and Kudo 2006) and cytosolic(c) PGES(Pini et al. 2005; Tanioka et al. 2000), however mPGES-1 is the dominant source of PGE2 biosynthesis(Cheng et al. 2006), as assessed by excretion of its major urinary metabolite, in mice and humans. While cPGES is co-expressed with COX-1, mPGES-1 is often functionally co-regulated with COX-2, such as in the vasculature during zebra fish development (Pini et al. 2005) and in models of inflammation (Claveau et al. 2003). mPGS-1 also predominantly colocalizes with COX-1 in renal distal convoluted tubule and in medullary collecting ducts (Schneider et al. 2004).
Initially, mPGES-1 deletion in mice was found to modulate experimentally evoked pain and inflammation to a degree indistinguishable from treatment with tNSAIDs non- selective for inhibition of COXs(Kamei et al. 2004; Trebino et al. 2003). Although many small molecule inhibitors of mPGES-1 fail to block the rodent enzyme, one inhibitor has been used in mice in which the human enzyme has been overexpressed and shown to inhibit inflammatory pain (Xu et al. 2008). mPGES-1 deletion was shown to upregulate the anti-inflammatory nuclear receptor, PPARγ(Kapoor et al. 2006), which may also contribute to the efficacy of this strategy. While these results suggested that mPGES-1 might indeed be a promising alternative drug target to COX-2, enzyme deletion has been less impressive in some rodent models of analgesia(Scholich and Geisslinger 2006). This might reflect the contribution of (i) continuing synthesis of PGE2 by other synthases in mPGES-1−/− mice or (ii) the importance of PGI2 as a mediator of pain(Chen et al. 2008). Indeed, rediversion of accumulated PGH2 substrate to PGI2 synthase can augment PGI2 biosynthesis in mPGES-1−/− mice(Cheng et al. 2006).
Other areas of potential therapeutic application of mPGES-1 inhibitors have been explored. mPGES-1 was also identified as the central switch during immune-induced pyresis and as a target for the treatment of fever(Engblom et al. 2003; (Xu et al. 2008). Enzyme deletion delays and ameliorates the expression of experimental autoimmune encephalomyelitis, thought to be a model of multiple sclerosis (Kihara et al. 2009). By contrast, a note of caution was sounded by a recent provocative study, in which mPGES-1 derived PGE2, acting via EP2 and EP4 receptors mediates resolution of lipopolysaccharide (LPS)-induced spinal neuroinflammation after initial LPS priming(Brenneis et al. 2011).
mPGES-1 may have therapeutic potential in the chemoprevention of certain cancers (Radmark and Samuelsson 2010), including intestinal tumors(Nakanishi et al. 2008), prostate and lung cancers(Hanaka et al. 2009). Several small molecule inhibitors have been developed and initial experience of human tolerability of some compounds has been acquired. However, it remains a challenge to select an initial target for proof of clinical efficacy. Will these drugs be less likely to confer a cardiovascular hazard than NSAIDs selective for inhibition of COX-2? However, if so, will they be less efficacious than such drugs as analgesics? Might an alternative clinical target offer a more promising route to initial drug approval?
Cardiovascular consequences of mPGES-1 deletion
(i) Thrombosis and blood pressure
It has long been known that NSAID consumption is associated with hypertension and that the magnitude of this response is quite variable(Chan et al. 2009). It is thus unsurprising that disruption of COX-2 dependent formation of PGI2 and/or PGE2 will elevate blood pressure in mice and that this effect is highly conditioned by genetic background(Yang et al. 2005). Similarly, genetic deletion or pharmacological inhibition of COX-2 or disruption of the PGI2 receptor predisposes to accelerated thrombogenesis in rodent models of induced thrombosis(Cheng et al. 2006),consistent with the predisposition to myocardial infarction and stroke observed in placebo controlled trials of pdNSAIDs selective for inhibition of COX-2(Bresalier et al. 2005; Grosser et al. 2006; Solomon et al. 2005). Accelerated thrombogenesis is not observed in mPGES-1 mice which exhibit augmented formation of PGI2 coincident with suppression of PGE2(Cheng et al. 2006). Effects on both prostanoids may be relevant to this phenotype: PGI2 restrains thrombogenesis while low concentrations of PGE2 activate platelet aggregation via the EP3 receptor(Fabre et al. 2001).
The differential impact of mPGES-1 vs COX-2 inhibition on blood pressure is more nuanced. As mentioned, NSAIDs may elevate blood pressure and evidence in rodents suggests that this relates to inhibition of COX-2 and the selectivity with which this is attained(Qi et al. 2002). We and others have reported that deletion of mPGES-1fails to elevate blood pressure in mice fed either a normal or high salt diet (Cheng et al. 2006; Francois et al. 2007). Hypertension induced by infusion of angiotensin II in hyperlipidemic mice is also uninfluenced by mPGES-1 deletion(Harding et al. 2011; Wang et al. 2008). In contrast to these observations, Jia et al reported that mPGES-1 deletion augmented the hypertensive response to both a more intensive salt loading regimen and to angiotensin II infusion in normolipidemic mice (Jia et al. 2006). This discrepancy may reflect a differences in genetic background amongst the strains used in these experiments and differences in the experimental protocols. However, they suggest that any differences in the hypertensive effects of NSAIDs and mPGES-1 inhibitors may turn out to be relative rather than absolute. In contrast to COX-2 deficient mice, no impairment in renal development is observed in mPGES-1 deficient mice. PGE2 and PGI2 promote natriuresis, and thus, may both contribute to blood pressure homeostasis regulated by mPGES-1.
(ii) Vascular inflammation
Inflammation is a hallmark of atherosclerosis, however its functional contribution to disease evolution and, in particular to plaque destabilization and vascular occlusive thrombosis remains speculative. Indeed, the only placebo controlled trials of anti-inflammatory drugs in which cardiovascular endpoints have been assessed have been those involving NSAIDs. Various mutant mice have been developed which result in hyperlipidemia, permitting assessment of drugs or gene deletions on atherogenesis. While the lesions in these mice variably approximate the human condition, they differ in that they are largely resistant to plaque destabilization and consequent thrombosis. Given these caveats, there has been interest in assessing the impact of NSAIDs on atherogenesis in mice, as several clinical trials of COX-2 inhibitors in patients selected to be at low cardiovascular risk resulted in a delayed detection of a drug related increase in myocardial infarction and stroke, consistent with an impact on atherogenesis (Grosser et al. 2006). Indeed, deletion of the PGI2 receptor promotes initiation and early development of atherosclerosis in hyperlipidemic mice(Egan et al. 2004). By contrast COX-2 inhibitors or enzyme deletion have been shown variously to retard, accelerate, or leave unaltered, development of atherosclerosis in mouse models (Egan et al. 2005; Linton and Fazio 2004). These conflicting results may result from differences in drug specificity or dosing strategy, residual developmental effects of COX-2 deletion or the contrasting effects of COX-2-derived prostanoids that is elaborated in a cell-specific manner over the course of the disease. In any event, the effect of mPGES-1 deletion is quite distinct. Here, atherogenesis in LDL receptor deleted hyperlipidemic mice is retarded by coincident disruption of mPGES-1. Again this may reflect both suppression of PGE2 and augmentation of PGI2. Aside from the impact of IP deletion on atherogenesis, deletion of EP4 in macrophages by fetal liver cell transplantation restrains early atherogenesis in LDLR−/− mice(Babaev et al. 2008), suggesting that, as with thrombogenesis, the bivalent effects on formation of these two prostanoids may both have been mechanistically relevant. A note of caution in these studies is that the substrate rediversion products consequent to mPGES-1 deletion differs between cell types. Thus, in the setting of atherogenesis, the consequences of vascular smooth muscle cell deletion of mPGES-1 predominates, with augmented formation of PGI2. However, in lesional macrophages, the dominant rediversion products are PGE2 and TxA2, both of which may promote atherogenesis(Babaev et al. 2008; Kobayashi et al. 2004), albeit the evidence for myeloid EP4 receptor activation and atherogenesis is contradictory(Tang et al. 2011). Given that adults treated with mPGES-1 inhibitors are likely to have pre-existent atherosclerosis, the more convincing proof of concept would be the induction of lesional regression by a small molecule inhibitor in hyperlipidemic mice expressing the human enzyme with already established atherosclerosis. Lacking this evidence, it is conceivable that administration of an mPGES-1 inhibitor to patients with established atherosclerosis might exacerbate the disease, due to substrate rediversion in lesional macrophages.
Abdominal aortic aneurysm (AAA) is an inflammatory disorder, characterized by localized connective tissue degradation and smooth muscle cell (SMC) apoptosis, leading to aortic dilatation and rupture. Although both often coexist, the pathology of AAA differs from that atherosclerosis (Golledge et al. 2006; Sakalihasan et al. 2005) Human aortic aneurysm biopsies stain strongly for COX-2 ex vivo and a preliminary cohort study in patients suggest that use of tNSAIDs retarded aneurysmal growth (Walton et al. 1999). Deletion or selective inhibition of COX-2, but not inhibition of COX-1 decreases AAA formation in hyperlipidemic mice(King et al. 2006). We reported that deletion of mPGES-1 also retards formation of AAA induced by an angiotensin II infusion in LDLR−/− mice. This occurs concomitant with suppression of aortic and systemic indices of oxidative stress and matrix metalloproteinase 2 expression, themselves previously implicated in the pathogenesis of AAA(Sakalihasan et al. 1996; Thomas et al. 2006). Deletion of mPGES-1 inhibited production of PGE2, but also resulted in substrate rediversion to augment production of PGI2 and PGD2, both of which might upregulate antioxidant enzymes and restrain oxidant stress(Alvarez-Maqueda et al. 2004; Egan et al. 2004).
A third vascular phenotype associated with evidence of inflammation is the response to injury. Both COXs and PGs differentially modulate the response to vascular injury. For example, wire induced vascular proliferation is enhanced in mice that are genetically deficient in the IP, while deletion of the TxA2 receptor (TP) depresses this response (Cheng et al. 2002). There are conflicting reports of the impact of disrupting COX-2 on vascular remodeling. For example, pharmacological suppression of COX-2 derived PGI2 with nimesulide promotes adverse vascular remodeling in a flow-induced injury model, an effect replicated by deletion of the IP(Rudic et al. 2005). However, COX-2 inhibition by celecoxib reduces neointimal hyperplasia in balloon-injured carotid arteries in rats and rabbits(Wang et al. 2005; Yang et al. 2004). It is unclear whether this discrepancy reflects off target effects of celecoxib or species differences in the response to vascular injury. Furthermore, despite the risk of myocardial infarction conferred by celecoxib in placebo controlled trials(Solomon et al. 2008), preliminary evidence suggests that in patients who underwent angioplasty and received platelet inhibitors to limit this risk, in-stent late luminal loss is reduced by this COX- selective inhibitor(Koo et al. 2007). Deletion of mPGES-1 in mice attenuates neointimal hyperplasia after vascular wire-injury(Wang et al. 2011) (Figure 3). Again, both suppression of PGE2 and rediversion of the accumulated PGH2 substrate to PGI2 seem mechanistically relevant. Both modulate the injury induced upregulation of tenascin-C — an abundant extracellular matrix glycoprotein, which affords a scaffold along which vascular smooth muscle cells migration to proliferate in the neointima.
Fig. 3. Deletion of mPGES-1 reduced neointimal formation.

Mice were subjected to wire injury in femoral artery and assessed 4 weeks later. Wild type (WT) mice developed extensive neointima as compared to sham operated control mice (A and D), while this mPGES-1−/− (KO) mice demonstrate significantly reduced neointima area (A), ratio of intima to media (B) and lumen occlusion (C and D). Representative H&E staining of cross sections from sham operated or wire-injured arteries is shown (D). N denotes neointima; M denotes media; ▶ indicates internal elastin; ◀ indicates external elastin. Scale bar denotes 20 μm. This figure is reproduced with permission from Circulation(Wang et al. 2011).
(iii)Cardiac function
An increased incidence of congestive cardiac failure has been observed in placebo controlled trials of NSAIDs (Grosser et al. 2006). Although this may in part reflect NSAID induced hypertension, it appears that COX-2 dependent formation of PGI2 in cardiomyocytes affords cardioprotection. Thus, selective deletion of COX-2 in cardiomyocytes results in mild heart failure and cardiac fibrosis in mice(Wang et al. 2009) demonstrating the direct role of COX-2-derived prostanoids in cardiac function. The primary COX-2-derived mediators implicated in cardioprotection are PGI2 and PGE2, which, acting on the IP or the EP3, respectively (Dowd et al. 2001; Shinmura et al. 2005), can protect against oxidative injury in cardiac tissue. Wu et al(Wu et al. 2009a) found that celecoxib decreased survival in mice after acute MI, while mPGES-1 deletion did not change the survival rate due to increased PGI2 signaling (Wu et al. 2009b). On the other hand, Degousse et al observed that deletion of mPGES-1 leads to eccentric cardiac myocyte hypertrophy, left ventricle (LV) dilation, and impaired LV contractile function after acute MI. This adverse LV remodeling contrasts with the favorable PGI2 dependent vascular remodeling in mPGES-1 knockout mice and is due to suppression of PGE2 formation by inflammatory cells in the infarct and peri-infarct regions(Degousee et al. 2008). Others reported that depletion of mPGES-1 impairs the compensatory hypertrophic response to prolonged angiotensin II infusion and reduces the ejection fraction (Harding et al. 2011). These studies raise the possibility of adverse cardiac effects of mPGES-1 inhibitors in patients who had recently suffered a myocardial infarction or exhibit cardiac decompensation. In contrast with the results following myocardial ischemia or infarction, deletion of mPGES-1 attenuates brain injury and promotes functional recovery after experimentally induced stroke(Kapoor et al. 2006).
Summary
The mPGES-1 enzyme represents an intriguing target for drug development. Given the limitations of presently available data on analgesic efficacy in animals we will not know how the analgesic efficacy of its inhibition compares with NSAIDs until such studies are performed in humans. Will augmented PGI2 formation dilute the analgesic potency of mPGES-1 inhibitors and if so, will this only apply to certain types of pain? Limited evidence, at the preclinical level, suggests an improved adverse effect profileof this therapeutic strategy compared to a tNSAID(Xu et al. 2008). Preclinical studies favor the cardiovascular profile of mPGES-1 inhibitors. However, while they may confer limited cardiovascular hazard per se, their use may be injudicious in patients who have suffered a myocardial infarction or are in heart failure. A comparative evaluation of their impact relative to NSAIDs selective for inhibition of COX-2 on blood pressure in mildly hypertensive individuals would seem a priority early in clinical development. Local delivery of mPGES-1 inhibitors from endovascular stents might limit the consequences of vascular injury in patients subject to angioplasty while limiting systemic adverse effects and merits consideration as a route to initial drug approval. Similarly, an impact of mPGES-1 inhibition on progression to surgery in patients with AAA might represent an orphan indication. Meantime, further preclinical experimentation with tool compounds will provide valuable information concerning substrate rediversion to PGD2, and its implications for patients with asthma(Wang et al. 2010), the relative GI tolerability of mPGES-1 inhibitors versus NSAIDs and whether the major products formed via substrate rediversion change with functional consequence during chronic therapy where lesional infiltrations alter during the natural history of disease.
Acknowledgments
Funding Sources
This work was supported by American Heart Association [0735397N to Dr. Wang] and the National Institutes of Health [HL083799 to Dr. FitzGerald]. Dr. FitzGerald is the McNeil Professor of Translational Medicine and Therapeutics.
Footnotes
Conflict of Interest Disclosures
Dr. FitzGerald has consulted in the past year for Astra Zeneca, Daiichi Sankyo, Logical Therapeutics, Lilly and Nicox on NSAIDs and related compounds.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alvarez-Maqueda M, El Bekay R, Alba G, Monteseirin J, Chacon P, Vega A, Martin-Nieto J, Bedoya FJ, Pintado E, Sobrino F. 15-deoxy-delta 12,14-prostaglandin J2 induces heme oxygenase-1 gene expression in a reactive oxygen species-dependent manner in human lymphocytes. J Biol Chem. 2004;279:21929–37. doi: 10.1074/jbc.M400492200. [DOI] [PubMed] [Google Scholar]
- Babaev VR, Chew JD, Ding L, Davis S, Breyer MD, Breyer RM, Oates JA, Fazio S, Linton MF. Macrophage EP4 deficiency increases apoptosis and suppresses early atherosclerosis. Cell Metab. 2008;8:492–501. doi: 10.1016/j.cmet.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneis C, Coste O, Altenrath K, Angioni C, Schmidt H, Schuh CD, Zhang DD, Henke M, Weigert A, Brune B, et al. Anti-inflammatory Role of Microsomal Prostaglandin E Synthase-1 in a Model of Neuroinflammation. J Biol Chem. 2011;286:2331–42. doi: 10.1074/jbc.M110.157362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K, Lines C, Riddell R, Morton D, Lanas A, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352:1092–102. doi: 10.1056/NEJMoa050493. [DOI] [PubMed] [Google Scholar]
- Chan CC, Reid CM, Aw TJ, Liew D, Haas SJ, Krum H. Do COX-2 inhibitors raise blood pressure more than nonselective NSAIDs and placebo? An updated meta-analysis. J Hypertens. 2009;27:2332–41. doi: 10.1097/HJH.0b013e3283310dc9. [DOI] [PubMed] [Google Scholar]
- Chen M, Boilard E, Nigrovic PA, Clark P, Xu D, Fitzgerald GA, Audoly LP, Lee DM. Predominance of cyclooxygenase 1 over cyclooxygenase 2 in the generation of proinflammatory prostaglandins in autoantibody-driven K/BxN serum-transfer arthritis. Arthritis Rheum. 2008;58:1354–65. doi: 10.1002/art.23453. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Austin SC, Rocca B, Koller BH, Coffman TM, Grosser T, Lawson JA, FitzGerald GA. Role of prostacyclin in the cardiovascular response to thromboxane A2. Science. 2002;296:539–41. doi: 10.1126/science.1068711. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, Fitzgerald GA. Cyclooxygenases, microsomal prostaglandin E synthase-1, and cardiovascular function. J Clin Invest. 2006;116:1391–9. doi: 10.1172/JCI27540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claveau D, Sirinyan M, Guay J, Gordon R, Chan CC, Bureau Y, Riendeau D, Mancini JA. Microsomal prostaglandin E synthase-1 is a major terminal synthase that is selectively up-regulated during cyclooxygenase-2-dependent prostaglandin E2 production in the rat adjuvant-induced arthritis model. J Immunol. 2003;170:4738–44. doi: 10.4049/jimmunol.170.9.4738. [DOI] [PubMed] [Google Scholar]
- Degousee N, Fazel S, Angoulvant D, Stefanski E, Pawelzik SC, Korotkova M, Arab S, Liu P, Lindsay TF, Zhuo S, et al. Microsomal prostaglandin E2 synthase-1 deletion leads to adverse left ventricular remodeling after myocardial infarction. Circulation. 2008;117:1701–10. doi: 10.1161/CIRCULATIONAHA.107.749739. [DOI] [PubMed] [Google Scholar]
- Dowd NP, Scully M, Adderley SR, Cunningham AJ, Fitzgerald DJ. Inhibition of cyclooxygenase-2 aggravates doxorubicin-mediated cardiac injury in vivo. J Clin Invest. 2001;108:585–90. doi: 10.1172/JCI11334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan KM, Lawson JA, Fries S, Koller B, Rader DJ, Smyth EM, FitzGerald GA. COX-2-derived prostacyclin confers atheroprotection on female mice. Science. 2004;306:1954–7. doi: 10.1126/science.1103333. [DOI] [PubMed] [Google Scholar]
- Egan KM, Wang M, Fries S, Lucitt MB, Zukas AM, Pure E, Lawson JA, FitzGerald GA. Cyclooxygenases, thromboxane, and atherosclerosis: plaque destabilization by cyclooxygenase-2 inhibition combined with thromboxane receptor antagonism. Circulation. 2005;111:334–42. doi: 10.1161/01.CIR.0000153386.95356.78. [DOI] [PubMed] [Google Scholar]
- Fabre JE, Nguyen M, Athirakul K, Coggins K, McNeish JD, Austin S, Parise LK, FitzGerald GA, Coffman TM, Koller BH. Activation of the murine EP3 receptor for PGE2 inhibits cAMP production and promotes platelet aggregation. J Clin Invest. 2001;107:603–10. doi: 10.1172/JCI10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg L, Leeb L, Thoren S, Morgenstern R, Jakobsson P. Human glutathione dependent prostaglandin E synthase: gene structure and regulation. FEBS Lett. 2000;471:78–82. doi: 10.1016/s0014-5793(00)01367-3. [DOI] [PubMed] [Google Scholar]
- Francois H, Facemire C, Kumar A, Audoly L, Koller B, Coffman T. Role of microsomal prostaglandin E synthase 1 in the kidney. J Am Soc Nephrol. 2007;18:1466–75. doi: 10.1681/ASN.2006040343. [DOI] [PubMed] [Google Scholar]
- Garcia Rodriguez LA, Tacconelli S, Patrignani P. Role of dose potency in the prediction of risk of myocardial infarction associated with nonsteroidal anti-inflammatory drugs in the general population. J Am Coll Cardiol. 2008;52:1628–36. doi: 10.1016/j.jacc.2008.08.041. [DOI] [PubMed] [Google Scholar]
- Golledge J, Muller J, Daugherty A, Norman P. Abdominal aortic aneurysm: pathogenesis and implications for management. Arterioscler Thromb Vasc Biol. 2006;26:2605–13. doi: 10.1161/01.ATV.0000245819.32762.cb. [DOI] [PubMed] [Google Scholar]
- Grosser T, Fries S, Fitzgerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006;116:4–15. doi: 10.1172/JCI27291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosser T, Yu Y, FitzGerald GA. Emotion Recollected in Tranquility: Lessons Learned from the COX-2 Saga. Ann Rev Med. 2010;61:17–33. doi: 10.1146/annurev-med-011209-153129. [DOI] [PubMed] [Google Scholar]
- Hanaka H, Pawelzik SC, Johnsen JI, Rakonjac M, Terawaki K, Rasmuson A, Sveinbjornsson B, Schumacher MC, Hamberg M, Samuelsson B, et al. Microsomal prostaglandin E synthase 1 determines tumor growth in vivo of prostate and lung cancer cells. Proc Natl Acad Sci U S A. 2009;106:18757–62. doi: 10.1073/pnas.0910218106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding P, Yang XP, He Q, Lapointe MC. Lack of Microsomal Prostaglandin E Synthase-1 Reduces Cardiac Function Following Angiotensin II Infusion. Am J Physiol Heart Circ Physiol. 2011 doi: 10.1152/ajpheart.00772.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson PJ, Thoren S, Morgenstern R, Samuelsson B. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc Natl Acad Sci U S A. 1999;96:7220–5. doi: 10.1073/pnas.96.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jegerschold C, Pawelzik SC, Purhonen P, Bhakat P, Gheorghe KR, Gyobu N, Mitsuoka K, Morgenstern R, Jakobsson PJ, Hebert H. Structural basis for induced formation of the inflammatory mediator prostaglandin E2. Proc Natl Acad Sci U S A. 2008;105:11110–5. doi: 10.1073/pnas.0802894105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Z, Zhang A, Zhang H, Dong Z, Yang T. Deletion of microsomal prostaglandin E synthase-1 increases sensitivity to salt loading and angiotensin II infusion. Circ Res. 2006;99:1243–51. doi: 10.1161/01.RES.0000251306.40546.08. [DOI] [PubMed] [Google Scholar]
- Kamei D, Yamakawa K, Takegoshi Y, Mikami-Nakanishi M, Nakatani Y, Oh-Ishi S, Yasui H, Azuma Y, Hirasawa N, Ohuchi K, et al. Reduced pain hypersensitivity and inflammation in mice lacking microsomal prostaglandin e synthase-1. J Biol Chem. 2004;279:33684–95. doi: 10.1074/jbc.M400199200. [DOI] [PubMed] [Google Scholar]
- Kapoor M, Kojima F, Qian M, Yang L, Crofford LJ. Microsomal prostaglandin E synthase-1 deficiency is associated with elevated peroxisome proliferator activated receptor gamma: Regulation by prostaglandin E2 via the PI3 kinase and AKT pathway. J Biol Chem. 2006 doi: 10.1074/jbc.M610153200. [DOI] [PubMed] [Google Scholar]
- Kihara Y, Matsushita T, Kita Y, Uematsu S, Akira S, Kira J, Ishii S, Shimizu T. Targeted lipidomics reveals mPGES-1-PGE2 as a therapeutic target for multiple sclerosis. Proc Natl Acad Sci U S A. 2009;106:21807–12. doi: 10.1073/pnas.0906891106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King VL, Trivedi DB, Gitlin JM, Loftin CD. Selective cyclooxygenase-2 inhibition with celecoxib decreases angiotensin II-induced abdominal aortic aneurysm formation in mice. Arterioscler Thromb Vasc Biol. 2006;26:1137–43. doi: 10.1161/01.ATV.0000216119.79008.ac. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Tahara Y, Matsumoto M, Iguchi M, Sano H, Murayama T, Arai H, Oida H, Yurugi-Kobayashi T, Yamashita JK, et al. Roles of thromboxane A(2) and prostacyclin in the development of atherosclerosis in apoE-deficient mice. J Clin Invest. 2004;114:784–94. doi: 10.1172/JCI21446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo BK, Kim YS, Park KW, Yang HM, Kwon DA, Chung JW, Hahn JY, Lee HY, Park JS, Kang HJ, et al. Effect of celecoxib on restenosis after coronary angioplasty with a Taxus stent (COREA-TAXUS trial): an open-label randomised controlled study. Lancet. 2007;370:567–74. doi: 10.1016/S0140-6736(07)61295-1. [DOI] [PubMed] [Google Scholar]
- Linton MF, Fazio S. Cyclooxygenase-2 and inflammation in atherosclerosis. Curr Opin Pharmacol. 2004;4:116–23. doi: 10.1016/j.coph.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Murakami M, Kudo I. Prostaglandin E synthase: a novel drug target for inflammation and cancer. Curr Pharm Des. 2006;12:943–54. doi: 10.2174/138161206776055912. [DOI] [PubMed] [Google Scholar]
- Nakanishi M, Montrose DC, Clark P, Nambiar PR, Belinsky GS, Claffey KP, Xu D, Rosenberg DW. Genetic deletion of mPGES-1 suppresses intestinal tumorigenesis. Cancer Res. 2008;68:3251–9. doi: 10.1158/0008-5472.CAN-07-6100. [DOI] [PubMed] [Google Scholar]
- Pawelzik SC, Uda NR, Spahiu L, Jegerschold C, Stenberg P, Hebert H, Morgenstern R, Jakobsson PJ. Identification of key residues determining species differences in inhibitor binding of microsomal prostaglandin E synthase-1. J Biol Chem. 2010;285:29254–61. doi: 10.1074/jbc.M110.114454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pini B, Grosser T, Lawson JA, Price TS, Pack MA, FitzGerald GA. Prostaglandin E synthases in zebrafish. Arterioscler Thromb Vasc Biol. 2005;25:315–20. doi: 10.1161/01.ATV.0000152355.97808.10. [DOI] [PubMed] [Google Scholar]
- Qi Z, Hao CM, Langenbach RI, Breyer RM, Redha R, Morrow JD, Breyer MD. Opposite effects of cyclooxygenase-1 and -2 activity on the pressor response to angiotensin II. J Clin Invest. 2002;110:61–9. doi: 10.1172/JCI14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radmark O, Samuelsson B. Microsomal prostaglandin E synthase-1 and 5-lipoxygenase: potential drug targets in cancer. J Intern Med. 2010;268:5–14. doi: 10.1111/j.1365-2796.2010.02246.x. [DOI] [PubMed] [Google Scholar]
- Rudic RD, Brinster D, Cheng Y, Fries S, Song WL, Austin S, Coffman TM, FitzGerald GA. COX-2-derived prostacyclin modulates vascular remodeling. Circ Res. 2005;96:1240–7. doi: 10.1161/01.RES.0000170888.11669.28. [DOI] [PubMed] [Google Scholar]
- Sakalihasan N, Delvenne P, Nusgens BV, Limet R, Lapiere CM. Activated forms of MMP2 and MMP9 in abdominal aortic aneurysms. J Vasc Surg. 1996;24:127–33. doi: 10.1016/s0741-5214(96)70153-2. [DOI] [PubMed] [Google Scholar]
- Sakalihasan N, Limet R, Defawe OD. Abdominal aortic aneurysm. Lancet. 2005;365:1577–89. doi: 10.1016/S0140-6736(05)66459-8. [DOI] [PubMed] [Google Scholar]
- Samuelsson B, Morgenstern R, Jakobsson PJ. Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharmacol Rev. 2007;59:207–24. doi: 10.1124/pr.59.3.1. [DOI] [PubMed] [Google Scholar]
- Schneider A, Zhang Y, Zhang M, Lu WJ, Rao R, Fan X, Redha R, Davis L, Breyer RM, Harris R, et al. Membrane-associated PGE synthase-1 (mPGES-1) is coexpressed with both COX-1 and COX-2 in the kidney. Kidney Int. 2004;65:1205–13. doi: 10.1111/j.1523-1755.2004.00493.x. [DOI] [PubMed] [Google Scholar]
- Scholich K, Geisslinger G. Is mPGES-1 a promising target for pain therapy? Trends Pharmacol Sci. 2006;27:399–401. doi: 10.1016/j.tips.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Shinmura K, Tamaki K, Sato T, Ishida H, Bolli R. Prostacyclin attenuates oxidative damage of myocytes by opening mitochondrial ATP-sensitive K+ channels via the EP3 receptor. Am J Physiol Heart Circ Physiol. 2005;288:H2093–101. doi: 10.1152/ajpheart.01003.2004. [DOI] [PubMed] [Google Scholar]
- Solomon SD, McMurray JJ, Pfeffer MA, Wittes J, Fowler R, Finn P, Anderson WF, Zauber A, Hawk E, Bertagnolli M. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005;352:1071–80. doi: 10.1056/NEJMoa050405. [DOI] [PubMed] [Google Scholar]
- Solomon SD, Wittes J, Finn PV, Fowler R, Viner J, Bertagnolli MM, Arber N, Levin B, Meinert CL, Martin B, et al. Cardiovascular risk of celecoxib in 6 randomized placebo-controlled trials: the cross trial safety analysis. Circulation. 2008;117:2104–13. doi: 10.1161/CIRCULATIONAHA.108.764530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang EH, Shimizu K, Christen T, Rocha VZ, Shvartz E, Tesmenitsky Y, Sukhova G, Shi GP, Libby P. Lack of EP4 receptors on bone marrow-derived cells enhances inflammation in atherosclerotic lesions. Cardiovasc Res. 2011;89:234–43. doi: 10.1093/cvr/cvq262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanioka T, Nakatani Y, Semmyo N, Murakami M, Kudo I. Molecular identification of cytosolic prostaglandin E2 synthase that is functionally coupled with cyclooxygenase-1 in immediate prostaglandin E2 biosynthesis. J Biol Chem. 2000;275:32775–82. doi: 10.1074/jbc.M003504200. [DOI] [PubMed] [Google Scholar]
- Thomas M, Gavrila D, McCormick ML, Miller FJ, Jr, Daugherty A, Cassis LA, Dellsperger KC, Weintraub NL. Deletion of p47phox attenuates angiotensin II-induced abdominal aortic aneurysm formation in apolipoprotein E-deficient mice. Circulation. 2006;114:404–13. doi: 10.1161/CIRCULATIONAHA.105.607168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoren S, Weinander R, Saha S, Jegerschold C, Pettersson PL, Samuelsson B, Hebert H, Hamberg M, Morgenstern R, Jakobsson PJ. Human microsomal prostaglandin E synthase-1: purification, functional characterization, and projection structure determination. J Biol Chem. 2003;278:22199–209. doi: 10.1074/jbc.M303227200. [DOI] [PubMed] [Google Scholar]
- Trebino CE, Stock JL, Gibbons CP, Naiman BM, Wachtmann TS, Umland JP, Pandher K, Lapointe JM, Saha S, Roach ML, et al. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc Natl Acad Sci U S A. 2003;100:9044–9. doi: 10.1073/pnas.1332766100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton LJ, Franklin IJ, Bayston T, Brown LC, Greenhalgh RM, Taylor GW, Powell JT. Inhibition of prostaglandin E2 synthesis in abdominal aortic aneurysms: implications for smooth muscle cell viability, inflammatory processes, and the expansion of abdominal aortic aneurysms. Circulation. 1999;100:48–54. doi: 10.1161/01.cir.100.1.48. [DOI] [PubMed] [Google Scholar]
- Wang D, Patel VV, Ricciotti E, Zhou R, Levin MD, Gao E, Yu Z, Ferrari VA, Lu MM, Xu J, et al. Cardiomyocyte cyclooxygenase-2 influences cardiac rhythm and function. Proc Natl Acad Sci U S A. 2009;106:7548–52. doi: 10.1073/pnas.0805806106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Tarakji K, Zhou Z, Zhang M, Forudi F, Zhou X, Koki AT, Smith ME, Keller BT, Topol EJ, et al. Celecoxib, a selective cyclooxygenase-2 inhibitor, decreases monocyte chemoattractant protein-1 expression and neointimal hyperplasia in the rabbit atherosclerotic balloon injury model. J Cardiovasc Pharmacol. 2005;45:61–7. doi: 10.1097/00005344-200501000-00011. [DOI] [PubMed] [Google Scholar]
- Wang M, Cooper PR, Jiang M, Zhao H, Hui Y, Yao Y, Tate JC, Damera G, Lawson JA, Jester WF, et al. Deletion of Microsomal Prostaglandin E Synthase-1 Does Not Alter Ozone-induced Airway Hyper-responsiveness. J Pharmacol Exp Ther. 2010 doi: 10.1124/jpet.110.166678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Ihida-Stansbury K, Kothapalli D, Tamby MC, Yu Z, Chen L, Grant G, Cheng Y, Lawson JA, Assoian RK, et al. Microsomal prostaglandin e2 synthase-1 modulates the response to vascular injury. Circulation. 2011;123:631–9. doi: 10.1161/CIRCULATIONAHA.110.973685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Lee E, Song W, Ricciotti E, Rader DJ, Lawson JA, Pure E, FitzGerald GA. Microsomal prostaglandin E synthase-1 deletion suppresses oxidative stress and angiotensin II-induced abdominal aortic aneurysm formation. Circulation. 2008;117:1302–9. doi: 10.1161/CIRCULATIONAHA.107.731398. [DOI] [PubMed] [Google Scholar]
- Wu D, Mennerich D, Arndt K, Sugiyama K, Ozaki N, Schwarz K, Wei J, Wu H, Bishopric NH, Doods H. Comparison of microsomal prostaglandin E synthase-1 deletion and COX-2 inhibition in acute cardiac ischemia in mice. Prostaglandins Other Lipid Mediat. 2009a;90:21–5. doi: 10.1016/j.prostaglandins.2009.06.006. [DOI] [PubMed] [Google Scholar]
- Wu D, Mennerich D, Arndt K, Sugiyama K, Ozaki N, Schwarz K, Wei J, Wu H, Bishopric NH, Doods H. The effects of microsomal prostaglandin E synthase-1 deletion in acute cardiac ischemia in mice. Prostaglandins Leukot Essent Fatty Acids. 2009b;81:31–3. doi: 10.1016/j.plefa.2009.05.019. [DOI] [PubMed] [Google Scholar]
- Xing L, Kurumbail RG, Frazier RB, Davies MS, Fujiwara H, Weinberg RA, Gierse JK, Caspers N, Carter JS, McDonald JJ, et al. Homo-timeric structural model of human microsomal prostaglandin E synthase-1 and characterization of its substrate/inhibitor binding interactions. J Comput Aided Mol Des. 2009;23:13–24. doi: 10.1007/s10822-008-9233-4. [DOI] [PubMed] [Google Scholar]
- Xu D, Rowland SE, Clark P, Giroux A, Cote B, Guiral S, Salem M, Ducharme Y, Friesen RW, Methot N, et al. MF63 [2-(6-chloro-1H-phenanthro[9,10-d]imidazol-2-yl)-isophthalonitrile], a selective microsomal prostaglandin E synthase-1 inhibitor, relieves pyresis and pain in preclinical models of inflammation. J Pharmacol Exp Ther. 2008;326:754–63. doi: 10.1124/jpet.108.138776. [DOI] [PubMed] [Google Scholar]
- Yang HM, Kim HS, Park KW, You HJ, Jeon SI, Youn SW, Kim SH, Oh BH, Lee MM, Park YB, et al. Celecoxib, a cyclooxygenase-2 inhibitor, reduces neointimal hyperplasia through inhibition of Akt signaling. Circulation. 2004;110:301–8. doi: 10.1161/01.CIR.0000135467.43430.16. [DOI] [PubMed] [Google Scholar]
- Yang T, Huang YG, Ye W, Hansen P, Schnermann JB, Briggs JP. Influence of genetic background and gender on hypertension and renal failure in COX-2-deficient mice. Am J Physiol Renal Physiol. 2005;288:F1125–32. doi: 10.1152/ajprenal.00219.2004. [DOI] [PubMed] [Google Scholar]